
A cationic lipid with advanced membrane fusion performance for pDNA and mRNA delivery†
Yu
Wei‡
a,
Ting
He‡
a,
Qunjie
Bi
a,
Huan
Yang
a,
Xueyi
Hu
a,
Rongrong
Jin
 a,
Hong
Liang
*bc,
Yongqun
Zhu
d,
Rongsheng
Tong
*bc and
Yu
Nie
*a
a,
Hong
Liang
*bc,
Yongqun
Zhu
d,
Rongsheng
Tong
*bc and
Yu
Nie
*a
aNational Engineering Research Center for Biomaterials/College of Biomedical Engineering, Sichuan University, Chengdu 610064, China. E-mail: nie_yu@scu.edu.cn
bDepartment of Pharmacy, Sichuan Academy of Medical Sciences & Sichuan Provincial People's Hospital, School of Medicine, University of Electronic Science and Technology of China, Chengdu 610072, China. E-mail: lianghong3689@163.com; 2207132448@qq.com; Fax: +86-28-8541-0246; Tel: +86-28-8541-2848
cPersonalized Drug Therapy Key Laboratory of Sichuan Province, School of Medicine, University of Electronic Science and Technology of China, Chengdu 610072, China
dLife Sciences Institute and Innovation Center for Cell Signaling Network, Zhejiang University, Hangzhou 310058, China
First published on 7th February 2023
Abstract
The success of mRNA vaccines for COVID-19 prevention raised global awareness of the importance of nucleic acid drugs. The approved systems for nucleic acid delivery were mainly formulations of different lipids, yielding lipid nanoparticles (LNPs) with complex internal structures. Due to the multiple components, the relationship between the structure of each component and the overall biological activity of LNPs is hard to study. However, ionizable lipids have been extensively explored. In contrast to former studies on the optimization of hydrophilic parts in single-component self-assemblies, we report in this study on structural alterations of the hydrophobic segment. We synthesize a library of amphiphilic cationic lipids by varying the lengths (C = 8–18), numbers (N = 2, 4), and unsaturation degrees (Ω = 0, 1) of hydrophobic tails. Notably, all self-assemblies with nucleic acid have significant differences in particle size, stability in serum, membrane fusion, and fluidity. Moreover, the novel mRNA/pDNA formulations are characterized by overall low cytotoxicity, efficient compaction, protection, and release of nucleic acids. We find that the length of hydrophobic tails dominates the formation and stability of the assembly. And at a certain length, the unsaturated hydrophobic tails enhance the membrane fusion and fluidity of assemblies and thus significantly affect the transgene expression, followed by the number of hydrophobic tails.
Introduction
The great success of message RNA (mRNA) vaccines against the COVID-19 pandemic has highlighted the potential of nucleic acids (DNA or RNA) as therapeutics or vaccines in a wide range of diseases.1 Gene therapies largely depend on whether the delivery carriers can efficiently deliver nucleic acid to the host cells in vivo. Considering the potential safety risks, production cost, and large-scale production, non-viral carriers have become the hot carriers in the marketization of gene carriers.To date, lipid-based vehicles remain the first option to deliver genetic materials of interest. The mature technology and high biosafety promote its rapid product transformation.2,3 For example, Onpattro® where small interfering (siRNA) is encapsulated in lipid nanoparticles (LNPs), was initially authorized by the U.S. Food and Drug Administration for polyneuropathy in 2018.4 Recently, mRNA-LNPs based vaccines, Spikevax® (Moderna) and Comirnaty® (Pfizer/BioNTech) were approved for emergency prevention of COVID-19.5 Typically, (ionizable) cationic lipids are the core components of LNPs, accounting for around 50% of the total lipids (molar ratio) and significantly affecting carriers’ delivery performance.6 However, these (ionizable) cationic lipids could form stable lipid carriers only in combination with helper lipids (phospholipids, PEGylated lipids) and/or cholesterol to achieve enough stability and membrane fusion properties.7 With the multiple components in lipid carriers, it is challenging to evaluate the contribution of each lipid to the overall biological performance. In addition, it is labor-intensive, time-consuming, and less cost-effective to screen the optimal formulations.
The (ionizable) cationic lipids are generally amphiphilic, with an optimal balance between hydrophilic and hydrophobic segments. Notably, the hydrophilic head is important in gene transfection efficiency and cytotoxicity. Inspired by the cell-penetrating peptides (CPP) in viruses, several arginine-rich residues have been introduced into the hydrophilic segment of lipidic carriers. For instance, linear,8 dendritic,9 and clustered10 arginine all showed an enhanced transfection efficiency compared with controls. The guanidine of arginine could form bidentate hydrogen bonds with the negative phosphates, sulfates, and carboxylates on the cell surface and be driven into cells.11 Our groups also did some explorations in screening the types and structures of amino acids and found that the structure, stability, size, and construction of assemblies also significantly affected the transfection efficiency of lipoplexes, especially in the presence of serum. As well, the hydrophobic tail of lipids mainly affects the assembly, which has been widely studied with a focus on the length (C), unsaturation (Ω), and the number of alkyl chains (N). However, their structure–activity relationship of these tails is still unclear or even in debate.
For example, Nakamura et al. found that cationic lipids containing longer unsaturated tails (C18:Ω1) induced a significantly improved silencing efficiency than their counterparts with shorter saturated ones (C14:Ω0 and C16:Ω0).12 In contrast, J. Shi et al. reported that short saturated tails (C12:Ω0 and C14:Ω0) resulted in 2-fold higher pDNA transgene expression than longer unsaturated tails (C18:Ω1).13 In addition, the architecture of the hydrophobic tail was found to be important. Some researchers reported that double-tailed cationic lipids showed about 10–20 fold higher gene transfection than single-tailed lipids in DNA delivery.14 Interestingly, three-tailed lipids induced the most robust relative hit rate (%) than the double-tailed and four-tailed lipids in Cas9 mRNA delivery, because the large tails encouraged membrane fusion and phase transformation from lamellar phase to hexagonal (HII) phase.15 Although tremendous efforts have been placed to screen a relatively prominent material, a systemic and comprehensive comparison is also needed.
In this work, we synthesized a library of cationic lipids with the same cationic hydrophilic part and linker but varying hydrophobic part (alkyl chain length, number, and unsaturation degree). Such single-component cationic lipids were self-assembled into nanoparticles that were examined to deliver mRNA and plasmid DNA by studying their complexation ability, stability, conformation, cytotoxicity, as well as transgene efficiency both in vivo and in vitro.
Materials and methods
Experimental materials
Octanoic acid, lauric acid, palmitic acid, stearic acid, oleic acid, Boc-Arg(Pbf)-OH, triethylamine, di-tert-butyl decarbonate, and 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU) was obtained from LeYan (Shanghai, China). Methyl L-lysinate dihydrochloride and 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were obtained from Adamas (Shanghai, China). Dimethyl sulfoxide (DMSO), dimethylformamide (DMF), dichloromethane (DCM), and methanol (MeOH) were bought from Greagent (Shanghai, China). Trifluoroacetic acid (TFA) was obtained from TCI (Shanghai, China). N-Ethyldiisopropylamine (DIEA) was bought from Astatech (Chengdu, China). 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) was bought from Aladdin (Shanghai, China). 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI), 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO), and cystamine dihydrochloride were obtained from Macklin (Shanghai, China). Branched polyethyleneimine (PEI, average Mw of 25 kDa) and 1,6-diphenylhexa-1,3,5-triene (DPH) were obtained from Sigma-Aldrich (Shanghai, China). Agarose was purchased from Biowest (Burgos, Spain). Dithiothreitol (DTT) was purchased from Beyotime (Shanghai, China). 50 × tris-acetate-EDTA buffer was obtained from Solarbio (Beijing, China). D-Luciferin potassium salt and PicoGreen dsDNA Quantitation Reagent were purchased from Yeasen (Shanghai, China). Quant-iT RiboGreen RNA Assay was obtained from Thermo (Waltham, America). Fetal bovine serum (FBS), minimum essential medium (MEM), and phosphate-buffered saline (PBS) were bought from Hyclone (Logan, America). Human cell lines, including hepatoma (HepG2), embryonic kidney (HEK293), and cervical carcinoma (HeLa), were purchased from the Chinese Academy of Science Cell Bank (Shanghai, China). Cell counter kit-8 (CCK-8) was obtained from Beyotime (Shanghai, China). Lipofectamine 2000 was obtained from Invitrogen (Carlsbad, America). Before use, all buffers were filtered (0.22 μm) in deionized water during preparation. The Endo-Free Plasmid Maxi Kit from Qiagen (Hilden, Germany) was used to purify the plasmid encoding enhanced green fluorescent protein (pEGFP). The luciferase gene in the pGL3 promoter was cloned into pcDNA3.1 to obtain plasmid pCMV-Luc. mRNA encoding enhanced green fluorescent protein (mEGFP) was obtained from the Yongqun Zhu group (Zhejiang University, China).Chemical synthesis of a series of cationic lipids
Compound 1 (5.0 g, 4.2 mmol) was treated with a methanolic sodium hydroxide solution (0.8 g, 20.0 mmol) and stirred for 12 h. The organic phase was evaporated at low pressure, and dissolved in deionized water (10 mL), and the pH value was adjusted to 2–3 by diluted HCl (1.0 mol L−1). Compound 2 was extracted by DCM and dried using anhydrous MgSO4 (4.0 g, 79% yield).
Compound 4 (3.9 g, 9.5 mmol) was dissolved in MeOH (15 mL), followed by adding sodium hydroxide (1.0 mol L−1, 10 mL) aqueous solution overnight at 30 °C. The residues were dissolved in water (5 mL) after the reaction solvent was evaporated, with the pH value adjusted to 2–3 by diluted HCl (1.0 mol L−1). Compound 5 was extracted with DCM and dried with MgSO4 (2.7 g, 72% yield).
Oleic acid was used to synthesize di-oleic acid with the same steps as compound 4 and compound 5.
Compound 5 (2.7 g, 7.0 mmol), compound 3 (1.8 g, 7.0 mmol), DIEA (1.8 g, 14.0 mmol), and HBTU (5.2 g, 14.0 mmol) were dissolved in anhydrous DMF (50 mL) and reacted at 45 °C under nitrogen protection for 10 h. Under reduced pressure, the solvent was removed, and the solid was dissolved in DCM (200 mL). The solvent was dried with MgSO4 after being washed with NaCl saturated solution. The crude products were separated using a silica–gel chromatographic column (DCM/MeOH = 15/1, v/v) to get a white solid compound 6 (3.2 g, 75% yield).
Compound 6 (3.2 g, 5.2 mmol) was dissolved in DCM (2 mL) and reacted continuously for 4 h at 30 °C with TFA (2 mL) added. After the solution was evaporated at low pressure, the obtained products were precipitated with ethyl ether and centrifuged at 8000 rpm for 3 min. After residual ethyl ether was removed in a vacuum, a yellow powder compound 7 (1.7 g, 60% yield) was obtained.
200.0 mg white solid was dissolved in anhydrous DCM (1.5 mL), then TFA (1.5 mL) was added dropwise and reacted for 12 h at 30 °C. The residues were precipitated with ice ether and collected by centrifugation after removing the solution in a vacuum. A white powder RLS1 (85.0 mg, 74% yield) was obtained.
Lauric, stearic, palmitic, oleic, and di-oleic acid were substituted for octanoic acid to synthesize the other cationic lipids (RLS2–6).
RLS1 1H NMR (400 MHz, DMSO-d6) δ (ppm) 4.09 (m, 2H), 3.38 (s, 2H), 2.97 (d, 4H), 2.74 (d, 4H), 2.12 (h, 2H), 2.02 (t, 3H), 1.79 (d, 5H), 1.41 (m, 13H), 1.23 (s, 20H), 0.85 (t, 6H). MS (MALDI-TOF), m/z: [M + H+] = 973.4525.
RLS2 1H NMR (400 MHz, DMSO-d6) δ (ppm) 4.12 (s, 2H), 3.70 (s, 2H), 3.38 (s, 2H), 3.09 (d, 5H), 2.99 (s, 2H), 2.65 (m, 3H), 2.10 (t, 2H), 2.02 (t, 2H), 1.67 (s, 7H), 1.46 (s, 6H), 1.23 (s, 34H), 0.85 (t, 6H). MS (MALDI-TOF), m/z: [M + H+] = 1085.9045.
RLS3 1H NMR (400 MHz, DMSO-d6) δ (ppm) 4.62 (m, 2H), 3.83 (q, 3H), 3.72 (t, 3H), 3.40 (d, 2H), 2.96 (m, 2H), 2.84 (d, 4H), 2.06 (m, 2H), 2.02 (t, 2H), 1.72–1.67 (m, 8H), 1.45 (m, 12H), 1.23 (s, 52H), 0.85 (t, 6H). MS (MALDI-TOF), m/z: [M + H+] = 1197.7869.
RLS4 1H NMR (400 MHz, DMSO-d6) δ (ppm) 4.70 (d, 2H), 3.37 (m, 6H), 3.12 (s, 6H), 3.03 (s, 3H), 2.84 (d, 5H), 2.05 (d, 4H), 1.60 (s, 8H), 1.47 (s, 12H), 1.23 (s, 60H), 0.82 (m, 6H). MS (MALDI-TOF), m/z: [M + TFA]+ = 1366.8543.
RLS5 1H NMR (400 MHz, DMSO-d6) δ (ppm) 5.35 (m, 4H), 4.65 (m, 2H), 3.73 (s, 4H), 3.63 (m, 2H), 3.03 (s, 2H), 2.97 (m, 6H), 2.83 (d, 4H), 2.01 (m, 12H), 1.71 (s, 8H), 1.47 (s, 12H), 1.23 (s, 44H), 0.85 (t, 6H). MS (MALDI-TOF), m/z: [M + H+] = 1248.3542.
RLS6 1H NMR (400 MHz, DMSO-d6) δ (ppm) 5.31 (t, 6H), 4.12 (s, 4H), 3.10 (s, 5H), 2.98 (s, 5H), 2.73 (s, 4H), 2.23 (m, 19H), 1.56 (m, 8H), 1.47 (d, 18H), 1.24 (s, 88H), 0.85 (t, 12H). MS (MALDI-TOF), m/z: [M + H+] = 2035.0947.
Preparation of gene complexes and related characterization
Preparation and characterization of gene complexes
An equal volume of pDNA or mRNA (100 μg mL−1) was added to the formed assemblies with various concentrations to obtain the desired N/P ratios, followed by a 20 minutes incubation period at room temperature before use. The physicochemical properties of lipid/pDNA lipoplexes were characterized by DLS.Gene binding affinities
The gel retardation electrophoresis was used to assess the gene binding ability of lipid assemblies. Samples comprising 200 ng pDNA were packed into 1% agarose gel, and electrophoresis was performed at an electric field of 85 volts for 40 minutes in 1 × tris–acetate–EDTA running buffer (20 mL 50 × tris–acetate–EDTA buffer with deionized water up to 1000 mL). The pDNA bands were observed with gel red staining and the Molecular Imager ChemiDoc XRS+ (Bio-Rad, America).Lipoplexes containing pDNA and cationic lipid assemblies (1.0 mg mL−1) (N/P 0-40) were incubated with 10 mmol L−1 DTT at 37 °C for 2 h. The release of cargo was then evaluated using gel electrophoresis under reducing environments.
Encapsulation efficiency
The mRNA encapsulation efficiency was calculated using the Quant-iT RiboGreen RNA Assay.17 Firstly, the standard curve was drawn according to the reagent manual, using fluorescence intensity measurement by the BioTek Synergy H1 microplate reader (λex = 480 nm, λem = 520 nm). The free mRNA was obtained by ultrafiltration centrifugation (5000 rpm) and followed with RiboGreen staining. Finally, the encapsulation efficiency was calculated as follows (“W” represented weight):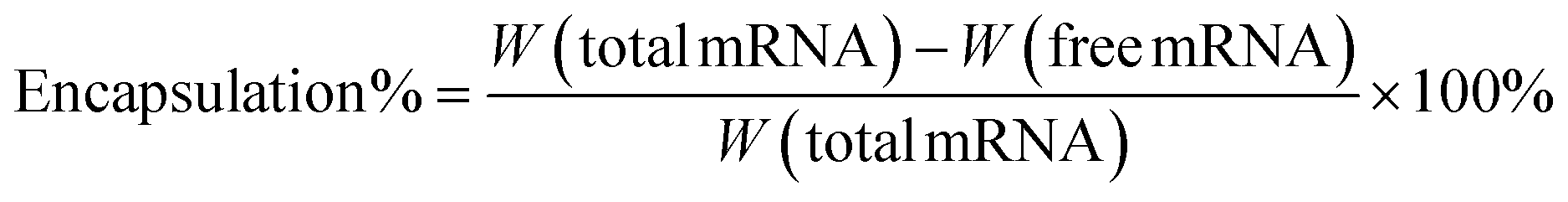
The encapsulation efficiency of pDNA was determined by PicoGreen reagent, which was similar to the mRNA encapsulation test.
Stability in serum
pDNA lipoplexes (N/P 20) were diluted to 1.0 mL with deionized water, with an ultimate pDNA concentration of 3.0 μg mL−1. After incubation with 10% FBS for 0, 15, 30, 45, and 60 min, DLS was used to determine particle size. To examine the dissociation of pDNA, lipoplexes were incubated with 10% FBS, followed by gel electrophoresis.Cell culture
HepG2, HeLa, and HEK293 cells were fostered in MEM medium included 10% FBS, 1% (v/v) penicillin/streptomycin (penicillin: 100 U mL−1, streptomycin: 100 μg mL−1), and 292 mg L−1L-glutamine at 37 °C, 5% CO2. Cells were detached from the bottom of the flask by incubating with trypsin-EDTA solution (0.25%/0.02%) when cell confluency was around 80–90%. For all in vitro studies, cell sowing density was 1 × 104 cells per well in 96-well plates before 24 hours until otherwise stated.In vitro pDNA or mRNA lipoplexes transfection
Before transfection, fresh MEM medium with or without 10% FBS was transferred to each well to replace the original cell culture medium. Lipoplexes (10 μL per well, N/P 5–40) were applied to cells in each well (200 ng pEGFP per well). As controls, PEI/pDNA (N/P 10) and lipofectamine 2000/pDNA (0.5 μL/200 ng) were exposed to cells.After 4 hours of incubation at 37 °C, the old medium was discarded, washed twice with PBS, and supplemented with MEM medium including 10% FBS. Cells were incubated for additional 44 hours. Positive EGFP cells were observed with an inverted fluorescence microscope (Olympus, Japan).
The transfection of mRNA in HeLa cells (200 ng per well) was similar to that of pDNA, and RLS5 was used for intracellular transfection experiments with N/P ratios of 5–20. A fluorescence microscope and IVIS Lumina Series III imaging system were used to observe cells expressing EGFP.
In vitro cytotoxicity assay
In order to monitor the cytotoxicity of the cationic lipoplexes, the CCK-8 assay was used. Lipoplexes with various concentrations were exposed to cells for 24 hours at 37 °C, 5% CO2. Then the serum-free medium was used to replace the original culture medium, and 10% CCK-8 was added for another 1 hour incubation. The results were measured by a microplate reader (Bio-Rad 550, USA) at 450 nm, and the cell viability was calculated using the following equation:where As was the absorbance of the experimental cells exposed to lipoplexes, Ab was the absorbance of the non-treated cells, and Ac was the absorbance of the blank well without cells at 450 nm.
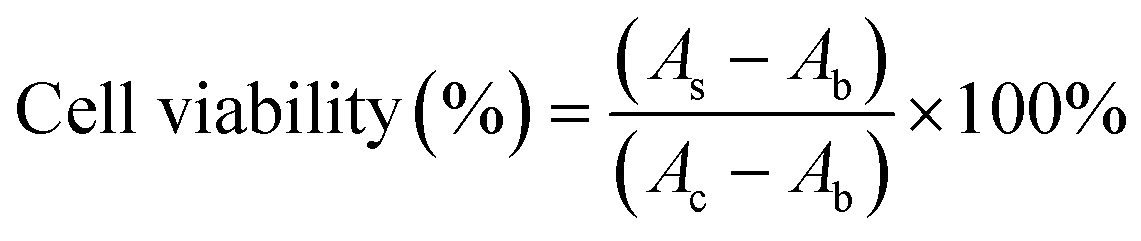
Membrane fusion experiments
8 nmol DiO dissolved in chloroform (100 μL) was mixed with 792 nmol cationic lipids in MeOH/chloroform (1/3, v/v, 1.0 mL), and dried by a rotary evaporator to formulate a thin film. PBS (pH 5.0) was added, vortexed for 5 min, and sonicated for 5 min to obtain DiO-labeled cationic lipid assemblies. The same method was used to obtain DiI-labeled DOPC vesicles.18 Both DiI-labeled DOPC vesicles and DiO-labeled cationic lipid assemblies were incubated in 96-well plates, and the fluorescence emission spectra (480–600 nm) of the mixture were evaluated at various times using BioTek Synergy H1 microplate reader at an excitation wavelength of 440 nm.100 μL DiI (5.0 μmol L−1) was added to 1 × 104 HeLa cells and co-incubated for 20 min before washing off the excess dye with PBS to obtain the DiI-labeled HeLa cells. DiO-labeled cationic lipid assemblies were added to DiI-labeled HeLa cells and incubated for 30 min, and the membrane fusion phenomenon was observed by a confocal laser scanning microscope (TCS SP 5, Leica Co., Germany). The cells were detached with trypsin and transferred into a 96-well plate. The signal was observed under the förster resonance energy transfer (FRET) channel using a BioTek Synergy H1 microplate reader (λex = 484 nm, λem = 565 nm).18
Membrane fluidity experiments
The membrane fluidity of six cationic assemblies was measured based on the fluorescence anisotropy of DPH (λex = 360 nm, λem = 430 nm), which was incorporated in the lipid assemblies. The fluorescence spectrophotometer (FP-6500, JASCO Co., Tokyo, Japan) with a polarizing plate was used to detect the fluorescence signal. 10 μL DPH (100 μmol L−1) in ethanol was added into lipid assembly suspension (1.0 mL, 0.3 mmol L−1). The following equation was used to calculate the polarity (P):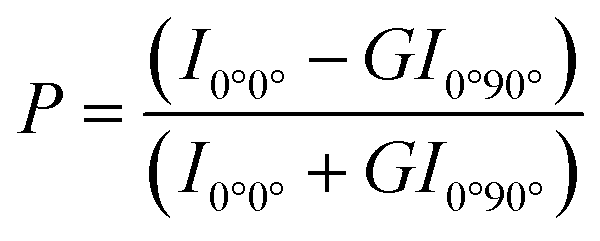
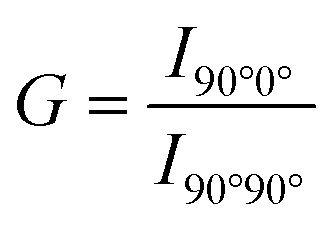
Membrane fluidity was expressed as 1/P because polarity was inversely proportional to fluidity.
Synchrotron-based small-angle X-ray scattering (SAXS)
SAXS experiments were executed at the Beijing Synchrotron Radiation Facility's (BSRF) beamline 1W2A (λ = 1.54 Å), with a Pilatus 1MF detector (169 mm × 179 mm). The detector was calibrated using a standard carbon fiber sample and a standard silver behenate sample. X-ray scattering intensity patterns were captured while the samples were exposed to the synchrotron beam for 20 s. The 2D scattering data were transformed into 1D profiles using the Fit2D program.19In vivo transfection
Results and discussion
Synthesis of six cationic lipids
According to our previous work, cationic lipids with dendritic arginine promoted cellular membrane penetration, and the linker of disulfide bonds induced a rapid release of pDNA under intracellular reduction conditions.9,20 Given that the hydrophobic portion of cationic lipids plays key roles in particle assembly, size, fluidity, as well as in vivo transfection,21,22 we synthesized six cationic lipids with different hydrophobic parts but sharing the same dendritic arginine-rich head and disulfide linker (Fig. 1). In detail, the hydrophobic parts differ in (1) lengths of the alkyl chain (numbers of carbon atoms, C = 8–18), (2) numbers of alkyl chains (dual or quadruple tails, N = 2, 4), and (3) degrees of unsaturation (Ω = 0, 1). MALDI-TOF-MS and 1H NMR were used to identify the molecular structure (Fig. S13–S18, ESI†).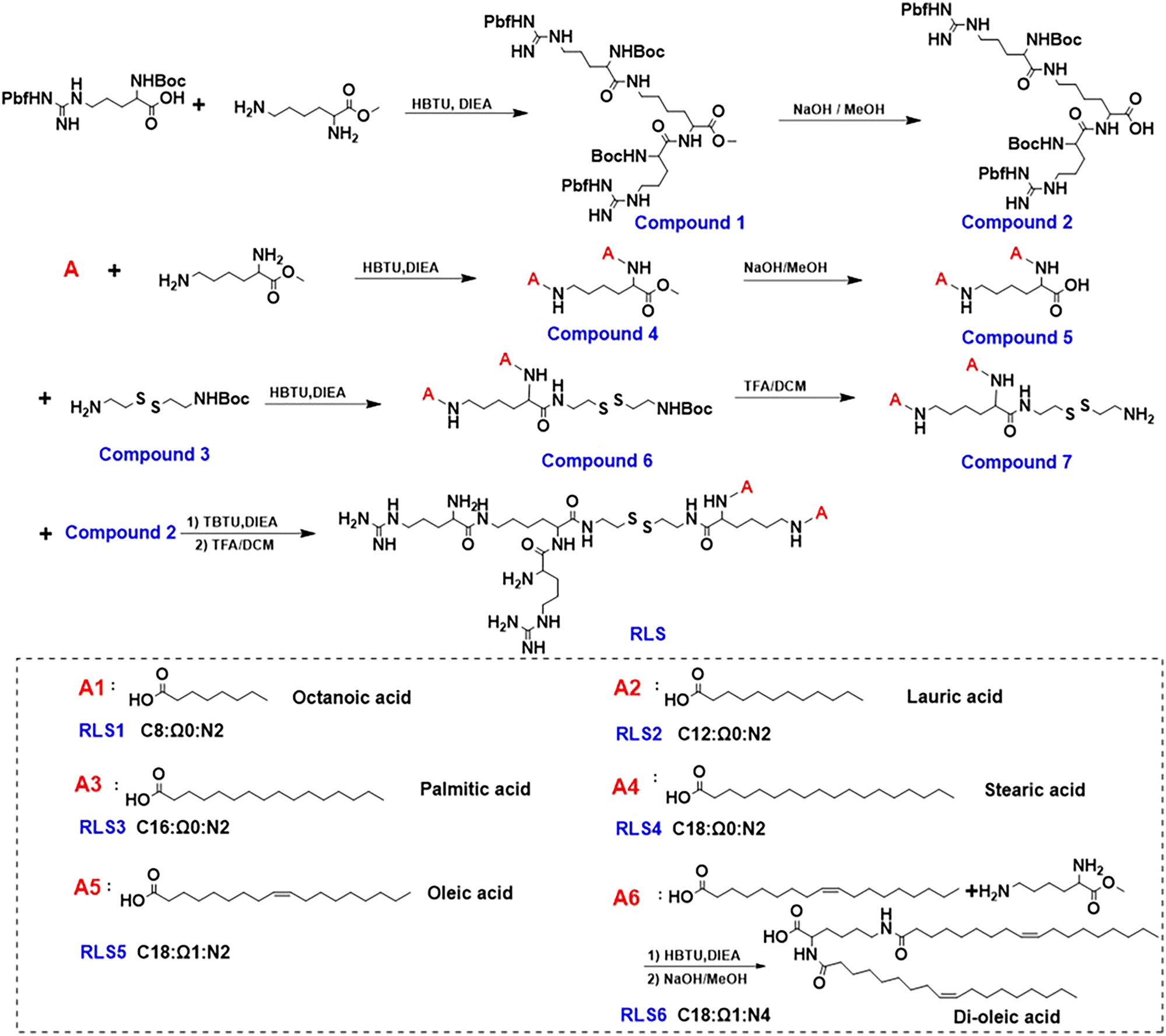 | ||
| Fig. 1 Synthesis of a series of cationic lipids with the same dendritic arginine peripheries and varied hydrophobic segments. C: length of alkyl chains; Ω: unsaturation of alkyl chains; N: number of alkyl chains. | ||
Characterization of lipid assemblies
First, lipid assemblies were characterized by DLS in terms of hydrodynamic diameters and ζ potential. As Fig. 2(A) displayed, all six cationic lipid assemblies were 140–220 nm in hydrodynamic diameter. For the lipid assemblies with the same saturated (Ω0) dual alkyl chains (RLS1–4), their Z-average particle sizes increased with prolonged alkyl chain length,13 ranging from 140 nm (C8), 164 nm (C12), 187 nm (C16), to 212 nm (C18) (Fig. 2(A) and Table S1, ESI†). In contrast, the particle size decreased with an increased unsaturation degree of cationic lipids comprising the same alkyl chain length and number (RLS4, C18:Ω0:N2 vs.RLS5, C18:Ω1:N2). This might be due to the folding effect of alkyl chains caused by the unsaturated bonds.23 In a direct comparison between RLS5 and RLS6 (C18:Ω1:N4) with the same alkyl chain length and unsaturation, lipids with 4 alkyl chains assembled into larger particles than their counterpart containing 2 alkyl chains. The TEM images (Fig. S5, ESI†) showed that the assemblies could form spherical shapes. The particle size measured by TEM was smaller than the results of DLS, which might be attributed to the fact that the particle size measured by DLS was obtained in the state of hydration.24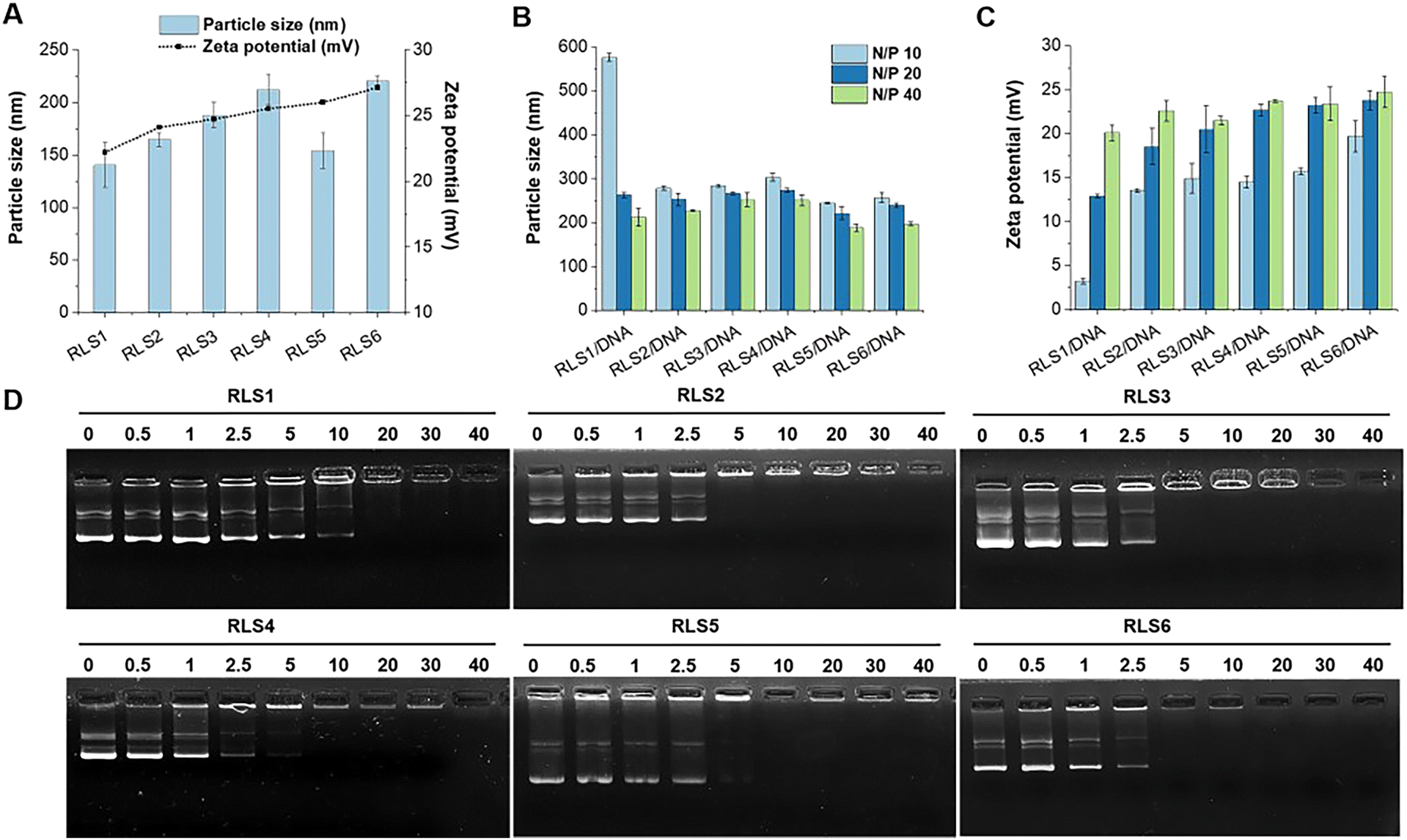 | ||
| Fig. 2 Particle size, zeta potential, and gene compression. (A) Particle size and zeta potentials of lipid assemblies. Particle size (B) and zeta potential (C) of lipid gene complexes at various N/P ratios. Data represent mean ± SD (n = 3). (D) Gel electrophoresis assay of cationic assemblies/pDNA complexes at different N/P ratios. | ||
The hydrophilic head mainly determined the ζ potential of cationic lipid assemblies. Thus, the ζ potentials of the six cationic lipid assemblies were all positive, between +21–27 mV (Fig. 2(A)).
Then, we investigated the particle size and ζ potential of pDNA complexes at various N/P ratios from 10 to 40 (Fig. 2(B) and (C)). The particle size of the lipoplexes decreased slightly as the N/P ratio increased. At a higher N/P ratio, pDNA could be more firmly compacted into particles with smaller sizes, which was consistent with the literature.25 In addition, almost all cationic lipid assemblies could compress pDNA below 300 nm at various N/P ratios, except for RLS1/pDNA (N/P 10, 576 nm). The results indicated that relatively high surface ζ potential required fewer assemblies for pDNA compression.
Next, the gel retardation method was used to test the compression capacity of the cationic lipid assemblies (Fig. 2(D)). We found that RLS1 with the shortest hydrophobic parts (C8) only fully compressed pDNA at a higher N/P ratio (20), compared to other lipids (RLS2–6) with longer hydrophobic parts that a full complexation was found at N/P 5. This might be due to the fact that the short hydrophobic length (C8) could not form a stable lipid assembly in the process of hydration.26 PicoGreen and RiboGreen reagents were used to assess the encapsulation efficiency of pDNA and mRNA, respectively. The results showed that the encapsulation efficiency of RLS1–6/pDNA (N/P 20) were 95.43%, 96.15%, 95.18%, 97.05%, 96.39%, and 98.44%, respectively. And the encapsulation efficiency of RLS5/mRNA (N/P 5) was 96.03% (Fig. S6 and Table S2, ESI†).
In order to achieve efficient gene delivery, sufficient intracellular release was of significance.27,28 We thus investigated pDNA release from lipoplexes in the presence of 10 mmol L−1 DTT by a retardation electrophoresis assay using agarose gel (Fig. S7, ESI†).29 The results demonstrated that pDNA was isolated from cationic lipids in an intracellular reduction environment, and free/naked pDNA bands could be observed after incubation for 2 hours. As previously reported, we believed that the disulfide bond breakage provided a prerequisite for releasing genes in carriers.20
Stability in serum
Under physiological conditions, the interaction between serum proteins and lipids affected the stability of lipoplexes. Negatively charged biomacromolecules (e.g., albumin) potentially bind onto the cationic lipoplexes, leading to aggregation, precipitation, or disintegration of the lipoplexes consequently resulting in transfection failure. Thus, serum resistance was essential for cationic lipoplexes.30 After incubation with serum, we evaluated the stability of pDNA lipoplexes (RLS1–6) in size and pDNA release (Fig. 3 and Fig. S8, ESI†). Obviously, lipoplexes based on RLS1, 2, and 3 aggregated after exposure to FBS, whereas other lipoplexes did not significantly differ along with incubation time. At the beginning (0 min), the particle size of all cationic lipoplexes was between 200–250 nm. With the increase of time, the size of RLS1–3 fluctuated obviously, and the fluctuation range decreased with the increase in the length of alkyl chains. After continuous incubation, the size of RLS1 reached almost 1 μm with poor dispersion (PDI = 0.72), and the maximum size of RLS2 and RLS3 was also up to about 500 nm (PDI = 0.45) and 400 nm (PDI = 0.48), respectively (Table S3, ESI†). In contrast, the particle size of RLS4–6 remained unchanged, representing good stability in serum. Gel electrophoresis assays (Fig. S8, ESI†) showed that partial pDNA was released from RLS1–3 lipoplexes, which might be the reason for the serious particle size variation. Gilmore et al. found that the serum stability of lipids was related to the force required to stretch the lipids laterally.31 We speculated that RLS4–6 showed good serum stability due to its long carbon chain and increased intermolecular van der Waals force, resulting in high stability of the whole lipid molecule and no deformation.32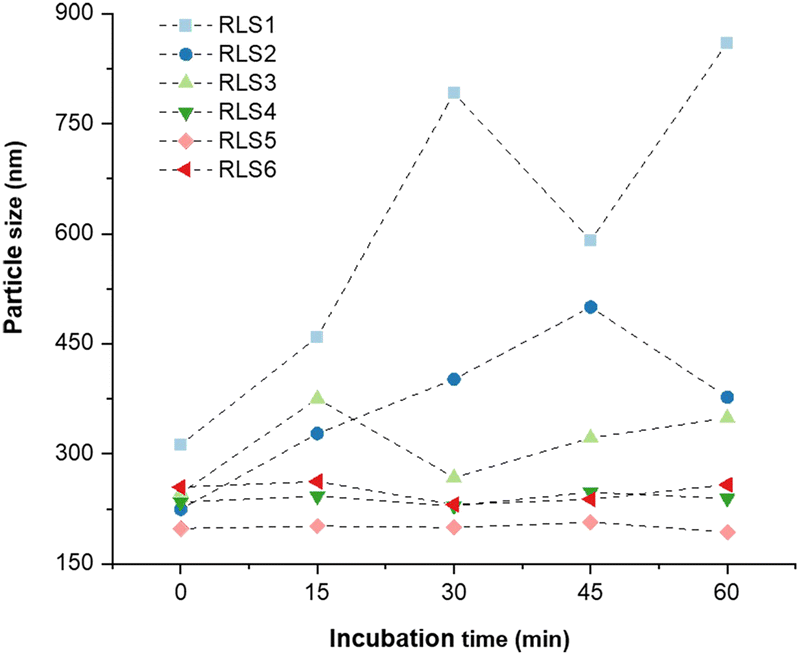 | ||
| Fig. 3 The particle size of lipid gene complexes after 0, 15, 30, 45, and 60 min of incubation with 10% FBS. | ||
Cytotoxicity of six cationic lipoplexes
The cytotoxicity of those cationic lipoplexes was assessed by CCK-8 assay. We found that over 90% of cells survived after exposure to all those six lipoplexes, regardless of cell lines (Fig. S9, ESI†). In general, six cationic lipids have satisfying cell survival viability, which might attribute to the introduction of disulfide bonds in the carrier. Our previous work has shown that the cytotoxicity of cationic lipid assemblies without disulfide bonds was much higher than that of cationic lipid assemblies containing disulfide bonds.20 The cytotoxicity of cationic lipids was mainly from the poor metabolism or decomposition of gene carriers by cellular enzymes. Gene carriers with disulfide bonds could be broken into fragments and easily excreted from the body without significant toxicity.33Transfection efficiency in vitro
Transfection efficiency of pEGFP in vitro was assessed on HEK293, HepG2, and HeLa cells at various N/P ratios with or without 10% FBS. Lipofectamine 2000 and PEI were used as controls (Fig. 4 and Fig. S10–S12, ESI†). The transfection efficiency of RLS1–3 was very low, and the gene expression of RLS4 with the longest saturated alkyl chains showed a slight increase (Fig. 4(A)). This trend was consistent in HEK293 and HepG2 cells (Fig. S10 and S11, ESI†). RLS5 and RLS6 showed far superior to RLS1–4 and the control groups in both HEK293 and HepG2 cells. The difference between RLS5 and RLS6 was displayed in HeLa cells (Fig. S12, ESI†). In the presence of FBS, RLS5 had significant transfection performance advantages over RLS6, whereas, without FBS, they achieved similar transfection efficiency. This indicated that an increased number of tails slightly decreased transfection efficiency. In short, the lipoplexes based on cationic lipids RLS5 and RLS6 with unsaturated alkyl chains had prominent transfection efficiency in different cells.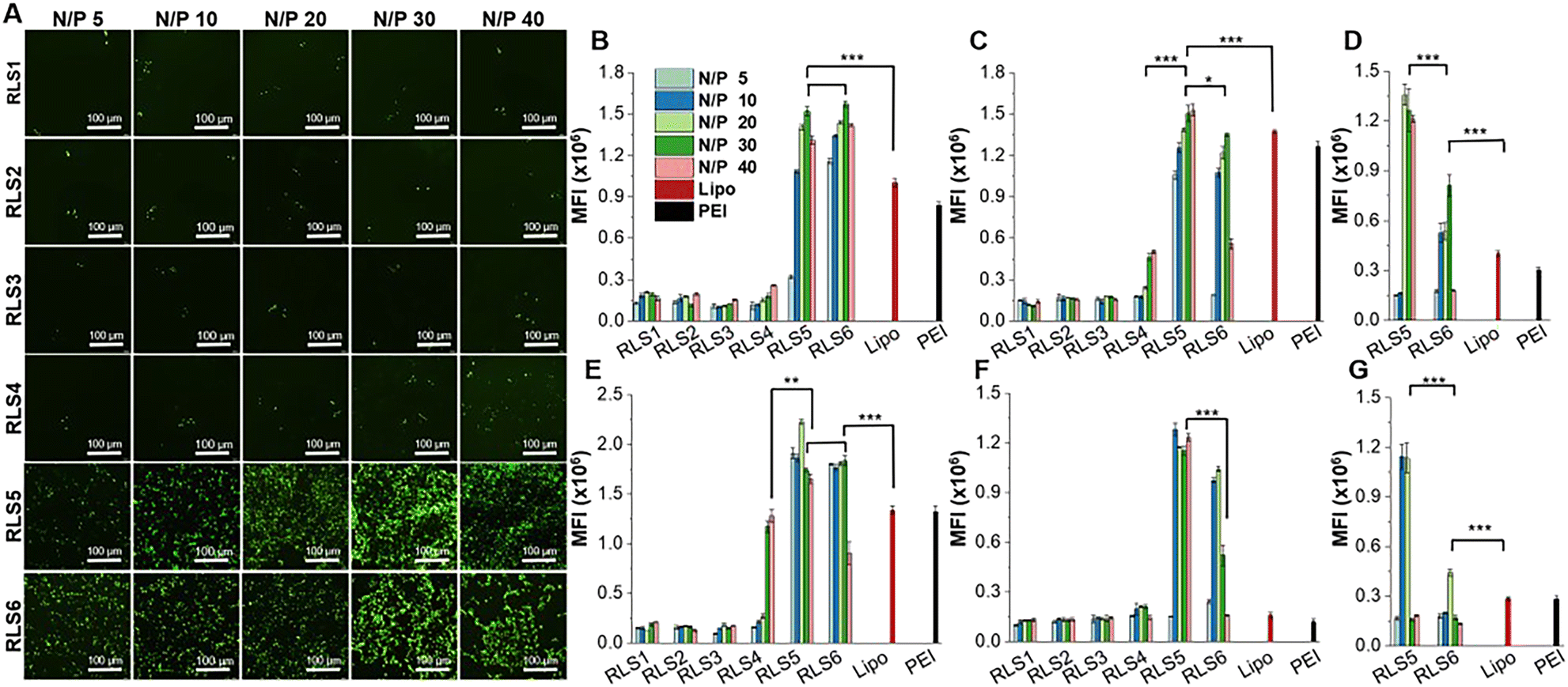 | ||
| Fig. 4 Transfection efficiency of pEGFP on various cells (HEK293, HepG2 and HeLa). (A) Fluorescence microscopy images of cells with pEGFP transfection on HEK293 cells with 10% FBS, scale bar was 100 μm. Quantitative transfection efficiency in different cells: HEK293 with 10% FBS (B) and without FBS (E), HepG2 with 10% FBS (C) and without FBS (F), HeLa with 10% FBS (D) and without FBS (G). (*p < 0.05, **p < 0.01, ***p < 0.001) | ||
Quantitative analysis by Imaging J software also confirmed the superiority of RLS5 and RLS6 in transfection, showing up to 7-fold and a 3.8-fold increase compared with that of RLS1–4 in the presence (Fig. 4(B) and (C)) and absence (Fig. 4(E) and (F)) of FBS. On HeLa cells, RLS5 resulted in 1.5-fold (with FBS) and 4-fold (without FBS) higher gene expression than that of RLS6 (Fig. 4(D) and (G)). Thus, we further evaluated the delivery efficiency of mRNA on HeLa cells. Excitingly, RLS5 showed superior green fluorescence protein expression at a very low N/P ratio (5) in the presence of FBS (Fig. 5(A) and (B)), which was 5-fold higher than lipofectamine 2000 (Fig. 5(C)).
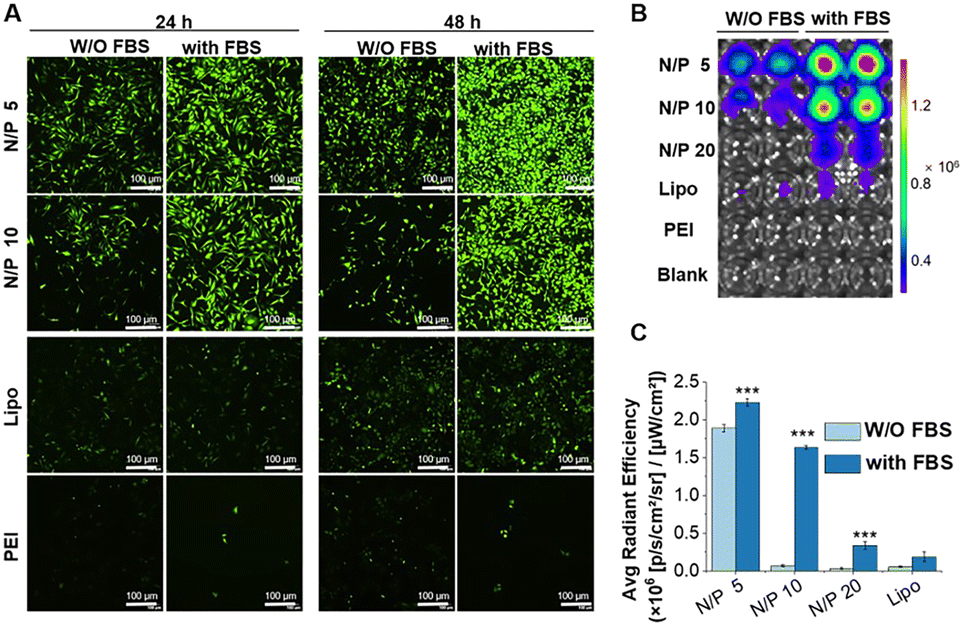 | ||
| Fig. 5 The transfection effect of mEGFP delivered by RLS5 in HeLa cells. The fluorescence images were observed by fluorescence microscope (A) and fluorescence imaging using IVIS Lumina Series III imaging system (B). (C) Quantitative transfection efficiency in HeLa cells. The scale bar was 100 μm. (***p < 0.001 vs. lipo with 10% FBS, W/O: without). | ||
While the transfection effect of four cationic lipids (RLS1–4) with the saturated hydrophobic part in different cells was not good, RLS4 (C18) with the longest carbon chain had relatively better transfection efficiency, which seemed to be related to its stability. In addition, we found that the unsaturation of the hydrophobic part of cationic lipids significantly affected the transfection efficiency, followed by the number of hydrophobic tails. The unsaturated structures disrupted the regular packing of the cellular membrane and facilitated the escape of nucleic acid within the cell.34,35 Meanwhile, an excessive number of hydrophobic parts might lead to more hydrophobicity and thus prevent the carriers from entering the cell surface.36
Much work has been carried out to optimize the hydrophobic part of lipids but without solid conclusions.24,37 Most studies found that cationic lipids with shorter saturated alkyl chains or unsaturated alkyl chains38 were crucial for acquiring splendid transfection efficiency since they were in charge of membrane fluidity. However, a degree of rigidity for the membrane was also needed to stabilize the lipid assemblies,39 and long-chain lipids would likely strengthen the stability.22 We assumed that the striking transfection effect of RLS5 was due to its long-chain stability and the introduction of unsaturated bonds that allowed it to have good fluidity. In order to explore the underlying mechanism leading to this result, we have carried out an in-depth exploration at the molecular level.
Study on the molecular mechanism
Gene delivery must overcome several biological barriers to achieve efficient transfection, including cellular attachment, plasma membrane penetration, endosomal degradation prevention, etc. The cellular entry mechanism of lipid carriers was still controversial. Some studies reported endocytic pathway was the primary means,40 and others suggested cellular membrane fusion was dominant.41 In addition, the pathway of endosomal escape (a critical step in gene release into the cytoplasm) of lipid gene carriers also has different interpretations. Nevertheless, the interaction of lipid carriers with biological membranes (cellular and endosomal membranes) was crucial for gene delivery, especially the stability and fluidity of lipid carriers. Therefore, we explored the membrane fusion, fluidity, and crystal conformation of six lipids for their stability and fluidity.Membrane fusion
The whole transfection process depends on “membrane interactions”, in which the membrane of the lipoplexes first interacts with the plasma membrane for internalization and then with the endosomal membrane to trigger the release of the nucleic acid cargo. Thus, we first investigated plasma membrane fusion using the FRET strategy with donor and acceptor fluorescent labeling. Gene complexes labeled with DiO as the donor was incubated with HeLa cells labeled with Dil as the acceptor to form FRET pairs (Fig. 6(A)). During the incubation, we could hardly detect red fluorescence in RLS1–3 groups under the FRET channel of the confocal laser scanning microscope (Fig. 6(B)). Because the distance between the two fluorescent labels was not close enough to produce the FRET phenomenon, indicating no plasma membrane fusion. Whereas cells incubated with RLS4–6 exhibited strong red fluorescence under FRET channels, resulting from the occurrence of membrane fusion. Quantitative data was showed in Fig. 6(C).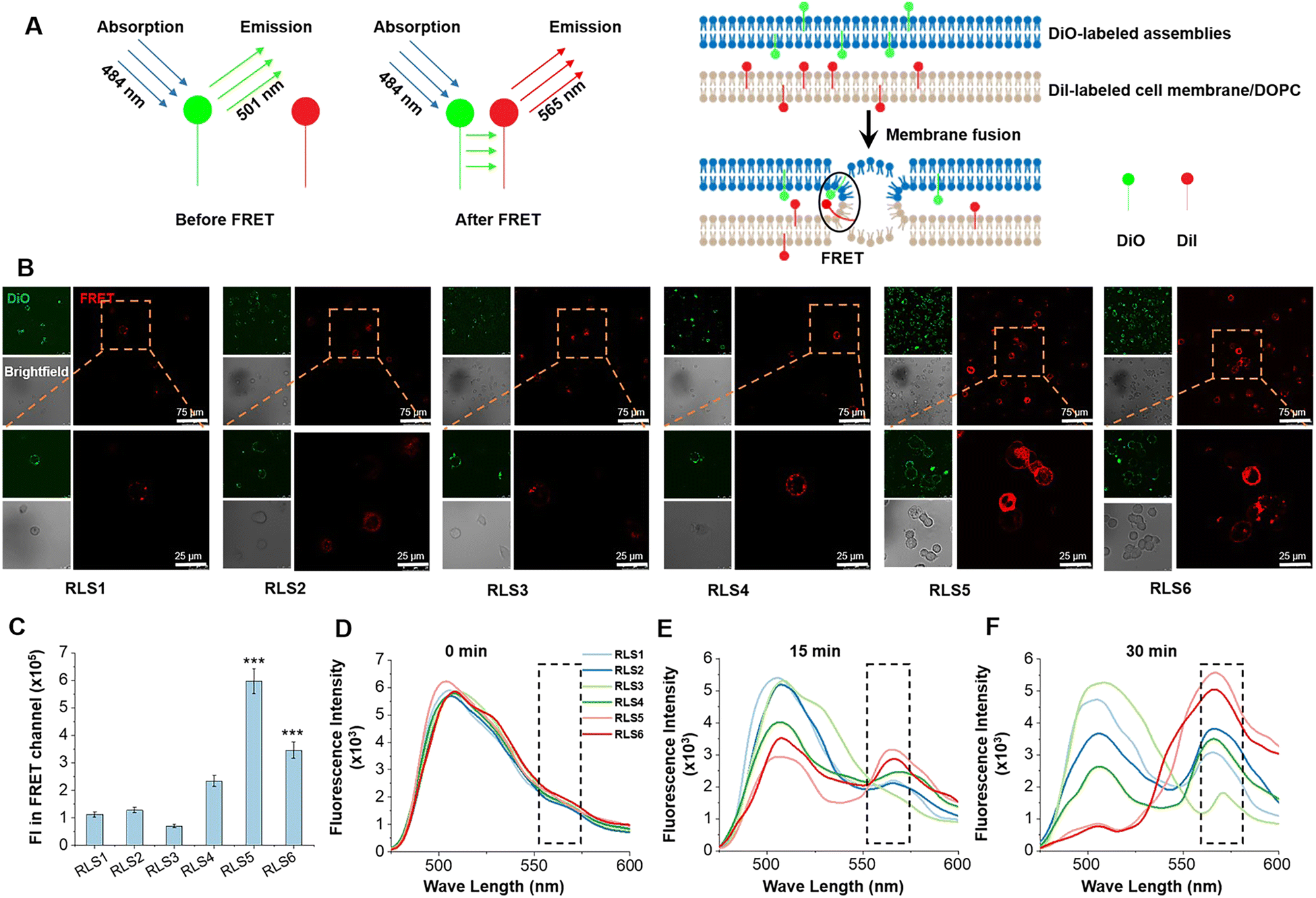 | ||
| Fig. 6 Membrane fusion effect of six cationic lipid assemblies in HeLa cells and simulated endosomal membrane. (A) Mechanism of FRET in membrane fusion. (B) The FRET effect in HeLa cells was observed by confocal laser scanning microscope after incubation for 30 min. Assemblies were labeled with DiO and cell membranes were labeled with DiI. The orange box indicated partial magnification. (C) FRET evaluation by measuring fluorescence intensity (FI). Data were the mean ± SD (n = 3, ***p < 0.001 vs.RLS4). (D)–(F) FRET emission peak (565 nm) under simulated conditions between DiO-labeled assemblies and DiI-labeled DOPC vesicles with different incubation times (0, 15, and 30 min). | ||
After endocytosis, the internalized substances were usually trapped in vesicles and eventually degraded within the endosomal compartments. Efficient endosomal escape was important for gene transfection. We further mimicked the endosomal/lysosomal membrane fusion using DiI-labeled DOPC vesicles in acidic conditions (pH 5.0). In RLS1–4, the fluorescence intensity of the donor at 501 nm did not decrease significantly, and the intensity of the acceptor at 565 nm did not increase significantly. It suggested that the fusion effect of these four cationic lipids was incomplete and slow. Whereas RLS5 and RLS6 showed a rapid increase in fluorescence intensity at the emission wavelength of 565 nm, indicating an excellent fusion with the simulated endosomal membrane (Fig. 6(D)–(F)). So, the transfection results were consistent with the fusion effect, which also confirmed that the satisfying transfection efficiency of RLS5 and RLS6 was mainly due to their complete and rapid membrane fusion with the endosomal membrane after the cytoplasm successfully.
Membrane fluidity
Membrane fluidity affected the fusion of endosomal membranes, which was necessary for the endosome escape of genes.42 As RLS5-6 showed the best membrane fusion ability, we further explored their fluidity. Membrane fluidity for various lipid assemblies was evaluated using a fluorescence probe DPH.43 The 1/P was the reciprocal polarization obtained by DPH fluorescence analysis, which reflected the fluidity of the membrane. Our experimental results showed that among the six lipids, only RLS5 (1/P = 9.2) and RLS6 (1/P = 9.6) had higher fluidity (Fig. 7(A)). It was probably due to the existence of unsaturated bonds increased fluidity by preventing the tight accumulation of lipids.44 In addition, a gradual decrease in fluidity could be seen with increasing alkyl chain length among the four cationic lipids containing saturated alkyl chains because short-chain lipids tended to separate in the bimolecular clusters or “rafts” form in the layer, resulting in an uneven distribution of lipids across the membrane. Although the fluidity of RLS1–4 was slightly different, these 1/P values were generally around 6. From the fluidity results, the length and number of alkyl chains in the hydrophobic part had no significant effects on lipid fluidity. However, increasing the unsaturation improved the overall fluidity of the lipids and thus significantly improved their gene transfection effects.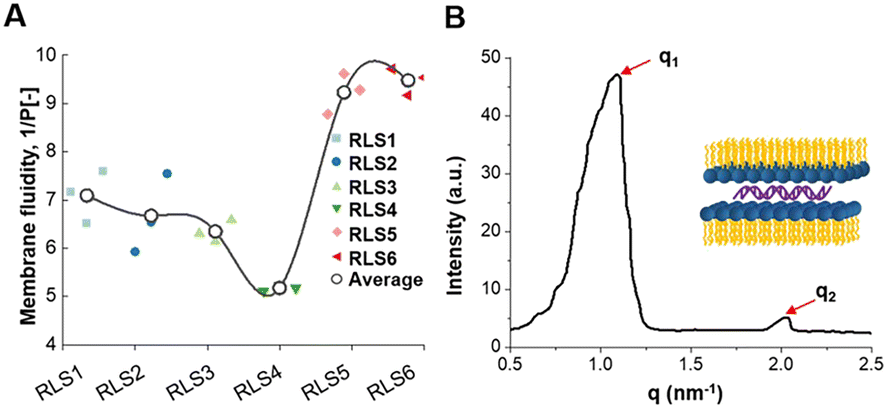 | ||
Fig. 7 Molecular mechanism exploration on cationic lipid assemblies. (A) Membrane fluidity (1/P [−]) of six lipid assemblies was performed by fluorescence spectrophotometer at room temperature 25 °C. (B) Small-angle X-ray scattering profiles of RLS5 at room temperature. The peak position ratio of q1 and q2 was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, which indicated the formation of well-ordered lamellar structures. :2, which indicated the formation of well-ordered lamellar structures. | ||
Crystal phase
In addition to the membrane fusion and fluidity discussed above, crystal conformation was an essential factor that should not be ignored. According to reports, the lamellar phase benefited the stability of lipid carriers, while the hexagonal (HII) phase benefited the fusion with biological membranes.45 Therefore, we utilized the SAXS technique to investigate the crystal phase of RLS5. The X-ray scattering patterns of RLS5 showed a peak ratio of 1:2 (Fig. 7(B)), which was characteristic of the lamellar phase. This lamellar phase probably made the delivery system less susceptible to dissociation in complicated biological environments, facilitating stability in serum and efficient gene transfection in vivo.
Through the above experiments, RLS5, with a stable lamellar phase, showed the best lipid membrane fusion and overall fluidity, which might be the reason for the outstanding in vitro transfection efficiency in serum.
Animal experiments
Further, we investigated the transfection effects in vivo. pCMV-Luc encoding luciferase was complexed into lipoplexes and intramuscularly injected into BALB/c nude mice. At day 1, 2, 3, 5, and 7 after administration, transgene expression was determined by intraperitoneal injection of D-luciferin potassium salt. As shown in Fig. 8(A) and (C), a notably higher bioluminescent signal was found in the mice injected with RLS5/pDNA compared to the mice treated with naked pDNA (5.5-fold) or PEI/pDNA (3.0-fold). This might be due to the superior serum stability of the lamellar phase and high fusion property with the endosome membrane in RLS5, resulting in the same satisfying transfection efficiency in vivo.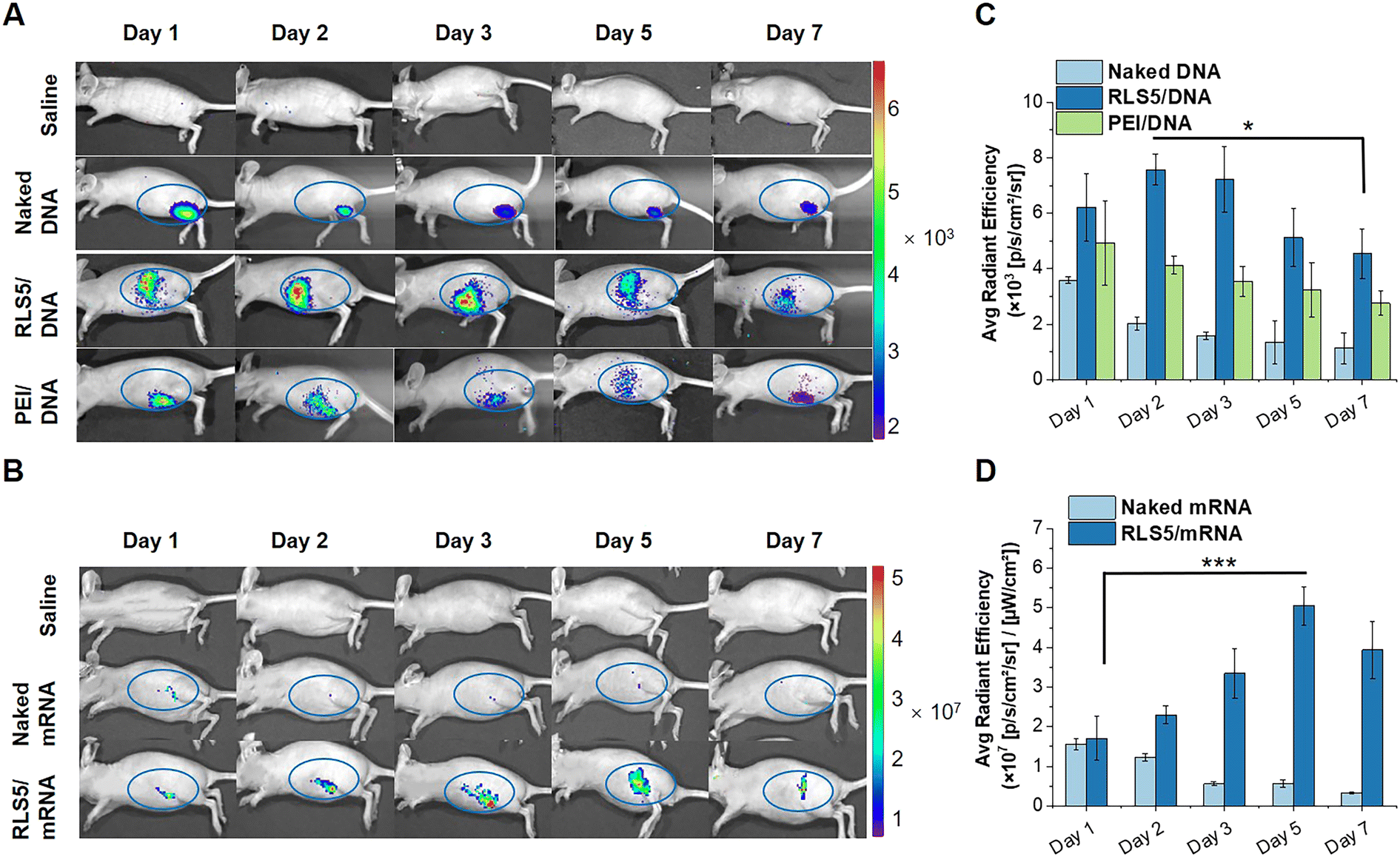 | ||
| Fig. 8 Transfection efficiency of pGL3 and mRNA in vivo after intramuscular injection. pGL3 (A) and mEGFP (B) in vivo expression efficiency at different time points after intramuscular injection by fluorescence imaging using IVIS Lumina Series III imaging system (the blue circle indicated the injection site). Quantitative fluorescence intensity in the blue circle of pGL3 (C) and mEGFP (D) expression in vivo. (*P < 0.05, ***P < 0.001). | ||
Likely, we examined the EGFP expression in nude mice through an intramuscular injection of RLS5/mEGFP as the same administration method as the mRNA vaccine. In the mice injected with naked mRNA, fluorescence was kept for 2 days post-administration at the intramuscular injection site (Fig. 8(B)). Notably, with RLS5/mRNA administration, the fluorescence gradually increased, peaked on day 5, and decreased on day 7. In the inter-group comparison, the average fluorescence intensity detected in the mice treated with RLS5/mRNA was comparable to that of naked mRNA on day 1, while increased up to 10-fold higher on day 5 (Fig. 8(D)). As the detection sensitivity and specificity of mEGFP were much lower than luciferase,46 there was a strong background signal existed in the study of mRNA transfection effect in vivo. To avoid interference, we only calculated the average fluorescence intensity in the blue circle for the quantitative analysis.
Overall, direct injection of pDNA/mRNA could lead to transgene expression, but the level was significantly low, which was generally considered insufficient to achieve a therapeutic effect.47 The presence of gene carriers of RLS5 was necessary to achieve efficient transgene expression in vivo for both pDNA and mRNA.
Conclusions
Our previous studies on the structure–activity relationship have pointed out the advantages of second-generation arginine as the hydrophilic part and disulfide bond as the linker in gene delivery systems. In this research, we further designed a series of single-component self-assembled cationic lipids (RLS1–6), studied the structure–activity relationship of the hydrophobic part, and explored the potential molecular mechanism affecting the various performance of gene carriers. The cationic lipids contain hydrophobic parts composed of alkyl chains with different lengths, unsaturation, and numbers. Research showed that the introduction of unsaturation could greatly improve the transfection effect, even significantly better than lipofectamine 2000. Mechanism exploration confirmed that the unsaturated bonds could dramatically improve the membrane fluidity and fusion of lipid nanoparticles, increasing the interaction with biological membranes to promote endocytosis and endosomal escape. During the screening, we found that the unsaturation of the alkyl chain had the greatest influence on the performance of lipid nanoparticles, followed by the number of hydrophobic tails. After screening, the optimized RLS5 with the lamellar phase was the best-performing cationic lipid, with a superior transfection efficiency of pDNA and mRNA in vivo, which has potential clinical translational value in the disease prevention and treatment. There are still some shortcomings in this study; for example, (1) more control materials need to be designed to further investigate the influence of alkyl chain unsaturation and quantity, (2) the in vivo behavior of lipid nanoparticles needs to be further investigated, such as tissue distribution, metabolic pathway, and long-term toxicity, etc. Nevertheless, this study provides reference and important guidance for the design and optimization of lipid gene carriers based on molecular characterization of lipid and cell membrane interactions. Our future studies will contribute to the further development of lipid pDNA and mRNA delivery systems.Author contributions
Yu Wei and Ting He: conceptualization, methodology, investigation, writing – original draft. Qunjie Bi, Huan Yang, and Xueyi Hu: part of cell and animal experiments. Yongqun Zhu and Rongsheng Tong: validation. Rongrong Jin: visualization, funding acquisition. Hong Liang and Yu Nie: writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (NSFC, No. 81873921 and 51903174), Sino-German Cooperation Group Project (GZ1512), Chengdu Science and Technology Program (2020-GH02-00007-HZ), Sichuan Science and Technology Program (23GJHZ0001), Miaozi Project in Science and Technology Innovation Program of Sichuan Province (2021024). Postdoctoral Science Foundation of Sichuan Province (TB2022047). Postdoctoral Science Foundation of Sichuan Academy of Medical Sciences & Sichuan Provincial People's Hospital (2022BH04). Open project foundation of Personalized Drug Therapy Key Laboratory of Sichuan Province (2022YB02). We acknowledged the Analytical & Testing Center, Sichuan University for sample analysis. In addition, we acknowledged Prof. Mattias Barz and Heyang Zhang (Leiden Academic Center for Drug Research, Leiden University) for their help in reviewing & editing.Notes and references
- D. Pushparajah, S. Jimenez, S. Wong, H. Alattas, N. Nafissi and R. A. Slavcev, Adv. Drug Delivery Rev., 2021, 170, 113–141 CrossRef CAS.
- Y. Klapper, O. A. Hamad, Y. Teramura, G. Leneweit, G. U. Nienhaus, D. Ricklin, J. D. Lambris, K. N. Ekdahl and B. Nilsson, Biomaterials, 2014, 35, 3688–3696 CrossRef CAS.
- E. S. Fried, J. Luchan and M. L. Gilchrist, Langmuir, 2016, 32, 3470–3475 CrossRef CAS.
- A. R. Nanna, A. V. Kel'in, C. Theile, J. M. Pierson, Z. X. Voo, A. Garg, J. K. Nair, M. A. Maier, K. Fitzgerald and C. Rader, Nucleic Acids Res., 2020, 48, 5281–5293 CrossRef CAS.
- A. Norman, C. Franck, M. Christie, P. M. E. Hawkins, K. Patel, A. S. Ashhurst, A. Aggarwal, J. K. K. Low, R. Siddiquee, C. L. Ashley, M. Steain, J. A. Triccas, S. Turville, J. P. Mackay, T. Passioura and R. J. Payne, ACS Cent. Sci., 2021, 7, 1001–1008 CrossRef CAS.
- M. Li, S. Li, Y. Huang, H. Chen, S. Zhang, Z. Zhang, W. Wu, X. Zeng, B. Zhou and B. Li, Adv. Mater., 2021, 33, e2101707 CrossRef.
- S. T. LoPresti, M. L. Arral, N. Chaudhary and K. A. Whitehead, J. Controlled Release, 2022, 345, 819–831 CrossRef CAS.
- Y. Zhang, T. Chu, L. Sun, X. Chen, W. Zhang, H. Zhang, B. Han, J. Chang, Y. Feng and F. Song, J. Biomed. Mater. Res., Part A, 2020, 108, 2409–2420 CrossRef CAS.
- H. Liang, A. Hu, X. Chen, R. Jin, K. Wang, B. Ke and Y. Nie, J. Mater. Chem. B, 2019, 7, 915–926 RSC.
- X. Yan, L. Lin, S. Li, W. Wang, B. Chen, S. Jiang, S. Liu, X. Ma and X. Yu, Chem. Eng. J., 2020, 395, 125171 CrossRef CAS.
- M. Hao, L. Zhang and P. Chen, Int. J. Mol. Sci., 2022, 23, 9038 CrossRef CAS.
- T. Nakamura, T. Nakade, K. Yamada, Y. Sato and H. Harashima, Int. J. Pharm., 2021, 609, 121140 CrossRef CAS.
- J. Shi, S. Yu, J. Zhu, D. Zhi, Y. Zhao, S. Cui and S. Zhang, Colloids Surf., B, 2016, 141, 417–422 CrossRef CAS.
- Y. Wu, L. Li, Q. Chen, Y. Su, P. A. Levkin and G. Davidson, ACS Comb. Sci., 2016, 18, 43–50 CrossRef CAS.
- S. Liu, Q. Cheng, T. Wei, X. Yu, L. T. Johnson, L. Farbiak and D. J. Siegwart, Nat. Mater., 2021, 20, 701–710 CrossRef CAS.
- V. A. Duong, T. T. L. Nguyen and H. J. Maeng, Molecules, 2020, 25, 4781 CrossRef CAS.
- H. Zhang, J. Leal, M. R. Soto, H. D. C. Smyth and D. Ghosh, Pharmaceutics, 2020, 12, 1042 CrossRef CAS.
- S. He, W. Fan, N. Wu, J. Zhu, Y. Miao, X. Miao, F. Li, X. Zhang and Y. Gan, Nano Lett., 2018, 18, 2411–2419 CrossRef CAS.
- F. G. Wu, H. Y. Sun, Y. Zhou, R. G. Wu and Z. W. Yu, RSC Adv., 2014, 4, 51171–51179 RSC.
- X. Chen, J. Yang, H. Liang, Q. Jiang, B. Ke and Y. Nie, J. Mater. Chem. B, 2017, 5, 1482–1497 RSC.
- D. Zhang, E. N. Atochina-Vasserman, D. S. Maurya, M. Liu, Q. Xiao, J. Lu, G. Lauri, N. Ona, E. K. Reagan, H. Ni, D. Weissman and V. Percec, J. Am. Chem. Soc., 2021, 143, 17975–17982 CrossRef.
- D. Zhi, S. Zhang, B. Wang, Y. Zhao, B. Yang and S. Yu, Bioconjugate Chem., 2010, 21, 563–577 CrossRef CAS.
- I. S. Zuhorn, V. Oberle, W. H. Visser, J. Engberts, U. Bakowsky, E. Polushkin and D. Hoekstra, Biophys. J., 2002, 83, 2096–2108 CrossRef CAS.
- H. J. Wang, Y. H. Liu, J. Zhang, Y. Zhang, Y. Xia and X. Q. Yu, Biomater. Sci., 2014, 2, 1460–1470 RSC.
- J. Qiu, L. Kong, X. Cao, A. Li, P. Wei, L. Wang, S. Mignani, A.-M. Caminade, J. P. Majoral and X. Shi, Nanomaterials, 2018, 8, 131 CrossRef.
- Z. Huang, Y. M. Zhang, Q. Cheng, J. Zhang, Y. H. Liu, B. Wang and X. Q. Yu, J. Mater. Chem. B, 2016, 4, 5575–5584 RSC.
- K. J. Whittlesey and L. D. Shea, Biomaterials, 2006, 27, 2477–2486 CrossRef CAS.
- M. O. Aviles, C. H. Lin, M. Zelivyanskaya, J. G. Graham, R. M. Boehler, P. B. Messersmith and L. D. Shea, Biomaterials, 2010, 31, 1140–1147 CrossRef CAS.
- H. Liang, X. Chen, R. Jin, B. Ke, M. Barz, H. Ai and Y. Nie, Small, 2020, 16, e1906538 CrossRef.
- Q. Jiang, D. Yue, Y. Nie, X. Xu, Y. He, S. Zhang, E. Wagner and Z. Gu, Mol. Pharmacol., 2016, 13, 1809–1821 CrossRef CAS.
- S. F. Gilmore, T. S. Carpenter, H. I. Ingolfsson, S. K. G. Peters, P. T. Henderson, C. D. Blanchette and N. O. Fischer, Nanoscale, 2018, 10, 7420–7430 RSC.
- M. Kanduc, E. Schneck and R. R. Netz, Langmuir, 2013, 29, 9126–9137 CrossRef CAS.
- S. Son, R. Namgung, J. Kim, K. Singha and W. J. Kim, Acc. Chem. Res., 2012, 45, 1100–1112 CrossRef CAS.
- S. Arpicco, S. Canevari, M. Ceruti, E. Galmozzi, F. Rocco and L. Cattel, Farmaco, 2004, 59, 869–878 CrossRef CAS.
- Y. Inoh, T. Furuno, N. Hirashima, D. Kitamoto and M. Nakanishi, Int. J. Pharm., 2010, 398, 225–230 CrossRef CAS.
- O. Paecharoenchai, N. Niyomtham, L. Leksantikul, T. Ngawhirunpat, T. Rojanarata, B. E. Yingyongnarongkul and P. Opanasopit, AAPS PharmSciTech, 2014, 15, 722–730 CrossRef CAS.
- C. H. Jones, C. K. Chen, A. Ravikrishnan, S. Rane and B. A. Pfeifer, Mol. Pharmaceutics, 2013, 10, 4082–4098 CrossRef CAS.
- H. Zhou, J. Yang, Y. Du, S. Fu, C. Song, D. Zhi, Y. Zhao, H. Chen, S. Zhang and S. Zhang, Bioorg. Med. Chem., 2018, 26, 3535–3540 CrossRef CAS.
- G. V. Srilakshmi, J. Sen, A. Chaudhuri, Y. Ramadas and N. M. Rao, Biochim. Biophys. Acta, Biomembr., 2002, 1559, 87–95 CrossRef CAS.
- N. Means, C. K. Elechalawar, W. R. Chen, R. Bhattacharya and P. Mukherjee, Mol. Aspects Med., 2022, 83, 100993 CrossRef.
- J. Zhang, S. Shrivastava, R. O. Cleveland and T. H. Rabbitts, ACS Appl. Mater. Interfaces, 2019, 11, 10481–10491 CrossRef CAS.
- Y. Tokudome, Y. Saito, F. Sato, M. Kikuchi, T. Hinokitani and K. Goto, Colloids Surf., B, 2009, 73, 92–96 CrossRef CAS.
- K. Hayashi, T. Shimanouchi, K. Kato, T. Miyazaki, A. Nakamura and H. Umakoshi, Colloids Surf., B, 2011, 87, 28–35 CrossRef CAS.
- A. Bouraoui, M. Berchel, R. Ghanem, V. Vie, G. Paboeuf, L. Deschamps, O. Lozach, T. Le Gall, T. Montier and P. A. Jaffres, Org. Biomol. Chem., 2019, 17, 3609–3616 RSC.
- T. Colombani, P. Peuziat, L. Dallet, T. Haudebourg, M. Mevel, M. Berchel, O. Lambert, D. Habrant and B. Pitard, J. Controlled Release, 2017, 249, 131–142 CrossRef CAS.
- T. Taheri, H. Saberi Nik, N. Seyed, F. Doustdari, M. H. Etemadzadeh, F. Torkashvand and S. Rafati, Exp. Parasitol., 2015, 150, 44–55 CrossRef CAS.
- L. M. Molle, C. H. Smyth, D. Yuen and A. P. R. Johnston, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2022, 14, e1809 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb02783f |
| ‡ Equal contribution to the manuscript as co-first authors. |
| This journal is © The Royal Society of Chemistry 2023 |