
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organized mineralized cellulose nanostructures for biomedical applications
Yanhuizhi
Feng
a,
Helmut
Cölfen
 *b and
Rui
Xiong
*c
*b and
Rui
Xiong
*c
aDepartment of Periodontology, Stomatological Hospital and Dental School of Tongji University, Shanghai Engineering Research Center of Tooth Restoration and Regeneration, Shanghai 200072, China
bPhysical Chemistry, Department of Chemistry, University of Konstanz, Universitätsstraße 10, Konstanz, Germany. E-mail: helmut.coelfen@uni-konstanz.de
cState Key Laboratory of Polymer Materials Engineering, Polymer Research Institute of Sichuan University, Chengdu, 610065, China. E-mail: rui.xiong@scu.edu.cn
First published on 21st February 2023
Abstract
Cellulose is the most abundant naturally-occurring polymer, and possesses a one-dimensional (1D) anisotropic crystalline nanostructure with outstanding mechanical robustness, biocompatibility, renewability and rich surface chemistry in the form of nanocellulose in nature. Such features make cellulose an ideal bio-template for directing the bio-inspired mineralization of inorganic components into hierarchical nanostructures that are promising in biomedical applications. In this review, we will summarize the chemistry and nanostructure characteristics of cellulose and discuss how these favorable characteristics regulate the bio-inspired mineralization process for manufacturing the desired nanostructured bio-composites. We will focus on uncovering the design and manipulation principles of local chemical compositions/constituents and structural arrangement, distribution, dimensions, nanoconfinement and alignment of bio-inspired mineralization over multiple length-scales. In the end, we will underline how these cellulose biomineralized composites benefit biomedical applications. It is expected that this deep understanding of design and fabrication principles will enable construction of outstanding structural and functional cellulose/inorganic composites for more challenging biomedical applications.
10th Anniversary StatementJournal of Materials Chemistry B is certainly among the first choices when publishing cutting-edge research in the field of bionic/bioinspired materials is considered. With the aging society as one of the grand human challenges, bio-inspired materials with medical applications are of rapidly increasing importance and need. Among other related topics, this journal contains a good mix of important research work, authoritative reviews and perspectives in this research field. Therefore, we congratulate the Journal of Materials Chemistry B for 10 years of successful publication and presentation of exciting research in this area and wish all success for further advancement of the journal. It will be nice to see further key findings in the field published here. |
1. Introduction
Natural structural mineralized materials created by living creatures are usually composed of stiff minerals and soft biopolymers that are organized into multi-dimensional hierarchical architectures.1,2 For instance, teeth and bones are essentially comprised of hydroxyapatite (HAp) and collagen with a highly aligned one-dimensional (1D) structure to obtain outstanding load bearing capability.3 Nacres utilize aragonite microplates and chitin/silk protein to build a two-dimensional (2D) brick-mortar architecture to overcome the trade-off between toughness and strength.4 Crustacean exoskeletons evolve amazing damage-tolerant properties through the growth of crystalline/amorphous calcium carbonate into chitin/silk protein three-dimensional (3D) twisted plywood structures.5,6 These complex structural designs and excellent biological interfaces endow natural structural materials with fascinatingly high strength, high stiffness, high toughness, and on-demanded functionalities.7 By learning these natural processes, the nucleation, growth, and assembly process of inorganic minerals can be regulated by artificial organic templates, and ordered nanostructures like cartilage, teeth and nacre and can be precisely synthesized, thereby realizing the excellent structural properties and functionalities of these materials.8–11 Thus, biomimetic mineralization is an important strategy to construct high-performance structural and functional materials,12–15 especially for biomedical applications, since natural biomineralized materials are usually used as the essential structural and functional tissues in living creatures.To mimic natural hierarchical architectures, the selection of organic building-blocks/templates is critical due to the importance of interface design and bio-enabled concerns in biomedical applications. Compared with synthetic polymers, biopolymers usually demonstrate the following advantages: (i) good biocompatibility, biodegradability, renewability, and rich surface chemistry, which are essentially important for the biomedical applications;16 (ii) self-assembly of ordered secondary nanostructures to greatly enrich the structural hierarchy;17 and (iii) chemical and structural ingredient similarity with these natural counterparts for obtaining good structural and functional properties.18
Cellulose is the most abundant naturally-derived biopolymer on earth with an annual production of 100 billion tons.19 It is synthesized by a large number of living organisms, including plants, algae/fungi, and bacteria, and comprises the major structural component of their cell walls.20 This naturally-occurring cellulose is in the form of nanofibers with linear molecular chains highly aligned together, thus gaining outstanding mechanical robustness that is comparable to Kevlar fibers.19 The structural hierarchy of cellulose at a multi-length scale ranging from the molecular level to nanoscale and microscale, and to the macroscopic level, facilitates the preparation of the various forms of cellulose at different length scales, including molecular solution, nanofibers/nanocrystals, and microfibers. These multi-scaled cellulose forms combined with tailorable surface chemistry are ideal bio-building blocks to construct mineralized materials with hierarchical organizations that closely mimic those that have been built by living creatures. The development of cellulose based mineralized materials from renewable and sustainable resources would have great potential as next-generation sustainable bio-enabled composites that have a low environmental impact and low safety risks to human health. Cellulose and cellulosic materials have distinguished properties for future multi-applications. (Table 1).
| Criteria | Molecular cellulose | Nanocellulose | Mineralized cellulose |
|---|---|---|---|
| Stability | Chemically modifiable21 | Chemically modifiable21 | Chemically modifiable22 |
| Flexibility | Good23 | Good | Good24 |
| Raw materials price | Low cost20 | Low cost20 | Low cost20 |
| Mass production capability | Commercially available25 | Commercially available25 | Non-commercially available26 |
Although studies to develop cellulose as a new advanced material have tremendously progressed in recent decades,27 controlling the biomineralization process as precisely as intricate biological systems remains a grand challenge. To apply cellulose based mineralized materials as ecofriendly high-performance biomedical materials, it is vital to understand how to utilize the surface chemistry and nanostructure characteristics of cellulose to regulate the biomineralized architectures with long-range order. The biomimetic hierarchical architectures are key in the optimal expression of minerals and cellulose properties, and further functionalities due to their synergistic toughening mechanisms. However, existing reviews rarely systematically discuss the related design and fabrication principles based on organized mineralized cellulose structures,28–30 and therefore we have written this review to address this issue. We summarize the chemistry and nanostructure characteristics of cellulose at multi-length scale and discuss how these favorable characteristics regulate the bio-inspired mineralization process for manufacturing highly ordered nanostructured biocomposites in multiple dimensions. The underlying mechanical and functional enhancement mechanisms are carefully discussed, and we highlight how these properties benefit biomedical applications (Fig. 1). It is expected that this deep understanding of design and fabrication principles will lead to the construction of outstanding structural and functional nanocellulose/inorganic composites for more challenging biomedical applications.
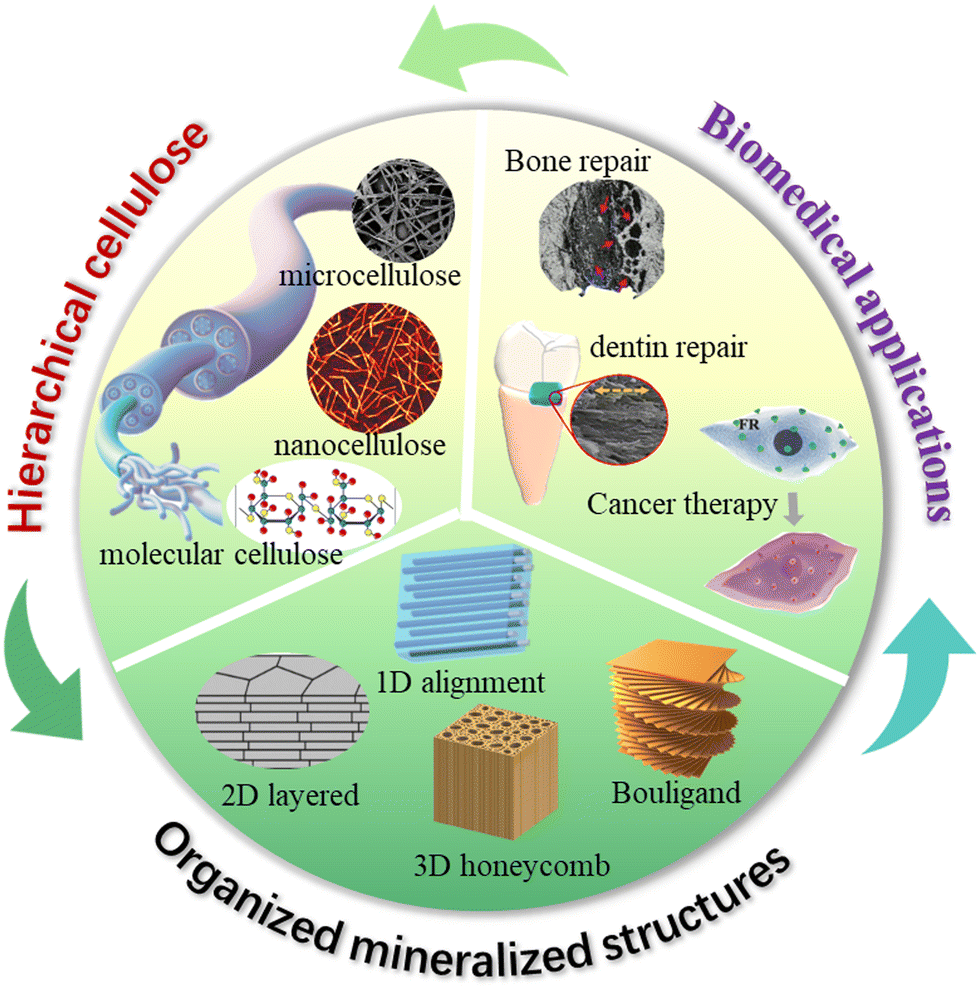 | ||
| Fig. 1 Overview of the organized mineralized cellulose structures for biomedical applications. | ||
2. Cellulose at the molecular-, nano-, and micro-scales
Cellulose is a linear polymer composed of several hundreds to thousands of glucose units that are joined by β-1,4 linkages. During biosynthesis, strong hydrogen bonding and van der Waals forces lead to parallel packing of multiple cellulose molecular chains to form elementary nanofibrils with a diameter of 2–3 nm, which further bundle into larger microfibrils with a diameter of 5–50 nm.31 The highly ordered anisotropic arrangement and strong intra- and inter-chain interactions make cellulose water-insoluble and provide cellulose fibers with a high axial strength and stiffness.32 The hierarchical structure of cellulose in nature is finely controlled through biosynthesis and assembly, resulting in an infinite number of structural differences across cellulosic species. These structural variations lead to numerous structural–functional diversities, such as the combination of lightweight and exceptional mechanical properties in wood, and brilliant structural colors in many flowers and fruits.7,33 Wood is the most typical example of the construction of cellulose into a complex hierarchical structure in nature. Native cellulose is a semicrystalline 1D fibril material consisting of crystalline (microfibers or rodlike entities) and amorphous regions, which are embedded in an amorphous matrix of hemicellulose and lignin to constitute the plant cell wall.34 These cellulose fibrils are usually oriented along the axial direction of wood to optimize the load bearing properties that could protect their living bodies against biological attack and external stress.35 Overall, cellulose possesses a structural hierarchy at different length scales, including linear chains at the molecular scale, nanofibers/crystals at the nanoscale, and microfibers at the microscale (Fig. 2(a)),36 which eventually build up the hierarchical structures of living organisms.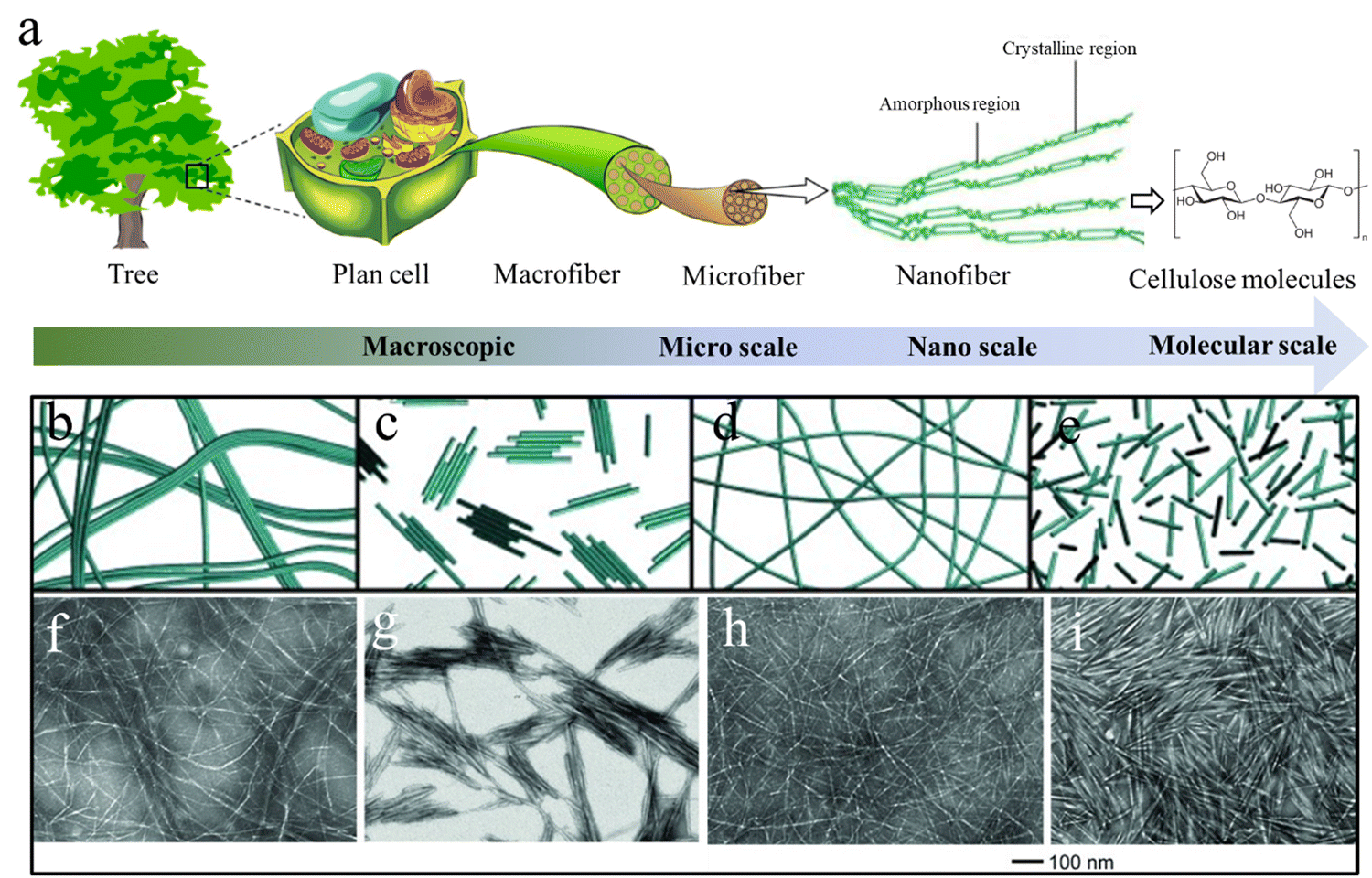 | ||
| Fig. 2 (a) The hierarchical cellulose structure in plants and trees at different length scales. Reprinted from ref. 36 with permission from DMPI, copyright 2020. (b)–(i) Schematic illustration and corresponding TEM images of nanocellulose materials prepared from wood pulp using different methods: (b) and (f) CNFs prepared by high-intensity ultrasonication; (c) and (g) CNCs prepared by HCl hydrolysis; (d) and (h) CNFs prepared by TEMPO pretreatment and ultrasonication; (e, i) CNCs prepared by H2SO4 hydrolysis. Reprinted from ref. 50 with permission from Wiley, copyright 2014. | ||
2.1 Molecular cellulose
Molecular cellulose mainly refers to the cellulose or cellulose derivatives solution, which can be utilized to fabricate regenerated cellulose. Regenerated cellulose is a class of cellulosic materials manufactured by cellulose dissolution and subsequent regeneration. Various forms of regenerated cellulose, including fibers, films, and hydrogels, can be fabricated by wet spinning, drop-casting, and molding of cellulose/cellulose derivative solutions, followed by a regeneration process of solvent evaporation or coagulation bath.37–39 Cellulose is insoluble in conventional polar/non-polar solvents due to the long-length molecular chains and strong chain interactions due to hydrogen bonds. Only some specific solvents, including NaOH/CS2, N-methylmorpholine-N-oxide (NMMO), LiCl/N,N-dimethylacetamide, ionic liquids (ILs) and alkali/urea etc., have been developed to dissolve cellulose through disrupting these hydrogen bonds.40 However, some of these solvents (such as NaOH/CS2 and NMMO) cause serious environmental problems so they have been gradually replaced by more “green” solvents (e.g. ILs and alkali/urea) in applications.41–44To make cellulose water soluble, esterification and etherification of cellulose is the main strategy. Many cellulose esters and ethers, such as methylcellulose (MC), hydroxypropyl cellulose (HPC), and carboxymethyl cellulose (CMC), are water-soluble polymers, which have already been commercially available for decades. The water solubility highly depends on the degree of substitution (DS) of hydroxyl groups on the cellulose chains. A high DS can significantly weaken the hydrogen bonding to give cellulose good water solubility and even solubility in polar organic solvents.45 Interestingly, some cellulose derivatives are temperature sensitive. For example, HPC aqueous solutions exhibit an unusual solubility behavior with good solubility in cold water, but insolubility in hot water, resulting in the gelation of cellulose chains.46 The transition temperature can be manipulated by controlling the ratio of hydroxypropyl substitutions. This unique temperature-sensitivity is highly attractive for the design and fabrication of temperature responsive biocomposites.
2.2 Nanocellulose
Nanocellulose is a form of one-dimensional nanosized cellulose fibers with a diameter <100 nm. It combines the desirable intrinsic properties of cellulose (e.g., renewability, biocompatibility, biodegradability, and low density) with the features of nanomaterials, including high strength (∼7 GPa) and stiffness (110–220 GPa), high-aspect-ratio, high specific surface area, reinforcing capabilities, and self-assembly behavior in aqueous media.47 These outstanding properties make nanocellulose a rising star in the materials science field including applications. Nanocelluloses can be categorized into cellulose nanofibers (CNFs), cellulose nanocrystals (CNCs), and bacterial cellulose (BC) depending on their morphologies and sources.Elementary CNFs are the smallest element next to cellulose molecules with a diameter of 2–3 nm and length of several-micrometers.48 They consist of fully extended cellulose chains closely packed along the longitudinal direction with alternatively highly ordered crystalline and amorphous regions. CNFs can be exfoliated from cellulosic sources through various intensive mechanical disintegration techniques, such as ultrasonication, a high-pressure homogenizer, grinder, ball mill etc.49 However, CNFs directly obtained from these techniques usually show a large diameter distribution and poor aqueous dispersibility (Fig. 2(b) and (f)),50 resulting in the semi-transmittance or non-transmittance of the dispersion and films. To prepare uniform elementary CNFs with good aqueous stability, pre-chemical treatments of cellulose have to be conducted to introduce significant amounts of charged groups on the cellulose microfiber surfaces. C6-carboxylation of cellulose by 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) pretreatments is the most widely used approach to gain homogeneous and stable CNF aqueous dispersions.51 The grafted negatively charged carboxyl groups during TEMPO treatment can produce strong electrostatic repulsion between cellulose microfibers.52 Consequently, using mild mechanical disintegration can break all the interfibrillar hydrogen bonds between cellulose microfibrils and obtain completely individualized elementary CNFs dispersed in water (Fig. 2(d) and (h)),50 which can significantly reduce energy consumption in the mechanical disintegration process.53
CNCs can form rigid rod-like nanocellulose with diameters in the range 3–50 nm and lengths of 50–500 nm depending on the cellulosic source and preparation methods.54 Strong acid hydrolysis of cellulose preferentially etches the amorphous regions of cellulose microfibers to liberate CNCs. The CNCs prepared by sulfuric acid hydrolysis show excellent aqueous stability due to the sulfate groups grafting during the sulfuric acid hydrolysis (Fig. 2(e) and (i)), while HCl treated CNCs usually aggregate together and show poor dispersion stability due to the absence of surface charges (Fig. 2(c) and (g)).50 In contrast to the uniform and smooth elementary CNFs, CNCs in most cases have spindle-like morphologies with larger diameters because of the presence of large crystal bundles. Intensive ultrasonication treatment of the CNC dispersion can separate their bundles into individual CNCs with a large decrease in the CNCs size.55 As a result, the white, turbid, or translucent CNC dispersion turned into a transparent blueish dispersion. Interestingly, CNCs are capable of self-organizing into a chiral nematic (cholesteric) liquid crystal (LC) phase with a helical arrangement when the CNC dispersion is above a critical concentration.56 Upon drying, the chiral nematic LC phase is saved in the solid-state film with a striking iridescent structural color because of Bragg reflection of visible light from dried films.57 Unlike the traditional structural colors from photonic crystals and layered structures, the more complex internal structure of CNC LC films adds the structural color sensitivity to circular polarization, which shares similarities with the brilliant structural color produced by the twisted plywood structure of fruits and beetles.58
BC can be produced by bacteria fermentation (such as A. xylinum, A. hansenii, and A. pasteurianus) with the form of hydrogel-like pellicles by cultivating in an aqueous culture medium for a few days.59,60 Compared to nanocellulose from plants, BC has a more crystalline structure, and is more chemically pure without hemicellulose or lignin that often exists in CNFs. The high polymerization degree and ultrafine network architecture also makes BC stronger and tougher than other types of nanocellulose based materials. However, the distinct ribbon-like 3D network structure with a large diameter of around 100 nm makes the dried BC film only semi-transparent.
