
Targeting endosomal receptors, a new direction for polymers in nanomedicine
Paulina D.
Ramirez-Garcia
*ac,
Nicholas A.
Veldhuis
 b,
Nigel W.
Bunnett
cd and
Thomas P.
Davis
*e
b,
Nigel W.
Bunnett
cd and
Thomas P.
Davis
*e
aDentistry Translational Research Center, New York University College of Dentistry, New York, 10010, USA. E-mail: pr2336@nyu.edu
bMonash Institute of Pharmaceutical Sciences, Monash University, Parkville, VIC 3052, Australia
cDepartment of Molecular Pathobiology, New York University College of Dentistry, New York, NY 10010, USA
dDepartment of Neuroscience and Physiology, Neuroscience Institute, New York University Langone School of Medicine, New York, NY 10010, USA
eAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, QLD 4072, Australia. E-mail: t.davis@uq.edu.au
First published on 22nd May 2023
Abstract
In this perspective, we outline a new opportunity for exploiting nanoparticle delivery of antagonists to target G-protein coupled receptors localized in intracellular compartments. We discuss the specific example of antagonizing endosomal receptors involved in pain to develop long-lasting analgesics but also outline the broader application potential of this delivery approach. We discuss the materials used to target endosomal receptors and indicate the design requirements for future successful applications.
10th Anniversary StatementCongratulations on a decade of excellence. This current contribution is a forward-looking new opportunity for materials scientists in assisting pharmacological solutions to health challenges and builds on the contribution of TPD to the journal going back to the distant days before the journal split into three a decade ago. TPD serving on the Editorial Board of J. Mater. Chem. and then J. Mater. Chem. B for many years and having published on numerous aspects of materials chemistry and nanomedicine over three decades as an academic. The strength of the journal is its focus on excellence in materials coupled with bioscience, bringing the best of interdisciplinary science to the forefront supported by an outstanding editorial and referee team committed to high quality. |
1. Conventional targets explored in nanomedicine
Since the discovery of living radical polymerization, there has been a drive to exploit the control achieved over macromolecular architectures to design and synthesize innovative materials at the nanoscale. In many cases, the application targeted by researchers is the drug delivery field, which has heavily relied on the development of new structures to exploit the enhanced permeability and retention effect (EPR). Under this principle, nanoparticles are postulated to ‘escape’ the vasculature and accumulate at tumor sites, altering biodistribution and optimizing the toxic effects of cancer medicines to target tumor cells.1 However, the heterogeneity of tumors and their multiple biological and pathophysiological barriers indicate that not all tumors benefit from EPR-based nanomedicines. A solution could rely on combination treatments to enhance the EPR effect.2The EPR effect has inspired many polymer chemists and dominated the field for more than a decade, resulting in a major focus within polymer nanoscience to provide potential treatments for solid tumors, largely to the neglect of other therapeutic opportunities. This approach has been used to justify many polymer-driven studies, where biological testing has focused on in vitro studies such as cell association, particle accumulation and nanotoxicity. These isolated in vitro studies can be misleading when attempting to translate findings to the clinic or predict their behavior in an in vivo setting. Moreover, when we look at the numerous in vivo studies conducted, there are still challenges regarding their accuracy in recapitulating true clinical situations.3,4 Despite a rapidly growing number of publications demonstrating clever nanocarrier designs with high potential for drug delivery, more or adequate testing continues to hamper the translation of polymeric nanomedicines.
In this short perspective, we aim to extend the vision of polymer scientists interested in therapeutic delivery to new targets beyond solid tumors and to exploit the need for nanoparticle delivery in unique pathophysiological environments to target a whole new range of medical conditions. Specifically, we describe the opportunity for intracellular targeting to deliver antagonists to G-protein coupled receptors (GPCRs) that drive pathologies from intracellular compartments – ideal targets for nanoparticle and bioconjugate delivery strategies in cells that constitutively internalize nanoparticulate materials into the endo-lysosomal network. We will introduce GPCRs and their role in pathophysiological processes, explain the role of receptor internalization, and give examples of the many GPCRs available as novel targets for a new approach to intracellular-targeted delivery. We will particularly focus on endosomal GPCRs involved in pain and give published examples that have shown the validity of the approach.
2. G protein-coupled receptors
G protein-coupled receptors (GPCRs) are the largest family of cell-surface receptors that mediate the communication between cells and the external environment. GPCRs respond to a wide variety of stimuli such as hormones, neurotransmitters, paracrine agents, light, odorants, and tastants.5 With almost 800 GPCR sequences in the human genome, these essential receptors are involved in diverse physiological and pathophysiological processes. GPCRs are also well-established therapeutic targets that account for more than 30% of drugs in the clinic.6,7Upon encountering a stimulus (ligand), GPCRs are activated to promote a cellular response by coupling to G proteins. G proteins are formed by a complex of three subunits: Gα, Gβ, and Gγ. In a resting state, the α-subunit remains bound to guanosine diphosphate (GDP) and upon ligand binding GPCR stimulation and conformational changes promote G protein engagement with GTP, resulting in. This GDP-GTP exchange is an essential process for the heterotrimeric G protein complex to dissociate from GPCRs, separating into Gα from Gβγ subunits to subsequently stimulate other downstream effector proteins. A key example is activation of the Gαs subunit, leading to increased adenylyl cyclase activity and production of the second messenger molecule cAMP, to promote protein kinase and transcriptional endpoints within cells (Fig. 1).8
