
Nanotubes and water-channels from self-assembling dipeptides
Ottavia
Bellotto
 a,
Paola
D’Andrea
b and
Silvia
Marchesan
*ac
a,
Paola
D’Andrea
b and
Silvia
Marchesan
*ac
aChem. Pharm. Sc. Dept., University of Trieste, Via Giorgieri 1, 34127 Trieste, Italy. E-mail: smarchesan@units.it
bLife Sc. Dept., University of Trieste, Via Weiss 2, 34128 Trieste, Italy
cINSTM, Unit of Trieste, Via Giorgieri 1, 34127 Trieste, Italy
First published on 8th February 2023
Abstract
Dipeptides are attractive building blocks for biomaterials in light of their inherent biocompatibility, biodegradability, and simplicity of preparation. Since the discovery of diphenylalanine (Phe-Phe) self-assembling ability into nanotubes, research efforts have been devoted towards the identification of other dipeptide sequences capable of forming these interesting nanomorphologies, although design rules towards nanotube formation are still elusive. In this review, we analyze the dipeptide sequences reported thus far for their ability to form nanotubes, which often feature water-filled supramolecular channels as revealed by single-crystal X-ray diffraction, as well as their properties, and their potential biological applications, which span from drug delivery and regenerative medicine, to bioelectronics and bioimaging.
 Ottavia Bellotto | Dr Ottavia Bellotto is a researcher at Dr Schär R&D center in Area Science Park (Trieste, Italy). In 2022, she was awarded her PhD in Chemistry with honours from the University of Trieste, supervised by Prof. Silvia Marchesan. Her doctoral research focussed on the design of self-assembling dipeptides for the development of nanotubes and hydrogel biomaterials. In 2018, she secured a 6-month fellowship funded by the National Cancer Institute (CRO) to develop biosensors based on antibodies and aptamers for Therapeutic Drug Monitoring. In 2017, she obtained her Master's degree in Medicinal Chemistry (University of Trieste), and she qualified as Pharmacist. |
 Paola D’Andrea | Prof. Paola D’Andrea is Associate Professor of Biochemistry at the University of Trieste, Department of Life Sciences. She carried out postdoctoral research at the Trinity College of Dublin (Ireland), at the Neuroscience Institute of the CNR in Milan, at the Department of Biotechnology (DIBIT) of the San Raffaele Hospital in Milan. She was visiting scientist at the Babraham Institute in Cambridge (UK) and at the Institut Pasteur of Paris (France). Her research activity has focused on the mechanisms of calcium signalling, on gap junctions in genetic diseases, and on cellular modifications induced by tailored biointerfaces mimicking the extracellular matrix. |
 Silvia Marchesan | Prof. Silvia Marchesan did her PhD at The University of Edinburgh (2008, UK), and postdoc research at the University of Helsinki (Finland), Monash University, and CSIRO (Melbourne, Australia). In 2013 she moved to the Center of Excellence for Nanostructured Materials at the University of Trieste (Italy) and she opened there the Superstructures Lab (https://www.marchesanlab.com) in 2015. In 2018, she became associate professor, got habilitated as full professor, and was selected by Nature Index as Rising Star in the natural sciences. In 2021–2022 (6 months) she has been Visiting Professor at the University of Cambridge (UK), Yusuf Hamied Department of Chemistry. |
10th Anniversary StatementNanotubes formed by minimalistic dipeptides have been raising researchers’ interest, not only for their inherent biocompatibility and biodegradability, but also for their emerging electronic properties. These nanotubes are being explored for diverse applications in light of their benign nature in terms of environmental impact. The roles of water in their inner cavities, and its impact on their physico-chemical properties, are a hot area of research. Our laboratories (https://www.marchesanlab.com) were opened in 2015, thanks to a starting grant on heterochiral tripeptides’ self-assembly and their design based on the introduction of D-amino acids at selected positions to modulate the resulting supramolecular nanostructures and materials. The same year, we reported in J. Mater. Chem. B our research demonstrating that a simple shift in the positioning of the D-stereoconfiguration along a tripeptide was sufficient to change the level of supramolecular order of the assemblies, hence, the physicochemical properties of the resulting hydrogels, and their rates of enzymatic hydrolysis to modulate their biodegradation. In 2020, Prof. Marchesan joined the Advisory Board of J. Mater. Chem. B, which offers an ideal platform for research on materials derived from, and inspired by, biomolecules for a range of applications that go well beyond medicine. |
Introduction
The most widely known nanotubes (NTs) are certainly carbon nanotubes, which, since their discovery by Ijima in 1991,1 have attracted great interest especially for their electronic properties towards advanced applications in areas as diverse as tissue engineering,2–4 energy5,6 and catalysis,7,8 conductive materials,9 and electrodes for interfacing with biological tissues.10 Besides inorganic alternatives,11 such as boron nitride12 and metal chalcogenides,13 peptide-based organic NTs are becoming popular alternatives.14,15 The reasons are varied, and certainly include the increasing chemical variety of non-natural building blocks being commercially available, the easy and modular nature of solid-phase peptide synthesis, and the inherent biocompatibility and potential biodegradability. The values of the energy band gap, calculated as the difference between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), can be even higher for peptide NTs relative to inorganic NTs (Fig. 1), demonstrating their high potential for uses in bioelectronics.16,17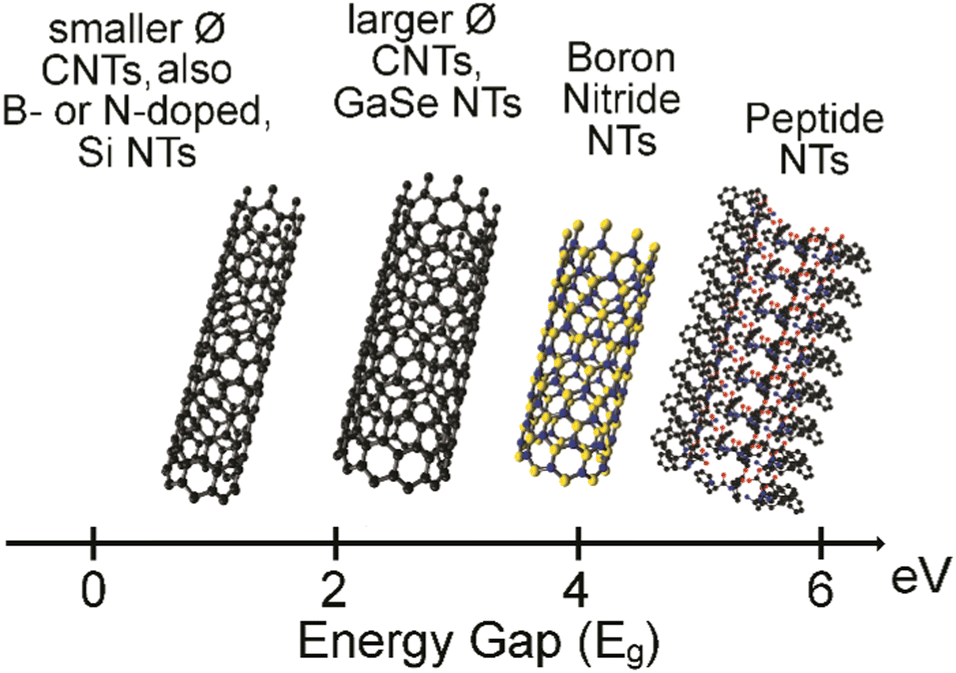 | ||
| Fig. 1 HOMO–LUMO energy band gap in electronvolt (eV) for CNTs, inorganic NTs, and peptide NTs.16,17 | ||
Polypeptides’ NTs were first reported using cyclic building blocks displaying alternating D- and L-amino acids by Ghadiri et al.,18 and, in recent years, supramolecular alternatives using simpler building blocks are becoming attractive for their lower cost and simplicity of preparation. Amongst the various options, Phe-Phe NTs reported by Reches and Gazit19 certainly stand out for robustness and interesting properties, and they are by far the most studied in this class.
Phe-Phe nanotubes (NTs) structure and properties
The Phe-Phe NT characteristic architecture features an inner hydrophilic cavity enclosed by six peptide molecules positioned in alternating planes and interacting head-to-tail, with peptide stacks held together by intermolecular H-bonds between amide groups.20 Remarkably, this structure revealed by single-crystal X-ray diffraction (XRD) data, was shown to correspond to that found in the NTs, as confirmed by spectral matching with powder XRD data of the NTs.21Phe-Phe NTs have been shown to be composed of nanocrystalline regions, which behave as quantum dots, as they display quantum confinement and exciton formation and luminescence at room temperature.22 Remarkably, the blue photoluminescence was maintained even after thermal dehydration above 140 °C into fibrils formed by their cyclic analogs. This behavior opens the avenue to their potential use in photonics and displays as non-toxic alternatives to luminescent materials.23
Another interesting feature is the light-induced ferroelectricity of Phe-Phe NTs. This phenomenon requires two essential features, which are spontaneous polarisation and the ability to switch between two states in an electric field. In particular, the H-bonding network between the termini of the dipeptide molecules within the NTs was shown to switch between the zwitterionic and neutral states. The process is aided by the photoluminescent character of the NTs, with excitation light inducing the reduction of the intrinsic polarisation field, indicating the phenomenon happens at the nanoscale.24
Phe-Phe NTs display high average point stiffness of 160 N m−1 and Young's modulus of 19–27 GPa, being amongst the stiffest biological materials known to date.25,26 The longitudinal rigidity has been attributed to the zipper-like arrangement of interlocked aromatic residues.27 Indeed, addition of further aromatic units using β,β-diphenylalanine enabled an increase to ∼70 GPa, which is comparable to aluminium.28 Conversely, the transversal rigidity has been attributed to the mobile water molecules inside the channels, with higher values corresponding to water-filled NTs.29 New advanced microscopy methods to measure piezoresponse, mechanical, and dielectric properties of dipeptide NTs at the nanoscale are continuously being developed.30
The inner content of water determines also the type of nanomorphology attained with Phe-Phe self-assembly that can be controlled by varying the amount of relative humidity.31 Interestingly, recent in silico investigations have suggested that the inner water clusters can acquire polar and chiral properties as a result of their interactions with the walls of the Phe-Phe NTs, which exert such effects thanks to their dipole-moments distribution.32
Phe-Phe NTs have also been shown to be able to reversibly switch between the NTs and nanovesicle morphologies, for instance by dilution and increased concentration, respectively.33 The morphological conversions have been shown to follow the Ostwald's rule of stages, with coalescence of monomers into nuclei, which then undergo ripening into NTs, as shown with Boc-Phe-Phe.34 Recent studies have unveiled how only certain peptide backbone conformations found in intermediate species are compatible with, and proceed towards, NT assembly.35 The use of doping additives and solvent choice offer further means to control the final nanomorphology of Phe-Phe assemblies.36
The role of water inside the channels
One of the fascinating features of this type of NTs is the nanotubular architecture that encloses water-filled channels. Two types of water molecules are present, i.e. those bound to the peptide walls of the channels, and those that are mobile in the inner cavity. The bound water molecules are necessary for the NTs’ structural integrity, as the NTs collapse without them. Conversely, the mobile water molecules are responsible for the physical properties of the NTs. Interestingly, a recent study showed that there are two types of diffusion regimes, with small clusters (<5 molecules per unit cell) displaying a ballistic type, and larger clusters displaying a Fickian type. The water diffusion coefficient was measured experimentally and found to be within range with other solid materials (Table 1).37| Material | D × 10−10 m2 s−1 | Channel ∅ nm | Temp. °C | Ref. |
| Phe-Phe NTs | 1.3–3.0 | 1.0 | 30–65 | 37 |
| Cyclo(WL)n | 0.6–1.3 | 0.6–1.0 | 37 | 38 |
| Cyclo(QaEa)4 | 4.4 | 1.0 | 25 | 39 |
| Gramicidin A | 0.3 | 0.5 | 27 | 40 |
| Aquaporin 1 | 4–8 | 1.0–1.2 | RT | 41 |
| Nafion | 0.1–19.2 | 3.2 | 23–70 | 42 and 43 |
| Zeolites | 0.5–1.3 | 0.3–1.0 | 25 | 44 |
| Hydroxyapatite | 10 | 2.0–24.0 | 37 | 45 |
| Bulk water | 21–26 | n.a. | 25 | 41 and 46 |
| Carbon NTs | 26–96 | 1.2–3.5 | −8 to 22 | 47 |
The radial growth and elongation of NTs during Phe-Phe self-assembly could be imaged by using liquid-cell transmission electron microscopy (LCTEM), which enables high resolution observation of solvated nanomaterials.48 TeraHertz spectroscopy revealed breathing motions of dipeptide sidechains within the NTs that can affect the NTs permeability to water through changes in both the pore diameter and the host-water potential.49
Water inside the channel was demonstrated to alter also the electronic properties of the peptide NTs, by reducing the band gap and increasing the probability of hopping, hence conductivity.50 Furthermore, addition of water molecules to the inner cavity of Phe-Phe NTs causes splitting of the valence band, which corresponds to a redshift and splitting of a UV photoluminescence peak.51
Peptide-water interactions are also important in the initial stages of self-assembly to direct it towards different nanomorphologies.52 Higher contents of free-water (i.e., lower ionic strength, lower solute concentrations) lead to preferred NT formation over nanowires, with the former being held by stronger H-bonds between dipeptide termini, and being more thermodynamically stable.53
The loss of water can be detected by low-frequency Raman spectroscopy, as it decreases the effect that water molecules have on the coupling between dipeptides, with an increase in bandwidth and energy separation between characteristic double-peak signals in the spectra.54 Water indeed plays a key role in the early nucleation stages of peptide self-assembly55,56 and NT elongation.57
Dipeptide NTs and their chemical structures
L-Dipeptides without derivatisations
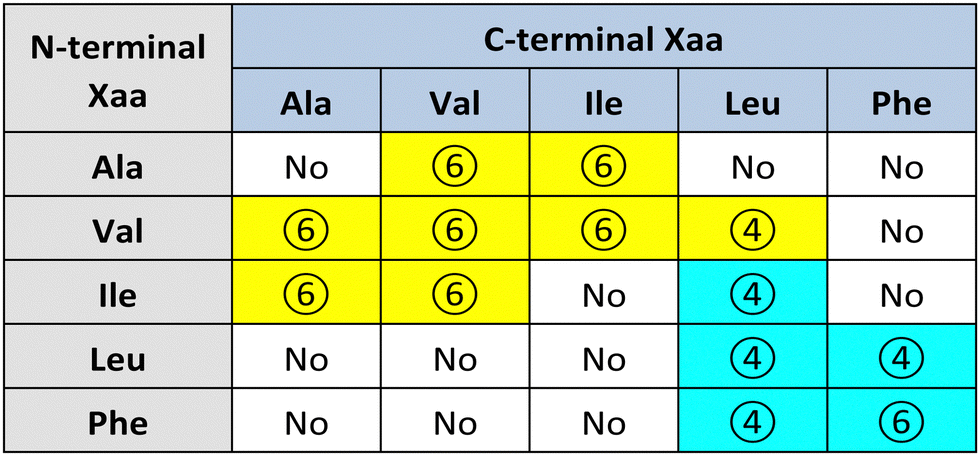
Görbitz first reported a series of hydrophobic L-dipeptide crystal structures featuring characteristic supramolecular nanotubes (Table 2). Interestingly, Val-Ala,58 Ala-Val,59 Ala-Ile, Ile-Ala, Val-Val, Val-Ile, Ile-Val,60 and Val-Leu61 had hydrophobic inner walls due to the presence of the amino acid sidechains (yellow cells in Table 2). The self-assembly of Ala-Val and Val-Ala into different nanomorphologies was studied by changing solvent systems.62
|
Conversely, Leu-Leu, Phe-Phe, Leu-Phe, Phe-Leu,20 Ile-Leu,63 (cyan cells in Table 2) and Phe-Trp64 displayed hydrophilic inner walls defined by the peptide backbones, with the sidechains defining the hydrophobic exterior of the nanotubes. The differences in packing and H-bonding networks are described in a detailed review.65 Trp-Tyr were also recently reported to form nanotubes, although no crystal structure was available.66