2.3 Cellulose microfibers
Original cellulose in plants usually exists in the form of cellulose microfibers, together with hemicellulose and lignin constituents in the cell walls of plants, which is one of the most ancient raw materials that human beings have developed for making paper. Through pulping processes, such as mechanical, semi-chemical or fully chemical methods (kraft and sulfite processes), individual cellulose microfibers can be isolated from plants. The original hollow structure of cellulose microfibers is usually flattened after the pulping process due to the collapse of the lumen during mechanical treatments and subsequent drying.61 The size of the microfibers depends on the cellulosic sources with a usual width of 10–50 microns and length of several millimeters. Compared with nanocellulose, cellulose microfibers are more cost-efficient and more porous. The large diameter size also makes them suitable for use as a bio-template for nano/micro-particle assembly or growth. Additionally, cellulose microfibers can be made into paper though well-established industrial paper-making processes for various applications, ranging from their ancient application for writing to emerging applications in flexible electronic devices, biomedicines, energy harvesting and storage.62–643. Mineralized cellulose materials at multiple dimensions
The structural diversity of cellulose provides numerous possibilities to arrange it with functional inorganic components to construct advanced structural and functional composites with high structural complexity. Considering the significant diversity of synthetic mineralized materials, appropriate classification of the material microstructures presents an important step toward a comprehensive understanding of design and fabrication strategies for architected materials. These efforts to produce mineralized cellulose composites can be categorized into three major groups (1D, 2D and 3D) according to the dimensional diversity of their architectures. In this section, we selectively focus on the construction of mineralized cellulose materials with well-defined order and the unique enhancement of their mechanical properties and functionalities.3.1 One dimensional (1D) individual fibril/aligned nanofibril structures
In situ growth. In situ growth of mineral NPs decorated on cellulose nanofibers ensures uniform NP distribution, thereby avoiding the traditional mixing stage with the potential risk of particle agglomeration. Cellulose fiber (CF) paper is a low-cost, sustainable, and flexible substrate that can be assembled from cellulose microfibers using paper-making techniques. The highly porous network, large surface area and rich surface chemistry of cellulose microfibers provide abundant bonding sites for direct mineralization. Great progress has been achieved in functionalizing CF paper by the in situ growth of inorganic particles to meet the requirements of specific applications. For instance, ultralong HAp nanowires (HAPNWs) with lengths larger than 10 μm have been demonstrated to radially grow on cellulose microfibers (Fig. 3(a)).65 This unique micro/nanoscale hierarchical structure can be easily customized by Eu3+/Tb3+ ion doping and poly(dimethylsiloxane) coating for added photoluminescence and superhydrophobicity. Additionally, zinc oxide (ZnO) nanowires can also be incorporated on the fiber surfaces to form a similar core/shell structure through well-established hydrothermal methods.66,67 Metal–organic frameworks (MOFs) are emerging nanomaterials that are of great interest with diverse applications in gas separation, catalysis, energy, environmental science, and biomedicine, due to their large specific surface areas, high porosities, and rich functionality. However, the difficulty in processing MOF crystals and controlling the MOF superstructure limits their applications. Li and Yu et al.68,69 have demonstrated that the 1D nanofibril morphology of CNF and the negatively charged surface chemistry could shift the balance of nucleation and growth towards synthesizing smaller MOF crystals, and further decreased their aggregation possibilities. Furthermore, CNF can provide extrinsic porosity, mechanical flexibility, and easy-processability for the resultant composites due to the hybrid core/shell structure.
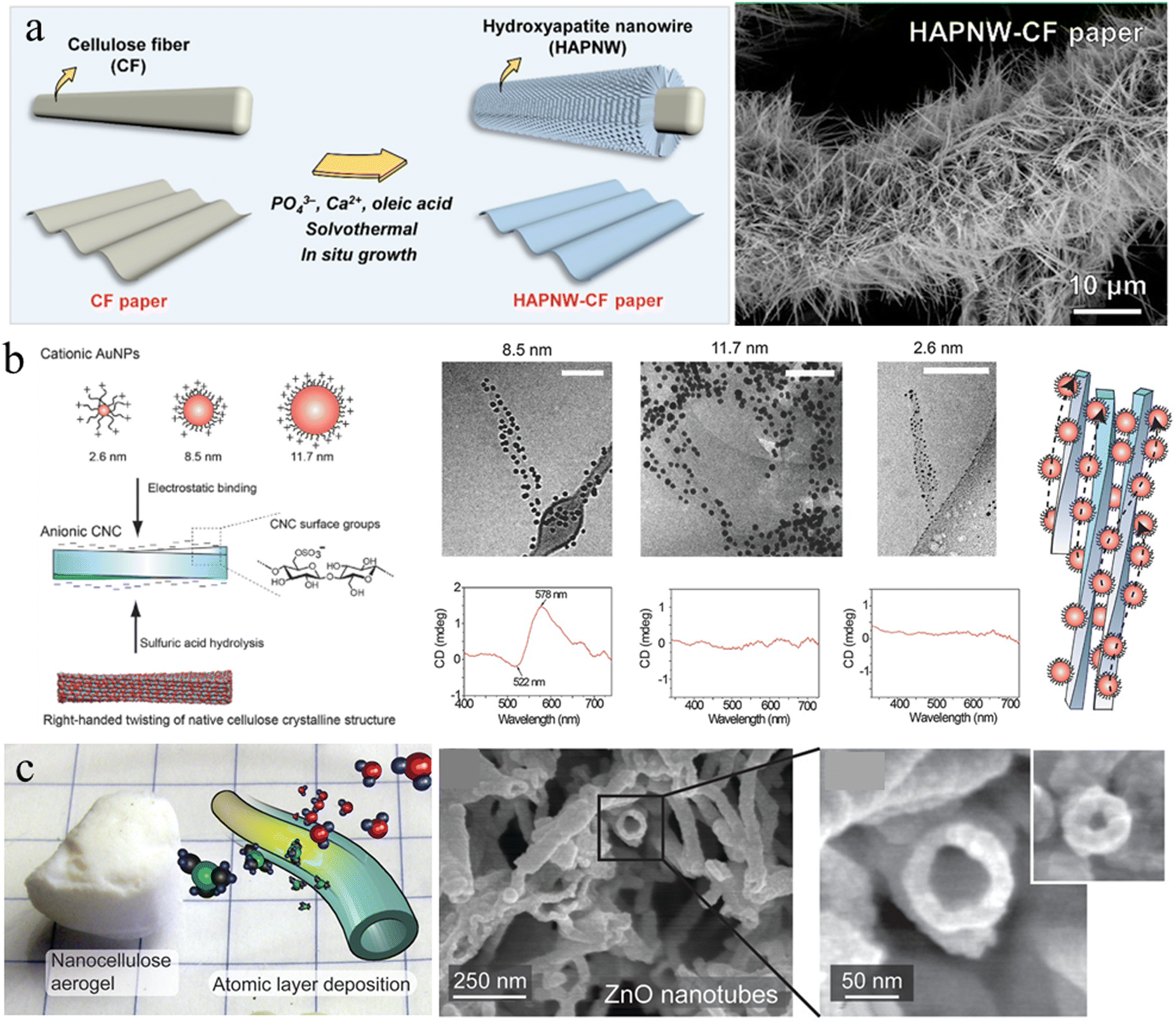 | ||
| Fig. 3 (a) Schematic representation of the in situ growth of HAp nanowires on a cellulose microfiber surface and the corresponding SEM image. Reprinted from ref. 65 with permission of the American Chemical Society, copyright 2021. (b) Scheme for electrostatic binding of plasmonic AuNPs onto CNCs for chiral plasmonics in dilute aqueous dispersion, and Cryo-TEM images and circular dichroism measurements upon electrostatic binding of AuNP onto CNC. Reprinted from ref. 81 with permission of Wiley, copyright 2016. (c) Inorganic hollow nanotubes prepared from the nanocellulose aerogel by the ALD and subsequent calcination. Reprinted from ref. 92 with permission of the American Chemical Society, copyright 2021. | ||
In addition to the nanowires and MOFs, various types of functional mineral nanospheres (e.g. noble metal NPs and magnetic NPs) have been widely in situ grown on cellulose nano/micro fiber surfaces for on-demand functionality. Interestingly, noble metal NPs, including gold, silver and palladium, could be directly deposited on nanocellulose using nanocellulose as a reducing agent and stabilizing template under hydrothermal conditions, due to the abundant electron-rich hydroxyl groups on the cellulose surface.70–74 This process is not only green without introducing extra toxic reducing agents, but also enables superior catalytic performance of the immobilized metal NPs.70 Conventional soft reducing or stabling agents of metal NPs, such as polyvinylpyrrolidone (PVP) and polyethylene glycol (PEG), usually densely wrap around the NPs surface, which significantly shields the interaction with other components, leading to deteriorated performance, such as catalysis and conductivity. By contrast, the reduction and stabilization of NPs by stiff nanocellulose could expose more active sites to external environments for favorable interaction and thus for enhanced performance. However, the approach is difficult to realize for high loading of metal NPs due to the limited reduction ability of nanocellulose. To achieve high decoration of NPs, additional reduction agents are usually needed to pre-assemble on the nanocellulose surface. For example, bio-inspired polydopamine (PDA) has been used to in situ grow and immobilize Ag NPs on the nanocellulose surface.75 Magnetic NPs have been in situ grown on cellulose fibril templates for adding magnetic functionalities, respectively.76–78 These inorganic-bio hybrids could be further assembled into flexible solid magnetic bio-films.79 In a recent study, a uniform layer of amorphous calcium carbonate in the form of small beads was demonstrated to grow on the CNF surface to form a core–shell structure using a binary water/organic solvent.80
Self-assembly. Interfacial interaction leading to self-assembly of nanocellulose with functional NPs is another approach to introduce NPs on the nanocellulose templates. The abundance of hydroxyl, carboxyl, and sulfate groups on the surface of the nanoparticle/fibril allows the generation of supramolecular interactions with functional NPs, such as hydrogen bonding, electrostatic interaction, and van der Waals forces. Majoinen et al., used the electrostatic interaction between cationic gold NPs (AuNPs) and anionic CNC to assemble AuNPs on the surfaces of individualized CNC colloidal rods (Fig. 3(b)).81 The right-handed twist along individual CNC templates and the chiral twisted assembly of AuNPs along their axis, led to a chiral plasmonic response on the nanoscale. CdSe/ZnS quantum dots (QDs) have also been demonstrated to be successfully decorated around carboxylated CNCs using a carbodiimide chemistry coupling approach.82 The QD–CNC hybrids exhibit colloidal stability in aqueous suspension and provide strong fluorescence to a CNC film with low NP loading, maintaining the overall physical properties or self-assembly of the CNCs.82 Anionic nanodiamond nanoparticles could also be used to successfully decorate cationic CNFs in hydrocolloidal form for greatly hardening cellulose materials.83 Additionally, nanocellulose was recently demonstrated to be an amphiphilic biofibril due to the highly ordered and dense packing of cellulose molecular chains.84–86 The OH groups decorate hydrophilic planes, while CH groups constitute hydrophobic planes.87 This unique amphiphilic behavior can be utilized to assemble and disperse hydrophobic nanocarbon materials and minerals, such as carbon nanotubes, nanodiamonds, graphene and boron nitride.88,89 These NPs stabilized by nanocellulose could be used to further construct robust and functional bio-enabled nanomaterials.
Atomic layer deposition (ALD). ALD is a sequential, self-limiting chemical vapor deposition method used to deposit an ultrathin, conformal and homogenous layer on various substrates, which enables precise control of the desired film thickness by the number of deposition cycles.90 Due to the easy operation, good reproducibility and high conformality of the as-deposited layer, ALD appears to be a promising technique for the deposition of inorganic layers on nanocellulose templates. Porous nanocellulose aerogels possess good mechanical strength and flexibility to ensure the structural integrity under the high vacuum conditions of ALD. Additionally, the complex porosity of the aerogels produces a large surface area for the gas phase of ALD reactions to fully saturate the nanocellulose network surface.91 Korhonen et al. reported that the highly porous percolating network of nanocellulose aerogel produced by supercritical drying is an ideal biological nanofibrillar template for ALD.92 Uniform oxide layers (titanium dioxide, zinc oxide, and aluminum oxide) have been deposited on the cellulose nanofiber surface to form an organic–inorganic core/shell structure with shell thickness down to <3 nm. Subsequent calcination at 450 °C to remove the cellulose core results in inorganic self-supporting aerogels consisting of hollow nanotubular networks (Fig. 3(c)). Similarly, Li et al. demonstrated that 3D TiO2 fiber-nanorod (NR) heterostructures can be prepared by the ALD assisted ZnO-coated CNF template and subsequent TiO2 NRs growth.93
Furthermore, progress has been achieved to assemble 1D ultralong HAp nanowires and soft polymers into highly aligned structures with good mechanical properties. Tooth enamel is the hardest tissue in the human body and it exhibits excellent resistance to vibrational damage due to parallelly oriented nanoscale HAp nanowires interconnected by a confined biomolecule matrix in the early tooth development stage. To mimic this enamel structure, excellent alignment of HAp nanowires in the poly(vinyl alcohol) (PVA) matrix has been realized thorough ice-templating and subsequent compression.100 Due to the versatility of these assembly techniques, the co-assembly of 1D mineral nanowires with nanocellulose is another promising strategy to produce aligned hybrid structures although there are only limited studies reporting this co-assembly. For example, silver nanowires have been co-assembled with bacterial cellulose to produce conductive fibers by wet spinning for pressure and proximity sensing, which could detect and recognize the weak signal of the human voice and pulse waves with high accuracy.101
3.2 Two dimensional (2D) layered structures
Nacre is one representative 2D biological structural material involving the coexistence of high strength and notable toughness, as well as brilliant iridescence.102,103 In a typical nacre nanoarchitecture, numerous hexagonal aragonite platelets with 0.5–1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) μm thickness and 10–20μm width are alternatively stacked with thin biopolymeric layers (5–30 nm thick) of silk protein/chitin (an interlamellar membrane) to form a 2D layered structure.104 This highly ordered layered structure combined with strong interfacial strength could greatly facilitate the pull-out and bridging between the mineral platelets, the shearing resistance caused by viscoelastic biopolymer glue and tablet interlocking.105,106 These hierarchical fracture mechanisms synergistically result in nacre being a tough and hard biological material, although its building blocks consist of only brittle minerals and weak polymers.107 Additionally, this 2D layered morphology is a photonic structure in the dimension of visible light wavelength that can produce multi-layer light interferences, leading to a view-angle-dependent iridescence or the metallic mirror-like luster of nacre.7,103 Such an outstanding combination of mechanical and photonic properties of nacre provides tremendous inspiration to construct corresponding biomimetic layered structures. To date, efforts to mimic the layered structure using cellulose can be mainly classified into two categories: self-assembly of cellulose and 2D nanomaterials, and layer-by-layer (LbL) growth.
μm thickness and 10–20μm width are alternatively stacked with thin biopolymeric layers (5–30 nm thick) of silk protein/chitin (an interlamellar membrane) to form a 2D layered structure.104 This highly ordered layered structure combined with strong interfacial strength could greatly facilitate the pull-out and bridging between the mineral platelets, the shearing resistance caused by viscoelastic biopolymer glue and tablet interlocking.105,106 These hierarchical fracture mechanisms synergistically result in nacre being a tough and hard biological material, although its building blocks consist of only brittle minerals and weak polymers.107 Additionally, this 2D layered morphology is a photonic structure in the dimension of visible light wavelength that can produce multi-layer light interferences, leading to a view-angle-dependent iridescence or the metallic mirror-like luster of nacre.7,103 Such an outstanding combination of mechanical and photonic properties of nacre provides tremendous inspiration to construct corresponding biomimetic layered structures. To date, efforts to mimic the layered structure using cellulose can be mainly classified into two categories: self-assembly of cellulose and 2D nanomaterials, and layer-by-layer (LbL) growth.
Conventional nacre-like composites are often simplified as binary composites, which adapt two components to mimic the mineral phases and polymer phase of nacres, respectively. However, nacre should be treated as a ternary composite because its organic phase consists of chitin nanofibers and silk-like proteins.115 The alternate stacking of aragonite platelets and nanofibrillar chitin network layers is glued by a soft protein adhesive, providing hierarchical interfacial strength for efficient load transfer.116,117 To mimic this ternary structure, the third soft polymeric components were usually added to the above binary combination. For example, Wang et al., combined PVA with the clay platelet/CNF to construct strong and transparent artificial nacre based on an evaporation-induced self-assembly technique (Fig. 4(a)).116 The strong interfacial strength combined with the synergistic toughening effect equips artificial nacre with an excellent balance of strength and toughness, superior to natural nacre and other conventional layered clay/polymer binary composites.118–120
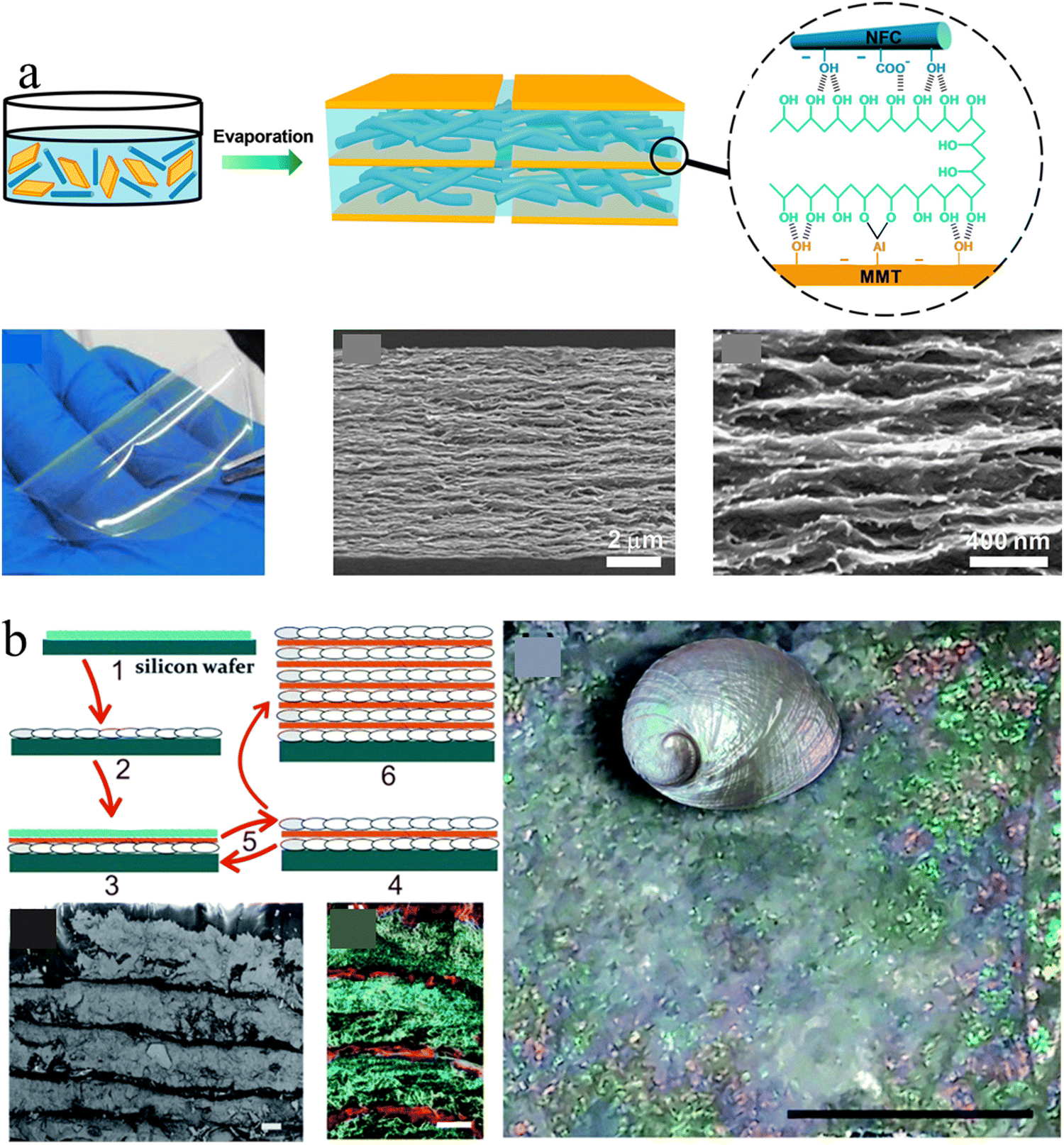 | ||
| Fig. 4 (a) Schematic illustration of a transparent artificial nacre film prepared from MMT platelets, CNF, and PVA by evaporation induced assembly, and the photo and cross-section SEM images of the corresponding film. Reprinted from ref. 116 with permission of the American Chemical Society, copyright 2014. (b) Schematic of the preparation protocol for the nacre-like material by the layer-by-layer deposition of nanocellulose and calcium carbonate, which exhibits a uniform layered structure and brilliant structural color like nacre. Reprinted from ref. 122 with permission from the Royal Society of Chemistry, copyright 2017. | ||
3.3 Three dimensional (3D) hierarchical architectures
Nature usually builds 3D structurally ordered and environmentally adaptive mineralized composite materials to survive in harsh environments, such as porous and twisted plywood structures, which are more complex than the simplified 1D and 2D structural motifs discussed above. These complex 3D architectures are primarily responsible for the excellent mechanical properties and multifunctionality of these hybrid materials. Porous biomineralized materials, such as the honeycomb-like structure of highly mineralized cuttlebone, typically have a light-weight feature as well as better crush performance and specific energy absorption performance that is comparable to metallic foams.124 The twisted plywood structure of the dactyl club of mantis shrimps has a bicontinuous structure with hydroxyapatite (∼88 vol%) integrated in an organic matrix. This special interpenetrated 3D structure provides an unusual combination of stiffness and damping.125 Thus, mimicking these the 3D architectures is remarkable as it presents new opportunities to construct high-performance synthetic materials.Nanocellulose is a highly attractive biodegradable reinforcing nanofiller for strong composites due to its ultrahigh stiffness and strength, high aspect ratio and abundant functional groups. The outstanding mechanical robustness of nanocellulose also could greatly increase the mechanical integrity of the 3D printed products. Additionally, nanocellulose also has strong shear thinning properties that are critical for 3D printing. Thus, nanocellulose has often been added into other biopolymer matrixes to prepare the bio-inks. Markstedt et al. combined the shear thinning properties of CNF and the fast cross-linking ability of alginate to prepare bio-inks for the 3D bio-printing of structures of the human ear and sheep meniscus.128 Usually, the strong shearing force during 3D printing can induce the alignment of nanocellulose along the printing nozzle. This alignment of nanocellulose not only greatly enhances the mechanical properties of the as-prepared products but also provides additional anisotropic functionalities. For example, Gladman et al., printed composite hydrogel architectures with the alignment of CNFs along pre-designed printing pathways. The resulting swelling anisotropies in water lead to shape transformations that occur on the order of minutes.129 Additionally, nanocellulose could be used as an individual building block to construct the 3D structures. Nanocellulose readily forms a hydrogel at high concentration due to chemical interactions of abundant hydroxyl groups and the physical fibril entanglement. The high zero shear viscosity and shear thinning features make nanocellulose hydrogels an ideal bioink for 3D printing of various aligned structures.130–132
The incorporation of mineral components into the 3D printed structures could be achieved through either pre-mixing with cellulose bioinks or the post templated guided growth. For example, HAp NPs are frequently added into the cellulose based bioinks to 3D print plausible scaffolds for bone tissue engineering.133–135 The addition of nano-sized HAp not only can enhance the mechanical properties of the scaffolds, but also provides the bioactivity and vice-versa to mimic the natural bone compositions.136,137 However, it is worth noting that the direct mixing of HAp powder with cellulose bio-inks usually results in a lack of homogeneity and restricted bioactivity. Therefore, in situ mineralization is another effective method to construct hierarchical mineralized composites.138,139 This strategy highly relies on the pre-printed cellulose scaffolds, where the surface groups of scaffolds serve as nucleation sites, and the porous scaffolds guide the mineral growth, eventually leading to biomimetic mineralized composites with predesigned microstructures.138 For instance, Abouzeid et al., reported the 3D printing of a CNF/alginate scaffold using CNF/alginate hydrogel partially crosslinked by calcium ions (Fig. 5(a)).140 The subsequent full crosslinking of the prepared scaffold could provide the scaffold with good rigidity and long-term stability for the biomimetic mineralization process. As a result, dense HAp successfully grew around the printed scaffolds using simulated body fluid.140 Moreover, Torres-Rendon et al. demonstrated a new reverse templating strategy to construct ordered CNF/chitin hydrogel scaffolds (Fig. 5(b)).141 In this approach, the pre-designed sacrificial resin templates were fabricated by lithographic 3D printing, which were then infiltrated by the CNF/chitin hydrogel. The removal of resin templates left the CNF/chitin hydrogel with a highly ordered replicated structure for bone tissue engineering.