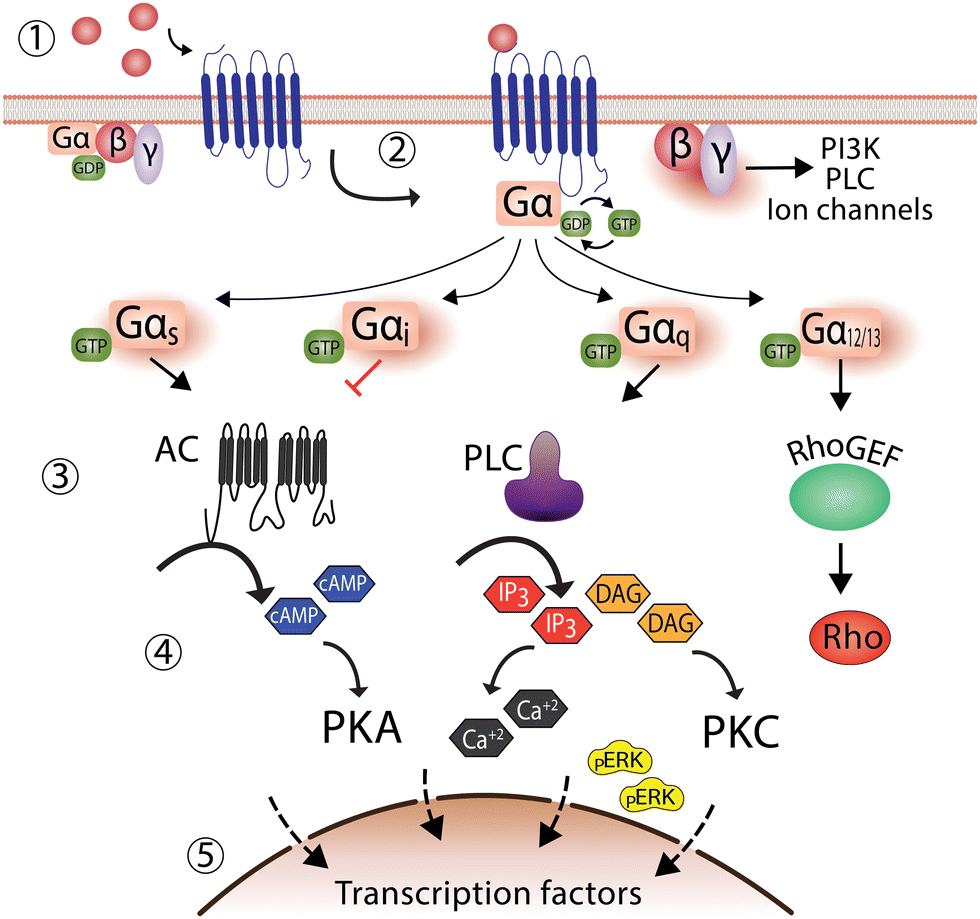 | ||
| Fig. 1 Classical G-protein dependent signaling of G protein-coupled receptors. (1) In the absence of an agonist, GPCRs remain inactivated, and G-protein is bound to GDP. (2) Followed agonist binding, G-protein exchange GDP by GTP, eliciting the dissociation of the G-protein trimeric complex into Gα and Gβγ, leading to the formation of a complex formed by the agonist bound GPCR and Gα subunit. (3) Several effectors are activated depending on the Gα subunit recruited. For instance, Gαs activates adenylate cyclase, while Gαi inhibits it. Similarly, the Gβγ subunit can also activate effectors such as phospholipase C. (4) Activation of effectors increases second messenger molecules, such as cAMP, calcium, and pERK. (5) The outcome is the activation of transcription factors in the nucleus with a concomitant cellular response. | ||
3. Endosomal signaling of receptors
Endocytosis was classically viewed as a critical mechanism for the desensitization and resensitization of receptors, leading to the termination of GPCR signals. In this process, activated receptors are internalized and dissociated from their ligands (desensitization), followed by sorting in endosomes to be recycled and returned to the plasma membrane (resensitization). Alternatively, receptors can also continue through the endosomal maturation pathway and undergo sorting into lysosomes, to terminate signalling via proteolytic degradation.9 However, it is now also more widely accepted that this organized network of dynamic intracellular membranes can also serve as sites for organization of signaling platforms or complexes. An increasing number of receptors have been reported to elicit endosomal signaling events distinct from those originating at the plasma membrane and regulated by separated mechanisms.10–13Although this perspective focuses on endosomal GPCRs, it is important to emphasize that endosomal signaling is not an event restricted to GPCRs. In fact, receptor Tyrosine kinases (RTKs) were the first receptor class described to signal from endosomes. First described for the epidermal growth factor receptor (EGFR) and Insulin receptor (IR),14,15 it was also later observed that upon nerve growth factor (NGF) stimulation, the tyrosine kinase A receptor (trkA) could also signal from endosomes.16 Subsequent work reinforced the concept of endosomal signaling by showing that another RTK, the platelet-derived growth factor receptor (PDGFR) could also elicit a biological response from endosomal membranes.17 Similarly, the Toll-like receptors TLR3, TLR4, and TLR9 are also shown to redistribute into endosomes for optimal innate immune responses.18–20
For GPCRs, initial evidence of endosomal signaling was observed by the activation of the α-factor receptor (Ste2p) in the mating response of Saccharomyces cerevisiae yeast.21 Later work demonstrated similar endosomal-mediated signaling responses via the parathyroid hormone receptor (PTHR) and V2 vasopressin receptor resulted in sustained cAMP production (V2R).22,23 The first evidence to suggest that endocytosis can promote acute signaling was demonstrated for the D1 dopamine receptor (DRD1).24 Similarly, studies on the β2-adrenergic receptor (β2AR) and thyroid-stimulating hormone receptor (TSHR) revealed that unique, sustained signaling profiles were associated with internalized receptors.25,26 Regarding pain transmission, the protease-activated receptor 2 (PAR2) expressed on doral root ganglion sensory neurons and the neurokinin 1 receptor (NK1R) within the spinal cord were the first receptors demonstrated to promote sustained neuroexcitability and nociceptive signals from endosomes.27,28
The discovery of receptors that signal from endosomes has been closely linked to the development of new tools to study these processes. Irannejad and colleagues,34 for example, were the first to use a nanobody that mimics the cognate Gαs protein subunit fused to GFP (Nb80-GFP). This nanobody enabled the direct visualization of activated β2AR by live confocal imaging.35 Nb80-GFP showed for the first time that stimulation of the β2AR initially promoted Gαs protein recruitment to the plasma membrane, followed by internalization and subsequent recruitment to endosomes.34 Since the development of nanobodies, we now have new biophysical tools that have allowed us to investigate and understand these events in much greater detail,29–31,36 helping to establish endosomal signaling as a platform for spatiotemporal regulation of cellular responses.32,33
4. The importance of compartmentalized signaling processes
In last two decades, the biological significance of receptor signaling from distinct locations beyond the plasma membrane has been revealed in much greater detail. This is largely owed to the availability of new, sensitive biophysical tools can simultaneously measure signalling in real time and in different locations, which have demonstarated that shifts in distribution of receptors to discrete subcellular locations (aka receptor compartmentalization) is an essential regulatory process that provides spatial and temporal separation of signals.9,37–39Fig. 2, for example, illustrates how a sustained signalling profile can be promoted exclusively from internalized receptor populations. Indeed, it is hypothesized that cells have evolved this compartmentalization of signals in a spatiotemporal manner, to increase signaling specificity and provide a high order of regulation, to ultimately enable a single receptor to initiate multiple signaling events within a cell.38 This spatial segregation serves to avoid unwanted crosstalk between different signaling cascades, especially since many signaling molecules are shared between different pathways. Beyond the endosomal network, other intracellular locations, such as the Golgi apparatus, mitochondria and nuclei are also key membranes for these compartmentalized signaling events. Each site presumably can also promote formation of unique protein complexes to allow receptors to initiate distinct signaling processes to achieve a physiological or disease-relevant outcomes.13,40–42 To this end, nanomaterials designed to accumulate in specific organelles have tremendous potential to deliver drugs in intracellular locations of pathophysiological relevance, to “fine-tune” cellular behavior. However, this application of nanomaterials is not only limited to spatial control – as discussed below, temporal regulation through sustained drug release may also be beneficial.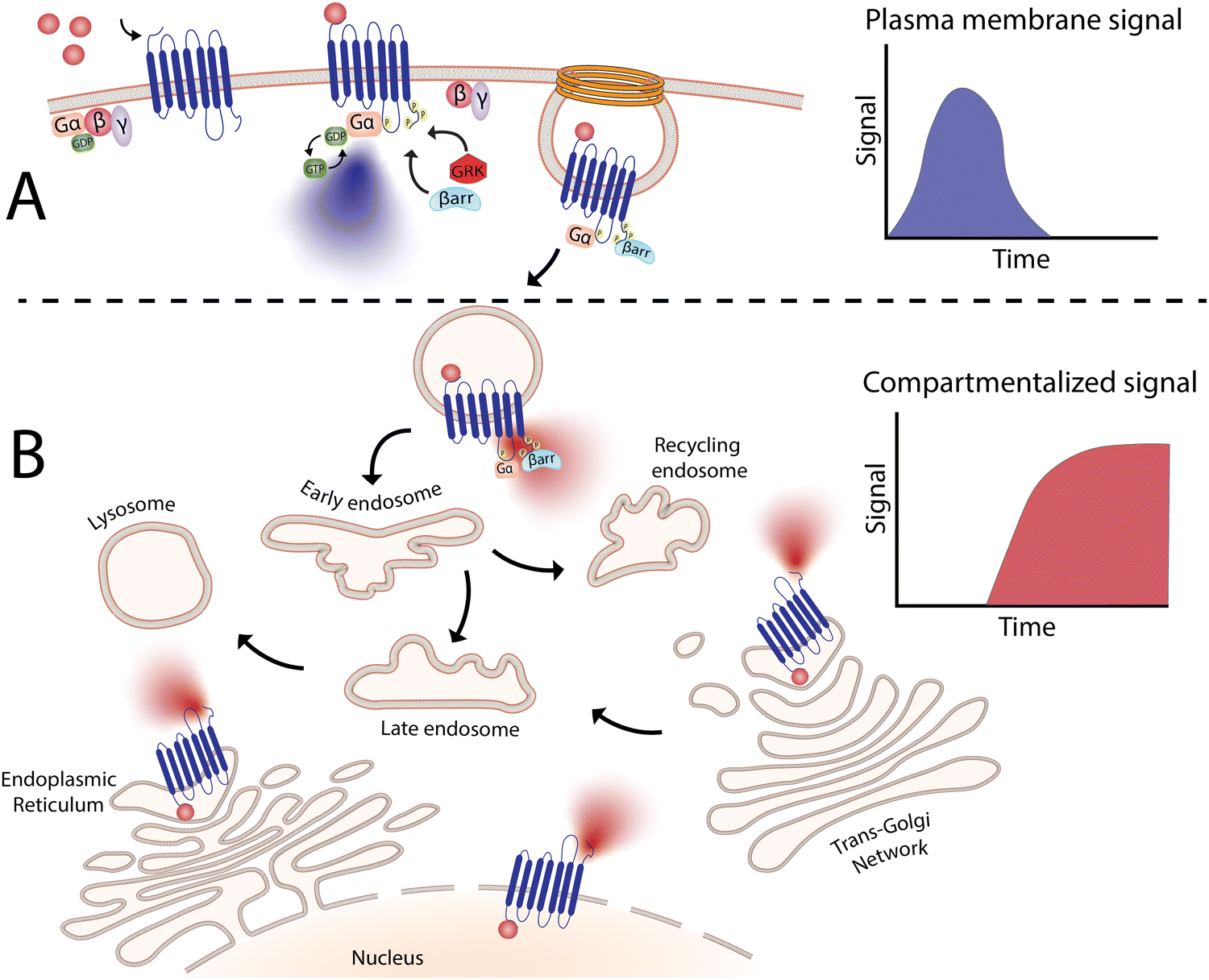 | ||