L-DOPA is another aromatic amino acid that has gained wide attention for its bioactivity, and inclusion into self-assembled nanostructures, as recently reviewed by Tomasini's group.67 However, when Phe was substituted with L-DOPA in the dipeptides DOPA–DOPA, nanofibrillar soft materials were obtained, with no evidence for nanotubes with a distinct inner cavity.68
L,D-Heterochiral dipeptides without derivatisations
Our group has developed expertise in the design of self-assembling minimalistic peptides whereby inclusion of L- and D-amino acids at specific positions plays a key role. In the case of hydrophobic tripeptides, heterochiral sequences can display an amphipathic conformation with hydrophobic sidechains being exposed on the opposite side relative to the hydrophilic bakbones.72 These peptides can also form supramolecular hydrogels at physiological conditions that generally display good cytocompatibility in vitro, with further advantages such as the possibility to fine-tune their biodegradation rate,73 and easily encode bioactive sequences to guide cell fate.74Recently, we explored also the case of hydrophobic heterochiral dipeptides’ self-assembly. In the case of D-Phe-L-Phe, surprisingly, NTs were formed with analogous architecture to the homochiral counterpart, and a water-filled inner cavity (Fig. 2). However, the differing stereoconfiguration of the heterochiral dipeptides, resulted in different spatial orientation of the sidechains, which could engage in intramolecular interactions at the expense of intermolecular ones. As a result, D-Phe-L-Phe formed 4 nm wide gelling nanotubes defined by two peptide layers, leading to transparent hydrogels with good cytocompatibility in cell culture. This is in marked contrast with the heterogeneous size distribution of NTs formed by homochiral Phe-Phe that display hierarchical bundling that proceeds uncontrolled, with detrimental effects for cytocompatiblity.75 Interestingly, substitution of 1 H with 1 atom of fluorine at the N-terminus did not impede NT formation, although it yielded to intermediate levels of bundling, depending on the position of fluorine on the aromatic ring.73 By contrast, the use of fluorophenylalanine at the C-terminus completely altered the supramolecular packing, leading to amphipathic layers – as opposed to water-channels – as revealed by single-crystal XRD.76
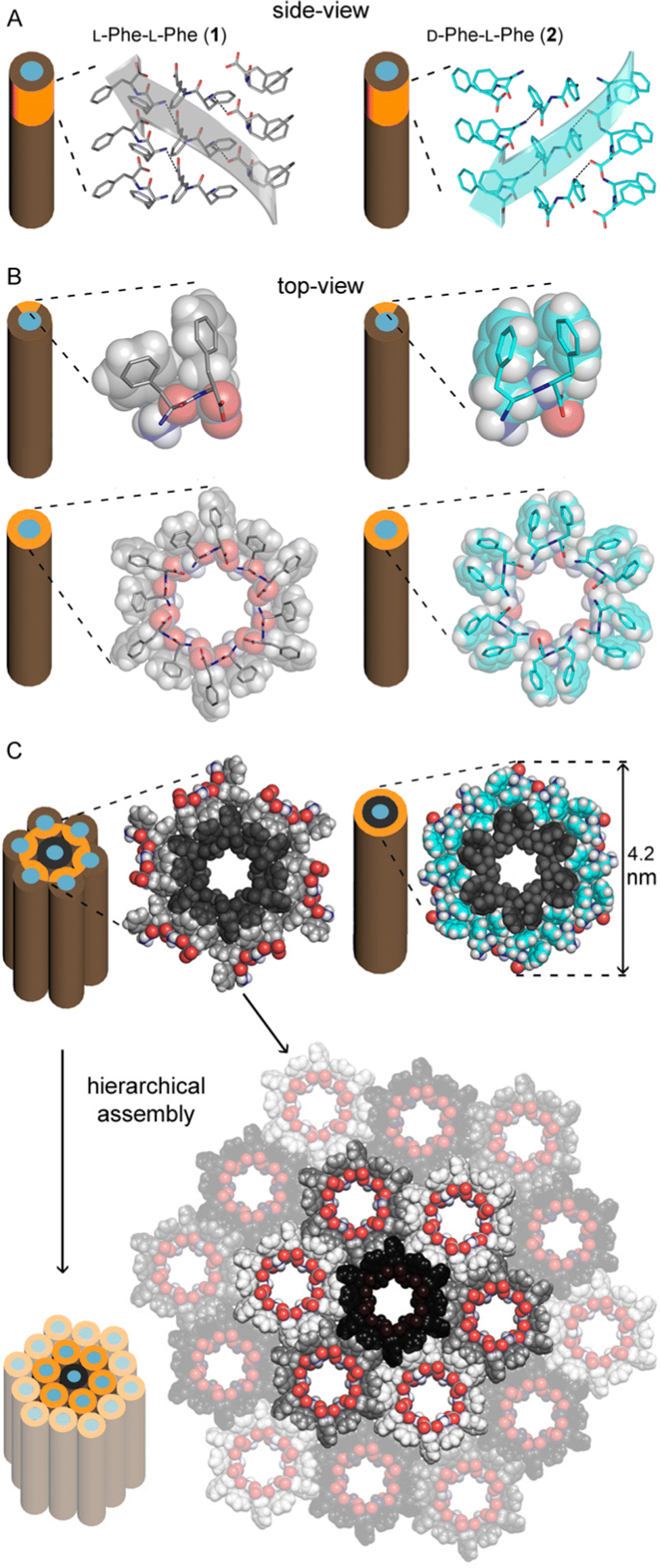 | ||
| Fig. 2 Single-crystal XRD structures of homochiral Phe-Phe201 (silver) and heterochiral D-Phe-L-Phe 2 (cyan) NTs. (A) Side-view of stacked peptides revealed opposite screw-sense for 1 and 2, dictated by the chirality of the N-terminal amino acids. (B) Top-view of nanotube inner cavities defined by the projection of six peptide molecules arranged head-to-tail for both 1 and 2. (C) Top-view of the nanotubes identified by the projection of 18 peptide molecules for 1 showed hierarchical bundling stabilized by aromatic zippers; instead, a space-fill view of heterochiral 2 revealed the outer diameter of a double layer of peptide molecules to agree with the diameter of fibrils measured by TEM.75 | ||
Substitution of one Phe with one of the hydrophobic aliphatic amino acids led to NTs too. We did not include Ala in the studies because our objective was to attain supramolecular hydrogels, and already the dipeptides Val-Phe and Phe-Val were at the limit of minimum hydrophobicity to yield stable gels.77 Studies of all the possible stereoisomer combinations of Xaa-Phe and Phe-Xaa (Xaa = Val, Ile, or Leu), revealed that heterochirality increased the dipeptides’ hydrophobicity. This fact was confirmed by increased retention times at the HPLC77–79 (which can be considered an experimental measure of hydrophobicity).80 Furthermore, the formation of water-filled channels by heterochiral Phe-Xaa sequences (cyan cells in Table 3) was supported by single-crystal XRD. Interestingly, those formed by D-Phe-L-Val featured a smaller inner cavity (∼0.5 nm) defined by four peptide molecules.77 Conversely, the NTs formed by D-Phe-L-Ile or by D-Phe-L-Phe featured a larger inner cavity (∼1.0 nm) defined by six dipeptide molecules.75,79 Therefore, it is possible to fine-tune the water-channel cavity by choosing differing peptide sequences. All these sequences gelled, but over various timescales eventually converted into crystals. Furthermore, they displayed good cytocompatibility in vitro as assessed by MTT metabolic assays and by live/dead assays (Fig. 3), but already after 24 hours the gels has partially dissolved, thus suggesting limited durability for practical use in cell culture.81 Noteworthily, the linear analog of Val (i.e., norvaline, Nva) in homo-and hetero-chiral Nva-Phe and Phe-Nva sequences, led in all cases to immediate crystallization in phosphate buffered solutions, and single-crystal XRD revealed an amphipathic layered packing devoid of channels.82 Ongoing studies in our laboratories are targeting the non-aromatic dipeptides of Table 3, for which no data is currently available pertaining their ability to form supramolecular channels, so as to provide a more complete picture.

|
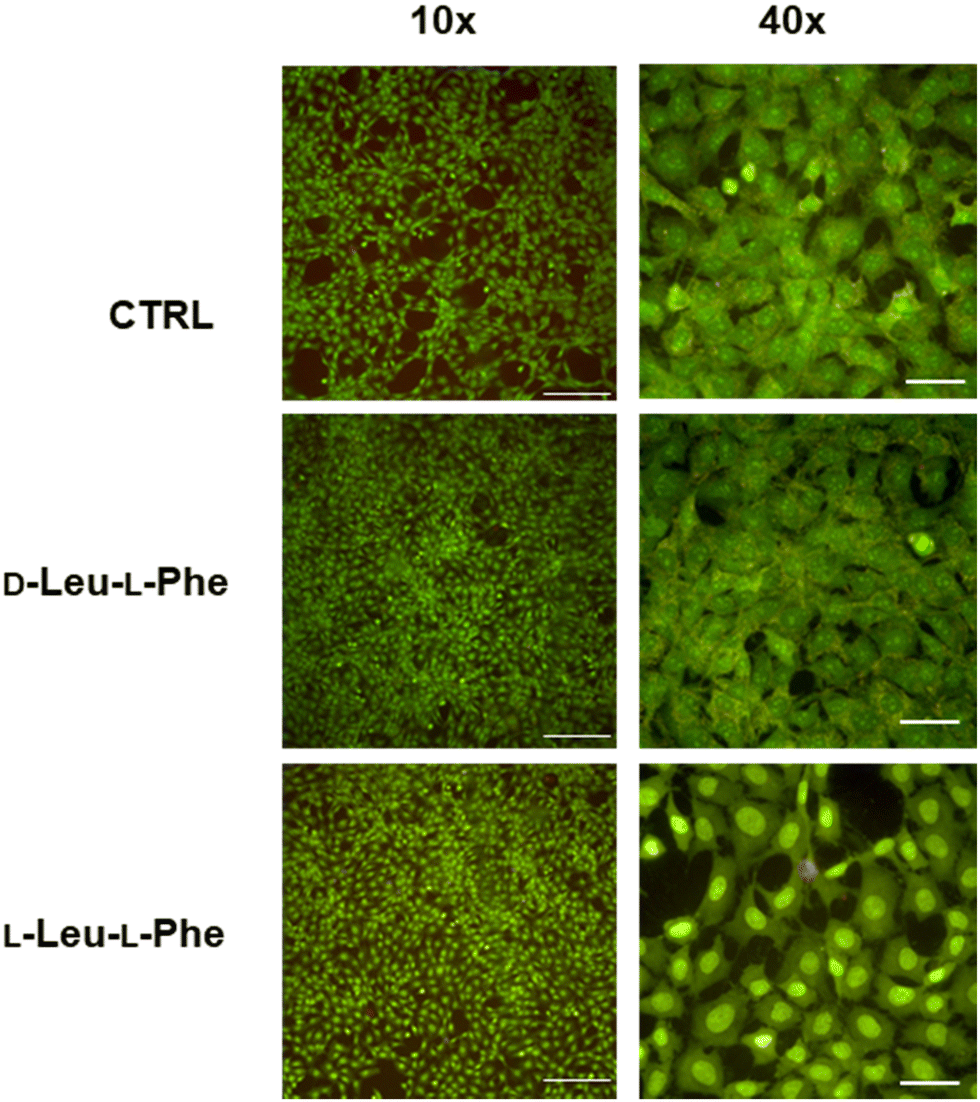 | ||
| Fig. 3 Live (green)/dead (red) assay for fibroblast cells grown for 24 hours on dipeptide hydrogels revealed excellent cell viability but also dissolution of the hydrogels. Scalebars = 200 μm (10×) or 50 μm (40×).81 | ||
N-Capped dipeptides
Various N-terminal caps, such as acetylation, were shown to enable Phe-Phe self-assembly into NTs.83 Boc-Phe-Phe can self-assemble into nanospheres or NTs depending on the solvents used, and the NTs can be inkjet printed to attain micropatterns.84 Boc-Phe-Phe NTs were also embedded in electrospun polymer fibers to maximise their piezoelectric response for biosensing and bioelectronic applications.85 Interestingly, the extent of the piezoelectric response depends also on the NT diameter.86 Adler-Abramovich et al. combined Boc-Phe-Phe with Phe-Phe into supramolecular copolymers to yield NTs whose dimensions could be controlled by changing the ratios between the two building blocks.87 Phe-Phe and Boc-Phe-Phe were also co-assembled into distinctive necklace nanoarchitectures as described by Reches’ group.88Boc-Ile-Ile-OMe was shown to self-assemble into NTs in methanol-containing solutions, with Boc playing a key role in directing the formation of the NT morphology.89 Likewise, a work led by Moretto reported NTs formed by Boc-Cys(Me)-Leu-OMe.90 One year later, NTs by Boc-Val-Ile-OMe, Boc-Val-Leu-OMe, Boc-Leu-Val-OMe, and Boc-Val-Val-OMe were described in solvent-water mixtures.91 Finally, heterochiral Boc-D-Leu-L-Leu-OMe and Boc-D-aIle-L-Ile-OMe also formed NTs with 6-fold symmetry analogously to Phe-Phe.92
Co-assembly of Fmoc-Phe-Phe with a phthalocyanine derivative resulted in the formation of helical NTs of 10–15 nm-diameter, depending on experimental conditions, which displayed intense circularly polarized luminescence. The authors envisaged potential applications in biomedicine or photocatalysis.93 Fmoc-Lys-Phe-OMe was also reported to self-assemble into NTs when templated by addition of anionic heparin.94
Phe-Phe has been N-capped with chromophores Cy-3 and Cy-5 to form NTs that accumulated selectively in the mitochondria of human-carcinoma cells, whose extremely negative inner membrane potential [(ΔΨm)cancer ≈ −220 mV] enabled their assembly leading to apoptosis.95 These NTs could thus be useful for theranostics, thanks to combined imaging and therapeutic activity.
Fmoc-Lys-Lys has been covalently functionalized on the amino sidechain with 5-fluorouracil to yield a hydrogel composed of 16 nm wide nanotubes, with 4 nm thick walls featuring a bylayered structure composed of two dipeptide molecules. The hydrogel enabled the anti-metabolite drug sustained release and displayed no cytotoxicity in vitro.96
Naphthalimide N-capped Phe-Tyr was recently shown to yield NTs too. In this case, a phosphorylated precursor was used to control self-assembly triggered by phosphatase-catalysed enzymatic hydrolysis. The NTs formed a luminescent hydrogel.97
Dehydro (Δ) dipeptides
Dehydro (Δ) amino acids display a double C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond between Cα and Cβ that restricts conformational freedom to enable the design of foldamers. Introduction of hydrophobic Δ residues in Pro-containing dipeptide sequences was found to influence turn conformations and their stability, which was reduced with increasing solvent polarity.98 In particular, the study of Ac-Pro-ΔXaa-NHCH3 revealed a tendency to adopt a β-turn in the decreasing order (Z)-ΔAbu > (E)-ΔAbu > ΔVal, with ΔAla not displaying such tendency. Furthermore, none of the folds was stable in strongly solvating media.99 Circular dichroism analysis of Phe-ΔPhe revealed a spectroscopic signature compatible with β-turns, and it formed proteolytically stable NTs. However, single-crystal XRD analyses revealed fine details over the supramolecular packing that was markedly different from Phe-Phe.100
C bond between Cα and Cβ that restricts conformational freedom to enable the design of foldamers. Introduction of hydrophobic Δ residues in Pro-containing dipeptide sequences was found to influence turn conformations and their stability, which was reduced with increasing solvent polarity.98 In particular, the study of Ac-Pro-ΔXaa-NHCH3 revealed a tendency to adopt a β-turn in the decreasing order (Z)-ΔAbu > (E)-ΔAbu > ΔVal, with ΔAla not displaying such tendency. Furthermore, none of the folds was stable in strongly solvating media.99 Circular dichroism analysis of Phe-ΔPhe revealed a spectroscopic signature compatible with β-turns, and it formed proteolytically stable NTs. However, single-crystal XRD analyses revealed fine details over the supramolecular packing that was markedly different from Phe-Phe.100
The dipeptide based on Phe and its dehydroanalog, ΔPhe, was used for similar applications as Phe-Phe NTs, as it maintained the ability to form NTs that were then loaded with the tumor-specific testis-cancer antigen MAGE-3 to enable its delivery for cancer immunotherapy. The NTs demonstrated good stability against protease-mediated hydrolysis, they accumulated into antigen-presenting-cells and successfully elicited T-cell activation and tumor inhibition in vivo.101 Phe-ΔPhe NTs were used also for the loading and release of curcumin as a model drug. Interestingly, simple variations of the sequence to Arg-ΔPhe, Val-ΔPhe, or Met-ΔPhe, hampered the NT formation in favour of spherical nanomorphologies.102
β- and γ-dipeptides
β- and γ-amino acids are characterized by one or two additional carbon atoms, respectively, between the amino and carboxylic functional groups. The position of the sidechain then determines the type of regioisomer, and the nomenclature of widely studied β-amino acids is reported in Fig. 4.103 Certain β-amino acids occur in nature as secondary metabolites or components of natural products.104,105 Amongst them, β-alanine is the most studied, and it occurs in natural bioactive dipeptides, such as carnosine.106 Self-assembly of this dipeptide was achieved when using more hydrophobic derivatives.107,108 β-Phe-Δ-Phe was designed to introduce flexibility at the N-terminus and rigidity at the C-terminus. This dipeptide maintained the ability of parent Phe-Phe to self-assemble into NTs, although with a different supramolecular packing that defined water channels of ∼0.5 nm in diameter with crystalline water inside. This nanomaterial was envisaged for drug delivery, thanks to its proteolytic stability and ability to encapsulate drugs (i.e., riboflavin, doxorubicin, chloroquine and chloramphenicol) without cytotoxicity in vitro.109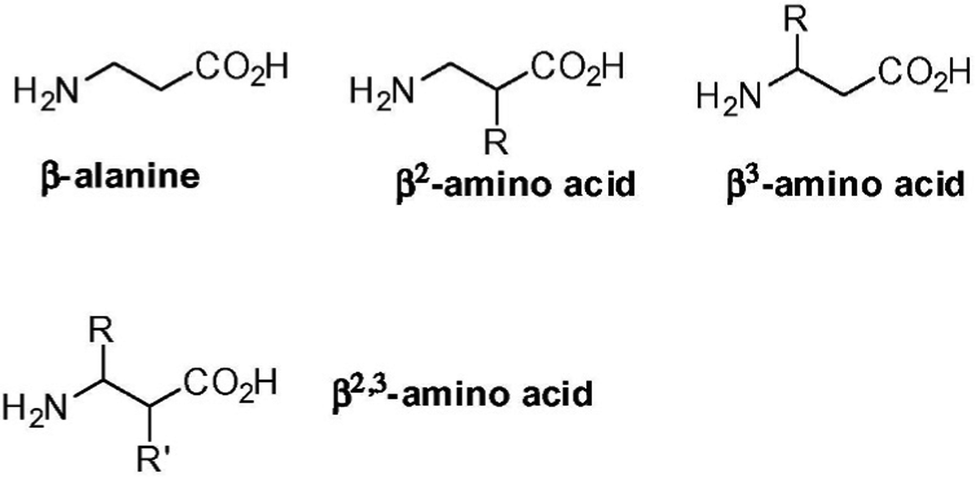 | ||
| Fig. 4 Nomenclature of β-amino acids. Reprinted from F. Clerici, E. Erba, M. L. Gelmi and S. Pellegrino, Tetrahedron Lett., 2016, 57, 5540, Copyright © 2016, with permission from Elsevier.103 | ||
Hybrid β,α-dipeptides featuring hydrophobic residues, and, in particular, a β-Ala at the N-terminus, followed by an α-amino acid (i.e., Phe, Ile, or Val) were reported to form NTs too, with good thermal and proteolytic stability.110 These NTs were used as scaffolds for the in situ production of gold nanoparticles on their surface.110 Alternatively, the decoration of β-Ala-α-Ile with fluorescent Au25 quantum clusters was exploited to the same end, by using electron-beam irradiation as a means to control the gold-nanoparticle growth and size distribution.111
By contrast, when both residues of hydrophobic dipeptides were composed of β- or γ-amino aicds (i.e., Boc-β3(R)Phe-β3(R)Phe and Boc-γ4(R)Phe-γ4(R)Phe) NTs were not formed, and other morphologies could be attained depending on experimental conditions, and including nanofibers and nanospheres.112
The dipeptide displaying Ala and the S,S (but not the R,R) stereoisomer of 3-amino-2-(2-fluorophenyl)-3-phenylpropanoic acid was shown to self-assemble into proteolytically stable NTs, thanks to the presence of the β-amino acid. Interestingly, the XRD structure of this dipeptide revealed a supramolecular helix with three peptide molecules per turn, with the screw axis corresponding to the crystallographic c axis. These NTs featured a hydrophobic inner channel with a diameter of 4 Å that was decorated by the methyl sidechains. No evidence for aromatic stacking was found in the crystal structure, suggesting that intermolecular H-bonds proved key in holding the NT architecture.113 Finally, also β-Ala-Ala and δ-Ava-Phe (Ava = aminovaleric acid) were reported to self-assemble into NTs.114
δ-Amino acids as dipeptide mimics and δ-dipeptides
δ-Amino acids are characterised by four atoms (typically carbon) between the amino and carboxylic functionalities and are seldom studied, possibly because of the synthetic steps needed and their limited solubility in water. They have been proposed as dipeptide mimics. In particular, β-Oxy-δ5-amino acids bear an oxygen atom in their backbone that improves their solubility.115 Inclusion of a cycle proved to be a successful strategy to restrict conformational freedom towards foldamers.116,117 The combination of both components in cyclohexyl ether δ-amino acids was successfully applied in building blocks to attain supramolecular channels with cation selectivity, thanks to the strategic disposition of the additional oxygen atom in the channel interior.118Cyclodipeptides (CDPs)
CDPs have attracted attention as building blocks for biocompatible nanostructures, since they are naturally occurring in a variety of food and beverages, and their rigidity and enhanced stability towards enzymatic degradation relative to their linear analogs, has enabled their use towards supramolecular nanostructures and soft materials for a variety of applications, as recently reviewed.119,120 NTs and microtubes were reported for homochiral and heterochiral cyclo(Tyr-Ala), respectively.121A different assembly was obtained using cyclodipeptides (CDPs) derivatised with long alkyl chains displaying photopolymerizable diacetylene functionalities for thermochromism and enable a lamellar structure to arise that rolled up into nanotubes (Fig. 5).122 Interestingly, these systems displayed a blue-to-red thermochromism upon heating from room temperature to 90 °C. Furthermore, appropriate choice of solvent polarity could be exploited to guide self-assembly towards different nanomorphologies.123
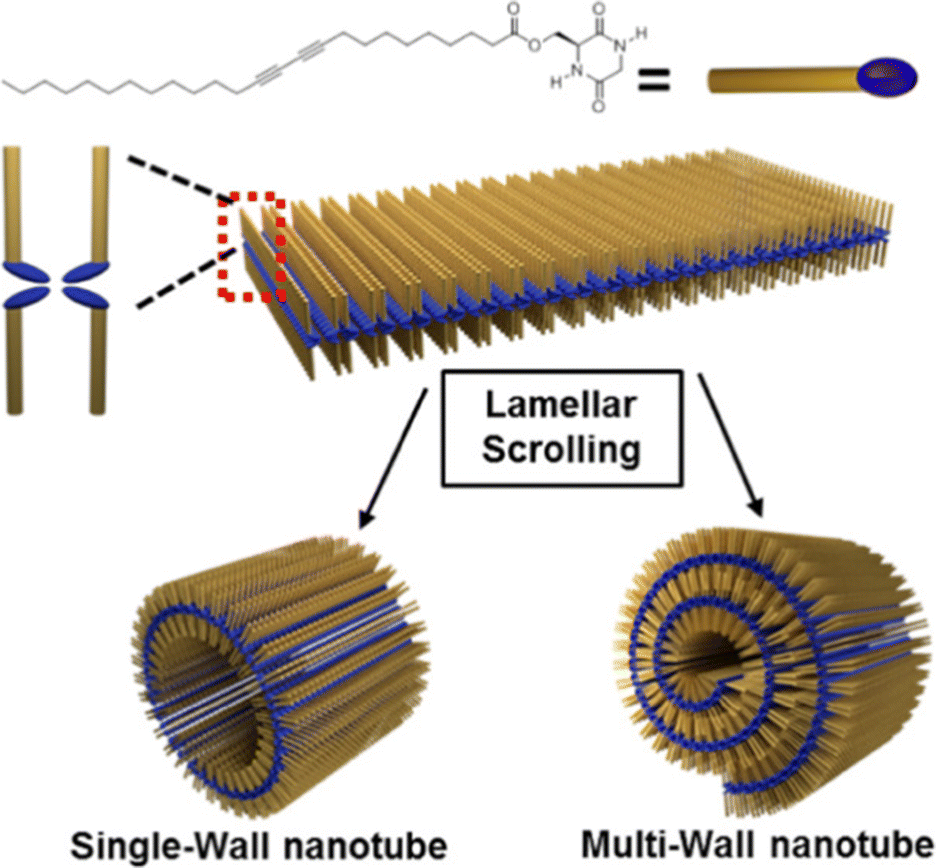 | ||
| Fig. 5 Cyclodipeptide derivative with an alkyl chain enabled the formation of lamellar structures that rolled up into NTs. Reproduced with permission from M. J. Seo et al. Langmuir, 2018, 34, 8365, Copyright © 2018, American Chemical Society.122 | ||
Cyclo(Gly-Asp) amidated through the sidechain with a long alkyl chain (C8–C18) self-assembled into various nanomorphologies, including NTs that were envisaged for drug loading and release, and studied for their co-assembly ability with nucleoside biomolecules.124
Dipeptides with non-natural aromatic structures
The key role played by the aromatic sidechains of Phe-Phe in the stability and physicochemical properties of the NTs125 has inspired the search for several analogs with differing aromatic moieties. Interestingly, the simple removal of the methylene units leading to Phg-Phg (Phg = phenylglycine), with the phenyl ring being directly attached to the peptide backbone, completely hindered the NT formation, leading to nanospherical morphologies instead.126 Conversely, dinaphthylalanine formed NTs,127 as well as Boc-Aib-m-ABA-OMe (Aib = α-aminoisobutyric acid; m-ABA = m-aminobenzoic acid).128 4′-(n-dodecyloxy)-1,1′-biphenyl-4-carbonyl Ala-Ala homo- and heterochiral isomers formed coiled nanoribbons that gave rise to NTs. Interestingly, the stereoconfiguration of the C-terminal Ala residue dictated the handedness of the coiled nanoribbons, and the stacking handedness of the biphenylene moiety. Conversely, the N-terminal Ala chirality determined the twist handedness of the aromatic groups.129 Another study showed that NTs with 6-fold symmetry could be obtained from the self-assembly of Boc-(S,S)c3diPhe-(R,R)c3diPhe-NHiPr, which is a dipeptide with disubstituted cyclopropyl sidechains with two phenyl rings each.130 This peculiar structure enabled the display of one of the aromatic units for each dipeptide in the inner cavity of the NT, in striking contrast with the typical NT architectures that feature hydrophilic peptide backbone at the NT interior and hydrophobic sidechains at the exterior.Applications
Clearly dipeptide NTs are ideally positioned for biological uses, spanning from drug delivery to cell culture and imaging. Control of self-assembly can be attained by using physiological or pathological triggers at the site of interest. A classical example is based on the enzymatic cleavage of a self-immolative unit, such as a dendrimer, to release the self-assembling dipeptide.131 C-terminal amidation of Phe-Phe was used to yield a cationic dipeptide that at physiological pH could self-assemble into NTs that could bind the polyanionic oligonucleotides and that, contrarily to the zwitterionic Phe-Phe, transitioned to vesicles when diluted to 7 mg ml−1. The vesicles could then be taken up by cells for oligonucleotide delivery.132 Flufenamic acid was used as a model drug that could be loaded in the hydrophobic walls of Phe-Phe NTs for sustained release over days.133 Crosslinking with glutaraldehyde has been proposed as a strategy to control the nanoarchitectures formed by Phe-Phe for drug delivery.134 The dipeptide Lys-Lys was conjugated through the amino sidechain to the anticancer drug camptothecin, which could be loaded at very high levels through hydrophobic interactions within the dipeptide NT walls, and slowly released through hydrolysis of a succinyl linker.135 In a similar approach, double conjugation of Lys-Lys with captothecin and 5-fluorouracil was used for the sequential release of the two drugs from the dipeptide NTs.136 Alternatively, the inclusion of bioactive residues in dipeptides could also exert bioactivity, as demonstrated with L-DOPA in (S)-isopropyl 2-(2-amino-2-methylpropanamido)-3-(3,4-dihydroxyphenyl)propanoate that displayed anti-Parkinsionan activity in a rat model after oral administration.137In light of the emergency we are facing in terms of emergence of antibiotic resistance, it is clear how the use of Phe-Phe NTs as antimicrobial agents is another hot topic. These NTs have been shown to eradicate Gram-positive bacterial biofilms, thanks to the formation of ion channels and/or surfactant-like activity, leading to biofilm matrix degradation and cell-membrane disruption, leading to cell death in S. aureus isolates.138 They inhibited bacterial growth and led to cell death also of E. coli, thanks to an increase in cell membrane permeation and depolarization.139
Further applications can be envisaged by decorating the NTs with biomolecules, for instance as shown with biotin that was successfully recognized by avidin.140S-Glycosylation has also been explored as an avenue to fine-tune self-assembly of Phe-Phe.141 For instance, addition of one, but not two, mannose units preserved Phe-Phe ability to form NTs, and recognition by lectins was envisaged to direct these materials towards biological use.142
Another growing field concerns bioelectronic applications towards sustainable and non-toxic alternatives to current systems in use. Several methods have been developed to orient and pattern dipeptide NTs into arrays for bioelectronics,143–147 also on electrodes.148,149 It was shown that even weak electric fields can facilitate the alignment of molecular dipole moments, accelerating the ordering of dipeptide and water molecules for self-assembly.150 Alignment of Phe-Phe NTs is key to achieve unidirectional polarization and maximise their piezoelectric response for power generation, i.e., for electrical energy production upon application of a mechanical force. Horizontally aligned Phe-Phe NT arrays have been used also as piezoelectric devices. Upon application of a force of 42 N, a peak voltage of 2.8 V was reached, with a current of 37.4 nA, and generated 8.2 nW that could power multiple liquid-crystal display panels.151 For comparison, with an applied force of 60 N, vertically aligned Phe-Phe NTs generated an open-circuit voltage of 1.4 V with a short-circuit current of 39.2 nA.152 Inclusion of porphyrins has been used as a strategy to slow down the biodegradation of Phe-Phe NTs in physiological conditions towards uses in implantable medical devices.153
The blue photoluminescence of dipeptide NTs has been envisaged as a biolabel, for applications in bioimaging and biosensing, thanks also to its persistence upon thermal treatment at temperatures in the 140–180 °C range, at which NTs undergo a transition to nanowires.154 Their variations in photoluminescence have been applied for temperature sensing, with precision of 1 °C in the range 5–65 °C.155 Dipeptide NTs have been studied also to develop biosensors for the electrochemical determination of dopamine.156 They have been used as antennas or photosensitisers to enhance lanthanide luminescence for the detection of the neurotoxin paraoxon too.157
Phe-Phe microtubes have been envisaged also as optical waveguides, as demonstrated by illuminating one end of the microstructures and observing a bright spot of luminescence at the other end. However, for practical applications, further improvement to reduce attenuation through the length of the tubes are needed, possibly by improving the crystallinity or finding suitable derivatives.158,159
Finally, Phe-Phe NTs have been considered as nanofillers for biomedical materials, as they demonstrated to increase toughness and adhesive strength of epoxy resins, whilst preserving the elongation and thermal properties of the polymer matrix.160
Conclusions and future perpectives
Dipeptide NTs have enjoyed two decades of intense research efforts worldwide since their early reports. Through the years, a plethora of NT-forming dipeptides have been reported, and their biological uses have been envisaged through a variety of triggers to control the NT formation, their uptake by cells, and their release of bioactive compounds. The understanding of the key structural features of the dipeptides leading to NTs is still far from complete, however. Nevertheless, Görbitz made a tremendous effort to map the NTs architectures attained by simple hydrophobic L-dipeptides, showing that the aliphatic sequences yield NTs with hydrophobic interiors, whilst the aromatic ones tend to yield NTs with water-filled inner cavities. Changes in amino acid sequence can also be used to control the inner cavity diameter, as some preferentially form those enclosed by four dipeptides (leading to a cavity of ∼0.5 nm), and others by six (leading to a cavity of ∼1 nm). Bigger channels (e.g., ∼2 nm) can be attained by switching to tripeptides.161 The control of NT growth for potential insertions into membranes could clearly open up new avenues, for instance to alter transport of guests selectively and find new means for therapeutic treatment.162 Amongst the various approaches to control NT length, microfluidics is emerging as one possible strategy.163 Alternatively, gases could be envisaged as channel guests, and one study appeared showing that porous dipeptide crystals have a remarkable selectivity for methane, carbon dioxide, and hydrogen absorption.164Other areas of great interest pertain bioelectronics, to find sustainable and biodegradable alternatives to currently used components. A key challenge in this field is its highly multidisciplinary nature, which requires expertise in fields as diverse as engineering, photophysics, chemistry, and nanotechnology to make a qualitative leap. Nevertheless, the allure of being able one day to use biodegradable components for our everyday commodity devices is a great driver to innovate in this field and it is reasonable to expect that implementation into commercial devices is on its way to be soon realized on a large scale for the benefit of society.
Author contributions
O. B. wrote the manuscript draft, under the supervision of P. D. A. and S. M. that guided the study, and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the University of Trieste for providing a PhD scholarship to O. B. and funding to S. M. through FRA2022.Notes and references
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- K. Aoki, N. Ogihara, M. Tanaka, H. Haniu and N. Saito, J. Mater. Chem. B, 2020, 8, 9227–9238 RSC.
- Z. Peng, T. Zhao, Y. Zhou, S. Li, J. Li and R. M. Leblanc, Adv. Healthcare Mater., 2020, 9, e1901495 CrossRef PubMed.
- S. Marchesan, L. Ballerini and M. Prato, Science, 2017, 356, 1010–1011 CrossRef CAS PubMed.
- L. Wieland, H. Li, C. Rust, J. Chen and B. S. Flavel, Adv. Energy Mater., 2021, 11, 2002880 CrossRef CAS.
- M. Melchionna and P. Fornasiero, ChemCatChem, 2017, 9, 3274–3284 CrossRef CAS.
- D. Iglesias and M. Melchionna, Catalysts, 2019, 9, 128 CrossRef.
- Z. Xu, W. Deng and X. Wang, Electrochem. Energy Rev., 2021, 4, 269–335 CrossRef CAS.
- J. S. Bulmer, A. Kaniyoor and J. A. Elliott, Adv. Mater., 2021, 33, 2008432 CrossRef CAS PubMed.
- C. Liang, Y. Liu, W. Lu, G. Tian, Q. Zhao, D. Yang, J. Sun and D. Qi, Nanoscale, 2022, 14, 3346–3366 RSC.
- M. Serra, R. Arenal and R. Tenne, Nanoscale, 2019, 11, 8073–8090 RSC.
- D. Zhang, S. Zhang, N. Yapici, R. Oakley, S. Sharma, V. Parashar and Y. K. Yap, ACS Omega, 2021, 6, 20722–20728 CrossRef CAS PubMed.
- M. Dai and R. Wang, Small, 2021, 17, 2006813 CrossRef CAS PubMed.
- I. W. Hamley, Angew. Chem., Int. Ed., 2014, 53, 6866–6881 CrossRef CAS PubMed.
- Q. Song, Z. Cheng, M. Kariuki, S. C. L. Hall, S. K. Hill, J. Y. Rho and S. Perrier, Chem. Rev., 2021, 121, 13936–13995 CrossRef CAS PubMed.
- G. Praveena, P. Kolandaivel, N. Santhanamoorthi, V. Renugopalakrishnan and S. Ramakrishna, J. Nanosci. Nanotechnol., 2007, 7, 2253–2259 CrossRef CAS PubMed.
- N. Santhanamoorthi, P. Kolandaivel, L. Adler-Abramovich, E. Gazit, S. Filipek, S. Viswanathan, A. Strzelczyk and V. Renugopalakrishnan, Adv. Mater. Lett., 2011, 2, 100–105 CrossRef CAS.
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324–327 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS PubMed.
- C. H. Görbitz, Chem. – Eur. J., 2001, 7, 5153–5159 CrossRef.
- C. H. Görbitz, Chem. Commun., 2006, 2332–2334 RSC.
- N. Amdursky, P. Beker, I. Koren, B. Bank-Srour, E. Mishina, S. Semin, T. Rasing, Y. Rosenberg, Z. Barkay, E. Gazit and G. Rosenman, Biomacromolecules, 2011, 12, 1349–1354 CrossRef CAS PubMed.
- A. Handelman, A. Natan and G. Rosenman, J. Pept. Sci., 2014, 20, 487–493 CrossRef CAS PubMed.
- Z. Gan, X. Wu, X. Zhu and J. Shen, Angew. Chem., Int. Ed., 2013, 52, 2055–2059 CrossRef CAS PubMed.
- N. Kol, L. Adler-Abramovich, D. Barlam, R. Z. Shneck, E. Gazit and I. Rousso, Nano Lett., 2005, 5, 1343–1346 CrossRef CAS PubMed.
- L. Niu, X. Chen, S. Allen and S. J. Tendler, Langmuir, 2007, 23, 7443–7446 CrossRef CAS PubMed.
- I. Azuri, L. Adler-Abramovich, E. Gazit, O. Hod and L. Kronik, J. Am. Chem. Soc., 2014, 136, 963–969 CrossRef CAS PubMed.
- V. Basavalingappa, S. Bera, B. Xue, J. O'Donnell, S. Guerin, P. A. Cazade, H. Yuan, E. U. Haq, C. Silien, K. Tao, L. J. W. Shimon, S. A. M. Tofail, D. Thompson, S. Kolusheva, R. Yang, Y. Cao and E. Gazit, ACS Nano, 2020, 14, 7025–7037 CrossRef CAS PubMed.
- P. Zelenovskiy, I. Kornev, S. Vasilev and A. Kholkin, Phys. Chem. Chem. Phys., 2016, 18, 29681–29685 RSC.
- A. Kalinin, V. Atepalikhin, O. Pakhomov, A. L. Kholkin and A. Tselev, Ultramicroscopy, 2018, 185, 49–54 CrossRef CAS PubMed.
- M. Wang, L. Du, X. Wu, S. Xiong and P. K. Chu, ACS Nano, 2011, 5, 4448–4454 CrossRef CAS PubMed.
- V. S. Bystrov and S. V. Filippov, J. Mol. Model., 2022, 28, 81 CrossRef CAS PubMed.
- X. Yan, Y. Cui, Q. He, K. Wang, J. Li, W. Mu, B. Wang and Z. C. Ou-Yang, Chem. – Eur. J., 2008, 14, 5974–5980 CrossRef CAS PubMed.
- A. Levin, T. O. Mason, L. Adler-Abramovich, A. K. Buell, G. Meisl, C. Galvagnion, Y. Bram, S. A. Stratford, C. M. Dobson, T. P. Knowles and E. Gazit, Nat. Commun., 2014, 5, 5219 CrossRef CAS PubMed.
- Q. Xiong, Y. Jiang, X. Cai, F. Yang, Z. Li and W. Han, ACS Nano, 2019, 13, 4455–4468 CrossRef CAS PubMed.
- A. C. C. Ribeiro, G. A. Souza, D. H. Pereira, D. S. Cordeiro, R. S. Miranda, R. Custódio and T. D. Martins, ACS Omega, 2019, 4, 606–619 CrossRef CAS PubMed.
- P. S. Zelenovskiy, E. M. Domingues, V. Slabov, S. Kopyl, V. L. Ugolkov, F. M. L. Figueiredo and A. L. Kholkin, ACS Appl. Mater. Interfaces, 2020, 12, 27485–27492 CrossRef CAS PubMed.
- J. Liu, J. Fan, M. Tang, M. Cen, J. Yan, Z. Liu and W. Zhou, J. Phys. Chem. B, 2010, 114, 12183–12192 CrossRef CAS PubMed.
- M. Engels, D. Bashford and M. R. Ghadiri, J. Am. Chem. Soc., 1995, 117, 9151–9158 CrossRef CAS.
- S. W. Chiu, E. Jakobsson, S. Subramaniam and J. A. McCammon, Biophys. J., 1991, 60, 273–285 CrossRef CAS PubMed.
- J. B. Heymann and A. Engel, News Physiol. Sci., 1999, 14, 187–193 CAS.
- Q. Zhao, P. Majsztrik and J. Benziger, J. Phys. Chem. B, 2011, 115, 2717–2727 CrossRef CAS PubMed.
- S. J. Paddison and R. Paul, Phys. Chem. Chem. Phys., 2002, 4, 1158–1163 RSC.
- T. Yamamoto, Y. H. Kim, B. C. Kim, A. Endo, N. Thongprachan and T. Ohmori, Chem. Eng. J., 2012, 181–182, 443–448 CrossRef CAS.
- T. Honório, T. Lemaire, D. D. Tommaso and S. Naili, Comput. Mater. Sci., 2019, 156, 26–34 CrossRef.
- J. H. Wang, J. Phys. Chem., 1965, 69, 4412 CrossRef CAS.
- J. Hassan, G. Diamantopoulos, L. Gkoura, M. Karagianni, S. Alhassan, S. V. Kumar, M. S. Katsiotis, T. Karagiannis, M. Fardis, N. Panopoulos, H. J. Kim, M. Beazi-Katsioti and G. Papavassiliou, J. Phys. Chem. C, 2018, 122, 10600–10606 CrossRef CAS.
- K. Gnanasekaran, J. Korpanty, O. Berger, N. Hampu, M. Halperin-Sternfeld, D. Cohen-Gerassi, L. Adler-Abramovich and N. C. Gianneschi, ACS Nano, 2021, 15, 16542–16551 CrossRef CAS PubMed.
- H. Zhang, K. Siegrist, D. F. Plusquellic and S. K. Gregurick, J. Am. Chem. Soc., 2008, 130, 17846–17857 CrossRef CAS PubMed.
- T. Andrade-Filho, F. F. Ferreira, W. A. Alves and A. R. Rocha, Phys. Chem. Chem. Phys., 2013, 15, 7555–7559 RSC.
- M. Wang, S. Xiong, X. Wu and P. K. Chu, Small, 2011, 7, 2801–2807 CrossRef CAS PubMed.
- C. Guo, Y. Luo, R. Zhou and G. Wei, ACS Nano, 2012, 6, 3907–3918 CrossRef CAS PubMed.
- J. Kim, T. H. Han, Y. I. Kim, J. S. Park, J. Choi, D. G. Churchill, S. O. Kim and H. Ihee, Adv. Mater., 2010, 22, 583–587 CrossRef CAS PubMed.
- X. Wu, S. Xiong, M. Wang, J. Shen and P. K. Chu, Opt. Express, 2012, 20, 5119–5126 CrossRef CAS PubMed.
- J. Wang, C. Yuan, Y. Han, Y. Wang, X. Liu, S. Zhang and X. Yan, Small, 2017, 13, 1702175 CrossRef PubMed.
- J. Anderson, P. T. Lake and M. McCullagh, J. Phys. Chem. B, 2018, 122, 12331–12341 CrossRef CAS PubMed.
- J. Wang, K. Liu, L. Yan, A. Wang, S. Bai and X. Yan, ACS Nano, 2016, 10, 2138–2143 CrossRef CAS PubMed.
- C. H. Görbitz and E. Gundersen, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 1764–1767 CrossRef.
- C. H. Görbitz, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 849–854 CrossRef PubMed.
- C. H. Görbitz, New J. Chem., 2003, 27, 1789–1793 RSC.
- C. H. Görbitz, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2018, 74, 311–318 CrossRef PubMed.
- H. Erdogan, E. Babur, M. Yilmaz, E. Candas, M. Gordesel, Y. Dede, E. E. Oren, G. B. Demirel, M. K. Ozturk, M. S. Yavuz and G. Demirel, Langmuir, 2015, 31, 7337–7345 CrossRef CAS PubMed.
- C. Görbitz and E. Gundersen, Acta Chem. Scand., 1996, 50, 537–543 CrossRef.
- C. Görbitz, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, o328–o330 CrossRef PubMed.
- C. H. Görbitz, Chem. – Eur. J., 2007, 13, 1022–1031 CrossRef PubMed.
- P. Macha, L. Perreault, Y. Hamedani, M. L. Mayes and M. C. Vasudev, ACS Appl. Bio Mater., 2018, 1, 1266–1275 CrossRef CAS PubMed.
- D. Giuri, P. Ravarino and C. Tomasini, Org. Biomol. Chem., 2021, 19, 4622–4636 RSC.
- G. Fichman, L. Adler-Abramovich, S. Manohar, I. Mironi-Harpaz, T. Guterman, D. Seliktar, P. B. Messersmith and E. Gazit, ACS Nano, 2014, 8, 7220–7228 CrossRef CAS PubMed.
- C. H. Görbitz, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, o371–o373 CrossRef PubMed.
- R. J. Fletterick, C. C. Tsai and R. E. Hughes, J. Phys. Chem., 1971, 75, 918–922 CrossRef CAS PubMed.
- C. Görbitz, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, o647–o650 CrossRef.
- A. V. Vargiu, D. Iglesias, K. E. Styan, L. J. Waddington, C. D. Easton and S. Marchesan, Chem. Commun., 2016, 52, 5912–5915 RSC.
- S. Marchesan, K. E. Styan, C. D. Easton, L. Waddington and A. V. Vargiu, J. Mater. Chem. B, 2015, 3, 8123–8132 RSC.
- M. C. Cringoli, C. Romano, E. Parisi, L. J. Waddington, M. Melchionna, S. Semeraro, R. De Zorzi, M. Grönholm and S. Marchesan, Chem. Commun., 2020, 56, 3015–3018 RSC.
- S. Kralj, O. Bellotto, E. Parisi, A. M. Garcia, D. Iglesias, S. Semeraro, C. Deganutti, P. D′Andrea, A. V. Vargiu, S. Geremia, R. De Zorzi and S. Marchesan, ACS Nano, 2020, 14, 16951–16961 CrossRef CAS PubMed.
- E. Scarel, O. Bellotto, P. Rozhin, S. Kralj, M. Tortora, A. V. Vargiu, R. De Zorzi, B. Rossi and S. Marchesan, Soft Matter, 2022, 18, 2129–2136 RSC.
- O. Bellotto, G. Pierri, P. Rozhin, M. Polentarutti, S. Kralj, P. D'Andrea, C. Tedesco and S. Marchesan, Org. Biomol. Chem., 2022, 20, 6211–6218 RSC.
- O. Bellotto, S. Kralj, R. De Zorzi, S. Geremia and S. Marchesan, Soft Matter, 2020, 16, 10151–10157 RSC.
- O. Bellotto, S. Kralj, M. Melchionna, P. Pengo, M. Kisovec, M. Podobnik, R. De Zorzi and S. Marchesan, ChemBioChem, 2022, 23, e202100518 CrossRef CAS PubMed.
- H. L. Bolt, C. E. J. Williams, R. V. Brooks, R. N. Zuckermann, S. L. Cobb and E. H. C. Bromley, Biopolymers, 2017, 108, e23014 CrossRef PubMed.
- O. Bellotto, PhD thesis, University of Trieste, 2022.
- E. Scarel, G. Pierri, P. Rozhin, S. Adorinni, M. Polentarutti, C. Tedesco and S. Marchesan, Chemistry, 2022, 4, 1417–1428 CrossRef CAS.
- M. Reches and E. Gazit, Isr. J. Chem., 2005, 45, 363–371 CrossRef CAS.
- L. Adler-Abramovich and E. Gazit, J. Pept. Sci., 2008, 14, 217–223 CrossRef CAS PubMed.
- R. M. F. Baptista, E. de Matos Gomes, M. M. M. Raposo, S. P. G. Costa, P. E. Lopes, B. Almeida and M. S. Belsley, Nanoscale Adv., 2019, 1, 4339–4346 RSC.
- A. Kholkin, N. Amdursky, I. Bdikin, E. Gazit and G. Rosenman, ACS Nano, 2010, 4, 610–614 CrossRef CAS PubMed.
- L. Adler-Abramovich, P. Marco, Z. A. Arnon, R. C. Creasey, T. C. Michaels, A. Levin, D. J. Scurr, C. J. Roberts, T. P. Knowles, S. J. Tendler and E. Gazit, ACS Nano, 2016, 10, 7436–7442 CrossRef CAS PubMed.
- S. Yuran, Y. Razvag and M. Reches, ACS Nano, 2012, 6, 9559–9566 CrossRef CAS PubMed.
- C. Subbalakshmi, S. V. Manorama and R. Nagaraj, J. Pept. Sci., 2012, 18, 283–292 CrossRef CAS PubMed.
- D. Mazzier, F. Carraro, M. Crisma, M. Rancan, C. Toniolo and A. Moretto, Soft Matter, 2016, 12, 238–245 RSC.
- C. Subbalakshmi, P. Basak and R. Nagaraj, Biopolymers, 2017, 108, e23033 CrossRef PubMed.
- B. Di Blasio, M. Saviano, V. Del Duca, G. De Simone, F. Rossi, C. Pedone, E. Benedetti and G. P. Lorenzi, Biopolymers, 1995, 36, 401–408 CrossRef CAS PubMed.
- X. Wang, L. Zhao, C. Wang, X. Feng, Q. Ma, G. Yang, T. Wang, X. Yan and J. Jiang, Small, 2022, 18, e2104438 CrossRef PubMed.
- Y. M. Abul-Haija and R. V. Ulijn, Biomacromolecules, 2015, 16, 3473–3479 CrossRef CAS PubMed.
- P. C. Saha, T. Bera, T. Chatterjee, J. Samanta, A. Sengupta, M. Bhattacharyya and S. Guha, Bioconjugate Chem., 2021, 32, 833–841 CrossRef CAS PubMed.
- Y. Sun, J. A. Kaplan, A. Shieh, H. L. Sun, C. M. Croce, M. W. Grinstaff and J. R. Parquette, Chem. Commun., 2016, 52, 5254–5257 RSC.
- R. D. Chakravarthy, M. Mohammed and H.-C. Lin, Chem. – Asian J., 2020, 15, 2696–2705 CrossRef CAS PubMed.
- M. Lisowski, G. Pietrzyński and B. Rzeszotarska, Int. J. Pept. Protein Res., 1993, 42, 466–474 CrossRef CAS PubMed.
- C. Pietrzyńaski, B. Rzeszotarska, E. Ciszak, M. Lisowski, Z. Kubica and G. Boussard, Int. J. Pept. Protein Res., 1996, 48, 347–356 CrossRef.
- M. Gupta, A. Bagaria, A. Mishra, P. Mathur, A. Basu, S. Ramakumar and V. S. Chauhan, Adv. Mater., 2007, 19, 858–861 CrossRef CAS.
- P. Verma, S. Biswas, N. Yadav, A. Khatri, H. Siddiqui, J. J. Panda, B. S. Rawat, P. Tailor and V. S. Chauhan, Mol. Pharming, 2021, 18, 3832–3842 CrossRef CAS PubMed.
- S. Alam, J. J. Panda, T. K. Mukherjee and V. S. Chauhan, J. Nanobiotechnol., 2016, 14, 26 CrossRef PubMed.
- F. Clerici, E. Erba, M. L. Gelmi and S. Pellegrino, Tetrahedron Lett., 2016, 57, 5540–5550 CrossRef CAS.
- G. Lelais and D. Seebach, Biopolymers, 2004, 76, 206–243 CrossRef CAS PubMed.
- F. Kudo, A. Miyanaga and T. Eguchi, Nat. Prod. Rep., 2014, 31, 1056–1073 RSC.
- A. A. Boldyrev, G. Aldini and W. Derave, Physiol. Rev., 2013, 93, 1803–1845 CrossRef CAS PubMed.
- V. Castelletto, C. J. C. Edwards-Gayle, F. Greco, I. W. Hamley, J. Seitsonen and J. Ruokolainen, ACS Appl. Mater. Interfaces, 2019, 11, 33573–33580 CrossRef CAS PubMed.
- V. Castelletto, G. Cheng, B. W. Greenland, I. W. Hamley and P. J. Harris, Langmuir, 2011, 27, 2980–2988 CrossRef CAS PubMed.
- S. Parween, A. Misra, S. Ramakumar and V. S. Chauhan, J. Mater. Chem. B, 2014, 2, 3096–3106 RSC.
- S. Guha and A. Banerjee, Adv. Funct. Mater., 2009, 19, 1949–1961 CrossRef CAS.
- P. Ramasamy, S. Guha, E. S. Shibu, T. S. Sreeprasad, S. Bag, A. Banerjee and T. Pradeep, J. Mater. Chem., 2009, 19, 8456–8462 RSC.
- B. Dinesh, M. A. Squillaci, C. Ménard-Moyon, P. Samorì and A. Bianco, Nanoscale, 2015, 7, 15873–15879 RSC.
- A. Bonetti, S. Pellegrino, P. Das, S. Yuran, R. Bucci, N. Ferri, F. Meneghetti, C. Castellano, M. Reches and M. L. Gelmi, Org. Lett., 2015, 17, 4468–4471 CrossRef CAS PubMed.
- S. Guha, M. G. B. Drew and A. Banerjee, Chem. Mater., 2008, 20, 2282–2290 CrossRef.
- R. M. Reja, R. Patel, V. Kumar, A. Jha and H. N. Gopi, Biomacromolecules, 2019, 20, 1254–1262 CrossRef CAS PubMed.
- H.-D. Arndt, B. Ziemer and U. Koert, Org. Lett., 2004, 6, 3269–3272 CrossRef CAS PubMed.
- E. O’Reilly, L. Pes, Y. Ortin, H. Müller-Bunz and F. Paradisi, Amino Acids, 2013, 44, 511–518 CrossRef PubMed.
- H.-D. Arndt, A. Knoll and U. Koert, Angew. Chem., Int. Ed., 2001, 40, 2076–2078 CrossRef CAS PubMed.
- S. Manchineella and T. Govindaraju, ChemPlusChem, 2017, 82, 88–106 CrossRef CAS PubMed.
- M. Scarel and S. Marchesan, Molecules, 2021, 26, 3376 CrossRef CAS PubMed.
- A. Jeziorna, K. Stopczyk, E. Skorupska, K. Luberda-Durnas, M. Oszajca, W. Lasocha, M. Gorecki, J. Frelek and M. J. Potrzebowski, Cryst. Growth Des., 2015, 15, 5138–5148 CrossRef CAS.
- M. J. Seo, J. Song, C. Kantha, M. I. Khazi, U. Kundapur, J. M. Heo and J. M. Kim, Langmuir, 2018, 34, 8365–8373 CrossRef CAS PubMed.
- M. I. Khazi, C. Balachandra, G. Shin, G. H. Jang, T. Govindaraju and J. M. Kim, RSC Adv., 2020, 10, 35389–35396 RSC.
- K. Pandurangan, B. Roy, K. Rajasekhar, Y. V. Suseela, P. Nagendra, A. Chaturvedi, U. R. Satwik, N. A. Murugan, U. Ramamurty and T. Govindaraju, ACS Appl. Bio Mater., 2020, 3, 3413–3422 CrossRef CAS PubMed.
- L. Adler-Abramovich, M. Reches, V. L. Sedman, S. Allen, S. J. B. Tendler and E. Gazit, Langmuir, 2006, 22, 1313–1320 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Nano Lett., 2004, 4, 581–585 CrossRef CAS.
- V. L. Sedman, S. Allen, X. Chen, C. J. Roberts and S. J. Tendler, Langmuir, 2009, 25, 7256–7259 CrossRef CAS PubMed.
- S. Kar and Y. Tai, Soft Matter, 2015, 11, 1345–1351 RSC.
- L. Zhang, J. Qin, S. Lin, Y. Li, B. Li and Y. Yang, Langmuir, 2017, 33, 10951–10957 CrossRef CAS PubMed.
- M. Crisma, C. Toniolo, S. Royo, A. I. Jiménez and C. Cativiela, Org. Lett., 2006, 8, 6091–6094 CrossRef CAS PubMed.
- L. Adler-Abramovich, R. Perry, A. Sagi, E. Gazit and D. Shabat, ChemBioChem, 2007, 8, 859–862 CrossRef CAS PubMed.
- X. Yan, Q. He, K. Wang, L. Duan, Y. Cui and J. Li, Angew. Chem., Int. Ed., 2007, 46, 2431–2434 CrossRef CAS PubMed.
- T. Zohrabi, N. Habibi, A. Zarrabi, M. Fanaei and L. Y. Lee, J. Biomed. Mater. Res., Part A, 2016, 104, 2280–2290 CrossRef CAS PubMed.
- A. Wu, Y. Guo, X. Li, Q. Li, G. Chen, H. Zang and J. Li, J. Colloid Interface Sci., 2023, 630, 161–169 CrossRef CAS PubMed.
- S. H. Kim, J. A. Kaplan, Y. Sun, A. Shieh, H. L. Sun, C. M. Croce, M. W. Grinstaff and J. R. Parquette, Chem. – Eur. J., 2015, 21, 101–105 CrossRef CAS PubMed.
- Y. Sun, C. M. Fry, A. Shieh, X. Cai, T. J. Reardon and J. R. Parquette, Org. Biomol. Chem., 2022, 20, 5254–5258 RSC.
- T. Zhou, R. C. Hider, P. Jenner, B. Campbell, C. J. Hobbs, S. Rose, M. Jairaj, K. A. Tayarani-Binazir and A. Syme, Bioorg. Med. Chem. Lett., 2013, 23, 5279–5282 CrossRef CAS PubMed.
- S. L. Porter, S. M. Coulter, S. Pentlavalli, T. P. Thompson and G. Laverty, Acta Biomater., 2018, 77, 96–105 CrossRef CAS PubMed.
- L. Schnaider, S. Brahmachari, N. W. Schmidt, B. Mensa, S. Shaham-Niv, D. Bychenko, L. Adler-Abramovich, L. J. W. Shimon, S. Kolusheva, W. F. De Grado and E. Gazit, Nat. Commun., 2017, 8, 1–10 CrossRef CAS PubMed.
- M. Reches and E. Gazit, J. Nanosci. Nanotechnol., 2007, 7, 2239–2245 CrossRef CAS PubMed.
- R. Roytman, L. Adler-Abramovich, K. S. Kumar, T. C. Kuan, C. C. Lin, E. Gazit and A. Brik, Org. Biomol. Chem., 2011, 9, 5755–5761 RSC.
- N. Gour, A. K. Barman and S. Verma, J. Pept. Sci., 2012, 18, 405–412 CrossRef CAS PubMed.
- L. Adler-Abramovich, D. Aronov, E. Gazit and G. Rosenman, J. Nanosci. Nanotechnol., 2009, 9, 1701–1708 CrossRef CAS PubMed.
- L. Adler-Abramovich, D. Aronov, P. Beker, M. Yevnin, S. Stempler, L. Buzhansky, G. Rosenman and E. Gazit, Nat. Nanotechnol., 2009, 4, 849–854 CrossRef CAS PubMed.
- J. Castillo, S. Tanzi, M. Dimaki and W. Svendsen, Electrophoresis, 2008, 29, 5026–5032 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Nat. Nanotechnol., 2006, 1, 195–200 CrossRef CAS PubMed.
- X. Liu, J. Danglad-Flores, S. Eickelmann, B. Sun, J. Hao, H. Riegler and J. Li, ACS Nano, 2022, 16, 10372–10382 CrossRef CAS PubMed.
- L. Adler-Abramovich, M. Badihi-Mossberg, E. Gazit and J. Rishpon, Small, 2010, 6, 825–831 CrossRef CAS PubMed.
- M. C. Vasudev, H. Koerner, K. M. Singh, B. P. Partlow, D. L. Kaplan, E. Gazit, T. J. Bunning and R. R. Naik, Biomacromolecules, 2014, 15, 533–540 CrossRef CAS PubMed.
- C. M. Kelly, T. Northey, K. Ryan, B. R. Brooks, A. L. Kholkin, B. J. Rodriguez and N. V. Buchete, Biophys. Chem., 2015, 196, 16–24 CrossRef CAS PubMed.
- J. H. Lee, K. Heo, K. Schulz-Schönhagen, J. H. Lee, M. S. Desai, H. E. Jin and S. W. Lee, ACS Nano, 2018, 12, 8138–8144 CrossRef CAS PubMed.
- V. Nguyen, R. Zhu, K. Jenkins and R. Yang, Nat. Commun., 2016, 7, 13566 CrossRef CAS PubMed.
- Y. Kim, H. Park, Y. Kim, C. Lee, H. Park and J.-H. Lee, ACS Appl. Mater. Interfaces, 2022, 14, 38778–38785 CrossRef CAS PubMed.
- A. Handelman, N. Kuritz, A. Natan and G. Rosenman, Langmuir, 2016, 32, 2847–2862 CrossRef CAS PubMed.
- Z. Gan, X. Wu, J. Zhang, X. Zhu and P. K. Chu, Biomacromolecules, 2013, 14, 2112–2116 CrossRef CAS PubMed.
- O. Matos Ide and W. A. Alves, ACS Appl. Mater. Interfaces, 2011, 3, 4437–4443 CrossRef PubMed.
- J. H. Kim, J. Ryu and C. B. Park, Small, 2011, 7, 718–722 CrossRef CAS PubMed.
- Q. Li, Y. Jia, L. Dai, Y. Yang and J. Li, ACS Nano, 2015, 9, 2689–2695 CrossRef CAS PubMed.
- Q. Li, H. Ma, A. Wang, Y. Jia, L. Dai and J. Li, Adv. Opt. Mater., 2015, 3, 194–198 CrossRef CAS.
- N. Even, L. Adler-Abramovich, L. Buzhansky, H. Dodiuk and E. Gazit, Small, 2011, 7, 1007–1011 CrossRef CAS PubMed.
- A. M. Garcia, D. Iglesias, E. Parisi, K. E. Styan, L. J. Waddington, C. Deganutti, R. De Zorzi, M. Grassi, M. Melchionna, A. V. Vargiu and S. Marchesan, Chem, 2018, 4, 1862–1876 CAS.
- G. Picci, S. Marchesan and C. Caltagirone, Biomedicines, 2022, 10, 885 CrossRef CAS PubMed.
- Z. A. Arnon, A. Vitalis, A. Levin, T. C. T. Michaels, A. Caflisch, T. P. J. Knowles, L. Adler-Abramovich and E. Gazit, Nat. Commun., 2016, 7, 13190 CrossRef CAS PubMed.
- A. Comotti, S. Bracco, G. Distefano and P. Sozzani, Chem. Commun., 2009, 284–286 RSC.
| This journal is © The Royal Society of Chemistry 2023 |