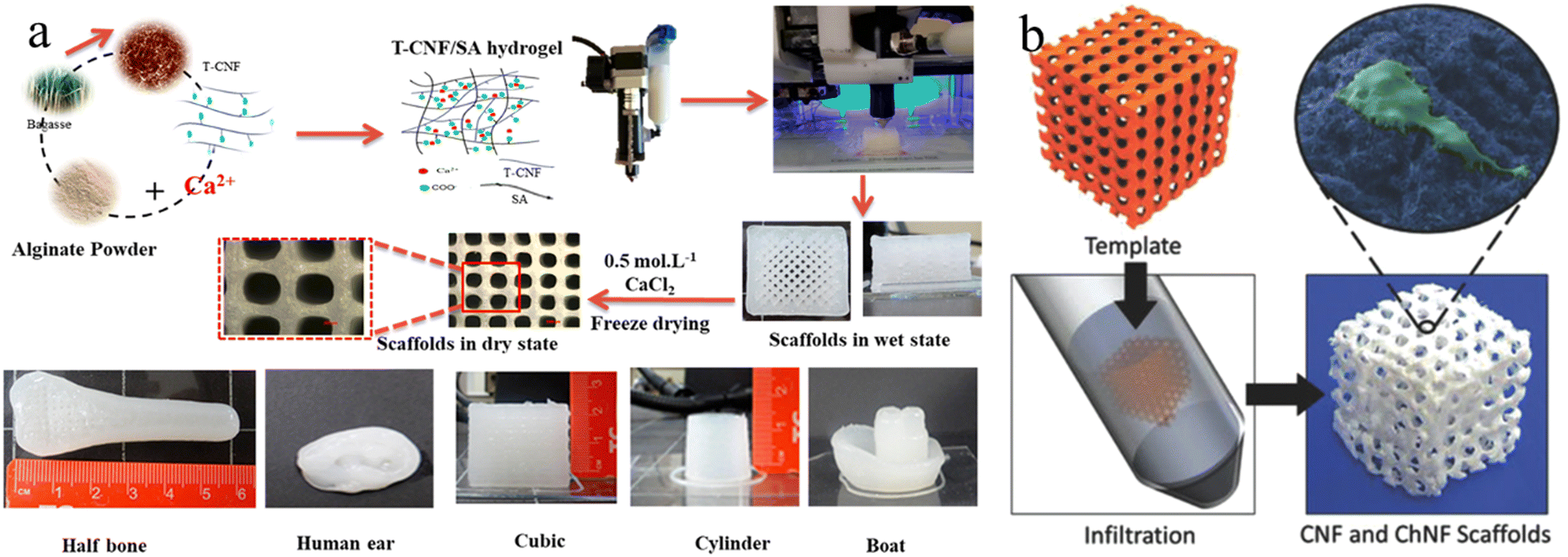 | ||
| Fig. 5 (a) Fabrication process for 3D printing CNF/alginate scaffolds and different printed scaffold forms. Reprinted from ref. 140 with permission of the American Chemical Society, copyright 2018. (b) The fabrication strategy of CNF/chitin hydrogel inverse scaffolds from 3D printed templates. Reprinted from ref. 141 with permission of Wiley, copyright 2015. | ||
This strategy was widely used for nanocellulose dispersions or with additional mineral NPs to produce anisotropic aerogels.145–151 Munier et al., reported that the unidirectional freezing of nanocellulose dispersions can lead to the good alignment of nanocellulose along the freezing direction.152 The resulting nanocellulose network is even sufficiently strong to resist melting. Chau et al., showed that nanocellulose aerogels with fibrillar, columnar, or lamellar structures can be constructed through the control of the composition of the nanocellulose dispersion and the freeze-casting temperature.151 Additionally, mineral NPs have often been added into the cellulose matrix to provide mechanical integrity and functionalities.145,153–155 For example, boron nitride nanosheets have been combined with an anisotropic aerogel to enhance the thermal management properties, such as high conductivity, thermal storage and flame resistance.156,157 In a recent work, Zhang et al., developed an interfacial engineering strategy to fabricate a mineralized bacterial cellulose aerogel with high compressibility and multifunctionality (Fig. 6).158 In this approach, nanocellulose with methyltrimethoxysilane (MTMS) was assembled into an anisotropic aerogel using the directional freezing technique, and subsequently polymethylsilsesquioxane (PMSQ) was mineralized as a coating on the bacterial cellulose networks to provide additional interfacial strength. As a result, this mineralized aerogel can achieve low density, mechanical robustness, ultrahigh compressibility, and super-hydrophobicity.
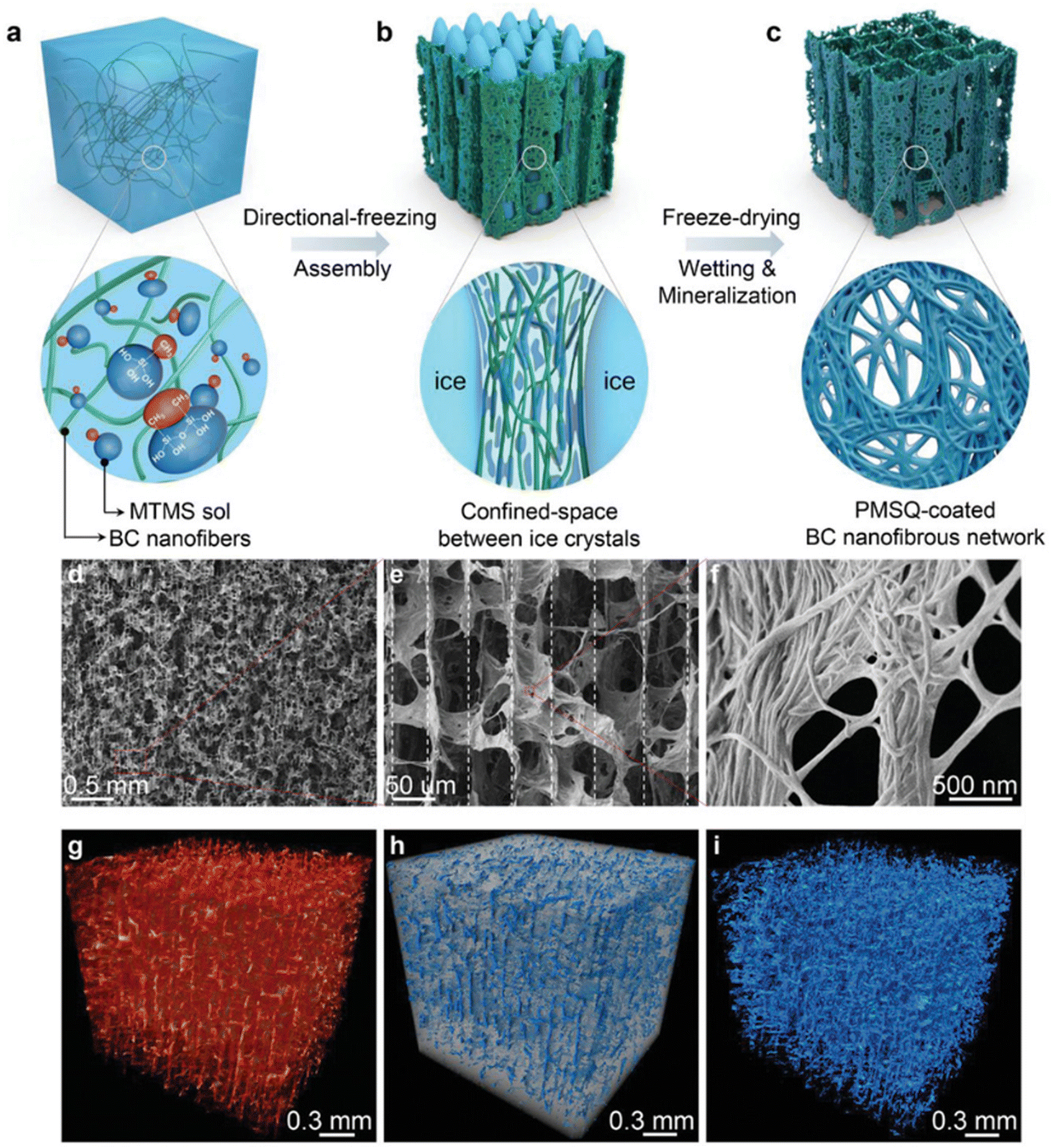 | ||
| Fig. 6 (a)–(c) The schematic illustration of the synthesis of anisotropic BC–PMSQ aerogel. (d)–(f) SEM images of the microscopic structure of the BC–PMSQ aerogel at different magnifications. (g)–(i) Reconstructed 3D micro-CT image of the representative BC–PMSQ hybrid aerogel, showing the two distinctive regions of the macropores and solid framework. Reprinted from ref. 158 with permission of Wiley, copyright 2021. | ||
Rod-like CNCs can form chiral nematic liquid crystal phases, which are similar to the natural Bouligand structure, in concentrated aqueous dispersions controlled by solution concentration.163 Liquid chiral nematic phases have long-range order with the pitch of helical structures in the range of tens of micrometers. However, the water evaporation causes the chiral pitch to reduce to the submicron range, leading to Bragg's light reflection in the visible spectrum and bright structural colors. This pitch distance can be adjusted by varying the sonication time, temperature, and the addition of salt and soft polymers for regulating the reflectivity.7,56,163,164 To obtain the bio-inorganic hybrid chiral structure, inorganic NPs usually co-assemble with CNCs into the chiral nematic liquid crystal phase, which is finally preserved in a solid film. To date, various functional NPs, such as gold nanoclusters, metal NPs, quantum dots and nanorods, have been added into the chiral CNC host matrix to construct functional chiral materials.165–171 In a recent study, Kang et al., used semiconducting quantum nanorods (QNRs) to co-assemble with CNC to prepare the chiral composites with a combination of tunable iridescence and active photoluminescence.165 The similar geometry and size between QNR and CNC allow the seamless co-assembly of highly ordered chiral nematic structures with unusually large photoluminescent asymmetry. Additional incorporation of elastic polymers further enhances the stretchability of the composites with stress induced dynamic photonic properties.
Moreover, this highly-ordered chiral cellulose nanostructure with confined interspace is also an ideal template for biomineralization with multiscale length hierarchy. Shopsowitz et al., developed photonic mesoporous silica films using the CNC chiral nematic liquid crystal structure as a biotemplate. They co-assembled silica precursors with CNC to form a chiral nematic phase, and the silica precursors can be hydrolyzed into silica by the acidic nature of the CNCs. With pyrolysis treatment, the CNC template can be removed to leave the mesoporous inorganic silica films in the free-standing state. Although the chiral nematic structure has promising toughening mechanisms, the prepared CNC chiral nematic composite films are usually very weak and brittle, which significantly limits their applications. Inspired by the natural molting process of crustaceans to produce a strong dactyl club and exoskeletons, our groups recently developed an artificial molting process to construct strong and tough mineralized CNC composites combined with tunable photonic properties (Fig. 7).172 The highly ordered chiral nematic structure of soft templates assembled from CNC and polyacrylic acid (PAA) can efficiently regulate the mineralization process of calcium carbonates. A short mineralization time results in a strong and flexible endocuticle-like structure, while prolonged mineralization causes a stiff and hard exo/endocuticle-like biphasic sandwich structure.172
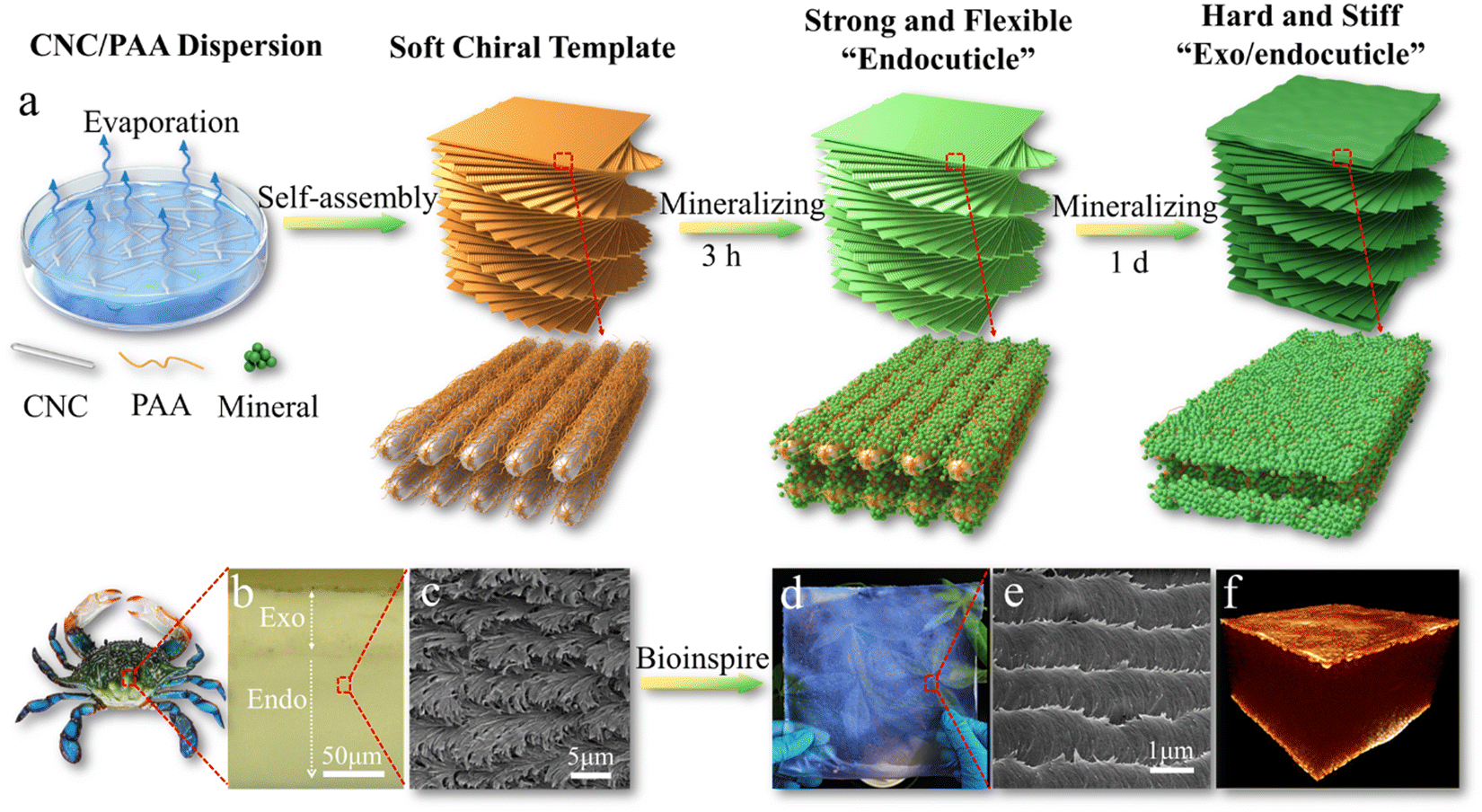 | ||
| Fig. 7 (a) Schematic illustration of mineralized CNC chiral composites with strong mechanical properties and bio-photonics; (b) the exo/endocuticle structure of crab exoskeletons; (c) the chiral structure of crab endocuticles. (d) A photo and (e) the biomimetic chiral structure of a large-area biomimetic iridescent film mineralized for 3 h. (f) X-ray tomography 3D image of the sandwiched structure of the biomimetic film mineralized for 1 d. Reprinted from ref. 172 with permission of Wiley, copyright 2021. | ||
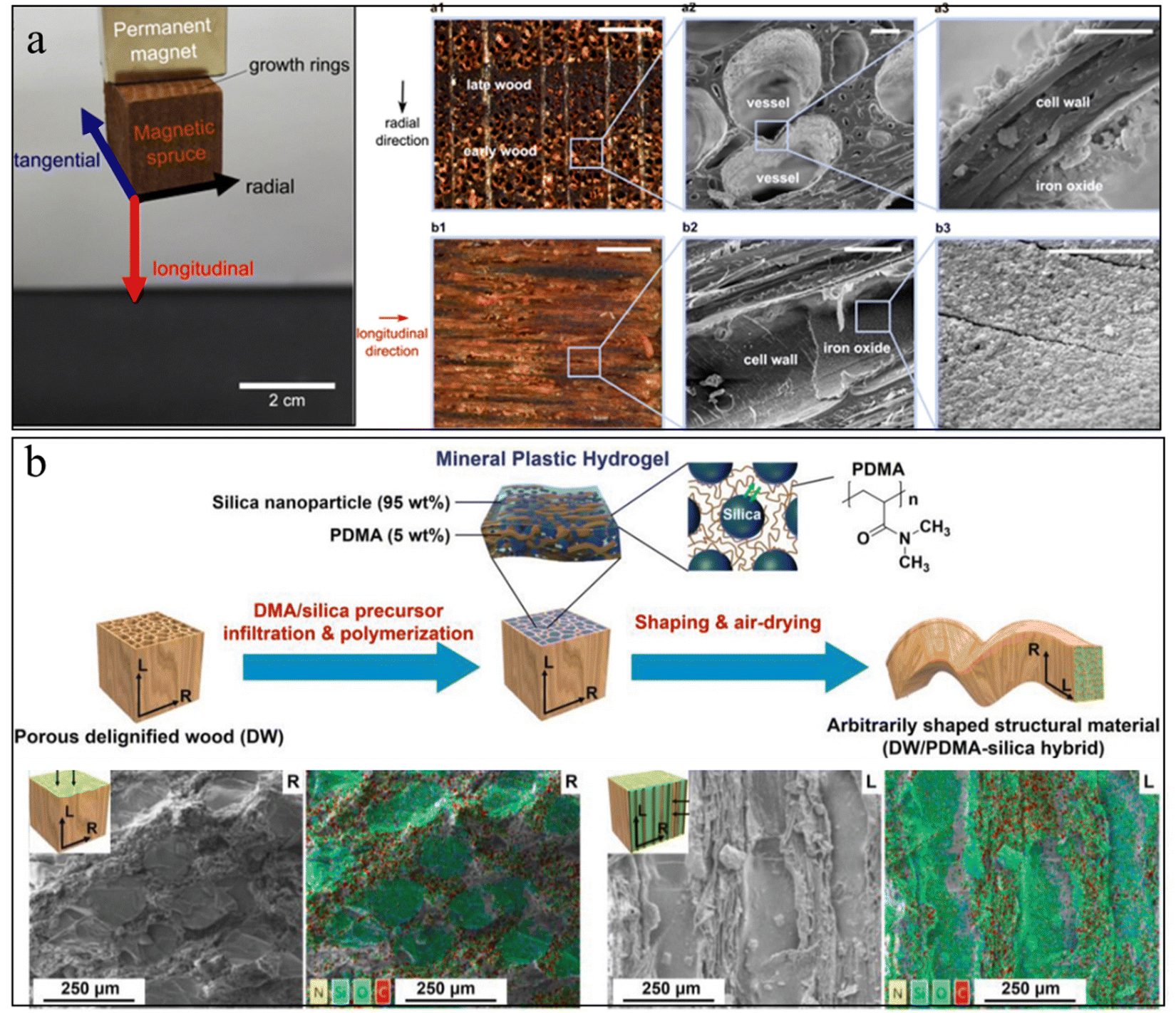 | ||
| Fig. 8 (a) Photo of magnetic wood by in situ growth of magnetic NPs inside the wood, and the corresponding SEM image of magnetic NPs distribution inside the wood. Reprinted from ref. 177 with permission of the American Chemical Society, copyright 2014. (b) Schematic illustration of wood–inorganic hybrid structural materials with the strategy of mineral plastic hydrogels. The corresponding SEM images show that the minerals filled the wood pores. Reprinted from ref. 182 with permission from Wiley, copyright 2020. | ||
However, these wood–mineral composites usually face issues of non-uniform mineral distribution and a small amount of mineral loading due to the hierarchical pore size and the closed pores caused by the sample preparation. To increase the porosity of the wood scaffold, delignification treatment has often been applied to the wood. This treatment could partially or fully remove the lignin from the wood scaffold, although it will also reduce the mechanical properties.179 Additional polymers can infiltrate into these delignified wood scaffolds to work as functional glues to enhance the mechanical properties, as well as to act as nucleation agents to regulate the uniform mineral growth.180,181 Zhang et al., prepared a mineral plastic hydrogel by infiltrating poly(N,N-dimethylacrylamide)/silica into a porous delignified wood scaffold (Fig. 8(b)).182 The as-prepared hydrogel exhibits good hydroplastic properties that allow manipulation of the hydrogel into arbitrary shapes. These designed shapes can be preserved after drying and good strength and toughness were obtained. In another example, Wang et al., infiltrated biocompatible alginate hydrogels into the microchannels of a delignified wood scaffold followed by in situ mineralization of HAp nanocrystals.183 The strong interfacial strength and the combination of HAp nanofillers with aligned cellulose scaffolds provide the mineralized composites with a high strength and modulus that are three orders of magnitude higher than those of conventional alginate hydrogels.