| Fig. 2 Compartmentalized signaling of G-protein coupled receptors. (A) Agonist binding results in the formation of a complex between Gα and agonist-bound GPCR, giving rise to classical G-protein-dependent signaling. (B) After GPCR activation, receptors are internalized in a process thought to mediate only desensitization and resensitization of receptors. However, signaling molecules can also be recruited into intracellular compartments where receptors can continue to signal. This signaling differs from the signals elicited by plasma membrane receptors in terms of duration and outcome. This spatiotemporal regulation allows receptors to produce highly complex signaling processes where the same receptor can now trigger diverse physiological processes mediated by signals elicited from different locations. | ||
5. Endosomal receptors involved in pain
Cancer has been the focus of nanomedicine for a long time. However, cancer is associated with high levels of pain that are often untreatable, reducing the quality of life of cancer patients tremendously. Each year, 14 million new cancer cases are diagnosed worldwide, with 52–77% of patients reporting pain triggered by the disease or its treatment. These numbers increase to 60–90% for patients with advanced cancer, with as many as 50% of patients manifesting inadequate analgesia.43,44 Moreover, chronic pain affects 28.4% of adults in the USA (79.6 million),45 with costs fluctuating between $560 and $635 billion annually.46 In Australia, pain was reported to affect 3.24 million people, with an estimated cost of $73.2 billion per year.47,48 Surveys have identified that 17–30% of adults suffer pain at any given time with increasing prevalence linked to advancing age.49–53It has been demonstrated that GPCRs can trigger intracellular signals associated with pain transmission, with receptors on its majority described to signal from endosomes.37,54 A key exception is the metabotropic glutamate receptor 5 (mGluR5), which show no apparent endosomal signalling and instead signals from the nucleus.54,55 A wide variety of receptors, have been reported to signal from other intracellular compartments such as Golgi, mitochondria, and endoplasmic reticulum (Table 1). However, these beyond the scope of this perspective, especially due to the fact that many nanparticulate systems can naturally accumulate within endosomes, even in the absence of ligand-directed nanoparticle uptake.56 We therefore focus on GPCRs that are associated with pain transmission and are also known to signal from endosomes – PAR2, the calcitonin receptor-like receptor (CLR), NK1R, and the delta (δ) opioid receptor (DOR).56 Preclinical work examining these receptors shows promise in the development of improved analgesics.
| Subcellular localization | Receptor |
|---|---|
| Endosomes | β2-adrenergic receptor (β2AR) |
| Angiotensin receptor 1 (AT1R) | |
| Calcitonin receptor-like receptor (CLR) | |
| Calcium-sensing receptor (CaSR) | |
| Dopamine D1 receptor (D1DR) | |
| Delta opioid receptor (DOR) | |
| 5-Hydrohytryptamine receptor 2 (5-HTR2) | |
| Mu opioid receptor (MOR) | |
| Neurokinin 1 receptor (NK1R) | |
| Parathyroid hormone receptor (PTHR) | |
| Protease-activated receptor-2 (PAR2) | |
| Vasopressin type 2 receptor (V2R) | |
| Endoplasmic reticulum | G protein-coupled estrogen receptor 1 (GPR30) |
| Golgi | β1-adrenergic receptor (β1AR) |
| Dopamine D1 receptor (D1DR) | |
| Mu opioid receptor (MOR) | |
| Sphingosine-1-phosphate 1 receptor (S1P1R) | |
| Thyroid-stimulating hormone receptor (TSHR) | |
| Mitochondria | 5-Hydrohytryptamine receptor (5-HTR3 & 5-HTR4) |
| Angiotensin II receptor type 1 (AT1R) | |
| Angiotensin II receptor type 2 (AT2R) | |
| Cannabinoid type 1 receptor 1 (CB1R) | |
| Melatonin MT1 receptor (MT1R) | |
| Purinoceptor 1 like receptor (P2Y1) | |
| Purinoceptor 2 like receptor (P2Y2) | |
To provide a non-exhaustive overview of these receptors, PAR2 is present in pain-sensing (nociceptive) neurons and promotes neurogenic inflammation and pain.57,58 This receptor is activated by proteases released after injury and inflammation, plays a role in oral cancer pain,59 and has been demonstrated to produce signalling from endosomed in sensory neurons.27,60 A number of studies have demonstrated that this is of particular relevance to the contribution of PAR2 endosomal signalling to chronic pain associated with irritable bowel syndrome61 and also colitis-evoked pain.62
CLR forms a complex with the receptor activity modifying protein 1 (RAMP1).63 When the body is exposed to noxious stimuli such as heat of chemical irritants, activation of sensory neurions leads to release of the neuropeptide calcitonin gene-related peptide (CGRP), which activates CLR/RAMP1 to endothelial cells to facilitating edema and inflammation.64 CGRP and CLR/RAMP1 receptors are also involved in migraine pain, and it was recently demonstrated that CLR/RAMP1 activity within the endosomes of Schwan cells contributes migraine and may be a valuable, unique therapeutic target.65
NK1R is predominantly distributed in endothelial cells of the airways, myenteric neurons and also in neurons of the central nervous system. This receptor is activated by the neuropeptide substance P (SP) to elicit plasma leakage, inflammation, and pain transmission.66–68 SP release and NK1R stimulation initiate receptor internalization on spinal neurons.69,70 From this location, the receptor elicits an endosomal signal that mediates pain transmission in acute, inflammatory, and neuropathic preclinical pain models.71
Opioid receptors are expressed in the central, peripheral, and enteric nervous systems, and unlike PAR2, CLR, and NK1R, their activation leads to pain relief. There are three classes of opioid receptors: mu (μ, MOR), delta (δ, DOR), and kappa (κ, KOR). These are activated by endogenous opioids, such as enkephalins and endorphins, and by synthetic compounds, like morphine and fentanyl.72 The most commonly used opioids act on the MOR, leading to analgesia and also side effects such as respiratory depression, addiction, and tolerance. The analgesic actions of KOR are mediated by agonists that engage Gαi signaling, whereas internalization of the receptor favors the side effects of dysphoria and sedation.73 In inflamed colons, activation of endosomal DOR was found to reduce inflammatory pain in mouse models.74
6. Utilising nanoparticle-based drug delivery to target endosomal receptors
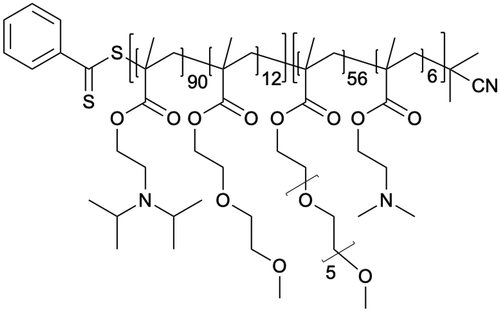
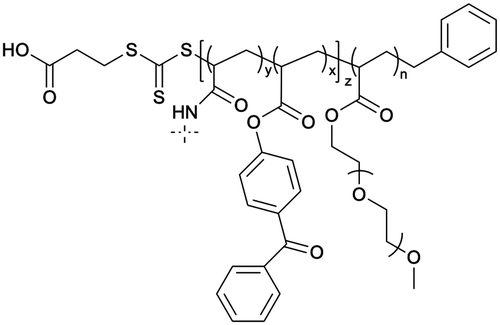
The targeting of plasma membrane-localized receptors depends on drugs distributing in such a way that sufficient levels of therapeutic agent remain in the cell exterior and engage with a receptor, before diffusing across membranes and throughout cells. However, several biological barriers make delivery more challenging when the targeted receptor has moved away from the cell surface, and instead, is “hidden” inside the cell. In particular, the therapeutic agent must cross the plasma membrane and sufficiently accumulate within the endosomal network to allow efficacious blockade of the endosomal signaling without disrupting endosomes – a feat that that is unlikely, especially for lipophilic small molecules that can indiscriminately distribute across all membranes. In contrast, nanoparticle-based drug delivery may offer significant advantages. Engineering nanomaterials with tunable drug release properties, for example, has been a common approach for delivery of cytotoxic agents to tumors while shielding healthy tissue, as a strategy to improve efficacy and safety profiles of chemotherpautic agents.2 With a focus in this perspective on internalized receptors, these same systems are also advantageous for shielding drug cargo from cell surface receptors and promoting localized delivery and controlled release of cargo, as a strategy to enhance endosomal accumulation of drugs to control endosomal receptors, as is discussed in more detail below (Table 2).| Nanoparticle | Polymer/lipid | M n (g mol−1) | PDI |
|---|---|---|---|
| DIPMA-AP77 |
|
17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 581 581 |
1.36 |
| DIPMA-MK320765 | |||
| VBA-AP78 |

|
115000 |
1.22 |
| Benzo-AP78 |
|
111000 |
1.33 |
| DADLE-LipoMSN-DADLE74 | Core: mesoporous silica | — | — |
| Coating: liposome assembly | |||
| 1,2-Dioleoyl-3-trimethylammonium propane:cholesterol:dioleoylphosphatidylethanolamine:1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] |
6.1. The use of pH-responsive materials to control internalized receptors
The need to precisely control the spatiotemporal delivery of drugs has led to the development of a wide variety of stimulus-responsive delivery systems that can promote drug release at specific sites when exposed to altered tissue environments specific to disease states.79,80 A common feature of pathologies such as cancer or chronic inflammation, for example, include highly localized acidic environments that have been widely exploited via application of pH-sensitive drug delivery systems.81,82 Interestingly, when considering strategies that may be valuable for selective targeting of endosomal receptors discussed above, the endo-lysosomal network is also a highly acidified micro-environment that can be exploited as a trigger for effective and selective intracellular drug release or accumulation.83Block copolymer materials synthesized by two-step sequential polymerization by the reversible addition-fragmentation chain (RAFT) method have played an essential role in exploring the design and development of stimulus-responsive nanoparticulate delivery systems.84–86 The chemical diversity of monomers that are utilized as the building blocks of copolymers offers a myriad of possibilities to tune the physicochemical properties of nanoparticles (size, morphology, stability, and surface properties) and subsequently design customized drug delivery systems.87 With respect to targeting endosomal receptors, polymeric pH-responsive nanoparticles made with block copolymers that incorporated units of P(PEGMA-co-DMAEMA) in the outer hydrophilic shell and pH-sensitive (∼pKa 6.1) monomers of 2-[N,N-(diisopropylamino)ethyl] methacrylate (DIPMA) within the hydrophobic portion, provides one of the first demonstrations of utilising the acidity of endosomes as a trigger for release of drugs that specifically modulate GPCR activity.77 These micellar-based systems were further loaded with the lipophilic NK1R antagonist, aprepitant, as an approach for targeting the pain-transmitting endosomal pools of the NK1R (Fig. 3).77 These nanoparticles were rapidly internalized by dynamin and clathrin-dependent endocytosis and showed a fast release of aprepitant into endosomes within five minutes of addition in vitro. DIPMA nanoparticles abolished the NK1R endosomal signaling in vitro. A single intrathecal dose showed superior analgesic properties compared to free aprepitant on in vivo models of acute, inflammatory, and neuropathic pain.77 This work was instrumental in demonstrating the potential of drug delivery systems to target endosomal receptors selectively. These findings were later endorsed using DIPMA nanoparticles to successfully target endosomal CLR/RAMP1 in Schwan cells in preclinical models of migraine pain.65
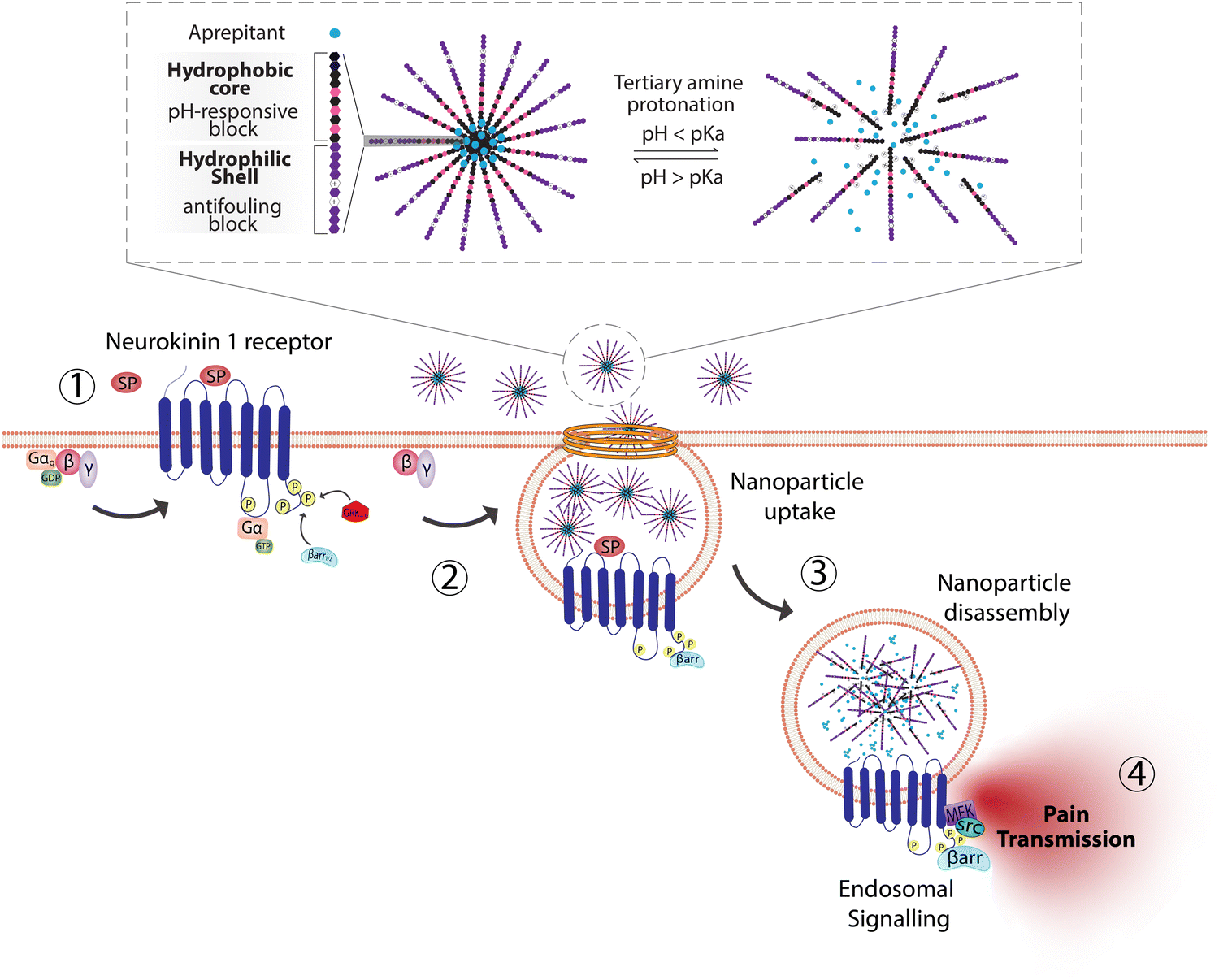 | ||
| Fig. 3 pH-responsive nanoparticles for endosomal targeting of the NK1R. pH-responsive nanoparticles were designed using amphiphilic diblock copolymers formed by four monomers. The hydrophobic portion of the diblock copolymer is formed by di(ethylene glycol) methyl ether methacrylate (DEGMA) and the pH-responsive monomer 2-(diethylamino)ethyl methacrylate (DIPMA). The hydrophilic portion is formed by poly(ethylene glycol) monomethyl ether methacrylate (PEGMA) and a positively charged monomer, 2-(dimethylamino)ethyl methacrylate (DEAEMA). The antagonist aprepitant was physically entrapped in the core of the nanoparticles. (1) The NK1R is activated by substance p (SP) and (2) couples to G-protein, followed by (3) rapid endocytosis. (4) From this location, NK1R transmits pain. Nanoparticles are passively endocytosed, and the acidity of endosomes triggers the disassembly of nanoparticles and release of aprepitant from the core. Released aprepitant can now antagonize the endosomal signal produced by the NK1R to decrease pain transmission. | ||
In addition to pH-responsive nanoparticles – a number of other polymeric nanoparticles have been designed88 using different mechanisms for pH release, including pH-sensitive crosslinking in nanoparticles and nanostars89,90 and hydrolysis of acetylated dextran.91,92 Interestingly, nanostars loaded with the NK1R antagonist, aprepitant, provided sustained release of aprepitant, which produced longer-lasting analgesia than DIPMA nanoparticles,78 indicating that the kinetics of release within endosomes plays a vital role in the duration of the analgesic effect.
6.2. Other types of nanoparticles
Mesoporous silica nanoparticles have also been used to deliver DADLE, a DOR agonist. DADLE was incorporated in the core, for endosomal delivery, and on the surface, to provide active targeting toward DOR-positive neurons.74 These nanoparticles provided long-lasting inhibition of pain receptors on human colon biopsies and a superior analgesic effect compared to free DADLE in mice models of inflammatory pain.74 The properties of the nanoparticles discussed in this perspective are found in Table 3. In addition, the feasibility of dual-release mechanisms for multiple release profiles with pH control has been published,93 exploiting not just nanoparticle degradation but the use of pH-sensitive linkers to release drugs from the nano-scaffolds. Theranostic nanoparticles have also been described to induce changes in MRI signals on pH-induced drug release,62 all of which could be utilized for applications involving the release of antagonists to target endosomal signaling.| Nanoparticle | Diameter | ζ Potential (mV) | Drug | GPCR target |
|---|---|---|---|---|
| DIPMA-AP77 | 40.4 ± 5.1 | −0.2 ± 1.6 | Aprepitant | NK1R |
| DIPMA-MK320765 | 30.7 ± 1.3 | −1.3 ± 1.6 | MK3207 | CLR/RAMP1 |
| VBA-AP78 | 14.5 ± 1.9 | −5.8 ± 1.0 | Aprepitant -NH2 | NK1R |
| Benzo-AP78 | 21.1 ± 2.6 | −2.1 ± 0.3 | Aprepitant- NH2 | NK1R |
| DADLE-LipoMSN-DADLE74 | 206.5 ± 3.3 | 29.9 ± 3.07 | DADLE | DOR |
7. The future of endosomal signaling
The high level of regulation provided by compartmentalized signaling of receptors is an ideal opportunity for nanomaterials to demonstrate their potential to achieve selective drug delivery in pathologies beyond cancer. Here we discussed the potential applicability of nanomaterials to target locations within the cell, with particular focus on endosomes as an important target site within cells. Numerous GPCRs are known to be activated and rapidly internalize into this membrane network and are likely to continue signaling from this location, thus suggesting that endosomal receptors is a unique target location that could significantly benefit from the pH-responsive delivery offered by nanomaterials. Furthermore, it is also important to note that intracellular signaling events differ from those originating at the plasma membrane, which may explain the clinical failure of many drugs that may have limited access to intracellular locations. At present, most therapeutic drugs are designed to target receptors at the cell surface and do not consider subcellular locations that are harder to target when testing drugs that allowed to freely distribute within cells or tissues. Hence, altering the intracellular distribution of drugs could be viewed as a distinct and potentially valuable opportunity for nanomaterials, and may also offer a strategy for repurposing approved drugs that previously failed for specific conditions such has pain, where it is now appreciated the neuroexcitation and pain transmission is likely to be driven by receptors that have internalised. A key example is the NK1R antagonist aprepitant (Emend®), which is currently used for treating emesis and nausea but has failed in clinical trials for pain.94,95 While there are likely to be many factors for prior failures in pain-specific trials, it is also tempting to speculate that the limited capacity for aprepitant to accumulate in endomes and directly control endosomal pools of the NK1R may have also contributed to the lack of success. Together, nanomaterials may have valuable utility beyond their use in exploiting EPR effect, and instead offer potential benefits for targeting trafficking receptors, to “fine-tune” specific cellular processes. It is also noted that endosomes are but one of many distinct membranes within the cell where disease-relevant signaling complexes can form and be targeted. Indeed, a wide variety of receptors, have been reported to signal from compartments such as the Golgi network, mitochondria, and nuclei.41,42,54,96 Moreover, since nanomaterials are often endocytosed and retained within the endo-lysosomal network, lysosomes are also potential targets due to their involvement in several diseases classified as lysosomal storage diseases (LSDs). LSDs comprise more than 50 genetic disorders, mostly involving dysfunction of lysosomal hydrolases.97,98 Accumulation of macromolecules in endosomes and lysosomes in various LSDs is of potential interest for the delivery of enzyme replacement therapies,98,99 where nanomaterials could be of great advantage.Although we do not fully understand all the nanoparticle-specific characteristics that govern intracellular delivery- e.g., the optimum nanoparticle size and the optimal residence time in the endosomes have yet to be determined. Still, we know that high cellular uptake and localized accumulation in the desired site of action are essential to target unique intracellular signals and achieve efficient drug delivery. We also know that sustained drug release favors prolonged interaction between the drug and the target. This localized and sustained release of drugs is one of the main contributors to the superior biological actions of drug-loaded nanoparticles.
Lastly, we have exemplified the potential of nanoparticles for intracellular targeting using pain as the disease of interest. For this disease setting, pH was exploited as the environmental stimulus of choice to trigger drug release in endosomes where nociceptive receptors reside. Pain served as an example of the benefits of guiding the design of nanoparticles by a specific disease mechanism. Still, the same principle can be applied to many other diseases driven by intracellular receptors. If we first identify the target and understand the disease environment. Then, we can fully exploit the potential of nanomaterials and design nanoparticles with the specific properties required to deliver drugs to our target of interest. This disease mechanism-based strategy may help us achieve the selectivity necessary to improve drug efficacy to either design new therapies or improve the existing ones.
Conflicts of interest
There are no conflicts to declare.References
- H. Maeda, Macromolecular therapeutics in cancer treatment: The EPR effect and beyond, J. Controlled Release, 2012, 164, 138–144 CrossRef CAS PubMed.
- S. K. Golombek, et al., Tumor targeting via EPR: Strategies to enhance patient responses, Adv. Drug Delivery Rev., 2018, 130, 17–38 CrossRef CAS PubMed.
- J. W. Nichols and Y. H. Bae, EPR: Evidence and fallacy, J. Controlled Release, 2014, 190, 451–464 CrossRef CAS PubMed.
- S. Wilhelm, et al., Analysis of nanoparticle delivery to tumours, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- R. Fredriksson, M. C. Lagerström, L.-G. Lundin and H. B. Schiöth, The G-Protein-Coupled Receptors in the Human Genome Form Five Main Families. Phylogenetic Analysis, Paralogon Groups, and Fingerprints, Mol. Pharmacol., 2003, 63, 1256–1272 CrossRef CAS PubMed.
- J. P. Overington, B. Al-Lazikani and A. L. Hopkins, How many drug targets are there?, Nat. Rev. Drug Discovery, 2006, 5, 993–996 CrossRef CAS PubMed.
- M. C. Lagerström and H. B. Schiöth, Structural diversity of G protein-coupled receptors and significance for drug discovery, Nat. Rev. Drug Discovery, 2008, 7, 339–357 CrossRef PubMed.
- K. L. Pierce, R. T. Premont and R. J. Lefkowitz, Seven-transmembrane receptors, Nat. Rev. Mol. Cell Biol., 2002, 3, 639–650 CrossRef CAS PubMed.
- A. C. Hanyaloglu and M. Zastrow von, Regulation of GPCRs by Endocytic Membrane Trafficking and Its Potential Implications, Annu. Rev. Pharmacol., 2008, 48, 537–568 CrossRef CAS PubMed.
- S. Sposini and A. C. Hanyaloglu, Spatial encryption of G protein-coupled receptor signaling in endosomes; Mechanisms and applications, Biochem. Pharmacol., 2017, 143, 1–9 CrossRef CAS PubMed.
- G. W. Gould and J. Lippincott-Schwartz, New roles for endosomes: from vesicular carriers to multi-purpose platforms, Nat. Rev. Mol. Cell Biol., 2009, 10, 287–292 CrossRef CAS PubMed.
- J. E. Murphy, B. E. Padilla, B. Hasdemir, G. S. Cottrell and N. W. Bunnett, Endosomes: a legitimate platform for the signaling train, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17615–17622 CrossRef CAS PubMed.
- M. Miaczynska, L. Pelkmans and M. Zerial, Not just a sink: endosomes in control of signal transduction, Curr. Opin. Cell Biol., 2004, 16, 400–406 CrossRef CAS PubMed.
- P. C. Baass, G. M. D. Guglielmo, F. Authier, B. I. Posner and J. J. M. Bergeron, Compartmentalized signal transduction by receptor tyrosine kinases, Trends Cell Biol., 1995, 5, 465–470 CrossRef CAS PubMed.
- G. M. D. Guglielmo, P. C. Baass, W. J. Ou, B. I. Posner and J. J. Bergeron, Compartmentalization of SHC, GRB2 and mSOS, and hyperphosphorylation of Raf-1 by EGF but not insulin in liver parenchyma, EMBO J., 1994, 13, 4269–4277 CrossRef PubMed.
- M. L. Grimes, et al., Endocytosis of Activated TrkA: Evidence that Nerve Growth Factor Induces Formation of Signaling Endosomes, J. Neurosci., 1996, 16, 7950–7964 CrossRef CAS PubMed.
- S. Pennock and Z. Wang, Stimulation of Cell Proliferation by Endosomal Epidermal Growth Factor Receptor As Revealed through Two Distinct Phases of Signaling, Mol. Cell. Biol., 2003, 23, 5803–5815 CrossRef CAS PubMed.
- K. Honda, et al., Spatiotemporal regulation of MyD88–IRF-7 signalling for robust type-I interferon induction, Nature, 2005, 434, 1035–1040 CrossRef CAS PubMed.
- I. B. Johnsen, et al., Toll-like receptor 3 associates with c-Src tyrosine kinase on endosomes to initiate antiviral signaling, EMBO J., 2006, 25, 3335–3346 CrossRef CAS PubMed.
- J. C. Kagan, et al., TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta, Nat. Immunol., 2008, 9, 361–368 CrossRef CAS PubMed.
- J. E. Slessareva, S. M. Routt, B. Temple, V. A. Bankaitis and H. G. Dohlman, Activation of the Phosphatidylinositol 3-Kinase Vps34 by a G Protein α Subunit at the Endosome, Cell, 2006, 126, 191–203 CrossRef CAS PubMed.
- S. Ferrandon, et al., Sustained cyclic AMP production by parathyroid hormone receptor endocytosis, Nat. Chem. Biol., 2009, 5, 734–742 CrossRef CAS PubMed.
- T. N. Feinstein, et al., Noncanonical control of vasopressin receptor type 2 signaling by retromer and arrestin, J. Biol. Chem., 2013, 288, 27849–27860 CrossRef CAS PubMed.
- S. J. Kotowski, F. W. Hopf, T. Seif, A. Bonci and M. Zastrow von, Endocytosis Promotes Rapid Dopaminergic Signaling, Neuron, 2011, 71, 278–290 CrossRef CAS PubMed.
- Y. Daaka, et al., Essential Role for G Protein-coupled Receptor Endocytosis in the Activation of Mitogen-activated Protein Kinase*, J. Biol. Chem., 1998, 273, 685–688 CrossRef CAS PubMed.
- D. Calebiro, et al., Persistent cAMP-Signals Triggered by Internalized G-Protein–Coupled Receptors, PLoS Biol., 2009, 7, e1000172 CrossRef PubMed.
- K. A. DeFea, et al., beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2, J. Cell Biol., 2000, 148, 1267–1281 CrossRef CAS PubMed.
- K. A. DeFea, et al., The proliferative and antiapoptotic effects of substance P are facilitated by formation of a beta -arrestin-dependent scaffolding complex, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 11086 CrossRef CAS PubMed.
- M. Zaccolo, et al., A genetically encoded, fluorescent indicator for cyclic AMP in living cells, Nat. Cell Biol., 2000, 2, 25–29 CrossRef CAS PubMed.
- C. D. Harvey, et al., A genetically encoded fluorescent sensor of ERK activity, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19264–19269 CrossRef CAS PubMed.
- M. Stierl, et al., Light modulation of cellular cAMP by a small bacterial photoactivated adenylyl cyclase, bPAC, of the soil bacterium Beggiatoa, J. Biol. Chem., 2011, 286, 1181–1188 CrossRef CAS PubMed.
- R. Irannejad and M. Zastrow von, GPCR signaling along the endocytic pathway, Curr. Opin. Cell Biol., 2014, 27, 109–116 CrossRef CAS PubMed.
- N. G. Tsvetanova, R. Irannejad and M. Zastrow von, G Protein-coupled Receptor (GPCR) Signaling via Heterotrimeric G Proteins from Endosomes, J. Biol. Chem., 2015, 290, 6689–6696 CrossRef CAS PubMed.
- R. Irannejad, et al., Conformational biosensors reveal GPCR signalling from endosomes, Nature, 2013, 495, 534–538 CrossRef CAS PubMed.
- S. G. F. Rasmussen, et al., Structure of a nanobody-stabilized active state of the β2 adrenoceptor, Nature, 2011, 469, 175–180 CrossRef CAS PubMed.
- Y. Namkung, et al., Monitoring G protein-coupled receptor and β-arrestin trafficking in live cells using enhanced bystander BRET, Nat. Commun., 2016, 7, 12178 CrossRef PubMed.
- P. Geppetti, N. A. Veldhuis, T. Lieu and N. W. Bunnett, G Protein-Coupled Receptors: Dynamic Machines for Signaling Pain and Itch, Neuron, 2015, 88, 635–649 CrossRef CAS PubMed.
- N. J. Pavlos and P. A. Friedman, GPCR Signaling and Trafficking: The Long and Short of It, Trends Endocrinol. Metab., 2017, 28, 213–226 CrossRef CAS PubMed.
- D. Calebiro and A. Godbole, Internalization of GPCRs: implication in receptor function, physiology and diseases, Best Pract. Res., Clin. Endocrinol. Metab., 2018, 32, 83–91 CrossRef CAS PubMed.
- K. L. O’Malley, Y.-J. I. Jong, Y. Gonchar, A. Burkhalter and C. Romano, Activation of Metabotropic Glutamate Receptor mGlu5 on Nuclear Membranes Mediates Intranuclear Ca2+ Changes in Heterologous Cell Types and Neurons, J. Biol. Chem., 2003, 278, 28210–28219 CrossRef PubMed.
- C. A. Nash, W. Wei, R. Irannejad and A. V. Smrcka, Golgi localized β1-adrenergic receptors stimulate Golgi PI4P hydrolysis by PLCε to regulate cardiac hypertrophy, eLife, 2019, 8, e48167 CrossRef CAS PubMed.
- Y. Suofu, et al., Dual role of mitochondria in producing melatonin and driving GPCR signaling to block cytochrome c release, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E7997–E8006 CrossRef CAS PubMed.
- P. J. Christo and D. Mazloomdoost, Cancer Pain and Analgesia, Ann. N. Y. Acad. Sci., 2008, 1138, 278–298 CrossRef CAS PubMed.
- C. Cluxton, The Challenge of Cancer Pain Assessment, Ulster Med. J., 2019, 88, 43–46 Search PubMed.
- J. Dahlhamer, et al., Prevalence of Chronic Pain and High-Impact Chronic Pain Among Adults—United States, 2016, Morb. Mortal. Wkly. Rep., 2018, 67, 1001–1006 CrossRef PubMed.
- D. J. Gaskin and P. Richard, The economic costs of pain in the United States, J Pain, 2012, 13, 715–724 CrossRef PubMed.
- AE, F., AEP, L., Foundation, M. & Sydney, U. of. The high price of pain: the economic impact of persistent pain in Australia, 2007.
- D. A. Economics, The cost of pain in Australia A painful reality, 2019 Search PubMed.
- P. F. M. Verhaak, J. J. Kerssens, J. Dekker, M. J. Sorbi and J. M. Bensing, Prevalence of chronic benign pain disorder among adults: a review of the literature, Pain, 1998, 77, 231–239 CrossRef PubMed.
- F. M. Blyth, et al., Chronic pain in Australia: a prevalence study, Pain, 2001, 89, 127–134 CrossRef CAS PubMed.
- H. Breivik, B. Collett, V. Ventafridda, R. Cohen and D. Gallacher, Survey of chronic pain in Europe: Prevalence, impact on daily life, and treatment, Eur. J. Pain, 2006, 10, 287 CrossRef PubMed.
- C. J. Phillips, The Cost and Burden of Chronic Pain, Br. J. Pain, 2009, 3, 2–5 Search PubMed.
- C. B. Johannes, T. K. Le, X. Zhou, J. A. Johnston and R. H. Dworkin, The prevalence of chronic pain in United States adults: results of an Internet-based survey, J. Pain, 2010, 11, 1230–1239 CrossRef PubMed.
- K. Vincent, et al., Intracellular mGluR5 plays a critical role in neuropathic pain, Nat. Commun., 2016, 7, 10604 CrossRef CAS PubMed.
- K. Vincent, S. F. Wang, A. Laferriere, N. Kumar and T. J. Coderre, Spinal intracellular metabotropic glutamate receptor 5 (mGluR5) contributes to pain and c-fos expression in a rat model of inflammatory pain, Pain, 2017, 158, 705–716 CrossRef CAS PubMed.
- J. S. Retamal, P. D. Ramírez-García, P. A. Shenoy, D. P. Poole and N. A. Veldhuis, Internalized GPCRs as Potential Therapeutic Targets for the Management of Pain, Front. Mol. Neurosci., 2019, 12, 273 CrossRef CAS PubMed.
- M. Steinhoff, et al., Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism, Nat. Med., 2000, 6, 151–158 CrossRef CAS PubMed.
- N. Vergnolle, et al., Proteinase-activated receptor-2 and hyperalgesia: A novel pain pathway, Nat. Med., 2001, 7, 821–826 CrossRef CAS PubMed.
- D. K. Lam and B. L. Schmidt, Serine proteases and protease-activated receptor 2-dependent allodynia: a novel cancer pain pathway, Pain, 2010, 149, 263–272 CrossRef CAS PubMed.
- O. Déry, M. S. Thoma, H. Wong, E. F. Grady and N. W. Bunnett, Trafficking of proteinase-activated receptor-2 and beta-arrestin-1 tagged with green fluorescent protein. beta-Arrestin-dependent endocytosis of a proteinase receptor, J. Biol. Chem., 1999, 274, 18524–18535 CrossRef PubMed.
- N. N. Jimenez-Vargas, et al., Protease-activated receptor-2 in endosomes signals persistent pain of irritable bowel syndrome, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E7438–E7447 CrossRef CAS PubMed.
- R. Latorre, et al., Mice expressing fluorescent PAR2 reveal that endocytosis mediates colonic inflammation and pain, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(6), e2112059119 CrossRef CAS PubMed.
- S. Hilairet, S. M. Foord, F. H. Marshall and M. Bouvier, Protein-Protein Interaction and Not Glycosylation Determines the Binding Selectivity of Heterodimers between the Calcitonin Receptor-like Receptor and the Receptor Activity-modifying Proteins*, J. Biol. Chem., 2001, 276, 29575–29581 CrossRef CAS PubMed.
- F. A. Russell, R. King, S.-J. Smillie, X. Kodji and S. D. Brain, Calcitonin Gene-Related Peptide: Physiology and Pathophysiology, Physiol. Rev., 2014, 94, 1099–1142 CrossRef CAS PubMed.
- F. D. Logu, et al., Schwann cell endosome CGRP signals elicit periorbital mechanical allodynia in mice, Nat. Commun., 2022, 13, 646 CrossRef PubMed.
- J. M. Lundberg and A. Saria, Capsaicin-induced desensitization of airway mucosa to cigarette smoke, mechanical and chemical irritants, Nature, 1983, 302, 251–253 CrossRef CAS PubMed.
- D. P. Poole, et al., Inflammation-induced abnormalities in the subcellular localization and trafficking of the neurokinin 1 receptor in the enteric nervous system, Am. J. Physiol.: Gastrointest. Liver Physiol., 2015, 309, G248–G259 CrossRef CAS PubMed.
- P. W. Mantyh, et al., Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor, Science, 1997, 278, 275–279 CrossRef CAS PubMed.
- J. J. Bowden, et al., Direct observation of substance P-induced internalization of neurokinin 1 (NK1) receptors at sites of inflammation, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 8964–8968 CrossRef CAS PubMed.
- P. W. Mantyh, et al., Rapid endocytosis of a G protein-coupled receptor: substance P evoked internalization of its receptor in the rat striatum in vivo, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 2622–2626 CrossRef CAS PubMed.
- D. D. Jensen, et al., Neurokinin 1 receptor signaling in endosomes mediates sustained nociception and is a viable therapeutic target for prolonged pain relief, Sci. Transl. Med., 2017, 9, eaal3447 CrossRef PubMed.
- H. Pathan and J. Williams, Basic opioid pharmacology: an update, Br. J. Pain, 2012, 6, 11–16 CrossRef PubMed.
- T. F. Brust, et al., Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria, Sci. Signaling, 2016, 9, ra117 CrossRef PubMed.
- N. N. Jimenez-Vargas, et al., Endosomal signaling of delta opioid receptors is an endogenous mechanism and therapeutic target for relief from inflammatory pain, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 15281 CrossRef PubMed.
- B. Plouffe, A. R. B. Thomsen and R. Irannejad, Emerging Role of Compartmentalized G Protein-Coupled Receptor Signaling in the Cardiovascular Field, ACS Pharmacol. Transl. Sci., 2020, 3, 221–236 CrossRef CAS PubMed.
- I. Fasciani, et al., GPCRs in Intracellular Compartments: New Targets for Drug Discovery, Biomol., 2022, 12, 1343 CAS.
- P. D. Ramírez-García, et al., A pH-responsive nanoparticle targets the neurokinin 1 receptor in endosomes to prevent chronic pain, Nat. Nanotechnol., 2019, 14, 1150–1159 CrossRef PubMed.
- R. Latorre, et al., Sustained endosomal release of a neurokinin-1 receptor antagonist from nanostars provides long-lasting relief of chronic pain, Biomaterials, 2022, 285, 121536 CrossRef CAS PubMed.
- M. A. C. Stuart, et al., Emerging applications of stimuli-responsive polymer materials, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- V. Torchilin, Multifunctional and stimuli-sensitive pharmaceutical nanocarriers, Eur. J. Pharm. Biopharm., 2009, 71, 431–444 CrossRef CAS PubMed.
- D. Schmaljohann, Thermo- and pH-responsive polymers in drug delivery, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- W. Gao, J. M. Chan and O. C. Farokhzad, pH-Responsive Nanoparticles for Drug Delivery, Mol. Pharmaceutics, 2010, 7, 1913–1920 CrossRef CAS PubMed.
- K. Zhou, et al., Tunable, Ultrasensitive pH-Responsive Nanoparticles Targeting Specific Endocytic Organelles in Living Cells, Angew. Chem., Int. Ed., 2011, 50, 6109–6114 CrossRef CAS PubMed.
- N. Nishiyama and K. Kataoka, Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery, Pharmacol. Ther., 2006, 112, 630–648 CrossRef CAS PubMed.
- R. K. O’Reilly, C. J. Hawker and K. L. Wooley, Cross-linked block copolymer micelles: functional nanostructures of great potential and versatility, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- R. Duncan, The dawning era of polymer therapeutics, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS PubMed.
- S. Eetezadi, S. N. Ekdawi and C. Allen, The challenges facing block copolymer micelles for cancer therapy: In vivo barriers and clinical translation, Adv. Drug Delivery Rev., 2014, 91, 7–22 CrossRef PubMed.
- F. Li, et al., Stimuli-responsive nano-assemblies for remotely controlled drug delivery, J. Controlled Release, 2020, 322, 566–592 CrossRef CAS PubMed.
- S. J. Kim, D. M. Ramsey, C. Boyer, T. P. Davis and S. R. McAlpine, Effectively Delivering a Unique Hsp90 Inhibitor Using Star Polymers, ACS Med. Chem. Lett., 2013, 4, 915–920 CrossRef CAS PubMed.
- J. Liu, H. Duong, M. R. Whittaker, T. P. Davis and C. Boyer, Synthesis of Functional Core, Star Polymers via RAFT Polymerization for Drug Delivery Applications, Macromol. Rapid Commun., 2012, 33, 760–766 CrossRef CAS PubMed.
- H. T. T. Duong, et al., Functionalizing Biodegradable Dextran Scaffolds Using Living Radical Polymerization: New Versatile Nanoparticles for the Delivery of Therapeutic Molecules, Mol. Pharmaceutics, 2012, 9, 3046–3061 CrossRef CAS PubMed.
- K. E. Broaders, J. A. Cohen, T. T. Beaudette, E. M. Bachelder and J. M. J. Fréchet, Acetalated dextran is a chemically and biologically tunable material for particulate immunotherapy, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5497–5502 CrossRef CAS PubMed.
- H. T. T. Duong, C. P. Marquis, M. Whittaker, T. P. Davis and C. Boyer, Acid Degradable and Biocompatible Polymeric Nanoparticles for the Potential Codelivery of Therapeutic Agents, Macromolecules, 2011, 44, 8008–8019 CrossRef CAS.
- R. Hill, NK1 (substance P) receptor antagonists—why are they not analgesic in humans?, Trends Pharmacol. Sci., 2000, 21, 244–246 CrossRef CAS PubMed.
- R. Hargreaves, et al., Development of aprepitant, the first neurokinin-1 receptor antagonist for the prevention of chemotherapy-induced nausea and vomiting, Ann. N. Y. Acad. Sci., 2011, 1222, 40–48 CrossRef CAS PubMed.
- M. Stoeber, et al., A Genetically Encoded Biosensor Reveals Location Bias of Opioid Drug Action, Neuron, 2018, 98, 963–976.e5 CrossRef CAS PubMed.
- A. H. Futerman and G. Meer van, The cell biology of lysosomal storage disorders, Nat. Rev. Mol. Cell Biol., 2004, 5, 554–565 CrossRef CAS PubMed.
- E. J. Parkinson-Lawrence, et al., Lysosomal Storage Disease: Revealing Lysosomal Function and Physiology, Physiology, 2010, 25, 102–115 CrossRef CAS PubMed.
- A. Ballabio and V. Gieselmann, Lysosomal disorders: From storage to cellular damage, Biochim. Biophys. Acta, Mol. Cell Res., 2009, 1793, 684–696 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |