DOI:
10.1039/D3TB00055A
(Paper)
J. Mater. Chem. B, 2023,
11, 3797-3807
Dendritic polyglycerolsulfate-SS-poly(ester amide) micelles for the systemic delivery of docetaxel: pushing the limits of stability through the insertion of π–π interactions†
Received
10th January 2023
, Accepted 22nd March 2023
First published on 3rd April 2023
Abstract
Insufficient stability of micellar drug delivery systems is still the major limitation to their systematic application in chemotherapy. This work demonstrates novel π-electron stabilized polyelectrolyte block copolymer micelles based on dendritic polyglycerolsulfate-cystamine-block-poly(4-benzoyl-1,4-oxazepan-7-one)-pyrene (dPGS-SS-POxPPh-Py) presenting a very low critical micelle concentration (CMC) of 0.3 mg L−1 (18 nM), 55-fold lower than that of conventional amphiphilic block copolymer micelles. The drug loading capacities of up to 13 wt% allow the efficient encapsulation of the chemotherapeutic Docetaxel (DTX). The spherical morphology of the micelles was proven by cryogenic electron microscopy (cryo-EM). Gaussian Analysis revealed well-defined sizes of 57 nm and 80 nm in the unloaded/loaded state, respectively. Experiments by dynamic light scattering (DLS), ultraviolet-visible spectroscopy (UV-VIS), fluorescence spectroscopy, and cross-polarization solid-state 13C NMR studied the π–π interactions between the core-forming block segment of dPGS-SS-POxPPh-Py and DTX. The findings point to a substantial contribution of these noncovalent interactions to the system's high stability. By confocal laser scanning microscopy (CLSM), the cellular uptake of fluorescein-labelled FITC-dPGS-SS-POxPPh-Py micelles was monitored after one day displaying the successful cell insertion of the cargo-loaded systems. To ensure the drug release in cancerous cells, the disassembly of the micellar DTX-formulations was achieved by reductive and enzymatic degradation studied by light scattering and GPC experiments. Further, no size increase nor disassembly in the presence of human serum proteins after four days was detected. The precise in vitro drug release was also given by the high potency of inhibiting cancer cell growth, finding half-maximal inhibitory concentrations (IC50) efficiently reduced to 68 nM coming along with high viabilities of the empty polymer materials tested on tumor-derived HeLa, A549, and McF-7 cell lines after two days. This study highlights the substantial potential of micelles tailored through the combination of π-electron stabilization with dendritic polyglycerolsulfate for targeted drug delivery systems, enabling them to have a significant foothold in the clinical treatment of cancer.
1. Introduction
In 2022, the American Cancer Society estimated almost two million new cancer cases in the United States1 – another 600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 patients will die of cancer – making it the second leading cause of death after cardiac disorders. The current state of chemotherapy is based on administering hydrophobic small-molecule drugs in order to kill the cancer cells or stop them from dividing.2,3 Self-assembled carrier systems are often applied to ensure the solubility of these water-insoluble compounds in the formulation and bloodstream.4,5 The fact that the delivery system gets highly diluted after injection and its simultaneous opsonization by a biomolecular layer has posed a challenge to scientists in chemistry, pharmacy, medicine, and pharmacology for decades.6–10 Upon administration, the dilution in the bloodstream triggers the disassembly of the system by causing the concentration to fall below the critical micelle concentration (CMC), resulting in a loss of function. Additionally, the nonspecific interaction with blood serum proteins is known to alter the applied systems’ stability10 and targeting properties.11 Most developed micellar carrier systems do not match their high performances drawn from in vitro experiments if tested in vivo.12 Consequently, the pharmacokinetic and biodistribution of the administered drug delivery system is similar to that of a free drug giving a distribution all over the body with low accumulation on its site of action, e.g., the tumor. In addition, this uncontrolled distribution causes further suffering for the cancer patient who is already worn out by the actual disease. Thus, to ensure therapy's success, the critical challenge lies in systematically improving the screening of the stability and protein interactions to keep the systems in their active states.13 Recently, biodegradable micelles based on dendritic polyglycerolsulfate (dPGS) demonstrated their high therapeutic potential in vivo with selective accumulation in tumor tissue showing no toxicity to the organisms.14,15 However, remaining is the central question of whether the stability of this system can be further increased to boost its therapeutic potential.
000 patients will die of cancer – making it the second leading cause of death after cardiac disorders. The current state of chemotherapy is based on administering hydrophobic small-molecule drugs in order to kill the cancer cells or stop them from dividing.2,3 Self-assembled carrier systems are often applied to ensure the solubility of these water-insoluble compounds in the formulation and bloodstream.4,5 The fact that the delivery system gets highly diluted after injection and its simultaneous opsonization by a biomolecular layer has posed a challenge to scientists in chemistry, pharmacy, medicine, and pharmacology for decades.6–10 Upon administration, the dilution in the bloodstream triggers the disassembly of the system by causing the concentration to fall below the critical micelle concentration (CMC), resulting in a loss of function. Additionally, the nonspecific interaction with blood serum proteins is known to alter the applied systems’ stability10 and targeting properties.11 Most developed micellar carrier systems do not match their high performances drawn from in vitro experiments if tested in vivo.12 Consequently, the pharmacokinetic and biodistribution of the administered drug delivery system is similar to that of a free drug giving a distribution all over the body with low accumulation on its site of action, e.g., the tumor. In addition, this uncontrolled distribution causes further suffering for the cancer patient who is already worn out by the actual disease. Thus, to ensure therapy's success, the critical challenge lies in systematically improving the screening of the stability and protein interactions to keep the systems in their active states.13 Recently, biodegradable micelles based on dendritic polyglycerolsulfate (dPGS) demonstrated their high therapeutic potential in vivo with selective accumulation in tumor tissue showing no toxicity to the organisms.14,15 However, remaining is the central question of whether the stability of this system can be further increased to boost its therapeutic potential.
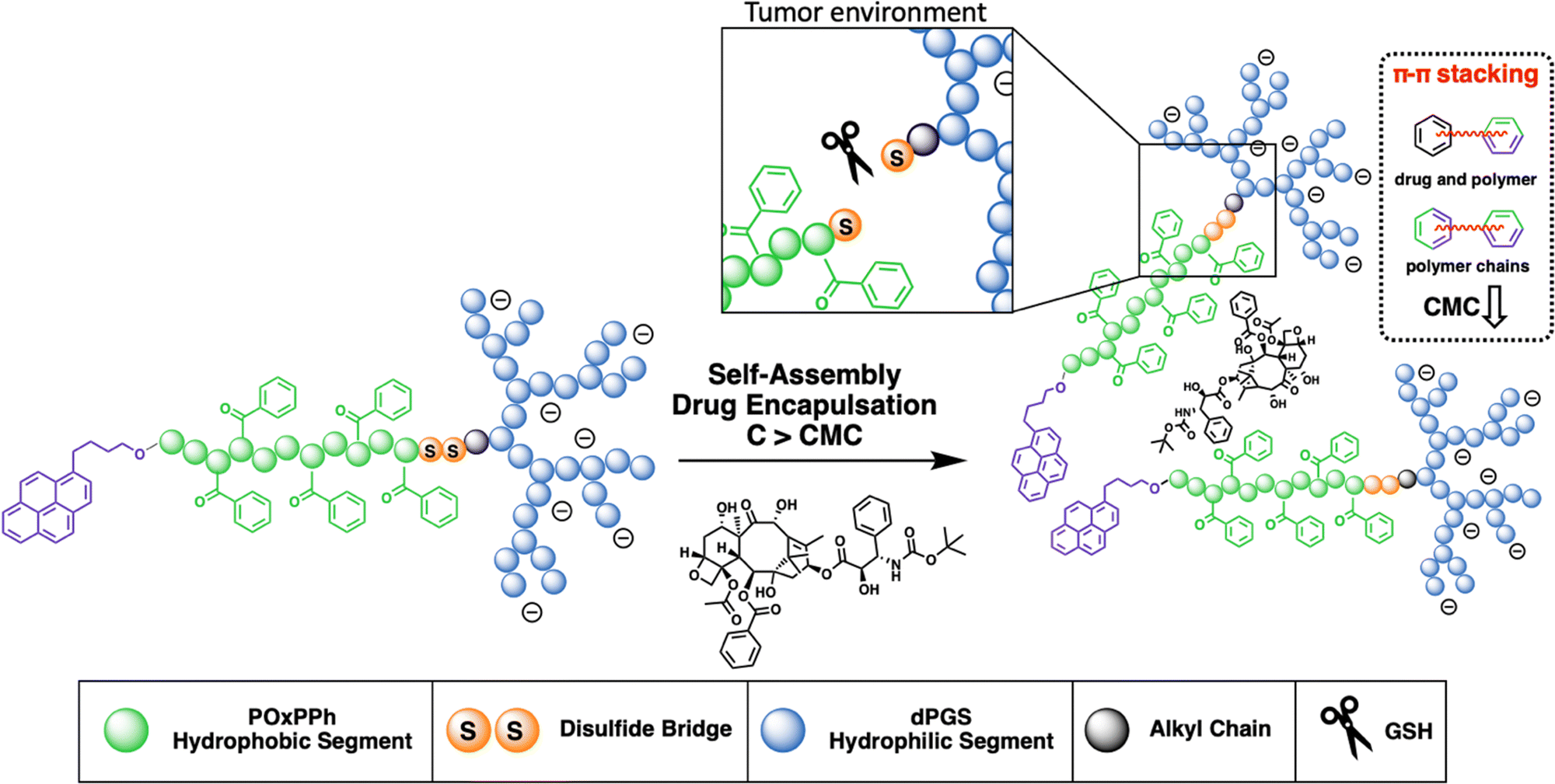
The development of micelles has attracted attention in both academia and industry.16 Yet, only minor progress has been reported in their clinical translation over the last few years.17–19 One of the early concepts, which has shown promising results, is the application of so-called π–π stabilized systems.20 This idea was applied in most of the very early systems that went into clinical trials; see NK91121 or NK105.22 This principle was adopted by Hennink et al., who again pointed out its high potential.23–25 In our study, we combine the efficiency of π-electron stabilization and the intrinsic targetability of dendritic polyglycerolsulfate towards inflammation and cancer. The selective accumulation of dPGS-based materials can be traced down to the binding of the sulfates with L-selectin,26–28 an overexpressed cell adhesion molecule in cancer progression. The hydrophobic π-electron-donating block is synthesized from a recently developed new class of monomers, the so-called N-acylated-1,4-oxazepan-7-one (OxP) monomers, a degradable analogue of poly(oxazoline).29 The hydrophilic segment is given as dPGS which is connected to the hydrophobic block via a disulfide bridge enabling the precise drug release in the reductive environment found in tumor tissue.30 This novel amphiphilic block copolymer is tailor-made for yielding micelles with (i) high stability; (ii) high drug loading capacity; (iii) a precise release of the payload; and (iv) excellent cell viability with substantial tumor growth inhibition (Fig. 1).
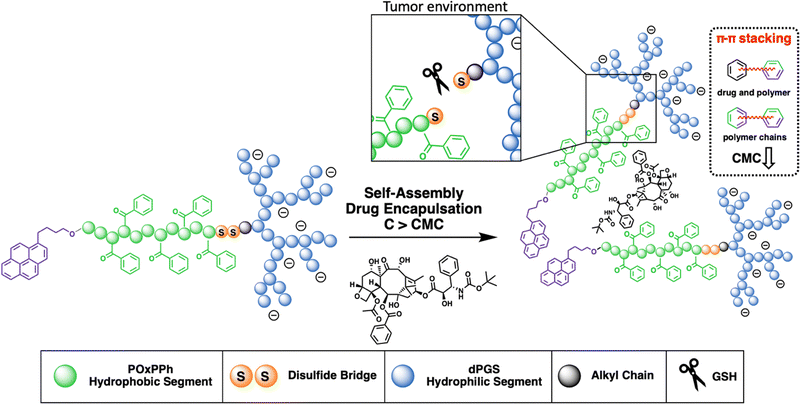 |
| | Fig. 1 Schematic illustration of dPGS-SS-POxPPh-Py amphiphiles undergoing self-assembly in an aqueous solution above their CMC, forming spherical micelles carrying a water-insoluble cargo, here docetaxel, in their hydrophobic inner-core, whereas the anionic-charged dPGS-shell (blue) facilitates the solubility of the system. The hydrophobic polymer, POxPPh (green), bears phenyl-rings on its side chain, facilitating π–π stacking in the micellar core between the polymer chains, the drug, and vice versa, decreasing CMC values and increasing drug loading capacities. The hydrophobic and hydrophilic segments are connected via a disulfide bridge (orange), enabling the polymer chain's reductive cleavage in tumor-mimicking GSH-rich environments. | |
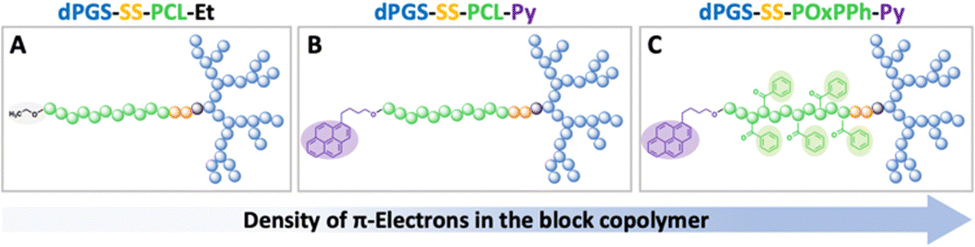
We designed a library of three novel block copolymer micelles (Fig. 2), each with varying amounts of π-electrons (i) dendritic polyglycerolsulfate-cystamine-block-poly(caprolactone)-ethyl (dPGS-SS-PCL-Et, Fig. 2(A)) having no π-electrons, (ii) dendritic polyglycerolsulfate-cystamine-block-poly(caprolactone)-pyrene (dPGS-SS-PCL-Py, Fig. 2(B)) with moderate π-electrons, and (iii) dendritic polyglycerolsulfate-cystamine-block-poly(4-benzoyl-1,4-oxazepan-7-one)-pyrene with high π-electron density (dPGS-SS-POxPPh-Py, Fig. 2(C)). The influence of π–π stacking in terms of self-assembly and stability was studied systematically by light scattering and cryo-TEM. The chemotherapeutic drug Docetaxel, which is part of first-line treatment regimens for numerous cancer types in the clinics, such as breast and non-small cell lung cancer, was used as the model compound. DLS measurements found decreased CMC values in the low nanomolar range with an increased density of π-electrons. Also, π-electron-bearing micelles exhibit higher drug-loading capacities than non-aromatic micelles. Furthermore, UV-VIS, fluorescence, and cross-polarization solid-state 13C NMR measurements unwrapped the structural morphology of the drug-loaded micelle core. The disassembly of the micelles was selectively triggered by reductive cutting of the disulfide bridge by GSH and by enzymatic cleavage of the esters by surface-immobilized lipases. The micelles were found to absorb no serum proteins on the micelle surface nor disassemble in their presence. FITC-labelled dPGS-SS-POxPPh-Py were successfully inserted into tumor-thrived cells shown by CLMS. As proof of concept, the polymers’ in vitro cell viability and in vitro cancer cell toxicity were evaluated, indicating low toxicity of the empty polymeric material while their DTX formulations exhibited highly potent anti-tumor performance. This study demonstrates the substantial potential of drug delivery systems achieved by the combination of π-electron stabilization and dendritic polyglycerolsulfate.
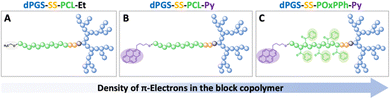 |
| | Fig. 2 Schematic overview of the amphiphilic block copolymer library with increasing π-electrons from left to right (A) dPGS-SS-PCL-Et with no π-electrons (B) dPGS-SS-PCL-Py with π-electrons on the chain-end attributed by pyrene (purple), and (C) dPGS-SS-POxPPh-Py with a high density of π-electrons in the presence of sidechain bearing phenyl rings (green) all over the hydrophobic segment and pyrene moiety on the chain end (purple). Notably, for dPGS-SS-POxPPh-Py, each repeating unit of the hydrophobic block carries a benzoyl group; this has been presented in a simplified manner for clarity. | |
2. Results and discussion
2.1 Synthesis and characterization of dPGS-SS-PCL-Et, dPGS-SS-PCL-Py, and dPGS-SS-POxPPh-Py
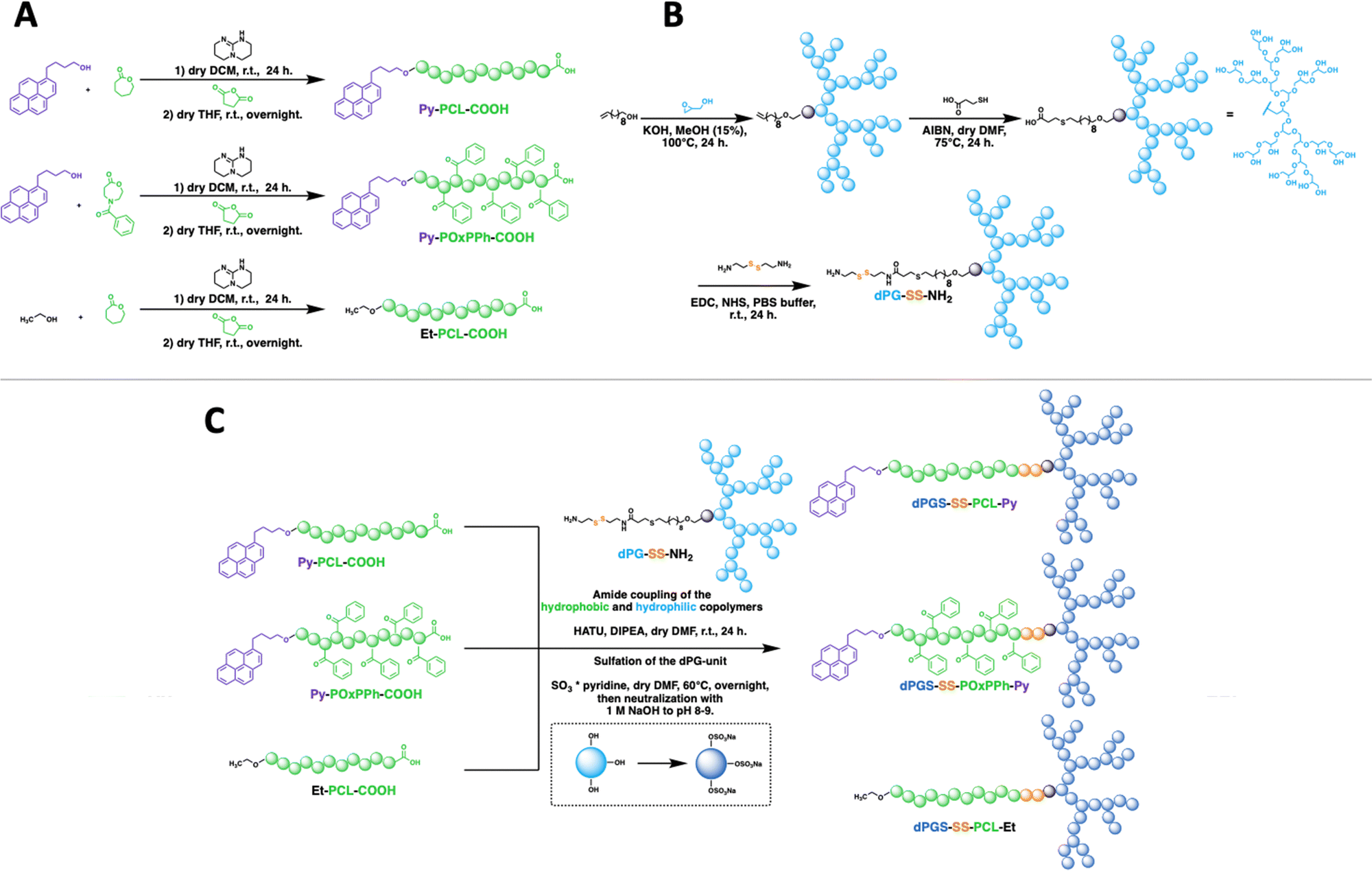
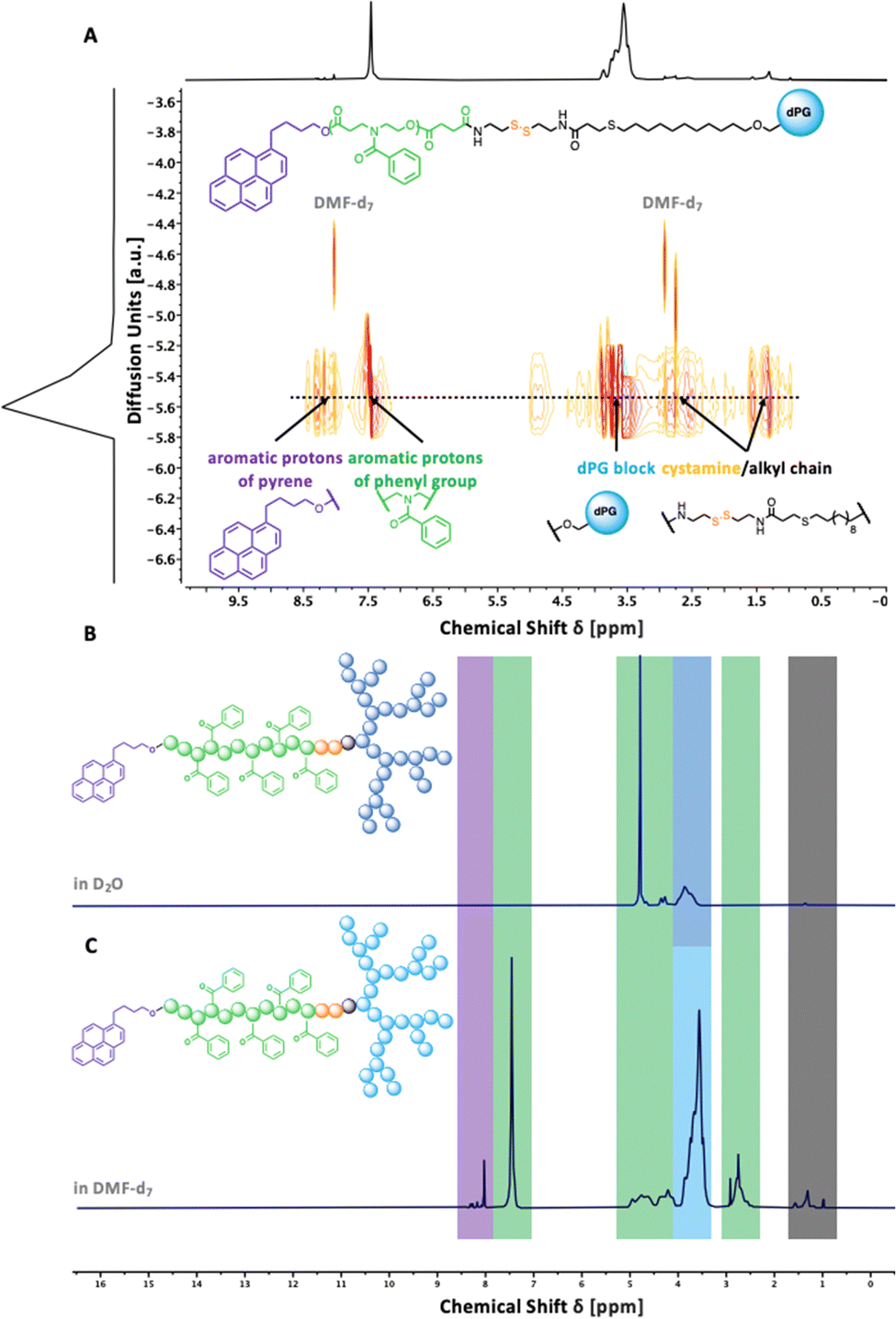
To understand the importance of π-electron-rich domains on the stability and efficacy of polymeric micelles, we have designed a library of three different amphiphilic block copolymers (Fig. 2) (i) with no π-electrons (dPGS-SS-PCL-Et, Fig. 2(A)); (ii) with π-electrons located on the chain end (dPGS-SS-PCL-Py, Fig. 2(B)); and (iii) with π-electron distributed along the hydrophobic polymer block (dPGS-SS-POxPPh-Py, Fig. 2(C)). The synthesis of 4-benzoyl-1,4-oxazepan-7-one (OxPPh) was performed following a Baeyer–Villiger oxidation of 1-benzoyl-4-piperidone (see ESI†). First, the hydrophobic segments Et-PCL-COOH, Py-PCL-COOH, and Py-POxPPh-COOH were synthesized starting from caprolactone (CL) or OxPPh using ethanol (Et-OH) or pyrene butanol (Py-OH) as initiators catalyzed by the organo-base 1,5,7-triazabicyclo(4.4.0)dec-5-en (TBD). Subsequently, the polymer chain ends were reacted with succinic anhydride quenching the polymerization and introducing a carboxylic acid (polymer-COOH) to the chain end (Fig. 3(A)). Since TBD has been demonstrated to be highly efficient in the polymerization of cyclic monomers,31 all polymers were obtained in their desired molecular weights of 7.8–7.9 kDa and low dispersity (Đ < 1.3, see ESI†). A 3-step synthesis formed the hydrophilic segment as follows: (i) anionic ring-opening polymerization of glycidol initiated by 10-undecenol; (ii) thiol-click reaction of mercaptopropionic acid; and (iii) amide-coupling of cystamine to introduce the reductive-sensitive disulfide bridge (Fig. 3(B)). The dPG-SS-NH2 was characterized by 1H NMR and GPC, revealing a molecular weight of 4.3 and 4.4 kDa, respectively, with a narrow dispersity of Đ = 1.5 (see ESI†). The amphiphilic block copolymers were obtained via an amide coupling procedure between Et-PCL7.8-COOH, Py-PCL7.9-COOH, and Py-POxPPh7.9-COOH with dPG4.3-SS-NH2 (numbers indicated the molecular weight in kDa) mediated by HATU/DIPEA in DMF at room temperature (Fig. 3(C)). The successful coupling of Py-OxPPh7.9-COOH and dPG4.3-SS-NH2 was proven by 1H diffusion-ordered spectroscopy (DOSY) NMR in DMF-d7, finding one significant diffusion species indicating the covalent attachment of the block copolymers with no free homopolymers (Fig. 4(A)). The ESI† shows the 1H NMR DOSY of the polymer blend with two different diffusion species of the mixed homopolymers (Fig. S11, ESI†). Lastly, the sulfation of the dPG4.3-SS-R yielded in dPGS8.6-SS-R (R = PCL7.8-Et, PCL7.9-Py, POxPPh7.9-Py), where it doubles the molecular weight of the dPG units by the mass increase of alcohols transformed to sulfate groups (see ESI†). From this point onwards, the characterization of the sulfated block copolymers is strongly limited due to the excessive amphiphilicity (see ESI†). In Fig. 4(B) and (C), the solution 1H NMR of dPG4.3-SS-POxPPh7.9-Py and dPGS8.6-SS-POxPPh7.9-Py is measured in DMF-d7 and D2O, respectively. Before sulfation, the NMR shows all characteristic structural motifs of the block copolymers. In contrast, the core-forming segments disappear after sulfation due to the micelle formation in D2O, the exclusive solvent for this polymer. However, the intact structure was proven via IR, GPC, Solid-State NMR (for more details see ESI†), and UV spectroscopy (Fig. 7).
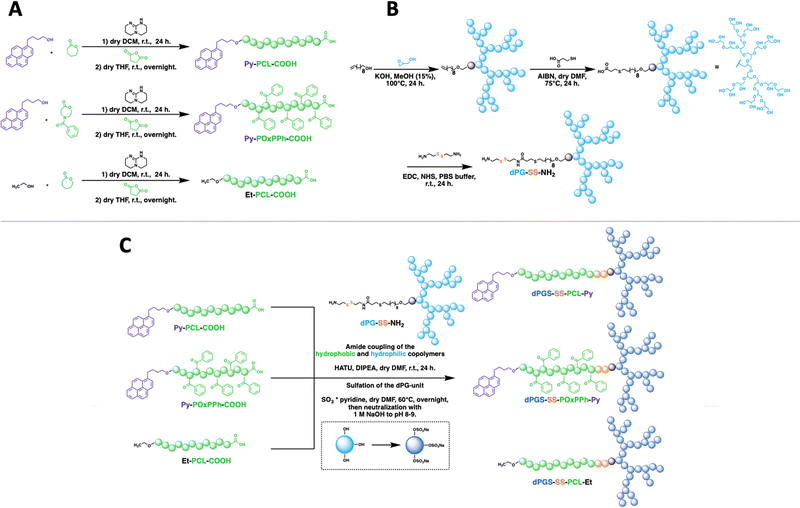 |
| | Fig. 3 Synthetic pathway to dPGS8.6-SS-POxPPh7.9-Py, dPGS8.6-SS-PCL7.9-Py, and dPGS7.8-SS-PCL7.8-Et. (A) Synthesis of the hydrophobic polyester segments Py-PCL-COOH, Py-POxPPh-Py, and Et-PCL-COOH by organo-catalyst TBD-mediated ring-opening polymerization of OxPPh or CL monomer initiated by pyrene butanol or ethanol with subsequent quenching via the addition of succinic anhydride. (B) Synthesis of the hydrophilic segment dPG-SS-NH2via an anionic ring-opening polymerization of glycidol initiated by 10-undecenol with following click reaction with mercaptopropionic acid and coupling of cystamine. (C) Synthesis of the amphiphilic block copolymers, first, via an amide coupling reaction mediated by HATU; the hydrophobic segments, Py-PCL-COOH, Py-POxPPh-COOH, and Et-PCL-COOH were coupled to the hydrophilic segment dPG-SS-NH2 (dPG unit represented as light blue branched architecture), leading to amphiphilic block copolymers. Subsequently, sulfation converts the hydroxy groups of the dPG unit to sulfates, as indicated by the dark blue colour of the branched dPG-architecture leading to the final products dPGS-SS-PCL-Py, dPGS-SS-POxPPh-Py, and dPGS-SS-PCL-Et. | |
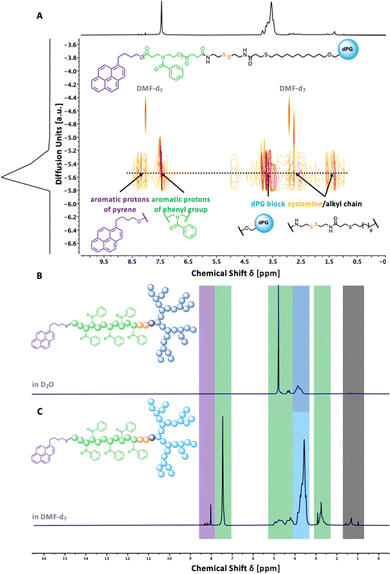 |
| | Fig. 4 (A) 1H-derived DOSY spectrum (500 MHz) in DMF-d7 after amide coupling of Py-POxPPh7.9-COOH and dPG4.3-SS-NH2 showing one significant diffusion species (dotted line) confirming successful coupling to a covalently connected amphiphilic block copolymer with no uncoupled educts. (B) 1H NMR spectrum of dPGS8.6-SS-POxPPh7.9-Py shows the signals of the shell-forming dPGS-segment in D2O. The signals of the core-forming segments disappear due to the formation of micelles in an aqueous solution. (C) 1H NMR spectrum of dPG4.3-SS-POxPPh7.9-Py showing all characteristic peaks of the pyrene butanol-initiator, hydrophobic POxPPh block, alkyl chain/disulfide linker, and of the hydrophilic dPG-segment in DMF-d7. | |
2.2 Formation and characterization of micelles: shape, size, charge, and stability
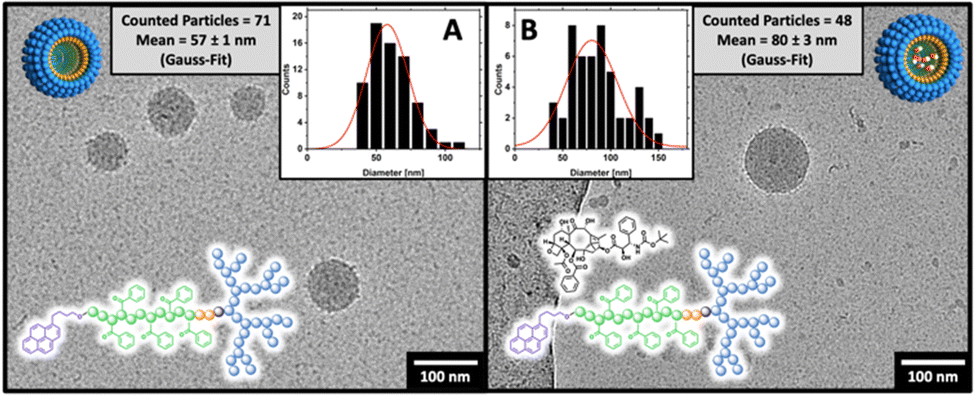
The amphiphilic block copolymers were used to form micelles following a nanoprecipitation protocol from acetone into PBS buffer (150 mM NaCl, pH 7.4) with subsequent organic solvent evaporation. Before light scattering experiments studied the formed micelles intensively, the morphology and size were analyzed by cryo-TEM, finding a spherical morphology (Fig. 5).
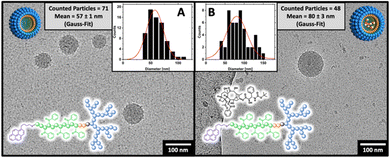 |
| | Fig. 5 cryo-TEM images of (A) empty and (B) docetaxel-loaded dPGS8.6-SS-POxPPh7.9-Py micelles in PBS at 1 mg mL−1 showing the spherical character of the aggregates. Gauss-analysis of multiple TEM measurements determined the particle size. The detected values show DLS-consistent sizes of 57 and 80 nm for empty/DTX-loaded micelles, respectively. (Scale bar: 100 nm.) | |
By Gaussian analysis of the detected particles, the mean particle size for dPGS8.6-SS-POxPPh7.9-Py was 57 nm in the unloaded state (Fig. 5(A)) and 80 nm upon loading with Docetaxel according to cryo-TEM (Fig. 5(B)). In DLS, the hydrodynamic diameter for dPGS8.6-SS-POxPPh7.9-Py micelles was found to be in similar ranges with 94 nm in the unloaded and 102 nm in the drug-loaded state. Notably, in the DLS measurements, the size is slightly larger due to the hydration shell around the particles, hence it is not present in cryo-TEM images. The sizes for dPGS8.6-SS-PCL7.9-Py, DTX@dPGS8.6-SS-PCL7.9-Py, and dPGS7.8-SS-PCL7.8-Et were found between 81–130 nm according to DLS. With increased π-electron domains on the polymer, the PDI detected by DLS starts to decrease from 0.17 for dPGS7.8-SS-PCL7.9-Et to 0.08 for DTX@dPGS8.6-SS-POxPPh7.9-Py. The surface charge of all micelles was measured and detected from −44 to −33 mV. For a complete characterization of all colloids and formulations, see Table 1.
Table 1 Characteristic of empty/DTX-loaded dPGS-SS-POxPPh-Py, dPGS-SS-PCL-Py, and dPGS-SS-PCL-Et block copolymers and their respective micelles in terms of molecular weight, drug loading efficiency, drug loading capacity, size, PDI, CMC, and surface potential
| Polymer |
Molecular weight Mn (kDa) |
Drug loading docetaxel |
Size (nm)c |
PDIc |
CMC at 37 °C in PBS |
ζ
(mV) |
| Theo. |
Calc.a |
DLEb (%) |
DLCb (wt%) |
mg L−1c |
nM |
|
Calculated MNMR,hydrophobicsegmentn + (2 × MNMR,hydrophilicsegmentn).
Docetaxel-loading, determined by HPLC.
Determined by light scattering in PBS at 37 °C.
Measured in 10 mM PB buffer at 37 °C at pH 7.4; n.s. = not significant; no detectable drug amount in the formulation.
|
| dPGS7.8-SS-PCL7.8-Et |
16.0 |
15.6 |
n.s. |
n.s. |
81.2 ± 1.1 |
0.17 |
2.1 |
133 |
−44 |
| dPGS8.6-SS-PCL7.9-Py |
16.0 |
16.5 |
— |
— |
111.9 ± 1.8 |
0.15 |
0.9 |
54 |
−34 |
| DTX@dPGS8.6-SS-PCL7.9-Py |
16.0 |
16.5 |
26 |
5 |
129.4 ± 18.8 |
0.13 |
1.0 |
55 |
— |
| dPGS8.6-SS-POxPPh7.9-Py |
16.0 |
16.5 |
— |
— |
93.7 ± 1.4 |
0.09 |
0.7 |
42 |
−39 |
| DTX@dPGS8.6-SS-POxPPh7.9-Py |
16.0 |
16.5 |
62 |
13 |
102.4 ± 7.6 |
0.08 |
0.3 |
18 |
— |
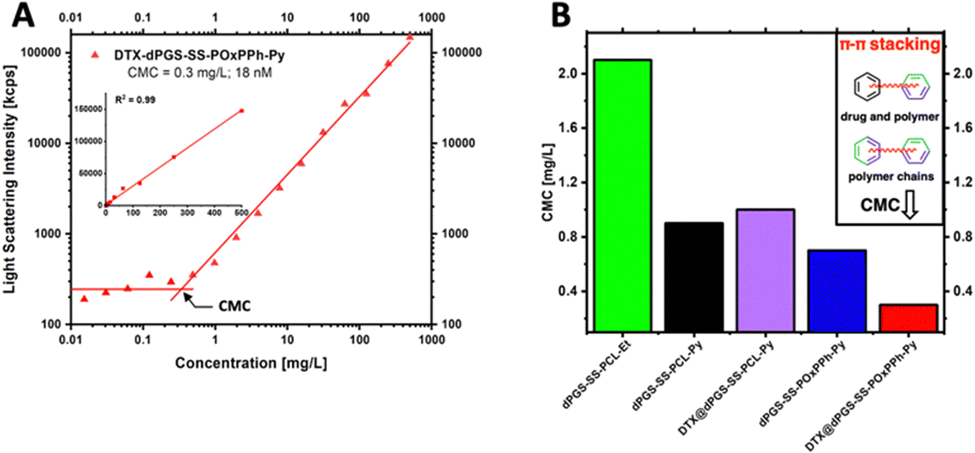
The first border of a self-assembled system is in the extreme dilution upon injection; thus, the stability of the micelles under physiological conditions (150 mM NaCl, pH 7.4, 37 °C) was systematically studied. To do so, a DLS-supported dilution experiment was conducted to monitor the evolution of the light scattering intensity, steadily decreasing the concentration of the amphiphiles in the solution. As the light scattering intensity is proportional to the concentration of a scattering species, it allows for the efficient determination of the CMC (Fig. 6(A)). Compared to other techniques, such as fluorescence, DLS allows the label-free determination of the CMC.32 Since encapsulation of hydrophobic cargos has been shown to alter the stability of polymeric micelles,33 fluorescence spectroscopy was not applicable in this study since this effect would interfere with the drug-loading stability studies by DTX. The analysis of the CMC of the block copolymer library revealed the trend of significantly decreased CMCs with increased π-electron-density of the systems following the order dPGS7.8-SS-PCL7.9-Et > dPGS8.6-SS-PCL7.9-Py > dPGS8.6-SS-POxPPh7.9-Py. For dPGS7.8-SS-PCL7.9-Et, the highest CMC of 2.1 mg L−1 (133 nM) was found. For dPGS8.6-SS-PCL7.9-Py, the implementation of the pyrene moiety decreased the CMC by 2-fold to 0.9 mg L−1 (54 nM). As expected, the dPGS8.6-SS-POxPPh7.9-Py, with a high π-electron density, shows a CMC of 0.7 mg L−1 (42 nM), which even can be further decreased upon loading with Docetaxel to 0.3 mg L−1 (18 nM) (Fig. 6(B)). Thus, also the drug contributes π-electron to the micellar core improving the CMC (see the structure of Docetaxel). The drug-loading of dPGS8.6-SS-PCL7.9-Py showed no influence on the CMC, most likely due to the insufficient loading capacity of only 5 wt% compared to 13 wt% for dPGS8.6-SS-POxPPh7.9-Py. The CMC determination of the other amphiphiles is shown in more detail in the ESI† (Fig. S34).
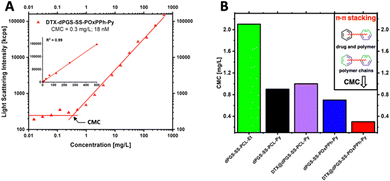 |
| | Fig. 6 (A) CMC determination of DTX-loaded dPGS8.6-SS-POxPPh7.9-Py micelles in PBS at 37 °C by a light scattering experiment with different concentrations ranging from 500 mg L−1 to 5 μg L−1 (inset: linear dependency of the count rate and concentration; R2 = 0.99); light scattering intensity below CMC was horizontalized according to ISLS ∼ cpolymer. (B) Influence of π-stabilization on dPGS-SS-PCL-Et, empty/DTX-loaded dPGS-SS-PCL-Py, and empty/DTX-loaded dPGS8.6-SS-POxPPh7.9-Py micelles showing significantly decreased CMC values for π-electron-rich systems. | |
As self-assembled systems are dynamic systems in an equilibrium between their monomeric and polymeric nature, it is essential to correlate the CMC to the minimum required dosage. For example, in Genexol-PM, a clinically approved nanomedicine based on PEG-b-PLA micelles, the formulation of one dosage contains 30 mg of taxane.34 For the DTX@dPGS8.6-SS-POxPPh7.9-Py systems, the DLC is determined as 13 wt%; thus, a theoretical dose of 230 mg of the polymeric material would be needed for a human application. Considering the total blood volume of an adult as 5 L, the final concentration of the amphiphile is 46 mg L−1, which still exceeds the CMC by 150-fold (CMC: 0.3 mg L−1). To our knowledge, the lowest CMC described yet is found at 2.7 nM achieved by so-called “sharp polarity contrast” micelles;35,36 however, typical polymer systems based on PEG-amphiphiles show CMCs in the 1000 nM range.37 All polymers included in this study, but especially DTX@dPGS8.6-SS-POxPPh7.9-Py, undercut this value underlining their high stability. In summary, we showed that implementing π-electrons onto a polymeric structure beneficially influences micellar stability.
2.3 Drug-loading with docetaxel: influence of π-electron density
By HPLC, the drug-loading capacity (DLC in wt%) and drug-loading efficiency (DLE in %) of Docetaxel in dPGS8.6-SS-PCL7.9-Py and dPGS8.6-SS-POxPPh7.9-Py micelles were determined. For micelles with a decent number of π-electrons in their core (dPGS8.6-SS-PCL7.9-Py), a low DLC of only 5 wt% (DLE: 26%) was detected. For dPGS7.8-SS-PCL7.9-Et, no significant encapsulation could be detected. The increase of the π-electron density in dPGS8.6-SS-POxPPh7.9-Py raises the DLC to 13 wt% and 62% loading efficiency (Table 1). This is due to the greater possibility of the drug Docetaxel interacting with the phenyl rings of the POxPPh7.9 segment, as shown in poly(β-benzyl malate)-b-polyethylene glycol systems interacting with Doxorubicin by Qiao et al.38
2.4 π–π-stacking in the micelle core: UV-VIS, cross-polarization solid-state 13C NMR, and fluorescence spectroscopy
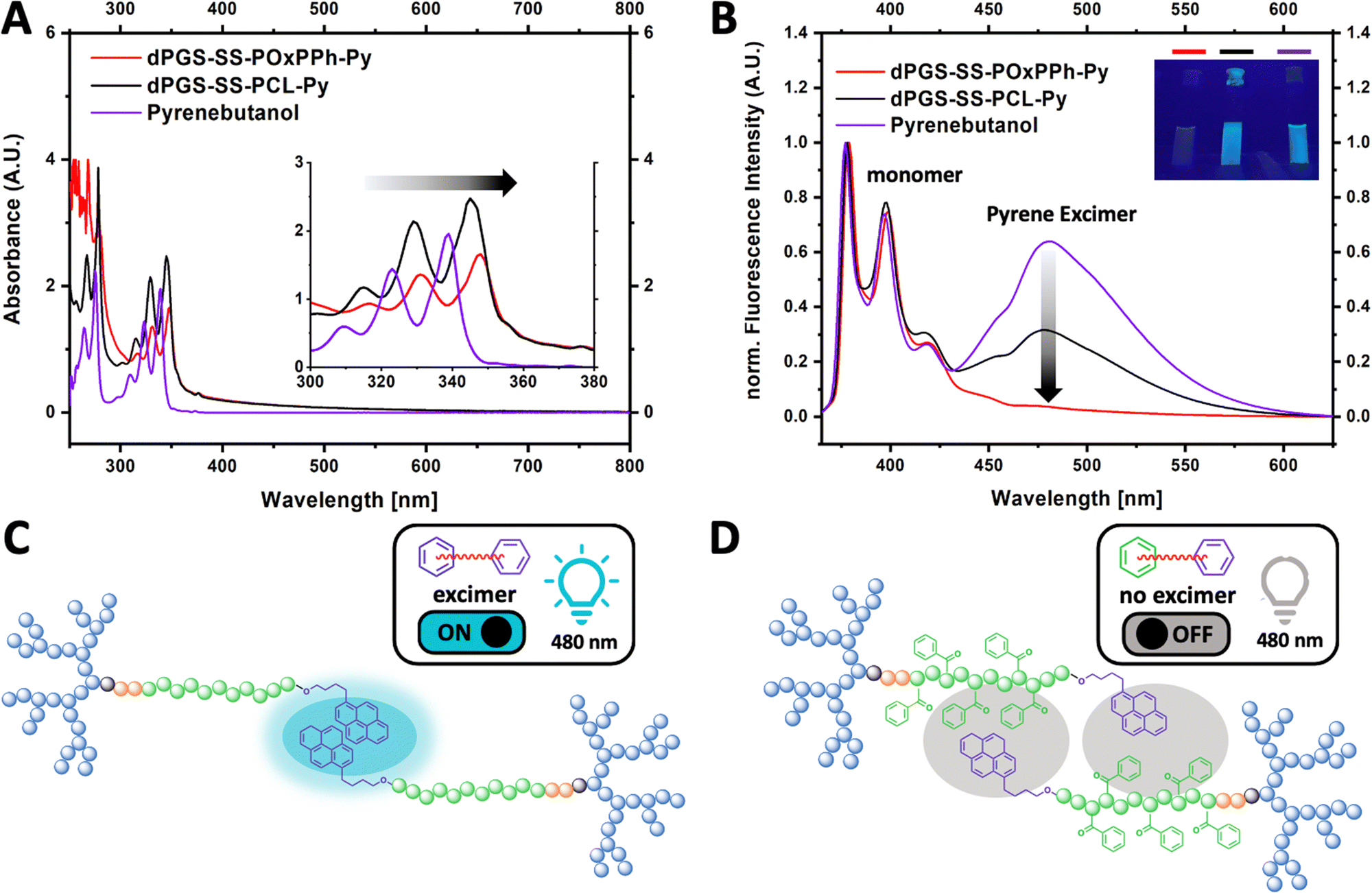
To classify π–π interactions, the structural behavior of the aromatic motifs was studied by UV-VIS and fluorescence spectroscopy (Fig. 7). The pyrene moiety on dPGS8.6-SS-POxPPh7.9-Py and dPGS8.6-SS-PCL7.9-Py chain ends allows the detection of π–π interaction. Due to the greater electron delocalization, the UV bands of dPGS8.6-SS-POxPPh7.9-Py and dPGS8.6-SS-PCL7.9-Py shifted to longer wavelengths (red-shift) compared to free pyrene butanol in PBS, which is also known as bathochromic effect (Fig. 7(A)).39 Further, pyrene displays the unique ability, besides its known fluorescence emission peaks (375–405 nm, monomer), to show an additional band at ca. 460 nm if a so-called excimer is formed.40 This phenomenon was used to obtain deeper insights into the structural orientation of the micellar core. Fig. 7(B) shows the fluorescence spectra of pyrene butanol, dPGS8.6-SS-POxPPh7.9-Py, and dPGS8.6-SS-PCL7.9-Py in PBS (λex 350 nm). For pyrene butanol, a strong, and for dPGS8.6-SS-PCL7.9-Py, a moderate excimer emission at 480 nm was observed. Thus, the excimer emission is called “turn ON”. In contrast, for dPGS8.6-SS-POxPPh7.9-Py, no significant band was detectable. The high π-electron density of dPGS8.6-SS-POxPPh7.9-Py forms a more randomly distributed micellar core by stacking the aromatic motifs. In dPGS8.6-SS-PCL7.9-Py, the π–π interaction is limited to the pyrene motifs only (Fig. 7(C)), whereas, in dPGS8.6-SS-POxPPh7.9-Py, the phenyl rings of the hydrophobic block OxPPh can undergo π–π interactions with the pyrene moiety quenching the excimer emission bringing the system in the “turn OFF” mode (Fig. 7(D)).
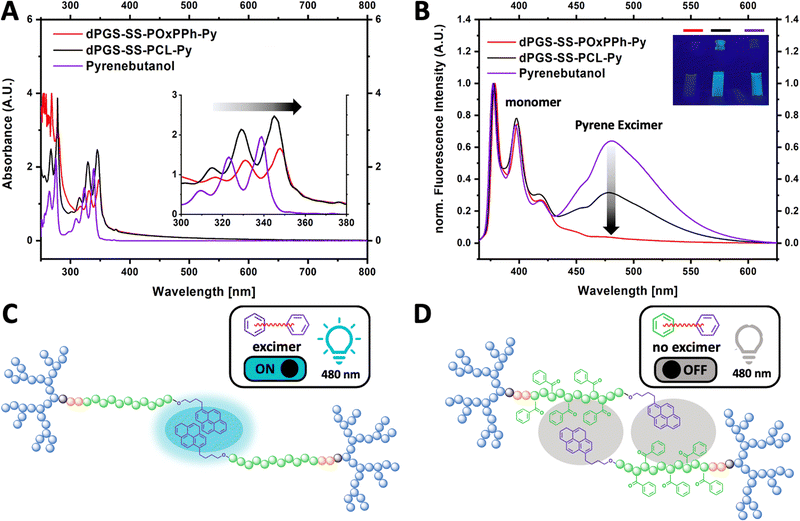 |
| | Fig. 7 (A) Absorbance spectra measured in PBS at 37 °C of dPGS8.6-SS-POxPPh7.9-Py (red), dPGS-SS-PCL-Py (black), and pyrene butanol (purple), inset showing red-shift of micellar formulations (B) normalized fluorescence spectra (λex 350 nm) in PBS at 37 °C of dPGS8.6-SS-POxPPh7.9-Py, dPGS8.6-SS-PCL7.9-Py, and pyrene butanol showing strong excimer formation for pyrene butanol, weak excimer for dPGS-SS-PCL-Py, and no significant excimer presence for dPGS8.6-SS-POxPPh7.9-Py (inset: dPGS8.6-SS-POxPPh7.9-Py, dPGS8.6-SS-PCL7.9-Py, and pyrene butanol under UV-light irradiation at 366 nm) (C) mechanistic illustration of the π–π interaction in dPGS8.6-SS-PCL7.9-Py micelles in an aqueous solution; in dPGS8.6-SS-PCL7.9-Py, the only interaction is between the pyrene moieties showing strong excimer formation (cyan circle); (D) in dPGS8.6-SS-POxPPh7.9-Py, the phenyl rings of the side chain interact with the pyrene suppressing excimer formation (grew circle) showing no band in the fluorescence at 480 nm with no emitted light (see inset in B). | |
As the light scattering experiments revealed a synergistic effect of DTX loading in dPGS8.6-SS-POxPPh7.9-Py on their CMC (Fig. 6(B)), the interaction of the cargo with the polymer was studied by solid-state NMR experiments. Docetaxel's low UV-VIS activity, no fluorescence, and its disappearance in solution NMR, if incorporated in micelles, exclude these techniques to check possible π–π interactions between the cargo and the polymeric material. In recent years, the use of cross-polarization solid-state 13C NMR has shown great potential to gain structural insights into a drug-loaded micelle core.41–43 For that, three formulations with (i) only drug; (ii) only polymer; and (iii) drug in micelles were prepared in aqueous solutions and subsequently freeze-dried. In the cross-polarization solid-state 13C NMR spectrum, the signals corresponding to the aromatic carbons of DTX between 120–145 ppm were found to shift slightly in the formulation compared to free DTX, exhibiting interactions between the cargo and the polymer (see ESI,† (Fig. S35 and S36)). However, as Docetaxel is a rather complex structure, this analysis remains challenging, only enabling little insights into the drug-loaded micelle core.
2.5
In vitro stability: serum–protein interaction and reductive/enzymatic degradation
After a potential administration into the bloodstream, micelles would interact with the blood's non-cellular components, leading to a biomolecular surface layer. This event is suspected as a cause of injected particles’ insufficient in vivo stability and to lower their targeting ability.44–46 In order to study the biomolecular layer and in vitro stability, the micelles were incubated with Human Serum Albumin (HSA), the most abundant serum protein in humans. By DLS, the size evolution of DTX@dPGS8.6-SS-POxPPh7.9-Py micelles was monitored in 10 mg mL−1 HSA in PBS at 37 °C (Fig. 8(A), red dots) over four days. To keep the viscosity of the solution low, the protein concentration was reduced from phycological concentrations of 45 mg mL−1 to 10 mg mL−1 to avoid distorting effects on the Brownian Motion. DTX@dPGS8.6-SS-POxPPh7.9-Py micelles without any additives in PBS at 4 °C (Fig. 8, purple squares) or 37 °C (Fig. 8, black squares) served as control (initial size for all samples 102 nm). The micelles were found to absorb no protein on their surface as the size was in line with the controls, with no significant size change over four days for all three samples ending up at 109 nm for the HSA samples, controls at 37 °C at 104 nm and 4 °C at 106 nm, respectively. Further, the light scattering intensity of serum-incubated micelles remained constant over four days, indicating stable particles in the presence of serum proteins (see ESI,† Fig. S32). These data are consistent with Isothermal titration calorimetry (ITC) experiments showing that dPGS and HSA are not interacting under physiological conditions (150 mM NaCl, 37 °C).47 This observation is critical as absorbed material would cover the dPGS units decreasing their targeting ability.
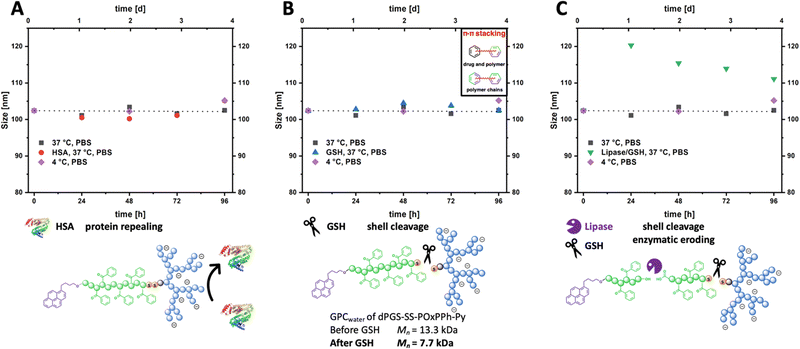 |
| | Fig. 8 Time-dependent DLS measurements on the evolution of the size of DTX@dPGS8.6-SS-POxPPh7.9-Py in different conditions in PBS at 37 °C, pH 7.4 (black) and 4 °C, pH 7.4 (purple) as control; (A) in the presence of HSA (red); (B) GSH, pH 5.0 (blue); (C) lipase/GSH, pH 5.0 (green); for four days, revealing stable particles without additives and in the presence of serum proteins as indicated by constant sizes. GSH incubation showed no significant changes in the size and light scattering intensity, indicating the stabilizing effect of π–π stacking on the aggregates. By GPC measurements, the cleavage of the disulfide by tracking the dPGS8.6-unit was confirmed. The hydrophobic POxPPh7.9-Py segment is retained on the column due to its limited water solubility. Incubation with additional lipase next to GSH caused an initial increase in the size attributed to a reordering of the degraded hydrophobic polyester polymer mediated by π–π stacking. Below are schematic illustrations of the protein-repealing and degradation mechanism of dPGS8.6-SS-POxPPh7.9-Py by lipase and GSH. The lipase degrades the ester moieties in the hydrophobic segment, and the GSH cleaves the disulfide bridge, destroying the block copolymer's amphiphilic nature. | |
In tumor cells, the GSH level is increased to 10 mM with only micromolar contents extracellular.48 So, the selective cleavage of the reductive-sensitive disulfide bridge was investigated in the presence of 10 mM GSH at 37 °C for four days (Fig. 8(B), blue triangles). Also, given the endocytosis pathway upon intracellular uptake, the micelles would be exposed to acidic conditions in the endosome and later lysosome; thus, pH was adjusted to acidic conditions. By DLS, no size change compared to the controls was detectable, ending at 103 nm. This can be attributed to the inter-and intramolecular π–π stacking of the POxPPh-Py segments leading to intact colloids in solution. However, by GPC experiments in water, the cleavage of the disulfide bridge was proven by detecting the cleaved dPGS8.6-S-R unit (GPC: Mn = 7.7 kDa, Fig. 8(B)). To break down these polymer aggregates, the enzymatic degradation of the ester-backbone was triggered by a surface-immobilized lipase (Novozyme 435) (Fig. 8(C), green triangle).49 The use of immobilized lipases does not affect the light scattering intensity as it would by adding non-immobilized enzymes into the solution. In the presence of additional enzymes next to GSH, the aggregates start to swell to 120 nm in the initial first 24 h with a subsequent decrease in size after four days at 112 nm. In line, the light scattering intensity of lipase-treated micelles constantly decreases over time, indicating the disassembly of the micelles. For a detailed discussion about the evolution of the light scattering intensity, see ESI† (Fig. S32). Based on these results, the micelles exhibited no interaction with serum proteins nor disassembly in their presence. They also revealed a prolonged degradation profile by selectively triggering the breaking points (ester, disulfide) in the amphiphilic polymer structure of dPGS8.6-SS-POxPPh7.9-Py.
2.6
In vitro cell viability, in vitro anti-cancer cell performance, and in vitro fate of dPGS-SS-micelles
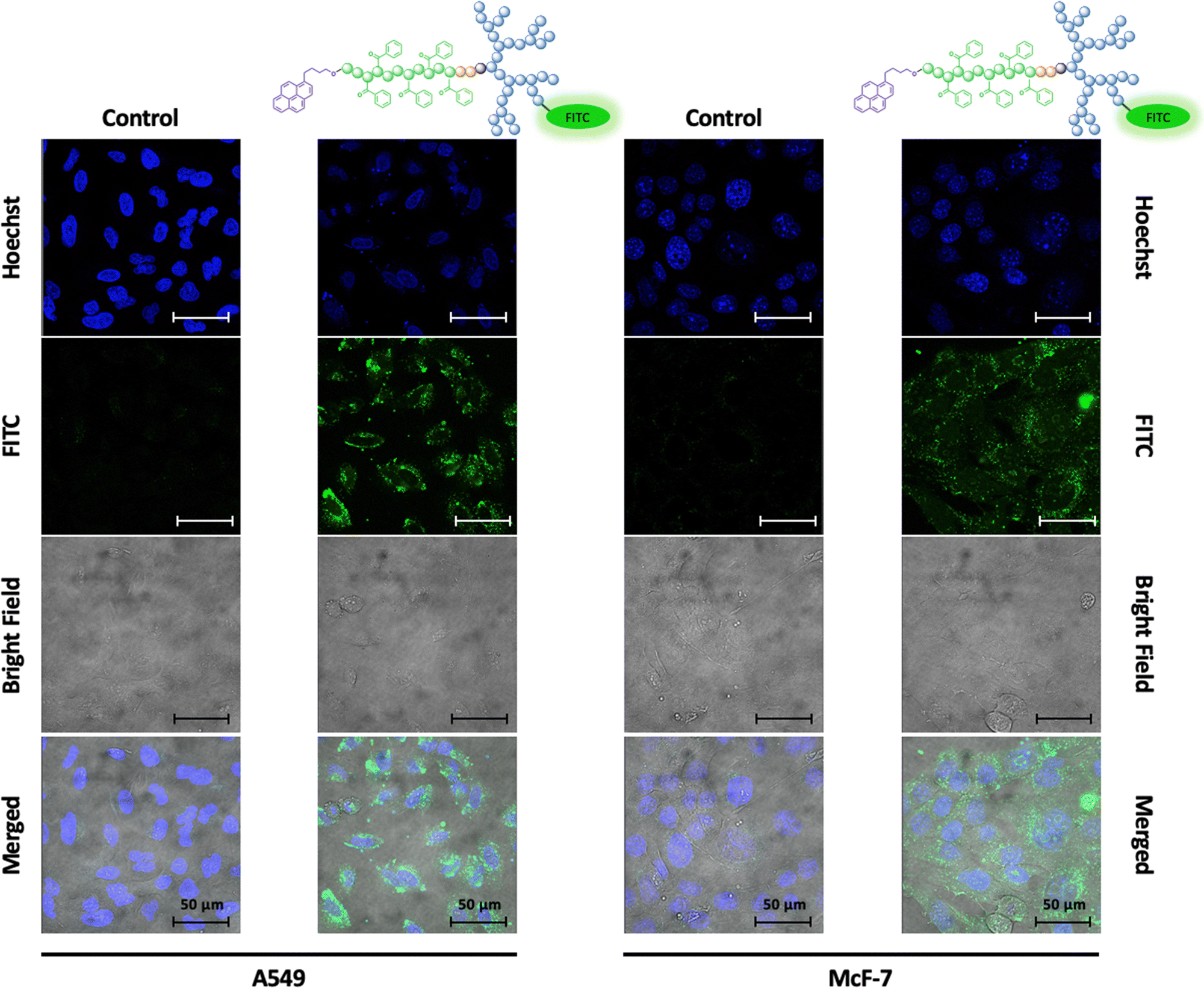
As given by the mode of action, the uptake of docetaxel into the cell is essential for unleashing its anti-mitotic activity.50 To visualize the fate of dPGS8.6-SS-POxPPh7.9-Py micelles in vitro, a fluorescence dye fluorescein isothiocyanate (FITC), was covalently attached to the hydrophilic shell leading to FITC-labelled dPGS8.6-SS-POxPPh7.9-Py. For the synthesis and characterization, see ESI.† By confocal laser scanning microscopy (CLSM), the cellular uptake of FITC-dPGS8.6-SS-POxPPh7.9-Py was monitored after 24 h incubation time on A549 (lung cancer) and McF-7 (breast cancer) cells (Fig. 9), depicting the successful insertion of the polymeric material into the cells.
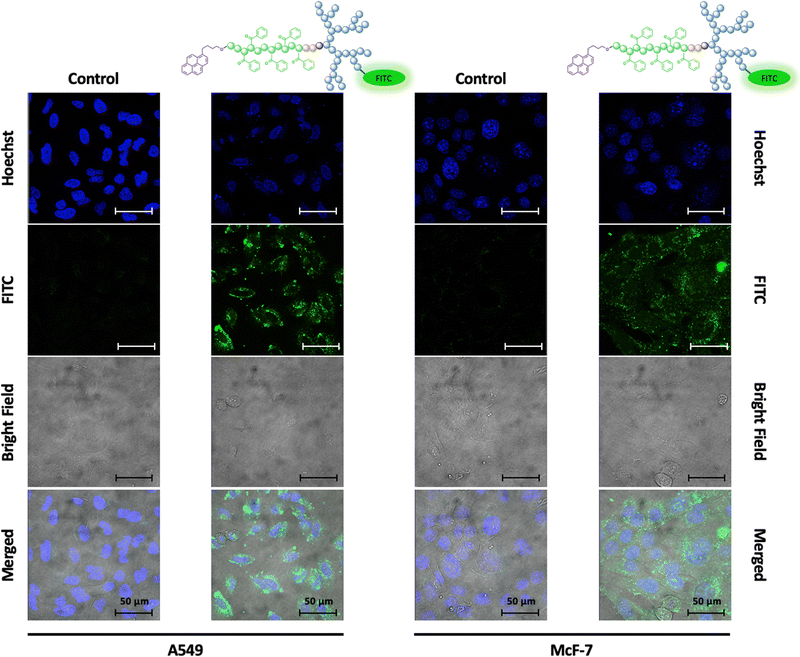 |
| | Fig. 9 Confocal laser scanning microscopy images of A549 (left) and McF-7 (right) tumor-derived cells incubated with and without FITC-dPGS8.6-SS-POxPPh7.9-Py micelles (incubation time 24 h); the polymeric material is successfully inserted into the cell as shown by the strong fluorescence signal of the covalently attached dye; scale bar: 50 μm, blue: Hoechst (Nuclei), green: FITC (fluorescent dye covalently attached to polymer). | |
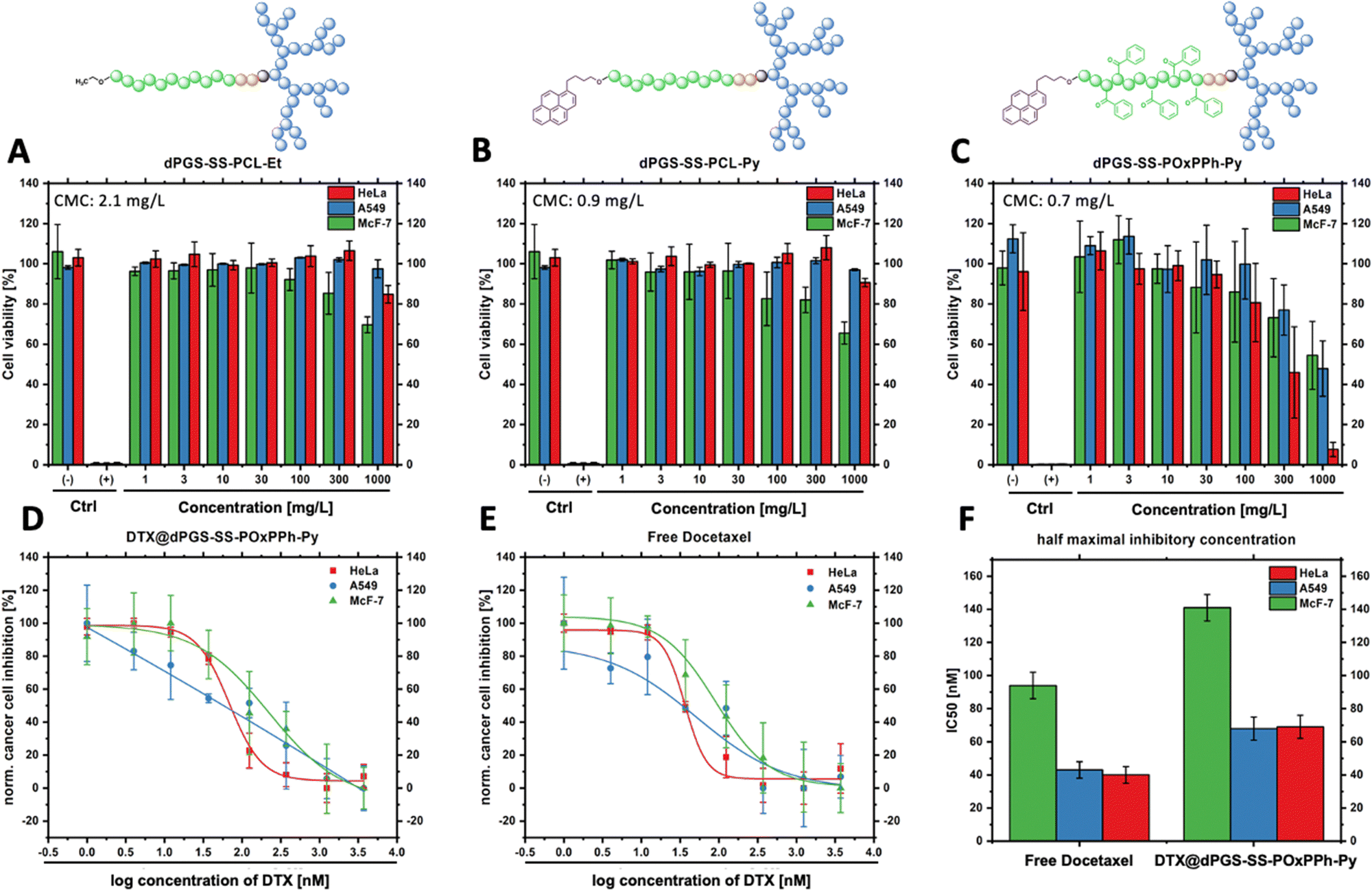
Knowing the capability of the micelle's cell insertion, the micelle's in vitro therapeutical performance was further studied. One of the early attempts to test drug delivery systems’ effectiveness starts with evaluating their cytotoxicity. Fig. 10 displays the in vitro performance of empty and DTX-loaded micelles. By CCK-8 assay, the in vitro cell compatibility of dPGS7.8-SS-PCL7.8-Et, dPGS8.6-SS-PCL7.9-Py, and dPGS8.6-SS-POxPPh7.9-Py was tested on McF7, HeLa (cervical cancer), and A549 tumor-derived cell lines after 48 h (Fig. 10(A)–(C)). No polymer showed significant toxicity in the range of its actual CMC. Even at elevated concentrations up to 100-fold higher than the CMC, the polymers showed no influence on the cell's viability. For dPGS8.6-SS-POxPPh7.9-Py, the cell viability falls below 50% at concentrations above 300 mg L−1 as tested on HeLa cells (46% cell viability, two days in vitro).
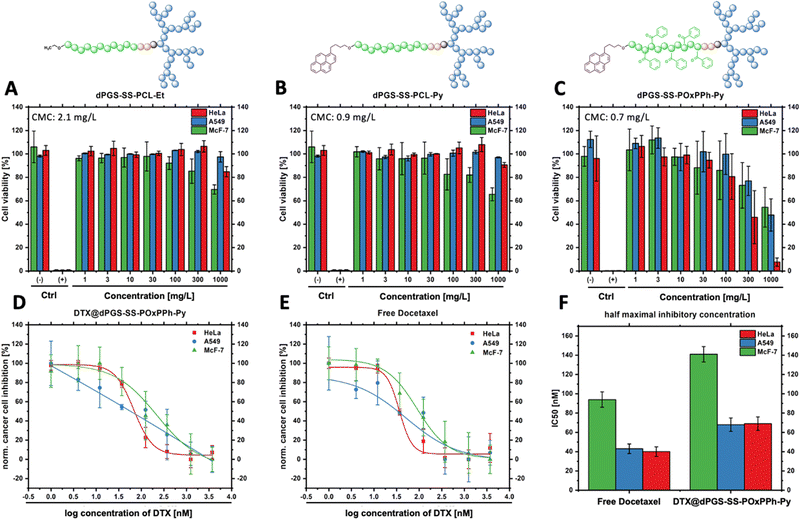 |
| | Fig. 10
In vitro cell compatibility studies on HeLa, A549, and McF-7 tumor-derived cell lines after 48 h incubation (n = 3, CCK-8, (−) PBS control, (+) SDS control) of (A) empty dPGS7.8-SS-PCL7.8-Et (A549 performed n = 2) (B) empty dPGS8.6-SS-PCL7.9-Py (A549 performed n = 2) (C) empty dPGS8.6-SS-POxPPh7.9-Py no polymer shows significant toxicity at a concentration of 100-fold higher than that of the CMC; in vitro dose-response curves of (D) DTX@dPGS8.6-SS-POxPPh7.9-Py (E) free docetaxel on HeLa, A549, and McF-7 cells after 48 h incubation; (F) anti-tumor performance of free Docetaxel and Docetaxel-loaded dPGS8.6-SS-POxPPh7.9-Py micelles, showing similar cancer cell inhibition (IC50) for the polymer-supported treatment and the free drug in vitro. | |
As the polymer dPGS8.6-SS-POxPPh7.9-Py was well-tolerated by HeLa, A549, and McF-7, the anti-tumor performance of free DTX and DTX-loaded dPGS8.6-SS-POxPPh7.9-Py micelles on these cell lines was investigated. Fig. 10(D) and (E) shows the dose-response curve of DTX@dPGS8.6-SS-POxPPh7.9-Py and free DTX after two days in vitro. After two days in vitro, the IC50 was calculated, and for free DTX, it exhibited values of 40 nM on HeLa, 43 nM on A549, and 94 nM on McF-7 cells. When incorporated in dPGS8.6-SS-POxPPh7.9-Py micelles, the DTX showed slightly increased IC50 values of 69 nM on HeLa, 68 nM on A549, and 141 nM on McF-7 (Fig. 10(F)). The phenomenon of minimal increased IC50 values of a polymer-supported treatment of taxane-based drugs is already known in the literature.51,52 It might be attributed to the extremely high stability of the system showing slow dissociation but still proving the drug release (see Fig. 7). The π-electron stabilized micelles can potentially overcome common issues such as toxin overload due to their slow degradation profile while remaining highly potent in vitro. Further, besides the already low IC50 of free DTX, its extreme hydrophobicity and off-side toxicity challenge its clinical administration; thus, drug delivery systems such as dPGS8.6-SS-POxPPh7.9-Py are essential for the successful medication of cancer patients with this class of drugs.
3. Conclusions
This work demonstrates the detailed characterization and substantial performance of π-electron stabilized dendritic polyglycerolsulfate amphiphilic block copolymers. The micelles show (i) very high stability in terms of extraordinarily low CMCs in the low nanomolar regime; (ii) no in vitro toxicity of the polymeric material; (iii) successful cell insertion shown by FITC-labelled micelles; and (iv) high efficacy in inhibiting cancer cell growth in vitro on several tumor-derived cell lines. Further, several spectroscopy techniques, such as UV-VIS, fluorescence, and cross-polarization solid-state 13C NMR, proved the presence of π–π interactions in the micellar core between the polymer chains and drug molecules, majorly contributing to the high stability of the system. Thus, drug delivery micelles based on dendritic polyglycerolsulfate-cystamine-block-poly(4-benzoyl-1,4-oxazepan-7-one)-pyrene (dPGS-SS-POxPPh-Py) may be new alternatives for chemotherapies and will be considered for in vivo investigations.
Author contributions
The manuscript was written entirely by D. B. All authors have approved the final version of the manuscript. D. B. synthesized and characterized the polymeric materials with assistance from J. H. P. The cryo-EM measurements were conducted by Dr M. D. and Dr K. L. All in vitro cell tests were done by E. Q. and Dr K. A. Dr M. S., Dr M. B., and Dr R. H. conceived and directed the project.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
We thank Cathleen Hudziak for conducting the GPC measurements. Marleen Selent gratefully contributed to the HPLC analysis. Eleonore Christmann-Oesterreich is acknowledged for performing the elemental analysis. The authors appreciate the access to the Core Facility BioSupraMol research facilities. Dr Andreas Schäfer significantly contributed to the solid-state NMR studies. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 434130070 within the GRK 2662.
References
- R. L. Siegel, K. D. Miller, H. E. Fuchs and A. Jemal, Ca-Cancer J. Clin., 2022, 72, 7–33 CrossRef PubMed.
- U. Anand, A. Dey, A. K. S. Chandel, R. Sanyal, A. Mishra, D. K. Pandey, V. De Falco, A. Upadhyay, R. Kandimalla, A. Chaudhary, J. K. Dhanjal, S. Dewanjee, J. Vallamkondu and J. M. Pérez de la Lastra, Genes Dis., 2022 DOI:10.1016/j.gendis.2022.02.007.
- L. Zhong, Y. Li, L. Xiong, W. Wang, M. Wu, T. Yuan, W. Yang, C. Tian, Z. Miao, T. Wang and S. Yang, Signal Transduction Targeted Ther., 2021, 6, 201 CrossRef PubMed.
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844–6892 CrossRef CAS PubMed.
- A. Varela-Moreira, Y. Shi, M. H. A. M. Fens, T. Lammers, W. E. Hennink and R. M. Schiffelers, Mater. Chem. Front., 2017, 1, 1485–1501 RSC.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- S. Zanganeh, R. Spitler, M. Erfanzadeh, A. M. Alkilany and M. Mahmoudi, Int. J. Biochem. Cell Biol., 2016, 75, 143–147 CrossRef CAS PubMed.
- M. Yang, E. Wu, W. Tang, J. Qian and C. Zhan, J. Mater. Chem. B, 2021, 9, 6713–6727 RSC.
- X. Sun, G. Wang, H. Zhang, S. Hu, X. Liu, J. Tang and Y. Shen, ACS Nano, 2018, 12, 6179–6192 CrossRef CAS PubMed.
- J. Lu, S. C. Owen and M. S. Shoichet, Macromolecules, 2011, 44, 6002–6008 CrossRef CAS PubMed.
- V. Mirshafiee, M. Mahmoudi, K. Lou, J. Cheng and M. L. Kraft, Chem. Commun., 2013, 49, 2557–2559 RSC.
- K. A. Whitehead, J. Matthews, P. H. Chang, F. Niroui, J. R. Dorkin, M. Severgnini and D. G. Anderson, ACS Nano, 2012, 6, 6922–6929 CrossRef CAS PubMed.
- M. Mahmoudi, Nat. Commun., 2022, 13, 49 CrossRef CAS PubMed.
- Y. Zhong, M. Dimde, D. Stöbener, F. Meng, C. Deng, Z. Zhong and R. Haag, ACS Appl. Mater. Interfaces, 2016, 8, 27530–27538 CrossRef CAS PubMed.
- D. Braatz, M. Dimde, G. Ma, Y. Zhong, M. Tully, C. Grötzinger, Y. Zhang, A. Mavroskoufis, M. Schirner, Z. Zhong, M. Ballauff and R. Haag, Biomacromolecules, 2021, 22, 2625–2640 CrossRef CAS PubMed.
- D. Braatz, M. Cherri, M. Tully, M. Dimde, G. Ma, E. Mohammadifar, F. Reisbeck, V. Ahmadi, M. Schirner and R. Haag, Angew. Chem., Int. Ed., 2022, 134, e202203942 CrossRef.
- D. Sun, S. Zhou and W. Gao, ACS Nano, 2020, 14, 12281–12290 CrossRef CAS PubMed.
- J. M. Metselaar and T. Lammers, Drug Delivery Transl. Res., 2020, 10, 721–725 CrossRef PubMed.
- H. He, L. Liu, E. E. Morin, M. Liu and A. Schwendeman, Acc. Chem. Res., 2019, 52, 2445–2461 CrossRef CAS PubMed.
- W.-R. Zhuang, Y. Wang, P.-F. Cui, L. Xing, J. Lee, D. Kim, H.-L. Jiang and Y.-K. Oh, J. Controlled Release, 2019, 294, 311–326 CrossRef CAS PubMed.
- Y. Masayuki, M. Mizue, Y. Noriko, O. Teruo, S. Yasuhisa, K. Kazunori and I. Shohei, J. Controlled Release, 1990, 11, 269–278 CrossRef.
- T. Hamaguchi, Y. Matsumura, M. Suzuki, K. Shimizu, R. Goda, I. Nakamura, I. Nakatomi, M. Yokoyama, K. Kataoka and T. Kakizoe, Br. J. Cancer, 2005, 92, 1240–1246 CrossRef CAS PubMed.
- Y. Shi, M. J. van Steenbergen, E. A. Teunissen, L. S. Novo, S. Gradmann, M. Baldus, C. F. van Nostrum and W. E. Hennink, Biomacromolecules, 2013, 14, 1826–1837 CrossRef CAS PubMed.
- Y. Shi, R. van der Meel, B. Theek, E. Oude Blenke, E. H. E. Pieters, M. H. A. M. Fens, J. Ehling, R. M. Schiffelers, G. Storm, C. F. van Nostrum, T. Lammers and W. E. Hennink, ACS Nano, 2015, 9, 3740–3752 CrossRef CAS PubMed.
- C. Liang, X. Bai, C. Qi, Q. Sun, X. Han, T. Lan, H. Zhang, X. Zheng, R. Liang, J. Jiao, Z. Zheng, J. Fang, P. Lei, Y. Wang, D. Möckel, J. M. Metselaar, G. Storm, W. E. Hennink, F. Kiessling, H. Wei, T. Lammers, Y. Shi and B. Wei, Biomaterials, 2021, 266, 120432 CrossRef CAS PubMed.
- Z. Tu, Y. Zhong, H. Hu, D. Shao, R. Haag, M. Schirner, J. Lee, B. Sullenger and K. W. Leong, Nat. Rev. Mater., 2022, 7, 557–574 CrossRef CAS PubMed.
- J. Dernedde, A. Rausch, M. Weinhart, S. Enders, R. Tauber, K. Licha, M. Schirner, U. Zügel, A. von Bonin and R. Haag, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19679–19684 CrossRef CAS PubMed.
- M. Weinhart, D. Gröger, S. Enders, J. Dernedde and R. Haag, Biomacromolecules, 2011, 12, 2502–2511 CrossRef CAS PubMed.
- X. Wang and N. Hadjichristidis, ACS Macro Lett., 2020, 9, 464–470 CrossRef CAS PubMed.
- R. Bej, P. Dey and S. Ghosh, Soft Matter, 2020, 16, 11–26 RSC.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS PubMed.
- B. M. Davis, J. L. Richens and P. O’Shea, Biophys. J., 2011, 101, 245–254 CrossRef CAS PubMed.
- S. C. Owen, D. P. Y. Chan and M. S. Shoichet, Nano Today, 2012, 7, 53–65 CrossRef CAS.
- S. C. Kim, D. W. Kim, Y. H. Shim, J. S. Bang, H. S. Oh, S. W. Kim and M. H. Seo, J. Controlled Release, 2001, 72, 191–202 CrossRef CAS PubMed.
- Y. Wen and J. Li, Nat. Biomed. Eng., 2018, 2, 273–274 CrossRef PubMed.
- Y. Lu, Z. Yue, J. Xie, W. Wang, H. Zhu, E. Zhang and Z. Cao, Nat. Biomed. Eng., 2018, 2, 318–325 CrossRef CAS PubMed.
- C. Oerlemans, W. Bult, M. Bos, G. Storm, J. F. W. Nijsen and W. E. Hennink, Pharm. Res., 2010, 27, 2569–2589 CrossRef CAS PubMed.
- Y. Qiao, C. Zhan, C. Wang, X. Shi, J. Yang, X. He, E. Ji, Z. Yu, C. Yan and H. Wu, J. Mater. Chem. B, 2020, 8, 8527–8535 RSC.
- A. T. Haedler, H. Misslitz, C. Buehlmeyer, R. Q. Albuquerque, A. Köhler and H.-W. Schmidt, ChemPhysChem, 2013, 14, 1818–1829 CrossRef CAS PubMed.
- J. Duhamel, Langmuir, 2012, 28, 6527–6538 CrossRef CAS PubMed.
- A.-C. Pöppler, M. Lübtow, J. Schlauersbach, J. Wiest, L. Meinel and R. Luxenhofer, Angew. Chem., Int. Ed., 2019, 58, 18540–18546 CrossRef PubMed.
- M. Grüne, R. Luxenhofer, D. Iuga, S. P. Brown and A.-C. Pöppler, J. Mater. Chem. B, 2020, 8, 6827–6836 RSC.
- M. Callari, P. L. De Souza, A. Rawal and M. H. Stenzel, Angew. Chem., Int. Ed., 2017, 56, 8441–8445 CrossRef CAS PubMed.
- M. Farshbaf, H. Valizadeh, Y. Panahi, Y. Fatahi, M. Chen, A. Zarebkohan and H. Gao, Int. J. Pharm., 2022, 614, 121458 CrossRef CAS PubMed.
- T. D. Langridge and R. A. Gemeinhart, J. Controlled Release, 2020, 319, 157–167 CrossRef CAS PubMed.
- R. Savić, T. Azzam, A. Eisenberg and D. Maysinger, Langmuir, 2006, 22, 3570–3578 CrossRef PubMed.
- Q. Ran, X. Xu, P. Dey, S. Yu, Y. Lu, J. Dzubiella, R. Haag and M. Ballauff, J. Chem. Phys., 2018, 149, 163324 CrossRef PubMed.
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071–5109 CrossRef CAS PubMed.
- C. Ortiz, M. L. Ferreira, O. Barbosa, J. C. S. dos Santos, R. C. Rodrigues, Á. Berenguer-Murcia, L. E. Briand and R. Fernandez-Lafuente, Catal. Sci. Technol., 2019, 9, 2380–2420 RSC.
- R. S. Herbst and F. R. Khuri, Cancer Treat. Rev., 2003, 29, 407–415 CrossRef CAS PubMed.
- A. W. G. Alani, Y. Bae, D. A. Rao and G. S. Kwon, Biomaterials, 2010, 31, 1765–1772 CrossRef CAS PubMed.
- A. W. Du, H. Lu and M. H. Stenzel, Biomacromolecules, 2015, 16, 1470–1479 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Rainer
Haag
*a and
Rainer
Haag