Besides, the study of mineralized cellulose mechanisms is a research hotspot. Generally, there are many techniques to explore the underlying mechanism of mineralized nanocellulose materials, such as small-angle scattering (SAS), quartz crystal microbalance with dissipation monitoring (QCM-D), cryogenic electron microscopy, tomography and computer simulations of materials. Computer simulations are a feasible and rapid way to study the reactions and structures of mineralized nanocellulose. By comparing the computational results and experimental data, researchers can predict hypothetical model structures and properties.184 However, simulations are suitable for specific length scales and it is hard to examine the integration of mineralized cellulose with complex structures like hierarchies.185 The common simulations methods include Ab inito electronic structures simulations, peridynamics simulations, phase field models simulations, the finite element method (FEM), all-atom MD stimulations, reactive molecular dynamics (MD) simulations, atomistic MD simulations, and coarse-grained (CG) simulations.186 All these simulations could be utilized solely or in combination to predict the structure and function across different length scales.
4. Biomedical applications
Bioinspired mineralization is a process in which researchers mimic biomineralization and utilize a biological matrix as a template to regulate the nucleation, orientation, growth, and self-assembly of inorganic minerals.187 This revolutionary approach exhibits tremendous potential to produce extraordinary biomaterials by integrating organic–inorganic composites with micro- or nano-scale replication of hierarchical natural structures (e.g., bone, dentin, and enamel), defining novel properties with coating biomolecules or chemicals (e.g., soft tissue engineering, anticancer, antibacterial, and drug delivery).Cellulose is a nature-derived polymer with excellent mechanical properties, fine tunability in functional groups modification and morphology, green properties (e.g., record biocompatibility, nontoxicity, and biodegradability), abundance, and accessibility.188 Cellulose, as a “multi-use mucilage,” is introduced to bioinspired mineralization and envisioned for the most promising future directions in the field of biomedical applications.189–193 Cellulose related materials exhibit extraordinary properties with bioinspired mineralization, especially in terms of physicochemical, mechanical, biodegradability and in vitro/in vivo properties along with non-mineralized cellulose (Table 2). However, there are only a few reported studies investigating the mineralized cellulose-based scaffolds for medical related tissue engineering so far. Thus, much of the work on this subject needs further effort. In this part, we summarize the biomedical applications of cellulose and its derivates via biomimetic mineralization. However, a more detailed insight into this topic is provided in dedicated reviews.20,25,194–198
| Properties/materials | Non-mineralized cellulose | Mineralized cellulose (synergistic type) | Ref. | |
|---|---|---|---|---|
| Hydrogel | CNF composite hydrogel | CNF composite hydrogel with bioactive glass | 199 | |
| Physicochemical | Viscosity (Pa s) | 176 ± 16 | 838 ± 123 | |
| Spreading ratio | 0.78 ± 0.11 | 0.89 ± 0.18 | ||
| Biological function | Ion release | (—) | Release of B, Si, Sr, Mg, Ca, and P | |
| In vitro cell biology | Good viability and proliferation ability in Saos-2 cells and hBMSCs | Slight decreased viability and proliferation ability, but higher osteogenic ability in Saos-2 cells and hBMSCs | ||
| Hydrogel | Cellulose hydrogels | Cellulose/HAP nanocomposite hydrogels | 200 | |
| Physicochemical | Recyclability | 96.58% | 78.60% | |
| Mechanical | Young's modulus (kPa) | 69 | 333 | |
| Compressive stress (kPa) | 100 | 401–570 | ||
| Cryogel | CNC cryogel | Mineralized CNC cryogel with SBF | 201 | |
| Physicochemical | Weight loss of the samples immersed in PBS for 21 days | 25.3 ± 2.0% | ∼47.9 ± 4.4% | |
| Mechanical | Young's modulus (kPa) | 4.1 ± 0.6 | ∼3.4 ± 0.3 | |
| Biological function | Cell viability of human adipose-derived stem cells (hASCs) | >90% lives three days after seeding | ||
| Metabolic activity and cell proliferation | Better | |||
| Osteogenesis ability of hASCs | (—) | Higher ALP activity and increased gene expression of RUNX2, COL1A1, BMP-2, DKK-1, and PDPN | ||
| Release of PDGF and VEGF (pg ml−1) | 50 for PDGF | 30 for PDGF | ||
| 40 for VEGF | 25 for VEGF | |||
| Bacterial cellulose | Aligned-BC | Mineralized aBC in situ | ||
| Physicochemical | Thermal stability | 225–350 °C | 350–480 °C | |
| Weight loss (%) between 150 and 450 °C | 90.37 ± 2.59 | 51.68 ± 2.06 | 202 and 203 | |
| T max (°C) at decomposition | 375.9 ± 3.2 | 351.4 ± 2.5 | ||
| Surface morphology | Less dense arrangement of fibrils | More dense arrangement of fibrils | ||
| Mechanical | Elastic modulus (Gpa) | 5 | 10 | |
| Hardness (Gpa) | 0.1 | 0.6 | ||
| Degradation | In vitro degradation with cellulosic enzyme | Degraded in 5 days | Slower degradation speed | |
| Biological function | In vitro cell biology | Better attachment and proliferation in the two-day cultivation of MC3T3-E1 | ||
| Cellulose nanofibers | Mineralized cellulose nanofibers with SBF | 204 | ||
| Physicochemical | Thickness (mm) | 0.092 ± 0.004 | 0.149 ± 0.013 | |
| Surface pH | 7.0 ± 0.23 | 6.6 ± 0.091 | ||
| Mechanical | Tensile strength (MPa) | 21.25 ± 1.41 | 26.25 ± 2.50 | |
| Strain at maximum load (%) | 16.33 ± 3.053 | 24.97 ± 2.95 | ||
| Modulus of elasticity (MPa) | 687.5 ± 102.1 | 815.9 ± 93.9 | ||
| Biological function | Ion release | Release of Ca, P and F | ||
| Ability to remineralize the demineralized teeth in vitro | Increase the Vickers hardness number from 41.2 ± 0.09 to 137.7 ± 0.1 | |||
4.1 Hard tissue engineering
Biomineralization in nature primarily involves the mineralization process of bone, teeth, and shells. Bone, a hierarchical–architecture nanocomposite material, is mainly made of inorganic hydroxyapatite nanocrystallites and a collagen-rich organic matrix.205,206 Bone can be considered as the self-assembly of insoluble collagen fibrils as a structural template mineralized with nanoscopic apatite platelets, thus exhibiting unique mechanical properties.206,207 Nanocellulose is promising as a base organic matrix for biomineralization, which applies to the tissue engineering of bone, dentin, and enamel repair since the multi-scale hierarchical anisotropic structure has been the greatest advantage for wood.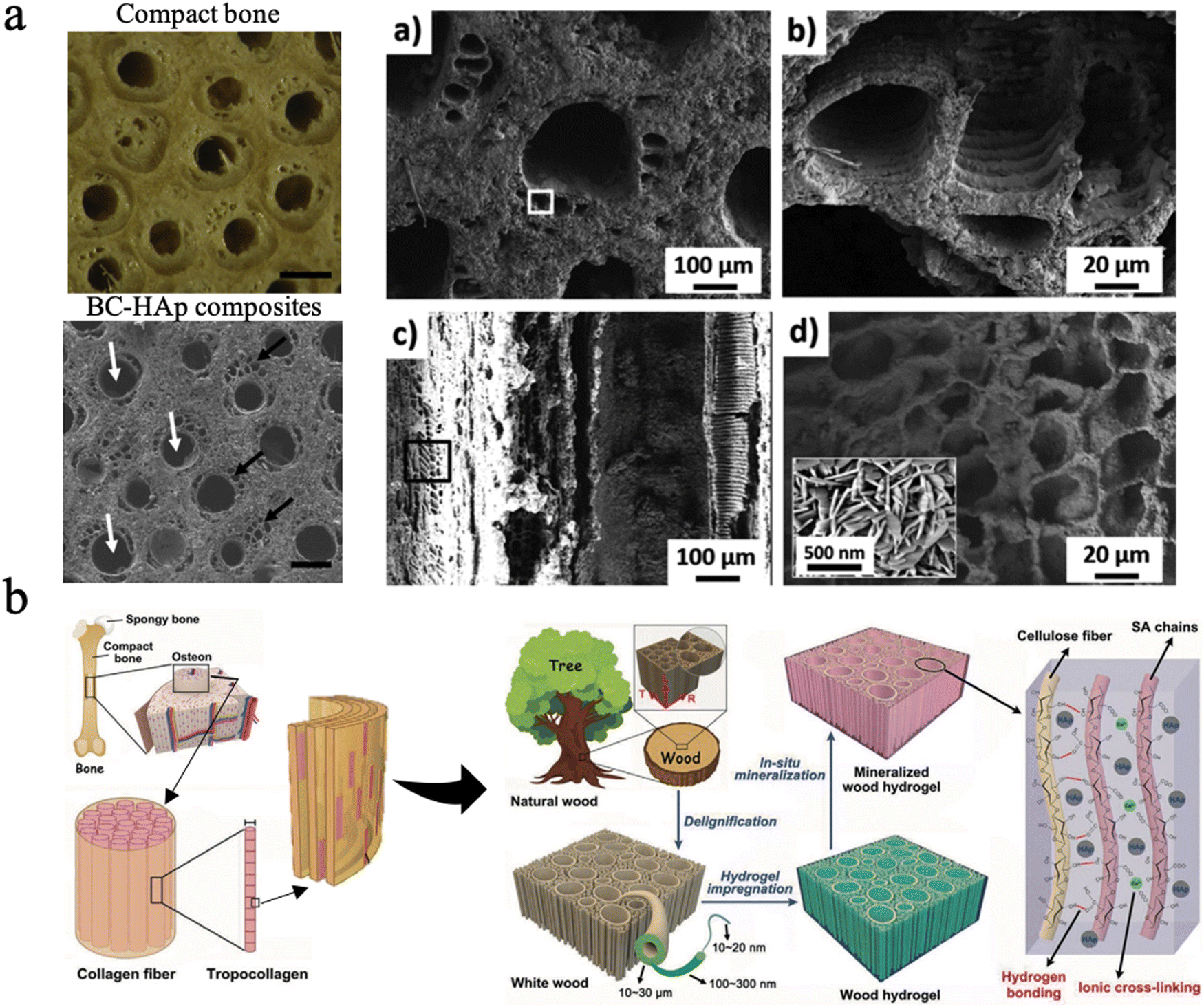 | ||
| Fig. 9 (a) The SEM picture of BC–Hap composites and the material microstructure at multiple scales, similar to the osteon structure. Reproduced from ref. 209 with permission of Elsevier, Copyright 2020. (b) Design strategy, fabrication and microscopic structure of the mineralized wood hydrogel composite, which is bioinspired by the hierarchical structure of natural bone. Reproduced from ref. 210 with permission of Wiley, Copyright 2021. | ||
Liu et al. designed a nanocellulose-reinforced hybrid membrane by assembling ultralong Hap nanobelts in a BC hydrogel.211 This hybrid membrane was proven to promote rapid new bone growth and the formation of blood capillaries, and nerve regeneration.211 Moreover, Paul et al. also mineralized a BC scaffold with HAp for bone healing applications in simulated body fluid. MSCs showed good adhesion and differentiation on mineralized BC with the results of alkaline phosphatase gene expression.202,212
The mineralization of cellulose or BC scaffolds improved their mechanical properties and impeded their applicability in hard tissue regeneration. It is vital that the mechanical strength of bone substitute scaffolds should match that of the surrounding bone tissue to prevent the occurrence of some clinical problems like stress shielding.193 Cortical bone has a modulus strength ranging from 3 to 17 GPa while cancellous bone has a modulus of 0.44 GPa, since bone strength changes extensively with age as well as the type of bone. In terms of tensile strength, cortical bone shows a tensile strength from 33 to 193 MPa and cancellous bone of approximately 6.8 MPa.213 Cheng et al. mineralized aligned BC through in situ mineralization using CaCl2 and K2HPO4 solutions to mimic the natural structure of bone. The BC composites exhibited an elastic modulus of 10.91 ± 3.26 GPa and hardness of 0.37 ± 0.18 GPa,214 which is close to that of cortical bone (Fig. 10(a)).
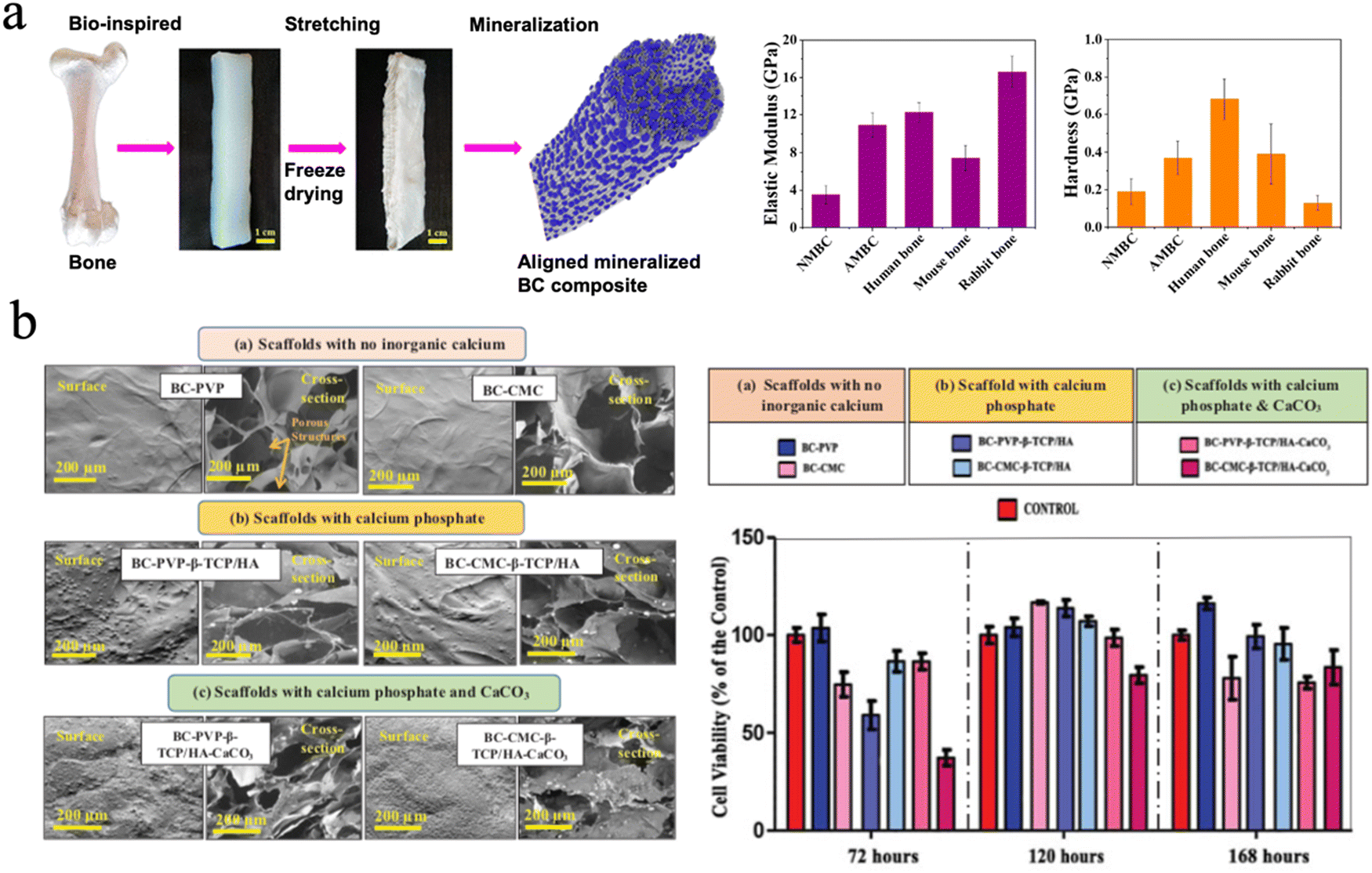 | ||
| Fig. 10 (a) The aligned mineralized BC composite mimicking the natural structure of bone exhibiting excellent mechanical strength. Reproduced from ref. 214 with permission of the American Chemical Society, copyright 2019. (b) SEM image of BC based hydrogel scaffolds with no inorganic calcium and different calcium fillers, and the cell viability profile of Lep-3 cells on different groups. Reproduced from ref. 223 with permission of Taylor & Francis, Copyright 2018. | ||
It is reported that the pore size of scaffolds impacts mesenchymal stem cell growth and adhesion, and neovascularization.217 The researchers focused on the rational design of pore sizes of cellulose-based scaffolds to balance the mechanical properties and the capability of promoting cell proliferation and differentiation.215,218 Favi et al. designed a microporous BC scaffold with well-defined honeycomb pore arrays inserted into nano-HAp. The BC-HAp scaffolds have a pore size of 300 μm and formed a well-designed port network to mimic the bone microstructure. The porous BC–HAp scaffold showed an ultimate tensile strength of 1.39 ± 0.28 MPa, a strain at break of 23.44 ± 2.52%, and an elastic modulus of 6.73 ± 0.7 MPa. The values for these scaffolds were significantly lower than that of compact human bone, but they are still mechanically suitable for cancellous bone. This study finally proves that the BC–HAp scaffolds exhibit good biocompatibility and adhesion for human-derived bone marrow mesenchymal stem cells as compared to native BC scaffolds.223
Huang et al. fabricated nanostructured macroporous BC scaffolds through an emulsion freeze-drying technique and modified them with gelatin (Gel) via the procyanidin (PA) crosslinking technique and HAp coating.219 They made three other scaffolds named BC, BC/gel, BC/PA/gel. BC scaffolds were prepared with crushed BC, agarose, and deionized water via emulsion freeze-drying and BC/gel scaffolds added one more component of gelatin. BC/PA/gel needs one more crosslinking step. The four scaffolds were then compared in terms of their biocompatibility and osteoinductivity. The numbers of macropores and nanopores in BC/gel, BC/PA/gel, and BC/PA/gel/HAp groups all showed a statistically significant increase compared with BC scaffolds, which was proven by the existence of a nanofibrous interconnected network structure. The mechanical results showed the BC/PA/gel/HAp scaffold displayed the highest Young's modulus, maximum load, and compressive strength. The adhesion, viability, proliferation, and osteogenic potential of hBMSCs were investigated after seeding on the different BC scaffolds. The results showed that the BC/PA/gel/HAp scaffold had the highest viability, best adhesion, and the most apparent proliferation and osteogenic differentiation among the four scaffolds. In vivo studies also revealed the same tendency of new bone formation in the BC/PA/gel/HAp sample. Therefore, porosity plays a role in promoting cell migration and new tissue formation.219
There are many forms of inorganic calcium minerals in nature. They can be simply divided into calcified minerals and noncalcified minerals. There is no doubt that calcium mineral is the main inorganic phase in natural bone.220 More minerals, such as bioactive glass221 and biphasic calcium phosphate222 were chosen to fabricate cellulose-based biomaterials for bone repair. Basu et al. focused on the biological efficiency of inorganic calcium-filled BC based hydrogel scaffolds for their future use in bone tissue engineering/bioengineering. They evaluated calcium phosphate, β-tricalcium phosphate (β-TCP) and HAp and calcium carbonate (CaCO3), which were incorporated into BC. All these hydrogel scaffolds facilitated human embryonic fibroblasts (Lep-3) and mouse bone explant cells (BEC) efficient growth and proliferation. However, it was found that BC-based β-TCP/HAp scaffolds show the lowest growth and proliferation of Lep-3 and BEC DNA strands, compared to BC-CMC based β-TCP/HAp scaffolds. BC-based β-TCP/HAp scaffolds facilitated notable cell adhesion and growth and were recommended as the most efficient bone tissue engineering scaffold among the four tested scaffolds (Fig. 10(b)).216,223
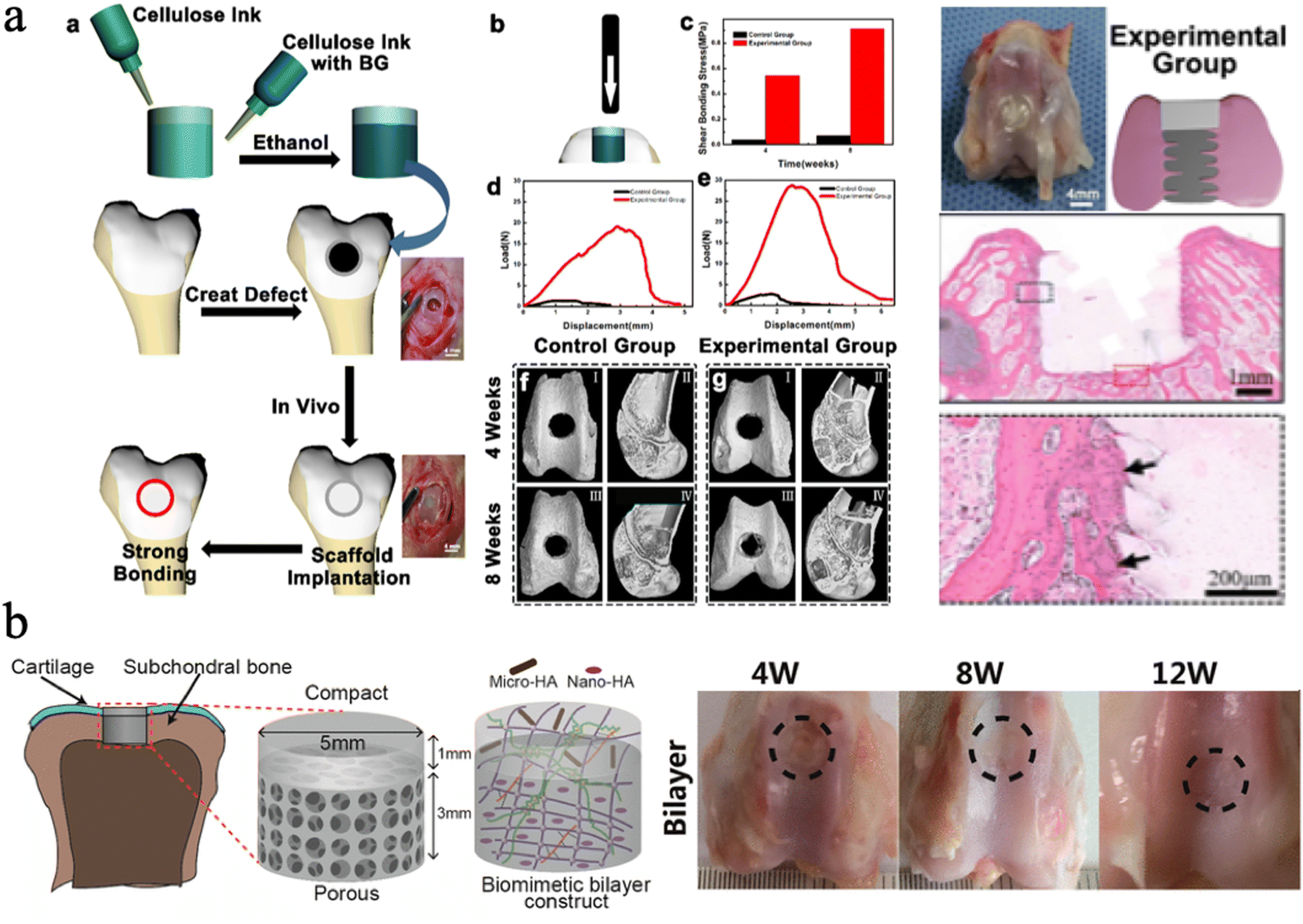 | ||
| Fig. 11 (a) The push-out test for cellulose ink with bioactive glass, and the histological images with gel plugs implanted into rabbit femurs. Reproduced from ref. 225 with permission of the American Chemical Society, Copyright 2022. (b) Schematic illustration of the bilayer BC based hydrogel, and the macroscopic images of knees repaired with the hydrogel. Reproduced from ref. 226 with permission of the American Chemical Society, copyright 2018. | ||
Moreover, different sizes of HAp nanoparticles can be chosen to assemble with nanocellulose for different hierarchal tissue regeneration. Zhu et al. incorporated CNFs into a double-network (DN) hydrogel scaffold based on covalently cross-linked poly (γ-glutamic acid) (PGA) and ionically cross-linked alginic acid, which was loaded with different sizes of HAp particles, Nano-HAp and Micro-HAp.226 They found that the Nano-HAp can enhance the compression modulus and osteogenesis, while Micro-HAp was added to promote cartilage matrix deposition. The results showed that the hydrogel exhibits a comparable compression modulus (0.322 MPa) to natural articular cartilage. In vivo experiments showed that the bilayer scaffolds with different sizes of HAp design have great osteochondral and bone tissue repair potential (Fig. 11(b)).226
Cellulose derivatives are also developed for strengthening properties and are beneficial for bone tissue scaffolding applications. Oliveria et al. demonstrate an injectable composite containing CaP glass-ceramic particles, dispersed within a (hydroxypropyl)methyl cellulose (HPMC) matrix, with the capacity to release calcium in a more sustained fashion.227 The results show that the composite could improve both bone formation and increase angiogenesis via a rat bone defect model.227 CNC with sulfate and phosphate half-ester surface groups were produced by chemical crosslinking and submerged in simulated body fluid solution with a 0.1 M CaCl2 pre-treatment. Sulfated CNC aerogels exhibited good compressive strength and long-term stability in liquid environments and showed their osteoconductive properties with a tremendous increase in bone volume by up to 50% compared to sham sites. Jafari et al. reported a novel epichlorohydrin (ECH)-crosslinked hydroxyethyl cellulose (HEC)/soy protein isolate (SPI) porous bi-component scaffold (EHSS) with HAp functionalization for bone defect repair via the combination of lyophilization and in situ biomimetic mineralization.228 The scaffold exhibited an interconnected porous structure, which is suitable for MC3T3-E1 cell attachment, proliferation, and growth. The scaffold had a similar Ca/P ratio to natural bone tissue and markedly promoted new bone formation and maturation in in vivo experiments.228 More recently, a novel nitrogen-doped multiwalled carbon nanotube/cellulose/nanoHAp nanocomposite scaffold was synthesized via a crosslinking/hydrothermal/freeze-drying method, resulting in layer-by-layer structures with functional and structural properties mimicking natural bone. Importantly, the resulting scaffold has favorable mechanical properties, high porosity, and interconnected architecture, thus providing a suitable microenvironment for bone mesenchymal stem cell (BMSC) proliferation, viability, mineralization, and osteoinduction in in vitro cultures. In vivo experiments confirmed that the scaffolds not only improved the interface bonding with bone tissue but also accelerated new bone formation and regeneration.229
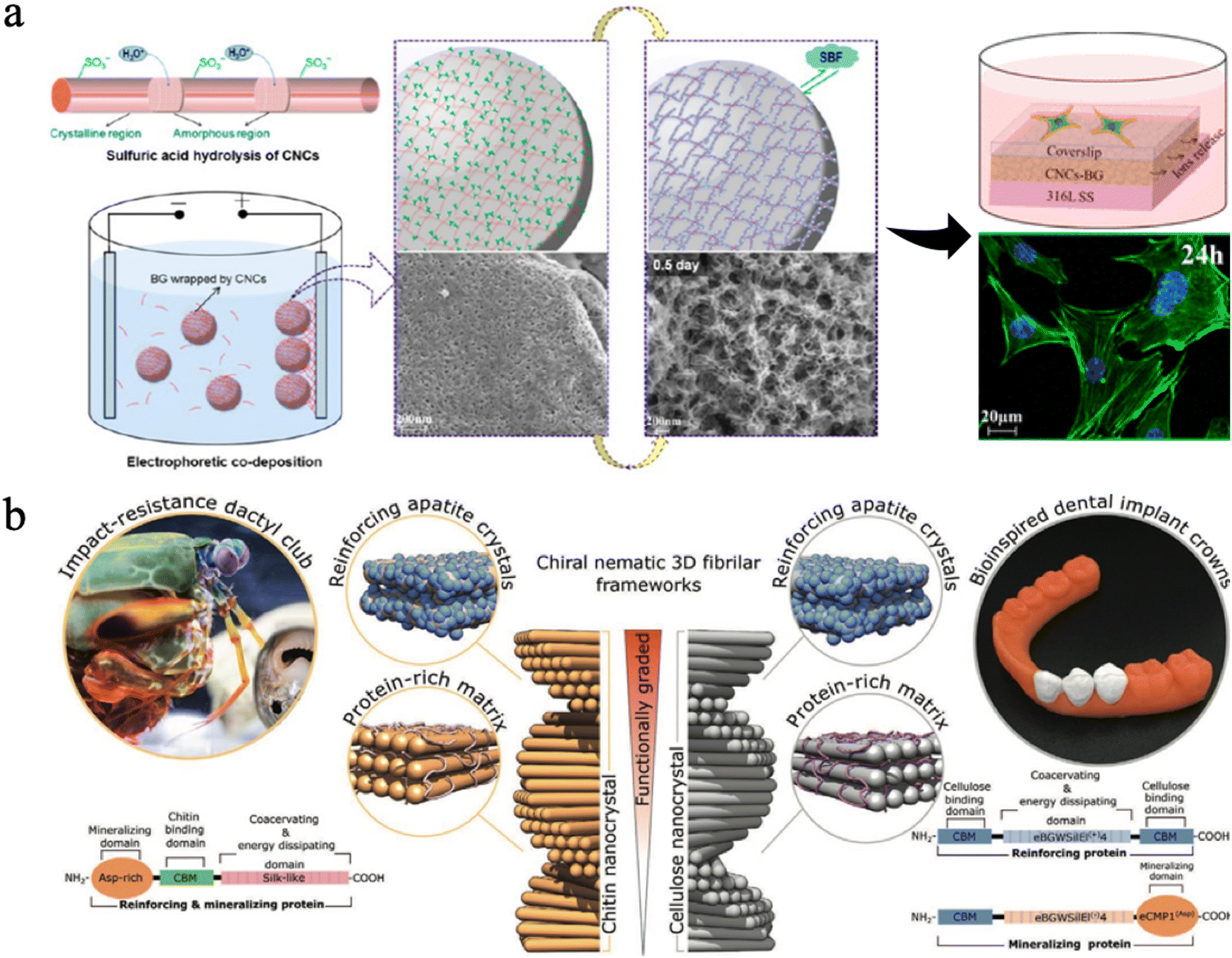 | ||
| Fig. 12 (a) Design strategy, fabrication of CNC-BG coatings, the SEM images, the confocal fluorescence images and schematic diagrams of cell skeletons on coatings. Reproduced from ref. 230 with permission of the American Chemical Society, copyright 2015. (b) A graded biologically inspired multiphase nanocomposite structure of CNCs mixed with genetically engineered proteins and apatite crystals. Reproduced from ref. 231 with permission from Wiley, copyright 2021. | ||
In another field, scientists utilized CNFs to assemble unique structures to mimic other natural hard tissues. These studies expanded the application of CNFs in hard tissue regeneration. Qi et al. fabricated highly compacted, aligned CNFs through a nonclassical mineralization process and achieved dentin-like ultrastructures.232 Zaki et al. reported innovative re-mineralizing oral films made from a blend of hydroxyethyl-cellulose (HEC) and CNF, loaded with nepheline fluorapatite glass powder.204 SEM results revealed that the film has a uniform glass powder particle distribution. They also measured surface hardness by evaluating the films’ effect on demineralized enamel micro-hardness. The results showed a significant increase in enamel micro-hardness compared to the demineralized specimen. The cellulose-derived films can be a candidate for remineralization of early demineralized tooth lesions.204
Some researchers found that nanocellulose could be organized in a pitch-graded chiral nematic structure to achieve great hardness properties (Fig. 12(b)).231 They combined assembly of CNCs with genetically engineered protein matrices, which can increase their interfacial strength and mediate nucleation and growth of apatite crystals. By mimicking the impact-resistant dactyl club of the stomatopod, scientists successfully produced this multi-phase nanocomposite with a CNC-based apatite. The resulting bioinspired composite exhibits a graded microstructure with a correlated mechanical gradient that closely resembles those observed in natural biomineralized composites such as human teeth,231 suggesting it has potential in dental applications.
4.2 Soft tissue engineering
Soft tissue engineering has been a research hotspot in tissue regeneration since soft tissue damage or soft tissue-related conditions such as muscle, blood vessels, skin, heart, and cartilage injuries became common in daily clinical work. Natural biopolymers such as polysaccharide-based bio-nanocomposites have extensively attracted scientists due to their great biomimicry and perfect ability to provide biological and physicochemical properties of extracellular matrices. Over recent decades, scientists tried to design new strategies to fabricate multifunctional soft tissue engineering substitutes for repairing damaged soft tissues, providing mechanical support, and killing bacteria simultaneously.190,191,233 To date, cellulose has been prepared into diverse forms and been modified with different inorganic agents or polymers to develop novel functional biomaterials for soft tissue regeneration.It is reported that mussels have extraordinary properties such as strong, tough, resistant to bacteria, stable over extreme environments. Xu et al. found that iron and the unique hierarchical structure play an essential role in the mussels’ mechanical properties.234 Hence, nanocellulose hybrids with metal or metal oxides or metal–organic-framework structures have also been extensively explored for soft tissue regeneration, especially for wound dressing, which is an intriguing research topic and always discussed in the field of biomaterials. Wang et al. designed a copper-containing mesoporous bioactive glass (Cu-MBG) and nanofibrillated cellulose (NFC) composite and co-cultured it with 3T3 fibroblasts, human umbilical vein endothelial cells (HUVECs), and E. coli. separately. The biocomposites showed a profound angiogenic effect on 3T3 fibroblasts and HUVECs and an inhibiting effect on the growth of E. coli. The biocomposite design based on nanocellulose not only resisted chronic bacterial infection but also promoted angiogenesis.235 Silver nanoparticles were synthesized into the matrix of CNCs to design wound dressing candidates. The dressings showed water absorption capacity and strong antibacterial activity and gained a synergistic effect on in vivo skin wound healing, and documented faster wound closure in treated mice.236
TEMPO-oxidized CNF (T-CNF) has low cytotoxicity and structural similarity to extracellular matrices in the form of hydrogels and thus stands out as a platform biomaterial in bioink formulation for 3D printing. Xu et al. fabricated a hydrogel composed of T-CNF and gelatin methacrylate (GelMA) via in situ crosslinking by Ca2+ and UV. The hydrogel showed non-cytotoxic and biocompatible features of 3T3 fibroblasts and promoted the proliferation of fibroblasts, which exhibits great potential in 3D printing for wound healing applications.189 In another study, Leppiniemi et al. fabricated a stable 3D hydrogel with T-CNF alginate glycerin via CaCl2 crosslinking.237 The T-CNF hydrogel can achieve the printed shape fidelity and stability as the printing paste. The described avidin-functionalized nanocellulose-alginate material provides a generic platform for the immobilization of bioactive components like growth factors or antimicrobial agents such as antibiotics. In addition, the buffer tests demonstrated that the 3D printing of porous structures reduced excess deformation of the objects, especially when the printed structure was cross-linked with CaCl2. Voids within the structure provide room for swelling in moist and wet conditions, thus reducing irritation and pain to accelerate wound healing.237
Bacterial cellulose is reported to be more suitable for skin tissue engineering and wound repair. For example, Wu et al. demonstrated a BC/lotus root starch/hydroxyapatite (BC/LRS-M/HAp) scaffold endowed with a nanoporous hierarchical structure.238 The researchers adopted a combinatorial approach to sequentially conduct BC surface modification by LRS, then constructed a 3D architecture by agarose porogen templating with HAp deposition on the BC/LRS-M via biomimetic mineralization. The target scaffold had a pore size of 300–500 μm, which was beneficial to cell adhesion and proliferation. Alkaline phosphatase (ALP) activity assay showed a significantly higher ALP activity by approximately 50% and 100% increments, respectively, on BC/LRS-M/HAp than the two control scaffolds. These results suggested that the BC/LRS-M/HAp scaffold could be a great articular cartilage tissue substitute.238
4.3 Cancer therapy
It is well known that the intersection between biology and materials science has provided extraordinary natural materials, which cannot be easily synthesized in the laboratory, although it is achieved in nature under ambient conditions without hazardous chemicals. To date, researchers mimicked the process of biomineralization to create abundant biomimetic materials for biological applications. In nature, the biological processes can be divided into biomineralization and remineralization. During remineralization, calcium ions could possibly be deposited anomalously in soft tissues, thus causing pathological diseases.239,240 However, scientists can promote cancer cell targeting calcification (CTCC) and accelerate cancer cell death to make it a new alternative cancer treatment. Tang et al. suggested this novel strategy, and this approach can effectively suppress tumor growth and secondary metastases without destroying normal cells compared to traditional chemotherapy (Fig. 13(a)).241,242 Epple et al. also fabricated calcium phosphate nanoparticles with an inner shell of CMC that incorporates a photoactive dye and an outer shell of poly(ethyleneimine) (PEI).243 The nanoparticles could be used for photodynamic therapy with HT29 cells (human colon adenocarcinoma cells), HIG-82 cells (rabbit synoviocytes), and J774A.1 cells (murine macrophages) and showed a similar killing efficiency to the pure photoactive dye.243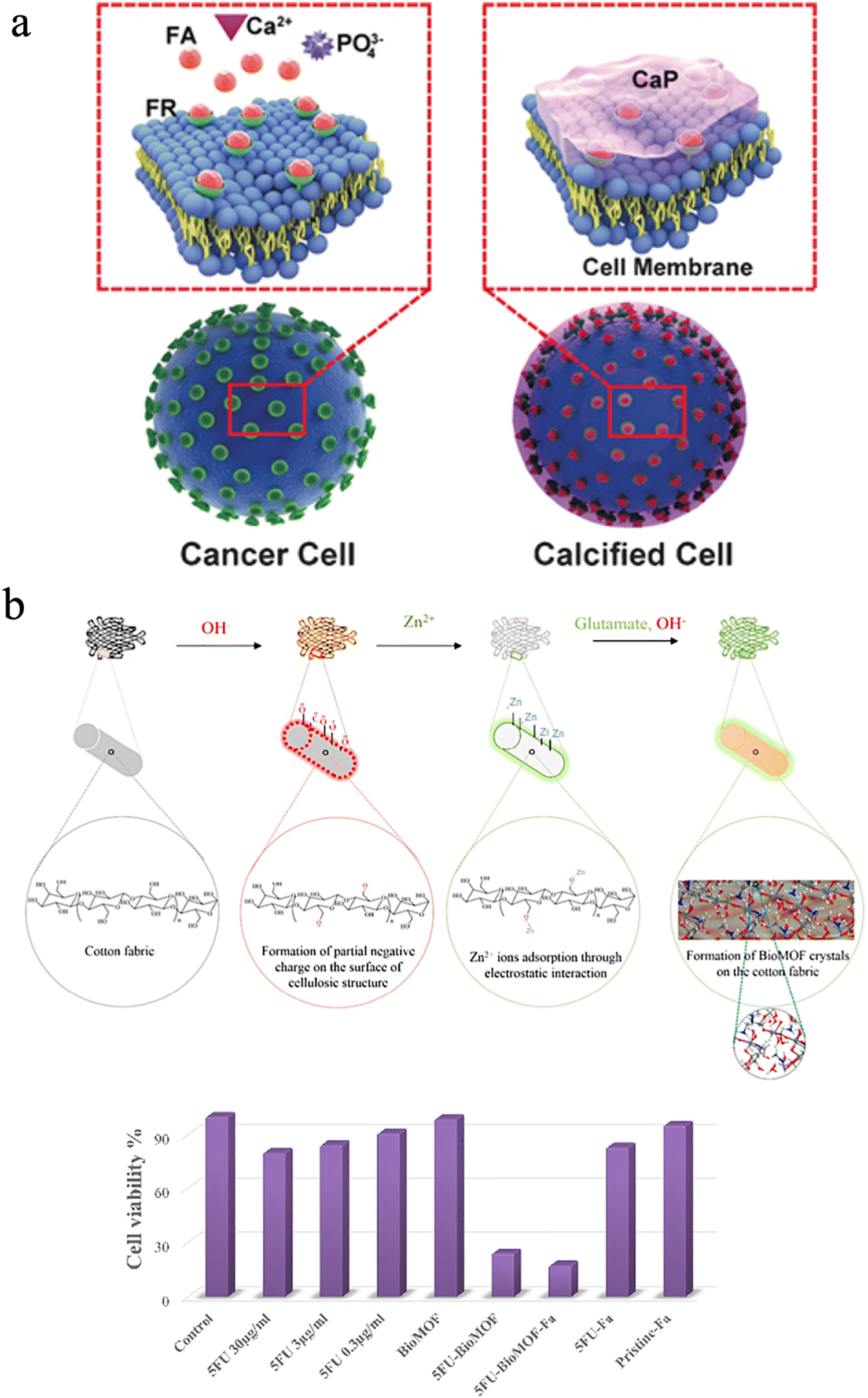 | ||
| Fig. 13 (a) A scheme of the drug-free tumor therapy strategy via biomineralization. Reproduced from ref. 242 with permission of Wiley, copyright 2016. (b) Schematic of in situ synthesis of a BioMOF on cotton fabric, and the corresponding cell viability of different groups. Reproduced from ref. 244 with permission of Elsevier, copyright 2019. | ||
Besides, the mineralization of structures with subcomponents such as metal oxides, silicates, and metal–organic frameworks can be integrated into one single material through chemical modification to cure cancers since nanocellulose has abundant hydroxyl groups. Noorian et al. developed a new kind of cellulose fabric combined with the zinc glutamate Bio Metal–Organic Framework (BioMOF) to control the delivery of nitric oxide (NO) and 5-fluorouracil (5FU) for skin cancer therapy.244 In this material, cellulose acted as a matrix-forming material for long-lasting sustained drug delivery (Fig. 13(b)).244 Fe3O4–Ag2O quantum dots also were decorated with CNFs for cancer drug delivery. The quantum dots play a role in connecting the cellulose fibers to the anticancer drug etoposide and methotrexate. These multiple hybrid materials have ferromagnetic properties and a constant speed of drug release.245 Besides, Xu et al. utilized carbon quantum dots and cellulose to improve anaerobic digestion.234
4.4 Drug, protein, and growth factor delivery
Nanocellulose is promising in medical applications for drug delivery because of its abundant origin, low cost, and biocompatibility. Cellulosic materials have already been made as commercial products for drug delivery. Different drug gradients have been selected to be modified with nanocellulose, thus achieving the goal for therapeutic treatment for different diseases. However, it is still challenging since complicated body environments (e.g., stomach, intestines, bone, cartilage) could limit the drug absorption capacity and curing diseases needs to achieve a long-time, sustainable, and defined amount of drug release.246 The question of how to achieve exact and efficient drug delivery systems with selective, stable, and controlled release of pharmaceuticals has been a research hotpot. Most specifically, mineralized cellulose-based hydrogel or aerogel scaffolds have been applied for drug delivery due to their high porosity, excellent network structure, and stable mechanical strength. Ryuji et al. made a BC/bone cement/antibiotics scaffold to achieve stable antibiotics release and simultaneously high strength. The results showed that the average values for compression strength, fracture toughness, and fatigue life of the cellulose antibiotic cement were 97%, 97%, and 78% of the values obtained for plain cement, respectively. In contrast to these excellent values, the traditional antibiotic cement exhibited only 79% of compression strength, 82% of fracture toughness, and 17% of the fatigue life of plain cement. Moreover, the cumulative elution over 35 days was 129% greater from the cellulose antibiotic cement than from the traditional antibiotic cement. The obtained BC/bone cement/antibiotics improved fatigue life and increased antibiotic elution.247Meanwhile, efforts are dedicated to mixing magnetic or gold nanoparticles as well as two-dimensional materials, into cellulose to obtain efficient drug delivery. Magnetic cellulose (m-CNCs) was synthesized and incorporated into alginate-based hydrogel beads as a potential drug delivery system. The study showed that m-CNCs could enhance mechanical strength and exhibit controlled release profiles by increasing the integrity of the alginate hydrogel beads and the swelling degree.248 Sekhar et al. developed a novel transdermal device fabricated from a polyelectrolyte complex (PEC) reinforced with nanogold-nanocellulose (GNP-NC) to enhance the skin permeation of diltiazem hydrochloride.249 More recently, Luo et al.250 fabricated a novel BC/graphene oxide (GO) nanocomposite drug nanocarrier by embedding GO into a 3D porous network of BC. Ibuprofen (IBU) as a model drug was loaded into the BC/GO nanocomposites. The formed IBU@BC/GO displays a more sustainable release behavior than IBU@BC, which suggests the critical role of GO in promoting the controlled release. Furthermore, in vitro cell viability experiments revealed that the BC/GO nanocarrier had many more living cells than BC. It is inferred that the BC/GO nanocarrier could be a great new drug delivery system.
More interestingly, Nishiguchi et al.251 reported an injectable, bioresponsive bone cement composed of bisphosphonate-modified nanocellulose (pNC) and β-tricalcium phosphate (β-TCP). Bisphosphonate is a class of bone resorption inhibitor drug for the treatment of osteoporosis and metastatic bone diseases.252 The pNC/β-TCP composites mimicked bone structures and possessed various favorable characteristics, including tunable viscoelastic properties, physical binding to the bone, apatite formation, and pH responsiveness. When the pH drops to ∼4.5, which corresponded to an osteoclast-induced pH decrease, the pNC-β-TCP composite degraded and released pNC, which suppressed osteoclast formation and pit formation. The injectable bone scaffold of pNC/β-TCP not only enhanced osteoblast differentiation but also regulated osteoclast activity, achieving a dual effect for the treatment of bone diseases and prevention of the locomotive syndrome.
Protein adsorption, protein, and growth factor delivery impact the properties of cellulose and expand the application of cellulose base materials. Fu et al. reported a facile and green sonochemical route for the rapid synthesis of cellulose/HA nanocomposites in NaOH/urea aqueous solution. The protein adsorption properties of the as-prepared cellulose/HA nanocomposites are investigated using hemoglobin (Hb) as a model protein. The adsorption of Hb on the surface of cellulose/HA nanocomposites was supported by FT-IR analysis. The peaks located at 1649 cm−1 and 1398 cm−1 (originating from Hb molecules) occurred after adsorption, indicating that the Hb molecules have been successfully adsorbed on the surface of cellulose/HA nanocomposites. The Hb adsorption properties of the as-prepared cellulose/HA nanocomposites were investigated at different initial Hb concentrations and different amounts of the sample. The amount of adsorbed Hb on the cellulose/HA nanocomposites increases with increasing initial concentration of Hb in the range of 0.2–4.0 mg mL−1 and reached 321.5 mg g−1 at a Hb initial concentration of 4.0 mg mL−1. The cellulose/HA nanocomposites still did not saturate adsorption at a high Hb concentration of 4.0 mg mL−1, indicating that the cellulose maintained a high protein adsorption capacity after HA mineralization. These results demonstrated that the cellulose/HA nanocomposites could be utilized for application in biomedical fields.253
Further research compared the protein adsorption and release ability of mineralized and nonmineralized CMC. Briefly, after calcium phosphate mineralization, the swelling behavior of the CMC-g-PDMAEMA hydrogels changes at pH 2.1 and 7.4. The swelling ratio was decreased after calcium phosphate mineralization. This may be due to the fact that inorganic calcium phosphate particles may act as additional crosslinking points between the polymeric chains which hinder the extension of the polymer chains and then suppress the swelling ratio. Moreover, the hybrid takes more time to attain the equilibrium state. Similarly, the BSA loading efficiency is lower in the PDMAEMA/calcium phosphate hybrid. The in vitro release of BSA from the CMC-g-PDMAEMA hydrogel shows an initial burst release which is possibly due to those BSA that are located near the hydrogel surface. However, the initial burst release of CMC-g-PDMAEMA hydrogel is reduced after biomimetic calcium phosphate mineralization. After the burst period, the hydrogel serves as a diffusion barrier and the drug is mainly released by the diffusion mechanism. Compared with the CMC-g-PDMEMA hydrogel, the release% was significantly decreased for the CMC-g-PDMAEMA/calcium phosphate hybrid at pH 2.1 and 7.4. For example, after 24 h, the release% decreased from 96% to 90% (at pH 2.1) and from 93% to 84% (at pH 7.4) after biomimetic calcium phosphate mineralization. These results suggest the role of mineralized calcium phosphate particles for reducing the permeability of the loaded protein and hence controlling the delivery of protein. These results offer promising multifunctional hybrid materials for bone tissue regeneration.254
Ribeiro et al. tested the release of the PDGF and VEGF, a chemotactic and a proangiogenic growth factor (GF), respectively, in mineralized and nonmineralized CNC cryogel. Contrary to what would be expected from samples with a higher weight loss, the immunodetection assay showed a decreased release of both GFs in the mineralized groups compared to the control. The more effective entrapment of these GFs in the cryogel scaffolds might result in either the microstructural alterations observed in the material or a favored interaction of these biomolecules with the mineral components. The prolonged bioactivity of the proposed mineralized cellulose cryogel scaffolds provides a new idea for the loading and delivery of proteins and growth factors.201
5 Conclusions and outlook
Considering all above-mentioned research, mineralized cellulose has been chosen as a safe, ideal medium to incorporate other active molecules, proteins, polymers, metals or metal oxides to achieve diverse medical applications. In this review, we grasped a comprehensive report of properties, synthesis methods of mineralized nanocellulose and the medical applications, especially in hard tissue engineering. For example, although plenty of studies have focused on hard tissue engineering, we need to incorporate all the important and necessary criteria (such as mechanical properties, natural tissue structure, biocompatibility and physiological properties) into one material. There are still many limitations for mineralized cellulose-related biomaterials to solve.First, nanocellulose-related tissue engineering substitutes are still a laboratory product. Many efforts need to be spent on fabricating a uniform, stable, green, biocompatible medical tissue engineering substitute since nanocellulose is reported to dissolve thoroughly in the human body.255 Second, the strategy of how to best speed up the integration of nanocellulose-related medical products with bone cartilage is still on the way. The underlying mechanism also needs to be explored. Third, mineralized nanocellulose possesses good chemical stability, mechanical tolerance and durability, which may result in complications in bioactive drug molecule delivery. How to balance these properties needs to be resolved to devise personal and intelligent designs for each disease.
Meanwhile, we are still a long way from achieving mineralized cellulose commercialization. There is no doubt that cellulose can be produced from cheap sources and waste materials and this has been commercialized by several companies. However, there are still some shortcomings hindering its clinical application and commercialization. First, the properties of cellulose need to be improved and the application scenarios need to be expanded. Besides, the current technologies for cellulose fabrication at the nanoscale are still expensive, require tedious efforts, and lack large-scale production for industrial and biomedical applications.256 Thus, it has been shown that modification of properties, increasing yields, a reduction in production costs, and choosing appropriate industrial fabrication lines are the main future goals of researchers.257 Furthermore, on the path to clinical translational research and clinical translation, the output of nanocellulose with high purity and the absence of endotoxins is highly needed for biomedical applications. Besides, the properties of cellulose should be improved by mixing with other materials, which will bring great possibility for their practical applications in a variety of fields. In addition, there are only limited studies on the toxicity of cellulose in the human body, and a great number of studies are still required to ensure that cellulose is not harmful to human beings.258 Creating controlled, reliable, and reproducible production techniques will be essential to the ultimate approval of new applications and will help to pave the way for greater acceptance of cellulose as the commercially available material for biomedical translations that it has the possibility to be.259
Abbreviations
| 1D | One-dimensional |
| HAp | Hydroxyapatite |
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| NMMO | N-Methylmorpholine-N-oxide |
| MC | Methylcellulose |
| HPC | Hydroxypropyl cellulose |
| CMC | Carboxymethyl cellulose |
| DS | Degree of substitution |
| CNFs | Cellulose nanofibers |
| CNCs | Cellulose nanocrystals |
| BC | Bacterial cellulose |
| TEMPO | 2,2,6,6-Tetramethylpiperidine-1-oxyl |
| LC | Liquid crystal |
| NPs | Nanoparticles |
| CF | Cellulose fiber |
| HAPNWs | HAp nanowires |
| ZnO | Zinc oxide |
| MOFs | Metal–organic frameworks |
| PVP | Polyvinylpyrrolidone |
| PEG | Polyethylene glycol |
| PDA | Polydopamine |
| AuNPs | Gold NPs |
| QDs | Quantum dots |
| ALD | Atomic layer deposition |
| NR | Nanorod |
| PVA | Poly(vinyl alcohol) |
| LbL | Layer-by-layer |
| MTM | Montmorillonite |
| AC | Aminoclays |
| PEI | Polyethylenimine |
| ILs | Ionic liquids |
| MTMS | Methyltrimethoxysilane |
| PMSQ | Polymethylsilsesquioxane |
| CD | Circular dichroism |
| QNRs | Quantum nanorods |
| PAA | Polyacrylic acid |
| SA | Sodium alginate |
| SBF | Simulated body fluids |
| CP | Calcium phosphate |
| Gel | Gelatin |
| PA | Procyanidin |
| β-TCP | β-Tricalcium phosphate |
| PGA | Poly (γ-glutamic acid) |
| HPMC | (Hydroxypropyl)methyl cellulose |
| BMSC | Bone mesenchymal stem cell |
| BG | Bioactive glass |
| HEC | Hydroxyethyl-cellulose |
| HUVECs | Human umbilical vein endothelial cells |
| T-CNF | TEMPO-oxidized CNF |
| CTCC | Cancer cell targeting calcification |
| NO | Nitric oxide |
| 5FU | 5-Fluorouracil |
| BioMOF | Bio metal–organic framework |
| m-CNCs | Magnetic cellulose |
| GNP-NC | Nanogold-nanocellulose |
| GO | Graphene oxide |
| IBU | Ibuprofen |
| pNC | Bisphosphonate-modified nanocellulose |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. X. gratefully acknowledges the funding from the Natural Science Foundation of China (Grant no. 52203142), The Open Project of State Key Laboratory of New Textile Materials and Advanced Processing Technologies (FZ2021003), and The Joint Project for Talent Innovation Sharing Alliance of Quanzhou (2021C064L). Y. F. gratefully acknowledges the funding from the Shanghai Pujiang Program (22PJD081) and the Fundamental Research Funds for the Centra Universities.References
- W. Li, H. Liu, Y. Mi, M. Zhang, J. Shi, M. Zhao, M. A. Ramos, T. S. Hu, J. Li, M. Xu and Q. Xu, Friction, 2022, 10, 80–93 CrossRef CAS.
- U. G. K. Wegst, H. Bai, E. Saiz, A. P. Tomsia and R. O. Ritchie, Nat. Mater., 2015, 14, 23–36 CrossRef CAS PubMed.
- E. A. Zimmermann and R. O. Ritchie, Adv. Healthcare Mater., 2015, 4, 1287–1304 CrossRef CAS PubMed.
- R. O. Ritchie, Nat. Mater., 2011, 10, 817–822 CrossRef CAS PubMed.
- J. C. Weaver, G. W. Milliron, A. Miserez, K. Evans-Lutterodt, S. Herrera, I. Gallana, W. J. Mershon, B. Swanson, P. Zavattieri, E. DiMasi and D. Kisailus, Science, 2012, 336, 1275–1280 CrossRef CAS PubMed.
- P. Romano, H. Fabritius and D. Raabe, Acta Biomater., 2007, 3, 301–309 CrossRef CAS PubMed.
- R. Xiong, J. Luan, S. Kang, C. Ye, S. Singamaneni and V. V. Tsukruk, Chem. Soc. Rev., 2020, 49, 983–1031 RSC.
- B. Yeom, T. Sain, N. Lacevic, D. Bukharina, S.-H. Cha, A. M. Waas, E. M. Arruda and N. A. Kotov, Nature, 2017, 543, 95–98 CrossRef CAS PubMed.
- N. Rauner, M. Meuris, M. Zoric and J. C. Tiller, Nature, 2017, 543, 407–410 CrossRef CAS PubMed.
- H.-L. Gao, S.-M. Chen, L.-B. Mao, Z.-Q. Song, H.-B. Yao, H. Cölfen, X.-S. Luo, F. Zhang, Z. Pan, Y.-F. Meng, Y. Ni and S.-H. Yu, Nat. Commun., 2017, 8, 287 CrossRef PubMed.
- L.-B. Mao, H.-L. Gao, H.-B. Yao, L. Liu, H. Cölfen, G. Liu, S.-M. Chen, S.-K. Li, Y.-X. Yan, Y.-Y. Liu and S.-H. Yu, Science, 2016, 354, 107–110 CrossRef CAS PubMed.
- M. J. Olszta, X. Cheng, S. S. Jee, R. Kumar, Y.-Y. Kim, M. J. Kaufman, E. P. Douglas and L. B. Gower, Mater. Sci. Eng., R, 2007, 58, 77–116 CrossRef.
- L. C. Palmer, C. J. Newcomb, S. R. Kaltz, E. D. Spoerke and S. I. Stupp, Chem. Rev., 2008, 108, 4754–4783 CrossRef CAS PubMed.
- A.-W. Xu, Y. Ma and H. Cölfen, J. Mater. Chem., 2007, 17, 415–449 RSC.
- F. C. Meldrum and H. Cölfen, Chem. Rev., 2008, 108, 4332–4432 CrossRef CAS PubMed.
- R. Xiong, A. M. Grant, R. Ma, S. Zhang and V. V. Tsukruk, Mater. Sci. Eng., R, 2018, 125, 1–41 CrossRef.
- S. Ling, W. Chen, Y. Fan, K. Zheng, K. Jin, H. Yu, M. J. Buehler and D. L. Kaplan, Prog. Polym. Sci., 2018, 85, 1–56 CrossRef CAS PubMed.
- S. Ling, D. L. Kaplan and M. J. Buehler, Nat. Rev. Mater., 2018, 3, 1–15 CrossRef PubMed.
- D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- B. Thomas, M. C. Raj, B. AK, H. RM, J. Joy, A. Moores, G. L. Drisko and C. Sanchez, Chem. Rev., 2018, 118, 11575–11625 CrossRef CAS PubMed.
- D. Klemm, D. Schumann, F. Kramer, N. Heßler, M. Hornung, H.-P. Schmauder and S. Marsch, in Polysaccharides II, ed. D. Klemm, Springer, Berlin, Heidelberg, 2006, pp. 49–96 Search PubMed.
- Y. Shchipunov and I. Postnova, Adv. Funct. Mater., 2018, 28, 1705042 CrossRef.
- H. Kim, J.-Y. Yi, B.-G. Kim, J. E. Song, H.-J. Jeong and H. R. Kim, PLoS One, 2020, 15, e0233952 CrossRef CAS PubMed.
- C. Pang, R. Shanks and F. Daver, J. Polym. Eng., 2014, 34, 141–144 CAS.
- E. J. Foster, R. J. Moon, U. P. Agarwal, M. J. Bortner, J. Bras, S. Camarero-Espinosa, K. J. Chan, M. J. D. Clift, E. D. Cranston, S. J. Eichhorn, D. M. Fox, W. Y. Hamad, L. Heux, B. Jean, M. Korey, W. Nieh, K. J. Ong, M. S. Reid, S. Renneckar, R. Roberts, J. A. Shatkin, J. Simonsen, K. Stinson-Bagby, N. Wanasekara and J. Youngblood, Chem. Soc. Rev., 2018, 47, 2609–2679 RSC.
- P. Makvandi, M. Ghomi, V. V. T. Padil, F. Shalchy, M. Ashrafizadeh, S. Askarinejad, N. Pourreza, A. Zarrabi, E. Nazarzadeh Zare, M. Kooti, B. Mokhtari, A. Borzacchiello and F. R. Tay, ACS Appl. Nano Mater., 2020, 3, 6210–6238 CrossRef CAS.
- K. De France, Z. Zeng, T. Wu and G. Nyström, Adv. Mater., 2021, 33, 2000657 CrossRef CAS PubMed.
- Q. Zhang, L. Zhang, W. Wu and H. Xiao, Carbohydr. Polym., 2020, 229, 115454 CrossRef CAS PubMed.
- L.-B. Mao, Y.-F. Meng, X.-S. Meng, B. Yang, Y.-L. Yang, Y.-J. Lu, Z.-Y. Yang, L.-M. Shang and S.-H. Yu, J. Am. Chem. Soc., 2022, 144, 18175–18194 CrossRef CAS PubMed.
- L. Valencia, R. Handa, S. Monti, A. B. Jasso-Salcedo, D. Georgouvelas, I. Magaña, R. D. de León, K. P. Velikov, A. P. Mathew and S. Kumar, J. Mater. Chem. A, 2022, 10, 9248–9276 RSC.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- H. P. S. Abdul Khalil, A. H. Bhat and A. F. Ireana Yusra, Carbohydr. Polym., 2012, 87, 963–979 CrossRef CAS.
- S. Vignolini, P. J. Rudall, A. V. Rowland, A. Reed, E. Moyroud, R. B. Faden, J. J. Baumberg, B. J. Glover and U. Steiner, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15712–15715 CrossRef CAS PubMed.
- H. Zhu, W. Luo, P. N. Ciesielski, Z. Fang, J. Y. Zhu, G. Henriksson, M. E. Himmel and L. Hu, Chem. Rev., 2016, 116, 9305–9374 CrossRef CAS PubMed.
- C. Chen, Y. Kuang, S. Zhu, I. Burgert, T. Keplinger, A. Gong, T. Li, L. Berglund, S. J. Eichhorn and L. Hu, Nat. Rev. Mater., 2020, 5, 642–666 CrossRef CAS.
- D. Miyashiro, R. Hamano and K. Umemura, Nanomaterials, 2020, 10, 186 CrossRef CAS PubMed.
- L. K. J. Hauru, M. Hummel, K. Nieminen, A. Michud and H. Sixta, Soft Matter, 2016, 12, 1487–1495 RSC.
- Q. Yang, H. Fukuzumi, T. Saito, A. Isogai and L. Zhang, Biomacromolecules, 2011, 12, 2766–2771 CrossRef CAS PubMed.
- J. Zhou, C. Chang, R. Zhang and L. Zhang, Macromol. Biosci., 2007, 7, 804–809 CrossRef CAS PubMed.
- S. Wang, A. Lu and L. Zhang, Prog. Polym. Sci., 2016, 53, 169–206 CrossRef CAS.
- H. Wang, G. Gurau and R. D. Rogers, Chem. Soc. Rev., 2012, 41, 1519–1537 RSC.
- A. Pinkert, K. N. Marsh, S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712–6728 CrossRef CAS PubMed.
- H. Tu, X. Li, Y. Liu, L. Luo, B. Duan and R. Zhang, Carbohydr. Polym., 2022, 296, 119942 CrossRef CAS PubMed.
- W. Ge, J. Shuai, Y. Wang, Y. Zhou and X. Wang, Polym. Chem., 2022, 13, 359–372 RSC.
- D. Wallick, in Microencapsulation in the Food Industry, ed. A. G. Gaonkar, N. Vasisht, A. R. Khare and R. Sobel, Academic Press, San Diego, 2014, pp. 181–193 Search PubMed.
- M. H. Godinho, D. G. Gray and P. Pieranski, Liq. Cryst., 2017, 44, 2108–2120 CAS.
- D. Klemm, F. Kramer, S. Moritz, T. Lindström, M. Ankerfors, D. Gray and A. Dorris, Angew. Chem., Int. Ed., 2011, 50, 5438–5466 CrossRef CAS PubMed.
- T. Rosén, H. He, R. Wang, C. Zhan, S. Chodankar, A. Fall, C. Aulin, P. T. Larsson, T. Lindström and B. S. Hsiao, ACS Nano, 2020, 14, 16743–16754 CrossRef PubMed.
- H. P. S. Abdul Khalil, Y. Davoudpour, Md. N. Islam, A. Mustapha, K. Sudesh, R. Dungani and M. Jawaid, Carbohydr. Polym., 2014, 99, 649–665 CrossRef CAS PubMed.
- W. Chen, Q. Li, Y. Wang, X. Yi, J. Zeng, H. Yu, Y. Liu and J. Li, ChemSusChem, 2014, 7, 154–161 CrossRef CAS PubMed.
- T. Saito, S. Kimura, Y. Nishiyama and A. Isogai, Biomacromolecules, 2007, 8, 2485–2491 CrossRef CAS PubMed.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- A. Isogai, Adv. Mater., 2021, 33, 2000630 CrossRef CAS PubMed.
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479–3500 CrossRef CAS PubMed.
- T. G. Parton, R. M. Parker, G. T. van de Kerkhof, A. Narkevicius, J. S. Haataja, B. Frka-Petesic and S. Vignolini, Nat. Commun., 2022, 13, 2657 CrossRef CAS PubMed.
- R. M. Parker, G. Guidetti, C. A. Williams, T. Zhao, A. Narkevicius, S. Vignolini and B. Frka-Petesic, Adv. Mater., 2018, 30, 1704477 CrossRef PubMed.
- C. Wang, C. Tang, Y. Wang, Y. Shen, W. Qi, T. Zhang, R. Su and Z. He, Curr. Opin. Solid State Mater. Sci., 2022, 26, 101017 CrossRef CAS.
- A. Tran, C. E. Boott and M. J. MacLachlan, Adv. Mater., 2020, 32, 1905876 CrossRef CAS PubMed.
- J. Wang, J. Tavakoli and Y. Tang, Carbohydr. Polym., 2019, 219, 63–76 CrossRef CAS PubMed.
- Y. Huang, C. Zhu, J. Yang, Y. Nie, C. Chen and D. Sun, Cellulose, 2014, 21, 1–30 CrossRef CAS.
- M. A. Hubbe, R. A. Venditti and O. J. Rojas, BioResources, 2007, 2, 739–788 CAS.
- S. Ge, L. Zhang, Y. Zhang, F. Lan, M. Yan and J. Yu, Nanoscale, 2017, 9, 4366–4382 RSC.
- Z. Weng, Y. Su, D.-W. Wang, F. Li, J. Du and H.-M. Cheng, Adv. Energy Mater., 2011, 1, 917–922 CrossRef CAS.
- A. T. Vicente, A. Araújo, M. J. Mendes, D. Nunes, M. J. Oliveira, O. Sanchez-Sobrado, M. P. Ferreira, H. Águas, E. Fortunato and R. Martins, J. Mater. Chem. C, 2018, 6, 3143–3181 RSC.
- F.-F. Chen, Z.-H. Dai, Y.-N. Feng, Z.-C. Xiong, Y.-J. Zhu and Y. Yu, ACS Nano, 2021, 15, 5355–5365 CrossRef CAS PubMed.
- T. J. Athauda, U. Butt and R. R. Ozer, RSC Adv., 2013, 3, 21431–21438 RSC.
- X. Yue, T. Zhang, D. Yang, F. Qiu and Z. Li, Cellulose, 2018, 25, 5951–5965 CrossRef CAS.
- L. Zhu, L. Zong, X. Wu, M. Li, H. Wang, J. You and C. Li, ACS Nano, 2018, 12, 4462–4468 CrossRef CAS PubMed.
- S.-C. Li, B.-C. Hu, L.-M. Shang, T. Ma, C. Li, H.-W. Liang and S.-H. Yu, Adv. Mater., 2022, 34, 2202504 CrossRef CAS PubMed.
- X. Wu, C. Lu, Z. Zhou, G. Yuan, R. Xiong and X. Zhang, Environ. Sci.: Nano, 2014, 1, 71–79 RSC.
- X. Wu, C. Lu, W. Zhang, G. Yuan, R. Xiong and X. Zhang, J. Mater. Chem. A, 2013, 1, 8645–8652 RSC.
- R. Xiong, C. Lu, Y. Wang, Z. Zhou and X. Zhang, J. Mater. Chem. A, 2013, 1, 14910–14918 RSC.
- R. Xiong, C. Lu, W. Zhang, Z. Zhou and X. Zhang, Carbohydr. Polym., 2013, 95, 214–219 CrossRef CAS PubMed.
- S. Ifuku, M. Tsuji, M. Morimoto, H. Saimoto and H. Yano, Biomacromolecules, 2009, 10, 2714–2717 CrossRef CAS PubMed.
- Z. Ma, J. Liu, G. Shen, X. Zheng, Y. Pei and K. Tang, Cellulose, 2021, 28, 6287–6303 CrossRef CAS.
- S. Galland, R. L. Andersson, M. Salajková, V. Ström, R. T. Olsson and L. A. Berglund, J. Mater. Chem. C, 2013, 1, 7963–7972 RSC.
- C. Katepetch and R. Rujiravanit, Carbohydr. Polym., 2011, 86, 162–170 CrossRef CAS.
- M. Sureshkumar, D. Yovita Siswanto and C.-K. Lee, J. Mater. Chem., 2010, 20, 6948–6955 RSC.
- Y. Li, H. Zhu, H. Gu, H. Dai, Z. Fang, N. J. Weadock, Z. Guo and L. Hu, J. Mater. Chem. A, 2013, 1, 15278–15283 RSC.
- Y.-M. Ju, Y. Zhao, Q.-F. Guan, S.-Y. Yang, W. Wang, B.-B. Yan, Y.-F. Meng, S.-C. Li, P.-P. Tang, L.-B. Mao and S.-H. Yu, Angew. Chem., Int. Ed., 2022, e202211254 CAS.
- J. Majoinen, J. Hassinen, J. S. Haataja, H. T. Rekola, E. Kontturi, M. A. Kostiainen, R. H. A. Ras, P. Törmä and O. Ikkala, Adv. Mater., 2016, 28, 5262–5267 CrossRef CAS PubMed.
- T. Abitbol, H. S. Marway, S. A. Kedzior, X. Yang, A. Franey, D. G. Gray and E. D. Cranston, Cellulose, 2017, 24, 1287–1293 CrossRef CAS.
- S. Morimune-Moriya, M. Salajkova, Q. Zhou, T. Nishino and L. A. Berglund, Biomacromolecules, 2018, 19, 2423–2431 CrossRef CAS PubMed.
- A. N. Fernandes, L. H. Thomas, C. M. Altaner, P. Callow, V. T. Forsyth, D. C. Apperley, C. J. Kennedy and M. C. Jarvis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, E1195–E1203 CrossRef PubMed.
- Y. Li, H. Zhu, F. Shen, J. Wan, S. Lacey, Z. Fang, H. Dai and L. Hu, Nano Energy, 2015, 13, 346–354 CrossRef CAS.
- R. Sinko, X. Qin and S. Keten, MRS Bull., 2015, 40, 340–348 CrossRef CAS.
- R. Xiong, H. S. Kim, L. Zhang, V. F. Korolovych, S. Zhang, Y. G. Yingling and V. V. Tsukruk, Angew. Chem., Int. Ed., 2018, 57, 8508–8513 CrossRef CAS PubMed.
- A. Hajian, S. B. Lindström, T. Pettersson, M. M. Hamedi and L. Wågberg, Nano Lett., 2017, 17, 1439–1447 CrossRef CAS PubMed.
- Q. Li, Z. Xue, J. Zhao, C. Ao, X. Jia, T. Xia, Q. Wang, X. Deng, W. Zhang and C. Lu, Chem. Eng. J., 2020, 383, 123101 CrossRef CAS.
- C. Marichy, M. Bechelany and N. Pinna, Adv. Mater., 2012, 24, 1017–1032 CrossRef CAS PubMed.
- M. E. Lamm, K. Li, J. Qian, L. Wang, N. Lavoine, R. Newman, D. J. Gardner, T. Li, L. Hu, A. J. Ragauskas, H. Tekinalp, V. Kunc and S. Ozcan, Adv. Mater., 2021, 33, 2005538 CrossRef CAS PubMed.
- J. T. Korhonen, P. Hiekkataipale, J. Malm, M. Karppinen, O. Ikkala and R. H. A. Ras, ACS Nano, 2011, 5, 1967–1974 CrossRef CAS PubMed.
- Z. Li, C. Yao, F. Wang, Z. Cai and X. Wang, Nanotechnology, 2014, 25, 504005 CrossRef PubMed.
- W. J. Landis, K. J. Hodgens, J. Arena, M. J. Song and B. F. McEwen, Microsc. Res. Tech., 1996, 33, 192–202 CrossRef CAS PubMed.
- S. Iwamoto, A. Isogai and T. Iwata, Biomacromolecules, 2011, 12, 831–836 CrossRef CAS PubMed.
- N. Mittal, F. Ansari, K. G. V, C. Brouzet, P. Chen, P. T. Larsson, S. V. Roth, F. Lundell, L. Wågberg, N. A. Kotov and L. D. Söderberg, ACS Nano, 2018, 12, 6378–6388 CrossRef CAS PubMed.
- A. Walther, J. V. I. Timonen, I. Díez, A. Laukkanen and O. Ikkala, Adv. Mater., 2011, 23, 2924–2928 CrossRef CAS PubMed.
- Y. Qi, Z. Cheng, Z. Ye, H. Zhu and C. Aparicio, ACS Appl. Mater. Interfaces, 2019, 11, 27598–27604 CrossRef CAS PubMed.
- Z. Cheng, Z. Ye, A. Natan, Y. Ma, H. Li, Y. Chen, L. Wan, C. Aparicio and H. Zhu, ACS Appl. Mater. Interfaces, 2019, 11, 42486–42495 CrossRef CAS PubMed.
- H. Zhao, S. Liu, Y. Wei, Y. Yue, M. Gao, Y. Li, X. Zeng, X. Deng, N. A. Kotov, L. Guo and L. Jiang, Science, 2022, 375, 551–556 CrossRef CAS PubMed.
- F. Guan, Y. Xie, H. Wu, Y. Meng, Y. Shi, M. Gao, Z. Zhang, S. Chen, Y. Chen, H. Wang and Q. Pei, ACS Nano, 2020, 14, 15428–15439 CrossRef CAS PubMed.
- F. Barthelat, Z. Yin and M. J. Buehler, Nat. Rev. Mater., 2016, 1, 1–16 Search PubMed.
- Z. Sun, T. Liao, W. Li, Y. Qiao and K. (Ken) Ostrikov, Adv. Funct. Mater., 2019, 29, 1901460 CrossRef.
- J. Gim, N. Schnitzer, L. M. Otter, Y. Cui, S. Motreuil, F. Marin, S. E. Wolf, D. E. Jacob, A. Misra and R. Hovden, Nat. Commun., 2019, 10, 4822 CrossRef PubMed.
- A. P. Jackson, J. F. V. Vincent, R. M. Turner and R. M. Alexander, Proc. R. Soc. London, Ser. B, 1988, 234, 415–440 Search PubMed.
- F. Barthelat, H. Tang, P. D. Zavattieri, C.-M. Li and H. D. Espinosa, J. Mech. Phys. Solids, 2007, 55, 306–337 CrossRef CAS.
- B. Wijerathne, T. Liao, K. (Ken) Ostrikov and Z. Sun, Small Struct., 2022, 3, 2100228 CrossRef CAS.
- P. Das, H. Thomas, M. Moeller and A. Walther, Sci. Rep., 2017, 7, 39910 CrossRef CAS PubMed.
- Y. Liu, S.-H. Yu and L. Bergström, Adv. Funct. Mater., 2018, 28, 1703277 CrossRef.
- H.-B. Yao, J. Ge, L.-B. Mao, Y.-X. Yan and S.-H. Yu, Adv. Mater., 2014, 26, 163–188 CrossRef CAS PubMed.
- K. Wu, J. Fang, J. Ma, R. Huang, S. Chai, F. Chen and Q. Fu, ACS Appl. Mater. Interfaces, 2017, 9, 30035–30045 CrossRef CAS PubMed.
- H. Tu, K. Xie, X. Lin, R. Zhang, F. Chen, Q. Fu, B. Duan and L. Zhang, J. Mater. Chem. A, 2021, 9, 10304–10315 RSC.
- W. Tian, A. VahidMohammadi, M. S. Reid, Z. Wang, L. Ouyang, J. Erlandsson, T. Pettersson, L. Wågberg, M. Beidaghi and M. M. Hamedi, Adv. Mater., 2019, 31, 1902977 CrossRef PubMed.
- S. Cao, L. Shi, M. Miao, J. Fang, H. Zhao and X. Feng, Electrochim. Acta, 2019, 298, 22–30 CrossRef CAS.
- Y. Levi-Kalisman, G. Falini, L. Addadi and S. Weiner, J. Struct. Biol., 2001, 135, 8–17 CrossRef CAS PubMed.
- J. Wang, Q. Cheng, L. Lin and L. Jiang, ACS Nano, 2014, 8, 2739–2745 CrossRef CAS PubMed.
- P. Laaksonen, A. Walther, J.-M. Malho, M. Kainlauri, O. Ikkala and M. B. Linder, Angew. Chem., Int. Ed., 2011, 50, 8688–8691 CrossRef CAS PubMed.
- A. Walther, I. Bjurhager, J.-M. Malho, J. Ruokolainen, L. Berglund and O. Ikkala, Angew. Chem., Int. Ed., 2010, 49, 6448–6453 CrossRef CAS PubMed.
- A. Walther, I. Bjurhager, J.-M. Malho, J. Pere, J. Ruokolainen, L. A. Berglund and O. Ikkala, Nano Lett., 2010, 10, 2742–2748 CrossRef CAS PubMed.
- H.-B. Yao, Z.-H. Tan, H.-Y. Fang and S.-H. Yu, Angew. Chem., Int. Ed., 2010, 49, 10127–10131 CrossRef CAS PubMed.
- J. J. Richardson, M. Björnmalm and F. Caruso, Science, 2015, 348, aaa2491 CrossRef PubMed.
- M. Farhadi-Khouzani, C. Schütz, G. M. Durak, J. Fornell, J. Sort, G. Salazar-Alvarez, L. Bergström and D. Gebauer, J. Mater. Chem. A, 2017, 5, 16128–16133 RSC.
- P. Tzeng, D. J. Hewson, P. Vukusic, S. J. Eichhorn and J. C. Grunlan, J. Mater. Chem. C, 2015, 3, 4260–4264 RSC.
- Z. Jia, Z. Deng and L. Li, Adv. Mater., 2022, 34, 2106259 CrossRef CAS PubMed.
- W. Huang, M. Shishehbor, N. Guarín-Zapata, N. D. Kirchhofer, J. Li, L. Cruz, T. Wang, S. Bhowmick, D. Stauffer, P. Manimunda, K. N. Bozhilov, R. Caldwell, P. Zavattieri and D. Kisailus, Nat. Mater., 2020, 19, 1236–1243 CrossRef CAS PubMed.
- K. Markstedt, J. Sundberg and P. Gatenholm, 3D Print. Addit. Manuf., 2014, 1, 115–121 CrossRef.
- J. S. Park, T. Kim and W. S. Kim, Sci. Rep., 2017, 7, 3246 CrossRef PubMed.
- K. Markstedt, A. Mantas, I. Tournier, H. Martínez Ávila, D. Hägg and P. Gatenholm, Biomacromolecules, 2015, 16, 1489–1496 CrossRef CAS PubMed.
- A. Sydney Gladman, E. A. Matsumoto, R. G. Nuzzo, L. Mahadevan and J. A. Lewis, Nat. Mater., 2016, 15, 413–418 CrossRef CAS PubMed.
- L. Dai, T. Cheng, C. Duan, W. Zhao, W. Zhang, X. Zou, J. Aspler and Y. Ni, Carbohydr. Polym., 2019, 203, 71–86 CrossRef CAS PubMed.
- G. Siqueira, D. Kokkinis, R. Libanori, M. K. Hausmann, A. S. Gladman, A. Neels, P. Tingaut, T. Zimmermann, J. A. Lewis and A. R. Studart, Adv. Funct. Mater., 2017, 27, 1604619 CrossRef.
- M. K. Hausmann, P. A. Rühs, G. Siqueira, J. Läuger, R. Libanori, T. Zimmermann and A. R. Studart, ACS Nano, 2018, 12, 6926–6937 CrossRef CAS PubMed.
- V. Saxena, A. Hasan and L. M. Pandey, Cellulose, 2021, 28, 9207–9226 CrossRef CAS.
- D. Kharaghani, D. Dutta, K. K. K. Ho, K.-Q. Zhang, W. Kai, X. Ren, M. D. P. Willcox and I. S. Kim, Cellulose, 2020, 27, 3319–3334 CrossRef CAS.
- A. M. Cakmak, S. Unal, A. Sahin, F. N. Oktar, M. Sengor, N. Ekren, O. Gunduz and D. M. Kalaskar, Polymers, 2020, 12, 1962 CrossRef CAS PubMed.
- S. Guerzoni, H. Deplaine, J. El Haskouri, P. Amorós, M. M. Pradas, U. Edlund and G. G. Ferrer, J. Bioact. Compat. Polym., 2014, 29, 15–31 CrossRef CAS.
- A. Salama, Int. J. Biol. Macromol., 2019, 127, 606–617 CrossRef CAS PubMed.
- A. Xin, Y. Su, S. Feng, M. Yan, K. Yu, Z. Feng, K. Hoon Lee, L. Sun and Q. Wang, Adv. Mater., 2021, 33, 2006946 CrossRef CAS PubMed.
- G. Chen, X. Liang, P. Zhang, S. Lin, C. Cai, Z. Yu and J. Liu, Adv. Funct. Mater., 2022, 32, 2113262 CrossRef CAS.
- R. E. Abouzeid, R. Khiari, D. Beneventi and A. Dufresne, Biomacromolecules, 2018, 19, 4442–4452 CrossRef CAS PubMed.
- J. G. Torres-Rendon, T. Femmer, L. De Laporte, T. Tigges, K. Rahimi, F. Gremse, S. Zafarnia, W. Lederle, S. Ifuku, M. Wessling, J. G. Hardy and A. Walther, Adv. Mater., 2015, 27, 2989–2995 CrossRef CAS PubMed.
- U. G. K. Wegst, M. Schecter, A. E. Donius and P. M. Hunger, Philos. Trans. R. Soc., A, 2010, 368, 2099–2121 CrossRef CAS PubMed.
- Q. Cheng, C. Huang and A. P. Tomsia, Adv. Mater., 2017, 29, 1703155 CrossRef PubMed.
- S. Deville, Adv. Eng. Mater., 2008, 10, 155–169 CrossRef CAS.
- B. Wicklein, A. Kocjan, G. Salazar-Alvarez, F. Carosio, G. Camino, M. Antonietti and L. Bergström, Nat. Nanotechnol., 2015, 10, 277–283 CrossRef CAS PubMed.
- Z. Zeng, T. Wu, D. Han, Q. Ren, G. Siqueira and G. Nyström, ACS Nano, 2020, 14, 2927–2938 CrossRef CAS PubMed.
- Y. Chen, D. Fan, S. Lyu, G. Li, F. Jiang and S. Wang, ACS Sustainable Chem. Eng., 2019, 7, 1381–1388 CrossRef CAS.
- P. Munier, V. Apostolopoulou-Kalkavoura, M. Persson and L. Bergström, Cellulose, 2020, 27, 10825–10836 CrossRef CAS.
- F. Carosio, L. Medina, J. Kochumalayil and L. A. Berglund, Adv. Mater. Interfaces, 2021, 8, 2101111 CrossRef CAS.
- K. J. De France, T. Hoare and E. D. Cranston, Chem. Mater., 2017, 29, 4609–4631 CrossRef CAS.
- M. Chau, K. J. De France, B. Kopera, V. R. Machado, S. Rosenfeldt, L. Reyes, K. J. W. Chan, S. Förster, E. D. Cranston, T. Hoare and E. Kumacheva, Chem. Mater., 2016, 28, 3406–3415 CrossRef CAS.
- P. Munier, K. Gordeyeva, L. Bergström and A. B. Fall, Biomacromolecules, 2016, 17, 1875–1881 CrossRef CAS PubMed.
- Z. Zeng, C. Wang, G. Siqueira, D. Han, A. Huch, S. Abdolhosseinzadeh, J. Heier, F. Nüesch, C. (John) Zhang and G. Nyström, Adv. Sci., 2020, 7, 2000979 CrossRef CAS PubMed.
- Z. Zeng, E. Mavrona, D. Sacré, N. Kummer, J. Cao, L. A. E. Müller, E. Hack, P. Zolliker and G. Nyström, ACS Nano, 2021, 15, 7451–7462 CrossRef CAS PubMed.
- S. Ahankari, P. Paliwal, A. Subhedar and H. Kargarzadeh, ACS Nano, 2021, 15, 3849–3874 CrossRef CAS PubMed.
- C. Liu, L. Wan, Q. Li, X. Sun, A. Natan, D. Cao, P. Luan and H. Zhu, ACS Appl. Polym. Mater., 2021, 3, 1358–1367 CrossRef CAS.
- L. Wan, C. Liu, D. Cao, X. Sun and H. Zhu, ACS Appl. Polym. Mater., 2020, 2, 3001–3009 CrossRef CAS.
- J. Zhang, Y. Cheng, C. Xu, M. Gao, M. Zhu and L. Jiang, Adv. Funct. Mater., 2021, 31, 2009349 CrossRef CAS.
- U. G. Wegst, H. Bai, E. Saiz, A. P. Tomsia and R. O. Ritchie, Nat. Mater., 2015, 14, 23 CrossRef CAS PubMed.
- Y. Yang, Z. Chen, X. Song, Z. Zhang, J. Zhang, K. K. Shung, Q. Zhou and Y. Chen, Adv. Mater., 2017, 29, 1605750 CrossRef PubMed.
- S.-M. Chen, H.-L. Gao, Y.-B. Zhu, H.-B. Yao, L.-B. Mao, Q.-Y. Song, J. Xia, Z. Pan, Z. He, H.-A. Wu and S.-H. Yu, Natl. Sci. Rev., 2018, 5, 703–714 CrossRef CAS.
- R. M. Erb, R. Libanori, N. Rothfuchs and A. R. Studart, Science, 2012, 335, 199–204 CrossRef CAS PubMed.
- R. Xiong, A. M. Grant, R. Ma, S. Zhang and V. V. Tsukruk, Mater. Sci. Eng., R, 2018, 125, 1–41 CrossRef.
- J. P. F. Lagerwall, C. Schütz, M. Salajkova, J. Noh, J. Hyun Park, G. Scalia and L. Bergström, NPG Asia Mater., 2014, 6, e80 CrossRef CAS.
- S. Kang, G. M. Biesold, H. Lee, D. Bukharina, Z. Lin and V. V. Tsukruk, Adv. Funct. Mater., 2021, 31, 2104596 CrossRef CAS.
- M. Xu, C. Ma, J. Zhou, Y. Liu, X. Wu, S. Luo, W. Li, H. Yu, Y. Wang, Z. Chen, J. Li and S. Liu, J. Mater. Chem. C, 2019, 7, 13794–13802 RSC.
- T.-D. Nguyen, W. Y. Hamad and M. J. MacLachlan, Adv. Funct. Mater., 2014, 24, 777–783 CrossRef CAS.
- T. Zhang, C. Tang, Y. Wang, C. Wang, Y. Zhang, W. Qi, R. Su and Z. He, Langmuir, 2022, 38, 4147–4155 CrossRef CAS PubMed.
- D. Qu, J. Zhang, G. Chu, H. Jiang, C. Wu and Y. Xu, J. Mater. Chem. C, 2016, 4, 1764–1768 RSC.
- A. Querejeta-Fernández, G. Chauve, M. Methot, J. Bouchard and E. Kumacheva, J. Am. Chem. Soc., 2014, 136, 4788–4793 CrossRef PubMed.
- Z. Cheng, Y. Ma, L. Yang, F. Cheng, Z. Huang, A. Natan, H. Li, Y. Chen, D. Cao, Z. Huang, Y. Wang, Y. Liu, R. Yang and H. Zhu, Adv. Opt. Mater., 2019, 7, 1801816 CrossRef.
- R. Xiong, W. Wu, C. Lu and H. Cölfen, Adv. Mater., 2022, 34, 2206509 CrossRef CAS PubMed.
- L. A. Berglund and I. Burgert, Adv. Mater., 2018, 30, 1704285 CrossRef PubMed.
- H. Zhou, D. Wen, X. Hao, C. Chen, N. Zhao, R. Ou and Q. Wang, Chem. Eng. J., 2023, 451, 138308 CrossRef CAS.
- C. Mai and H. Militz, Wood Sci. Technol., 2004, 37, 453–461 CrossRef CAS.
- S. Donath, H. Militz and C. Mai, Wood Sci. Technol., 2004, 38, 555–566 CrossRef CAS.
- V. Merk, M. Chanana, N. Gierlinger, A. M. Hirt and I. Burgert, ACS Appl. Mater. Interfaces, 2014, 6, 9760–9767 CrossRef CAS PubMed.
- V. Merk, M. Chanana, T. Keplinger, S. Gaan and I. Burgert, Green Chem., 2015, 17, 1423–1428 RSC.
- A. Kumar, T. Jyske and M. Petrič, Adv. Sustainable Syst., 2021, 5, 2000251 CrossRef CAS.
- W. Kong, C. Wang, C. Jia, Y. Kuang, G. Pastel, C. Chen, G. Chen, S. He, H. Huang, J. Zhang, S. Wang and L. Hu, Adv. Mater., 2018, 30, 1801934 CrossRef PubMed.
- Y. Li, E. Vasileva, I. Sychugov, S. Popov and L. Berglund, Adv. Opt. Mater., 2018, 6, 1800059 CrossRef.
- X. Zhang, B. Wu, S. Sun and P. Wu, Adv. Funct. Mater., 2020, 30, 1910425 CrossRef CAS.
- X. Wang, J. Fang, W. Zhu, C. Zhong, D. Ye, M. Zhu, X. Lu, Y. Zhao and F. Ren, Adv. Funct. Mater., 2021, 31, 2010068 CrossRef CAS.
- C. A. López, G. Bellesia, A. Redondo, P. Langan, S. P. S. Chundawat, B. E. Dale, S. J. Marrink and S. Gnanakaran, J. Phys. Chem. B, 2015, 119, 465–473 CrossRef PubMed.
- N. Mittal, F. Ansari, K. G. V, C. Brouzet, P. Chen, P. T. Larsson, S. V. Roth, F. Lundell, L. Wågberg, N. A. Kotov and L. D. Söderberg, ACS Nano, 2018, 12, 6378–6388 CrossRef CAS PubMed.
- D. Nepal, S. Kang, K. M. Adstedt, K. Kanhaiya, M. R. Bockstaller, L. C. Brinson, M. J. Buehler, P. V. Coveney, K. Dayal, J. A. El-Awady, L. C. Henderson, D. L. Kaplan, S. Keten, N. A. Kotov, G. C. Schatz, S. Vignolini, F. Vollrath, Y. Wang, B. I. Yakobson, V. V. Tsukruk and H. Heinz, Nat. Mater., 2023, 22, 18–35 CrossRef CAS PubMed.
- J. Wang, Q. Liu, Z. Guo, H. Pan, Z. Liu and R. Tang, ACS Biomater. Sci. Eng., 2022, 8, 484–492 CrossRef PubMed.
- K. Li, C. M. Clarkson, L. Wang, Y. Liu, M. Lamm, Z. Pang, Y. Zhou, J. Qian, M. Tajvidi, D. J. Gardner, H. Tekinalp, L. Hu, T. Li, A. J. Ragauskas, J. P. Youngblood and S. Ozcan, ACS Nano, 2021, 15, 3646–3673 CrossRef CAS PubMed.
- W. Xu, B. Z. Molino and F. Cheng, ACS Appl. Mater. Interfaces, 2019, 11, 8838–8848 CrossRef CAS PubMed.
- J. Li, R. Cha and K. Mou, Adv. Healthcare Mater., 2018, 7, 1800334 CrossRef PubMed.
- H. Shokrani, A. Shokrani and M. Jouyandeh, ACS Appl. Bio Mater., 2022, 5, 2107–2121 CrossRef CAS PubMed.
- C. Chen, W. Ding, H. Zhang, L. Zhang, Y. Huang, M. Fan, J. Yang and D. Sun, Carbohydr. Polym., 2022, 278, 118995 CrossRef CAS PubMed.
- M. Pang, Y. Huang, F. Meng, Y. Zhuang, H. Liu, M. Du, Q. Ma, Q. Wang, Z. Chen, L. Chen, T. Cai and Y. Cai, Eur. Polym. J., 2020, 122, 109365 CrossRef CAS.
- J. Ravoor, M. Thangavel and E. S. R, ACS Appl. Bio Mater., 2021, 4, 8129–8158 CrossRef CAS PubMed.
- W. Liu, H. Du and M. Zhang, ACS Sustainable Chem. Eng., 2020, 8, 7536–7562 CrossRef CAS.
- B. L. Tardy, B. D. Mattos, C. G. Otoni, M. Beaumont, J. Majoinen, T. Kamarainen and O. J. Rojas, Chem. Rev., 2021, 121, 14088–14188 CrossRef CAS PubMed.
- Q. F. Guan, Z. M. Han, Z. C. Ling, H. B. Yang and S. H. Yu, Acc. Mater. Res., 2022, 3, 608–619 CrossRef CAS.
- P. Makvandi, M. Ghomi and V. V. T. Padil, ACS Appl. Nano Mater., 2020, 3, 6210–6238 CrossRef CAS.
- M. Ojansivu, A. Rashad and A. Ahlinder, Biofabrication, 2019, 11, 035010 CrossRef CAS PubMed.
- G. Wang, T. Lu, X. Zhang, M. Feng, C. Wang, W. Yao, S. Zhou, Z. Zhu, W. Ding and M. He, Int. J. Biol. Macromol., 2021, 186, 377–384 CrossRef CAS PubMed.
- J. P. Ribeiro, R. M. A. Domingues, P. S. Babo, L. P. Nogueira, J. E. Reseland, R. L. Reis, M. Gomez-Florit and M. E. Gomes, Carbohydr. Polym., 2022, 292, 119638 CrossRef CAS PubMed.
- K. A. Zimmermann, J. M. LeBlanc, K. T. Sheets, R. W. Fox and P. Gatenholm, Mater. Sci. Eng., C, 2011, 31, 43–49 CrossRef CAS.
- Y. Hu, Y. Zhu and X. Zhou, J. Mater. Chem. B, 2016, 4, 1235–1246 RSC.
- D. Y. Zaki, E. M. Safwat, S. M. Nagi, H. N. Salem, T. M. Hamdy, L. M. Moharam, M. L. Hassan and E. M. A. Hamzawy, Carbohydr. Polym. Technol. Appl., 2021, 2, 100035 CAS.
- T. Dvir, B. P. Timko, D. S. Kohane and R. Langer, Nat. Nanotechnol., 2011, 6, 13–22 CrossRef CAS PubMed.
- S. Weiner and H. D. Wagner, Annu. Rev. Mater. Sci., 1998, 28, 271–298 CrossRef CAS.
- F. Nudelman, K. Pieterse, A. George, P. H. H. Bomans, H. Friedrich, L. J. Brylka, P. A. J. Hilbers, G. de With and N. A. J. M. Sommerdijk, Nat. Mater., 2010, 9, 1004–1009 CrossRef CAS PubMed.
- U. G. K. Wegst, H. Bai, E. Saiz, A. P. Tomsia and R. O. Ritchie, Nat. Mater., 2015, 14, 23–36 CrossRef CAS PubMed.
- S. Sprio, S. Panseri and M. Montesi, J. Eur. Ceram. Soc., 2020, 40, 1717–1727 CrossRef CAS.
- X. Wang, J. Fang, W. Zhu, C. Zhong, D. Ye, M. Zhu, X. Lu, Y. Zhao and F. Ren, Adv. Funct. Mater., 2021, 31, 2010068 CrossRef CAS.
- F. Liu, B. Wei, X. Xu, B. Ma, S. Zhang, J. Duan, Y. Kong, H. Yang, Y. Sang, S. Wang, W. Tang, C. Liu and H. Liu, Adv. Healthcare Mater., 2021, 10, 2001851 CrossRef CAS PubMed.
- J. Sundberg, C. Gotherstrom and P. Gatenholm, Biomed. Mater. Eng., 2015, 25, 39–52 CAS.
- T. Selorm and S. Prakit, Appl. Mater. Today, 2018, 11, 34–49 CrossRef.
- Z. Cheng, Z. Ye and A. Natan, ACS Appl. Mater. Interfaces, 2019, 11, 42486–42495 CrossRef CAS PubMed.
- P. M. Favi, S. P. Ospina, M. Kachole, M. Gao, L. Atehortua and T. J. Webster, Cellulose, 2016, 23, 1263–1282 CrossRef CAS.
- P. Basu, N. Saha, R. Alexandrova, B. Andonova-Lilova, M. Georgieva, G. Miloshev and P. Saha, Int. J. Mol. Sci., 2018, 19, 3980 CrossRef PubMed.
- N. Mitrousis, A. Fokina and M. S. Shoichet, Nat. Rev. Mater., 2018, 3, 441–456 CrossRef CAS.
- M. Mastrogiacomo, S. Scaglione, R. Martinetti, L. Dolcini, F. Beltrame, R. Cancedda and R. Quarto, Biomaterials, 2006, 27, 3230–3237 CrossRef CAS PubMed.
- Y. Huang, J. Wang, F. Yang, Y. Shao, X. Zhang and K. Dai, Mater. Sci. Eng., C, 2017, 75, 1034–1041 CrossRef CAS PubMed.
- C. Rey, C. Combes and C. Drouet, Osteoporosis Int., 2009, 20, 1013–1021 CrossRef CAS PubMed.
- M. Ojansivu, A. Rashad, A. Ahlinder, J. Massera, A. Mishra, K. Syverud, A. Finne-Wistrand, S. Miettinen and K. Mustafa, Biofabrication, 2019, 11, 035010 CrossRef CAS PubMed.
- M. Sukul, T. B. Nguyen, Y. K. Min, S. Y. Lee and B. T. Lee, Tissue Eng., Part A, 2015, 21, 1822–1836 CrossRef CAS PubMed.
- P. Basu, N. Saha and P. Saha, Int. J. Polym. Mater. Polym. Biomater., 2019, 68, 134–144 CrossRef CAS.
- R. E. Abouzeid, R. Khiari, D. Beneventi and A. Dufresne, Biomacromolecules, 2018, 19, 4442–4452 CrossRef CAS PubMed.
- J. Guo, Q. Li and R. Zhang, Biomacromolecules, 2022, 23, 877–888 CrossRef CAS PubMed.
- X. Zhu, T. Chen, B. Feng, J. Weng, K. Duan, J. Wang and X. Lu, ACS Biomater. Sci. Eng., 2018, 4, 3534–3544 CrossRef CAS PubMed.
- H. Oliveira, S. Catros and O. Castano, Acta Biomater., 2017, 54, 377–385 CrossRef CAS PubMed.
- H. Jafari, M. Shahrousvand and B. Kaffashi, ACS Biomater. Sci. Eng., 2018, 4, 2484–2493 CrossRef CAS PubMed.
- X. Zhang, X. Yin, J. Luo, X. Zheng, H. Wang, J. Wang, Z. Xi, X. Liao, J. O. Machuki, K. Guo and F. Gao, ACS Biomater. Sci. Eng., 2019, 5, 294–307 CrossRef CAS PubMed.
- Q. Chen, R. P. Garcia, J. Munoz, U. Perez de Larraya, N. Garmendia, Q. Yao and A. R. Boccaccini, ACS Appl. Mater. Interfaces, 2015, 7, 24715–24725 CrossRef CAS PubMed.
- P. Mohammadi, J. A. Gandier, Nonappa, W. Wagermaier, A. Miserez and M. Penttila, Adv. Mater., 2021, 33, e2102658 CrossRef PubMed.
- Y. Qi, Z. Cheng, Z. Ye, H. Zhu and C. Aparicio, ACS Appl. Mater. Interfaces, 2019, 11, 27598–27604 CrossRef CAS PubMed.
- P. Mujuan, H. Yinghong and M. Fansu, Eur. Polym. J., 2020, 122, 109365 CrossRef.
- Q. Xu, M. Xu, C. Lin, Q. Zhao, R. Zhang, X. Dong, Y. Zhang, S. Tian, Y. Tian and Z. Xia, Adv. Sci., 2019, 6, 1902043 CrossRef CAS PubMed.
- M. Wu, P. Wu and L. Xiao, Int. J. Biol. Macromol., 2020, 162, 1627–1641 CrossRef CAS PubMed.
- R. Singla, S. Soni and P. M. Kulurkar, Carbohydr. Polym., 2017, 155, 152–162 CrossRef CAS PubMed.
- J. Leppiniemi, P. Lahtinen and A. Paajanen, ACS Appl. Mater. Interfaces, 2017, 9, 21959–21970 CrossRef CAS PubMed.
- J. Wu, N. Yin and S. Chen, Cellulose, 2019, 26, 2513–2528 CrossRef CAS.
- L. L. Demer and Y. Tintut, Circulation, 2008, 117, 2938–2948 CrossRef PubMed.
- M. Parvinzadeh Gashti, M. Stir, M. Bourquin and J. Hulliger, Cryst. Growth Des., 2013, 13, 2166–2173 CrossRef CAS.
- S. Yao, B. Jin, Z. Liu, C. Shao, R. Zhao, X. Wang and R. Tang, Adv. Mater., 2017, 29, 1605903 CrossRef PubMed.
- R. Zhao, B. Wang and X. Yang, Angew. Chem., Int. Ed., 2016, 55, 5225–5229 CrossRef CAS PubMed.
- J. Klesing, A. Wiehe, B. Gitter, S. Grafe and M. Epple, J. Mater. Sci.: Mater. Med., 2010, 21, 887–892 CrossRef CAS PubMed.
- S. A. Noorian, N. Hemmatinejad and J. A. R. Navarro, J. Inorg. Biochem., 2019, 201, 110818 CrossRef CAS PubMed.
- A. Fakhri, S. Tahami and P. A. Nejad, J. Photochem. Photobiol., B, 2017, 175, 83–88 CrossRef CAS PubMed.
- M. M. Abeer, M. C. I. Mohd Amin and C. Martin, J. Pharm. Pharmacol., 2014, 66, 1047–1061 CrossRef CAS PubMed.
- R. Mori, T. Nakai and K. Enomoto, Clin. Orthop. Relat. Res., 2011, 469, 600–606 CrossRef PubMed.
- J. Supramaniam, R. Adnan, N. H. M. Kaus and R. Bushra, Int. J. Biol. Macromol., 2018, 118, 640–648 CrossRef CAS PubMed.
- T. S. Anirudhan, S. S. Nair and V. C. Sekhar, J. Membr. Sci., 2017, 539, 344–357 CrossRef CAS.
- H. Luo, H. Ao and G. Li, Curr. Appl. Phys., 2017, 17, 249–254 CrossRef.
- A. Nishiguchi and T. Taguchi, Biomacromolecules, 2019, 20, 1385–1393 CrossRef CAS PubMed.
- M. R. McClung, P. Geusens, P. D. Miller, H. Zippel, W. G. Bensen, C. Roux, S. Adami, I. Fogelman, T. Diamond, R. Eastell, P. J. Meunier, R. D. Wasnich, M. Greenwald, J. Kaufman, C. H. Chesnut and J. Reginster, N. Engl. J. Med., 2001, 344, 333–340 CrossRef CAS PubMed.
- L.-H. Fu, C. Qi, Y.-J. Liu, W.-T. Cao and M.-G. Ma, Sci. Rep., 2018, 8, 8292 CrossRef PubMed.
- A. Salama, M. El-Sakhawy and S. Kamel, Int. J. Biol. Macromol., 2016, 93, 1647–1652 CrossRef CAS PubMed.
- F. V. Ferreira, C. G. Otoni, K. J. De France, H. S. Barud, L. M. F. Lona, E. D. Cranston and O. J. Rojas, Mater. Today, 2020, 37, 126–141 CrossRef CAS.
- D. Klemm, E. D. Cranston, D. Fischer, M. Gama, S. A. Kedzior, D. Kralisch, F. Kramer, T. Kondo, T. Lindström, S. Nietzsche, K. Petzold-Welcke and F. Rauchfuß, Mater. Today, 2018, 21, 720–748 CrossRef CAS.
- S. Gorgieva and J. Trček, Nanomaterials, 2019, 9, 1352 CrossRef CAS PubMed.
- P. Shen, Q. Tang, X. Chen and Z. Li, Carbohydr. Polym., 2022, 290, 119462 CrossRef CAS PubMed.
- W. Liu, H. Du, M. Zhang, K. Liu, H. Liu, H. Xie, X. Zhang and C. Si, ACS Sustainable Chem. Eng., 2020, 8, 7536–7562 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |