
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the activity of glucose oxidase after exposure to organic solvents†
Vygailė
Dudkaitė
 a,
Visvaldas
Kairys
b and
Gintautas
Bagdžiūnas
*a
a,
Visvaldas
Kairys
b and
Gintautas
Bagdžiūnas
*a
aGroup of Supramolecular Analysis, Institute of Biochemistry, Life Sciences Centre, Vilnius University, Saulėtekio av. 7, LT-10257, Vilnius, Lithuania. E-mail: gintautas.bagdziunas@gmc.vu.lt
bDepartment of Bioinformatics, Institute of Biotechnology, Life Sciences Centre, Vilnius University, Saulėtekio av. 7, LT-10257, Vilnius, Lithuania
First published on 20th February 2023
Abstract
Long-term stability of enzymes in organic solvents is one of the most challenging problems in modern biotechnology and chemical industries. However, the resistance of enzymes to organic solvents is not very well understood so far. Herein, the effects of apolar, chlorinated, and polar organic solvents on the activity and structure of glucose oxidase from Aspergillus niger were systemically investigated using spectrophotometric activity assay of this enzyme and absorption and chiroptical spectroscopy. Molecular dynamics simulations and correlation of the activity with properties of the organic solvents were employed to understand the effects of organic solvents on the enzyme. The experimental and theoretical results showed that apolar solvents reduce the enzyme activity because they facilitate its aggregation through inter-enzymatic salt bridges. Moreover, polar solvents strongly coordinate with amino acid residues in the glucose binding pocket and prevent binding of the substrates. We found that this enzyme is stable in pure apolar and chlorinated solvents and these solvents can be used for the functionalization of its residues. This work provides an in depth understanding at the molecular level of the impact of various pure organic solvents on the structure and dynamics of glucose oxidase and the regulation of its catalytic activity.
Introduction
Investigation of enzyme catalytic activity in organic solvents as the non-natural media may help reveal new properties of the enzymes and could solve enzyme biotechnology problems such as low solubility of reactants in water and low thermostability.1,2 In addition, research in this direction may answer the question of the possibility of life in non-aqueous solutions.3 In many cases, organic solvents or their mixtures with water can inactivate enzymes. Therefore, developing strategies to enhance the lifetime of enzymes in the presence of organic solvents is an important task in modern biotechnology and bioorganic chemistry. To achieve this task, novel enzyme isolation strategies, modification of their structure to increase their resistance, and modification of solvents without a denaturing effect on the enzymes can be employed.4Glucose oxidase (GOx) from Aspergillus niger is one of the most widely investigated enzymes in the areas of biotechnology and bioelectrochemistry, because it oxidizes β-D-glucose, an important source of energy in all organisms, to gluconic acid and hydrogen peroxide.5 GOx as a relatively cheap, stable and selective (towards glucose) enzyme plays a key role in glucose monitoring with biosensors.6 In our laboratory, amperometric enzymatic biosensors based on immobilized GOx on their electrodes have been used to detect glucose.7–9 Recently, we developed a simple method for the functionalization of glucose oxidase with redoxable ferrocene groups in chloroform. The enzyme retained its activity after storage in this organic solvent and after the functionalization procedures.10 Therefore, expansion of the range of environmentally friendly organic solvents, in which the enzyme would maintain its activity, is an important task in bioconjugate chemistry. This task may also necessitate a more diverse range of organic synthesis methods. Furthermore, the stability of GOx-based electrodes is a major problem of these biosensors that can be solved by modification or storage of the enzyme in organic solvents.
The performance of an amperometric glucose sensor in butan-2-ol as a polar organic solvent has been evaluated by Iwuoha and Smyth.11 The authors found that this organic solvent does not alter the reactivity of the biosensor, but may even correct the deviation from the Michaelis–Menten kinetics. Vasileva and Godjevargova studied the relative activity and thermal stability of GOx immobilized onto an electrode in the presence of methanol, ethylene glycol, and glycerol. A concentration of 10 v/v% of glycerol at higher temperatures was found to stabilize the enzyme, but it was deactivated at higher concentrations of the organic solvent.12 Balistreri et al. found that GOx covalently immobilized onto mesocellular silica foams showed rather good stability after incubation in buffer/methanol or acetonitrile mixtures with less than 50 v/v% of the organic solvents.13
For understanding the physical basis of the structure and function of biological macromolecules, molecular dynamics (MD) simulations have frequently been employed.14 MD simulation is an efficient theoretical method for studying enzyme stability and dynamics in non-aqueous solvents.15 Recently, Milčić and colleagues provided experimental and computational insights into the inhibitory effect of dimethyl sulfoxide (DMSO) on halohydrin dehalogenase. Their MD simulations revealed that DMSO molecules replace water molecules, both on the surface and in the active site of the enzyme. According to the authors, the solvent molecules cause aggregation of the enzyme.16 Mohtashami et al. have theoretically shown that secondary structures of the Burkholderia cepacia lipase, Trametes versicolor laccase, and human lysozyme are the determining factors of their stability against both polar and apolar organic solvents, and β-structures are more prone to destabilization in organic solvents compared to α-helix structures.17 Yang and colleagues have demonstrated that a surfactant-solubilised subtilisin surface and its active site region are well hydrated in an aqueous medium, whereas with increasing polarity of the organic solvent, the hydration water is stripped from the enzyme surface. Moreover, penetration of polar solvent molecules by replacing mobile and weakly bound water molecules in the active site was observed.18 Similar results were obtained for a computational and direct evolution study of Bacillus subtilis lipase A.19 Organic solvent molecules stripped off essential water molecules from the surface of the enzyme and penetrated into the substrate binding cleft leading to inhibition and conformational change. By combining comprehensive experimental studies and MD simulations, Cui et al. have provided a rational strategy to improve lipase resistance to organic solvents: the introduction of positively charged and/or non-polar residues on the enzyme surface.20 They concluded that more favourable salt bridges as the oppositely charged acids approaching each other on the lipase surface result in greater organic solvent resistance. Recently, Ingenbosch et al. systematically investigated the activity and structure of lipase A from Bacillus subtilis in binary water–organic solvent mixtures and found that the modulation of the hydrogen-bonding network at the active site by the different solvent environments is a key structural feature regulating the enzyme activity.21
To study the thermostability and catalytic efficiency of GOx, the molecular dynamics approach has often been used. Using the quasi-elastic neutron scattering experiment and MD simulations, Tatke et al. demonstrated that MD is an excellent theoretical method to study the GOx structure.22 MD simulations showed that GOx does not tend to self-aggregate in buffer solutions.23 Tu et al. prepared mutants to improve the thermostability and catalytic efficiency of GOx.24 Based on their MD simulations, the substitution of glutamic acid with cysteine at position 82 facilitated enzyme stabilization by a change of loop conformation that caused Asn87 side chain rotation. Besides, the relationship between the catalytic efficiency of GOx and the conformation of histidine (His516), which is geometrically necessary for the proton transfer from glucose, has been observed by Petrovic et al.25 However, MD simulations of GOx in pure organic solvents are not available in the scientific literature.
In this work, we experimentally and theoretically investigated the activity and structural changes of GOx after treatment with apolar and polar organic solvents using an activity assay and molecular dynamics simulations. We found that only polar solvents cardinally alter the structure of the active centre of the enzyme. Moreover, we observed that organic solvents promote the formation of salt bridges on the surface of GOx. As a result, the enzyme aggregated into nanoparticles of a few hundred nanometers in diameter. The degree of aggregation correlated well with the decrease of the enzymatic activity. These results have never been described in the scientific literature before.
Results and discussions
Enzymatic activities of GOx after its exposure to solvents
To investigate the effects of organic solvents on the structure of glucose oxidase (GOx), 1.0 mg mL−1 concentration solutions of GOx in chloroform (CHF, relative permittivity, ε = 4.8), 1,2-dichloroethane (DCE, ε = 10.4), and dichloromethane (DCM, ε = 8.9) as the chlorinated apolar solvents, tetrahydrofuran (THF, ε = 7.5) and 1,2-dimethoxyethane (DME, ε = 7.2) as the water-soluble apolar solvents, toluene (PhMe, ε = 2.4) and ethyl acetate (EA, ε = 6.1) as the water non-soluble apolar solvents, ethanol (EtOH, ε = 25.3), acetonitrile (AN, ε = 36.6), N,N-dimethylformamide (DMF, ε = 38.3), and dimethyl sulfoxide (DMSO, ε = 47.2) as the polar solvents were prepared. We considered an organic solvent as apolar if its relative permittivity ε was less than 15. Unfortunately, the enzyme is completely insoluble in hexane and heptane. Before determination of kinetic parameters, the samples in organic solvents were homogenized using ultrasound and kept for 24 h in the refrigerator at 4 °C. The solvents were evaporated under vacuum without enzyme denaturation. However, this procedure failed for DMSO and DMF due to their high boiling points. Therefore, samples of the solutions of GOx in DMF and DMSO were used. Moreover, these pure polar solvents were tested through the activity determination experiments using the GOx solution in the buffer with pH 7.0. The GOx activity was examined using a coupled enzyme assay, in which GOx oxidizes glucose resulting in the production of H2O2 that reacts by catalyzing horseradish peroxidase (HRP) with o-dianisidine as the peroxidase substrate and colorimetric probe at 460 nm (Fig. 1a). The assay results were compared with the GOx activity in the buffer. The enzymatic activity of GOx was deduced from kinetic constants using the initial rate method. Fig. 1b shows the initial velocities of the glucose oxidation by GOx. These data and a Michaelis–Menten's model were used to estimate the kinetic parameters (kcat and KM) and the catalytic efficiencies (kcat/KM) of GOx after its exposure to the organic solvents.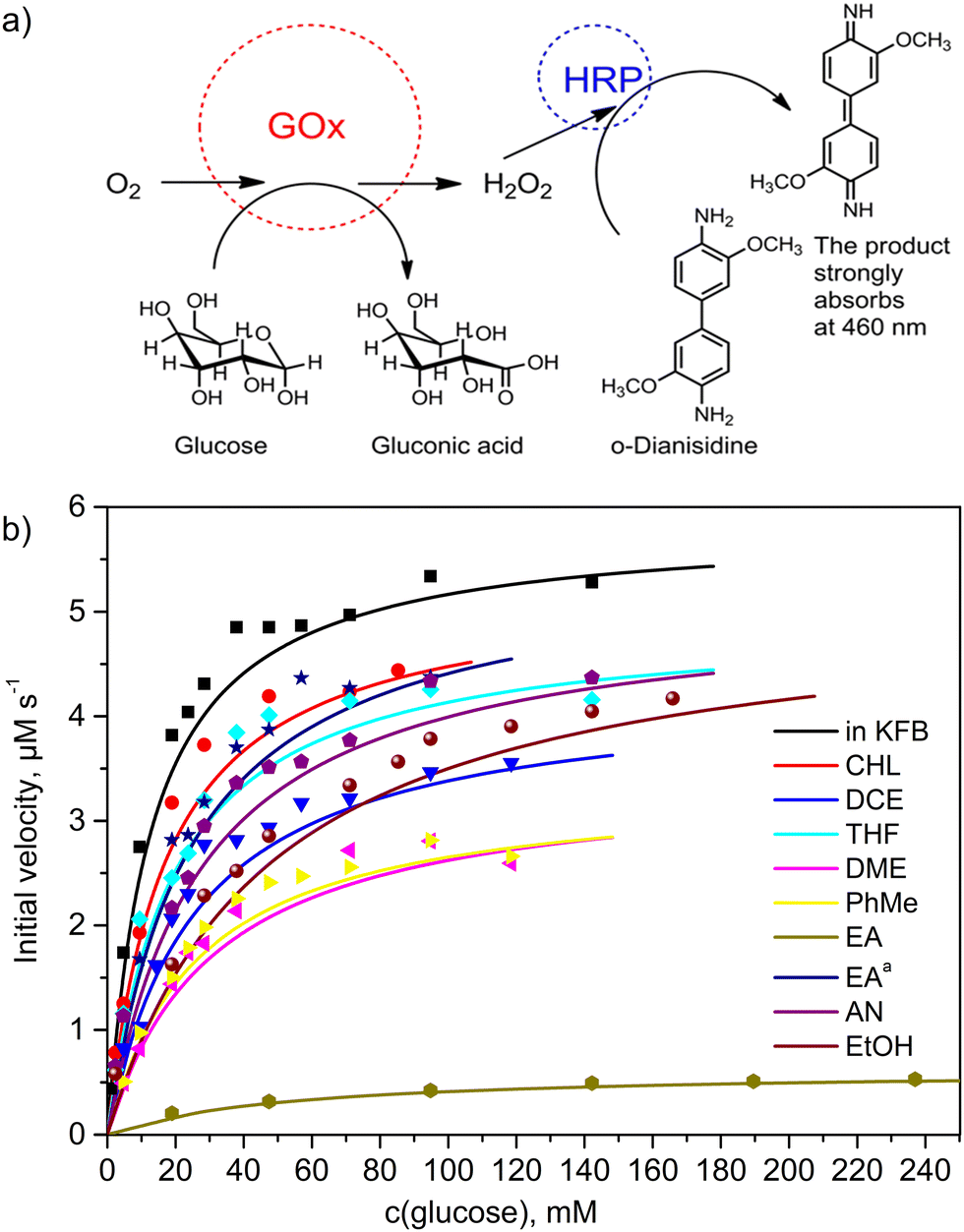 | ||
| Fig. 1 Kinetic parameters of GOx after its exposure to the organic solvents: (a) scheme of the activity determination and (b) the initial velocities of the glucose oxidation by GOx at 25 °C and pH 7.0, which was kept in the organic solvents for 24 h. | ||
It is well known that Michaelis's constant (KM) is defined as the substrate concentration at which the reaction rate is half of its maximal value. The rate constant kcat is defined as the number of catalytic cycles that each active site undergoes per second when GOx is saturated with glucose. The catalytic efficiency (kcat/KM) represents the overall ability of the enzyme to convert a substrate to a product.26 The calculated kinetic parameters are summarized in Table 1. The KM, kcat and kcat/KM parameters equal to 10.2 ± 0.8 mM, 97.4 ± 1.7 s−1 and 9.6 ± 0.2 mM−1 s−1, respectively, were measured in the buffer pH 7.0 solutions (Entry 1, Table 1). These values are very close to the literature data.27 Therefore, these values were used to compare with GOx kinetics results in other samples. After GOx exposure to CHF, DCM, and DCE as the chlorinated apolar solvents (Entries 2–4, Table 1), the catalytic efficiency (kcat/KM) of the CHF sample was close to the value of that parameter in the buffer. When GOx was exposed to THF and DME as the water-soluble apolar solvents with hydrogen bond acceptor properties, the enzyme showed a higher activity in THF than in DME (Entries 5 and 6, Table 1). It is worth noting that the DME molecule has a non-cyclic structure, and it is therefore more conformationally labile than THF. This feature of the DME molecule allows penetration into the enzyme structure by coordinating with amide groups. PhMe and EA as the water non-soluble apolar solvents demonstrated a strong decrease in the catalytic efficiency of GOx (entries 7–9, Table 1). However, upon removing acetic acid as an acidic impurity from EA using a mixture of NaHCO3 and Na2SO4 as the drying agent, and subsequent distillation, a much better activity of GOx was found (entry 9, Table 1). This suggests that acidic impurities play an important role in the stability of this enzyme in organic solvents. Although solutions of DMSO in water are widely used in enzyme biotechnology, the catalytic activity of GOx was absent in pure DMSO and DMF polar solvents. Moreover, after GOx exposure to the hydrogen bond acceptor and donor polar solvents AN and EtOH (entries 10, 11, Table 1), relatively low catalytic efficiencies of 3.9 ± 0.2 mM−1 s−1 and 2.3 ± 0.1 mM−1 s−1 were determined, respectively. According to our experiments, the best values of KM, kcat and kcat/KM (13.7 ± 1.6 mM, 87.4 ± 3.4 s−1 and 6.4 ± 0.2 mM−1 s−1, respectively) were determined after exposure to chloroform (entry 2, Table 1). Also, these parameters are comparable to those of chemically purified EA and THF. These solvents can be safely used when performing experiments with GOx, without much loss of catalytic activity.
| Entry | Solvent | K M (mM) | k cat (s−1) | k cat /K M (mM−1 s−1) | Salt bridges |
|---|---|---|---|---|---|
| a In ethyl acetate, traces of acetic acid were chemically removed. b Activity was too low to be measured. | |||||
| 1 | Water (KFB, pH 7.0) | 10.2 ± 0.7 | 97.4 ± 1.7 | 9.6 ± 0.2 | 23 ± 2 |
| 2 | CHF | 13.7 ± 1.6 | 87.4 ± 3.4 | 6.4 ± 0.2 | — |
| 3 | DCM | 12.4 ± 1.3 | 47.1 ± 3.4 | 3.8 ± 0.3 | 28 ± 2 |
| 4 | DCE | 20.8 ± 2.4 | 72.2 ± 3.4 | 3.5 ± 0.2 | 32 ± 2 |
| 5 | THF | 15.2 ± 2.3 | 82.3 ± 3.4 | 5.4 ± 0.2 | 34 ± 2 |
| 6 | DME | 25.0 ± 3.6 | 57.1 ± 3.4 | 2.3 ± 0.1 | — |
| 7 | PhMe | 21.1 ± 2.2 | 57.1 ± 1.7 | 2.7 ± 0.1 | 28 ± 2 |
| 8 | EA | 41.1 ± 3.4 | 10.1 ± 0.2 | 0.25 ± 0.01 | — |
| 9 | EAa | 19.5 ± 2.1 | 92.4 ± 3.4 | 4.7 ± 0.2 | 36 ± 2 |
| 10 | AN | 22.0 ± 2.7 | 85.7 ± 3.4 | 3.9 ± 0.2 | 34 ± 2 |
| 11 | EtOH | 37.4 ± 3.2 | 85.7 ± 1.7 | 2.3 ± 0.1 | 36 ± 2 |
| 12 | DMF | —b | — | — | 38 ± 2 |
| 13 | DMSO | —b | — | — | 37 ± 1 |
Optical properties and aggregation of GOx after its exposure to the solvent
To get more information on the structural changes after GOx exposure to the organic solvents, ultraviolet-visible (UV-Vis) absorption spectroscopy and circular dichroism (CD) spectroscopy were carried out (Fig. 2a and b). Prior to these experiments, the organic solvents were evaporated, and the residue was dissolved in the buffer. For comparison, the spectra of unaffected GOx in the buffer and flavin adenine dinucleotide (FAD) as a cofactor of this enzyme in the buffer were also recorded. First, the solution of unaffected GOx shows low intensity bands with an excitation coefficient of ∼104 M−1 cm−1 at 460 nm and 380 nm, which can be assigned to the absorption of FAD in the enzyme active centre. These bands are not active in the corresponding CD spectra due to a stretched conformation of the FAD cofactor in the active centre. Moreover, in the DCE, CHF, THF, and DME cases, these bands are widespread and have higher excitation coefficients. This phenomenon is characteristic of aggregated peptides.28 Second, high intensity UV-Vis bands at 278 nm (∼105 M−1 cm−1) and 207 nm (∼106 M−1 cm−1) with a shoulder at 225 nm can be assigned to an n → π* transition in the aromatic amino acid moieties with a charge transition from FAD to the indole moiety of tryptophane in the active centre of GOx, and the full allowed π → π* and intermolecular electronic transitions between chains of the aromatic residues of the enzyme, respectively.29,30 Moreover, the positive low intensity Cotton band at 273 nm and the negative high intensity Cotton bands at 217 nm and 210 nm correspond to these fully allowed transitions. Therefore, these bands have widely been applied to determine the changes in the tertiary structure of enzymes.31 | ||
| Fig. 2 Chiroptical and optical spectra of GOx and FAD in the buffer pH 7.0 solution: (a) CD spectra of GOx and FAD (inset: the extended CD spectra in the range of 260–290 nm) and (b) UV-Vis spectra of the enzyme after its exposure to organic solvents and subsequent solvent evaporation and dissolution in the buffer (the extinction coefficients are shown on a logarithmic scale). | ||
In the UV-Vis spectra, hypsochromic shifts of the UV-Vis bands at around 275 nm and 380 nm in the DCE, EA, and AN samples were observed. Moreover, the intensities of the corresponding CD bands at 275 nm of these samples were decreased (Fig. 2b). Therefore, one can guess that the orientation of FAD with respect to the nearby aromatic amino acids in the active centre was changed. Moreover, although the CD spectrum of FAD showed a lower intensity compared to the spectrum of the enzyme (Fig. 2a), the elimination of FAD from the active centre and the structural change were not observed in the samples. These spectroscopic results correlate well with the activity of the enzyme (Table 1). Unfortunately, we could not obtain the corresponding data on the enzyme dissolved in DMSO and DMF due to their high boiling points and their wide UV absorption cut-offs.
To explore the hypothesis of enzyme aggregation after exposure to organic solvents, the sizes of the enzyme particles were measured using a dynamic light scattering technique. Because GOx aggregation was predicted using the UV-Vis spectra (Fig. 2b), we used the dynamic light scattering technique to measure enzyme particle sizes in all investigated solvents. Furthermore, the samples of GOx in pure DMSO and DMF were also studied. As a reference, the ∼10 nm particle size was recorded for the unaffected GOx in the buffer solution (Fig. 3). This particle size well corresponded to the diameter of the native enzyme.10 However, for the samples of GOx affected with the chlorinated and apolar solvents, the particle sizes were in the 180–480 nm range (Fig. 3). Also, Fig. 3 shows that particles of various sizes from 80 nm to 600 nm were observed in the DMSO and DMF solutions. All the averaged particle sizes are summarized in Table S1 (ESI†). These results clearly confirm that the enzyme aggregates in the organic solvents. A comparison of our MD calculations of the GOx dimer in water, DMSO and DMF shows that in water the tendency of the dimer to dissociate is largest, in agreement with the experiment.
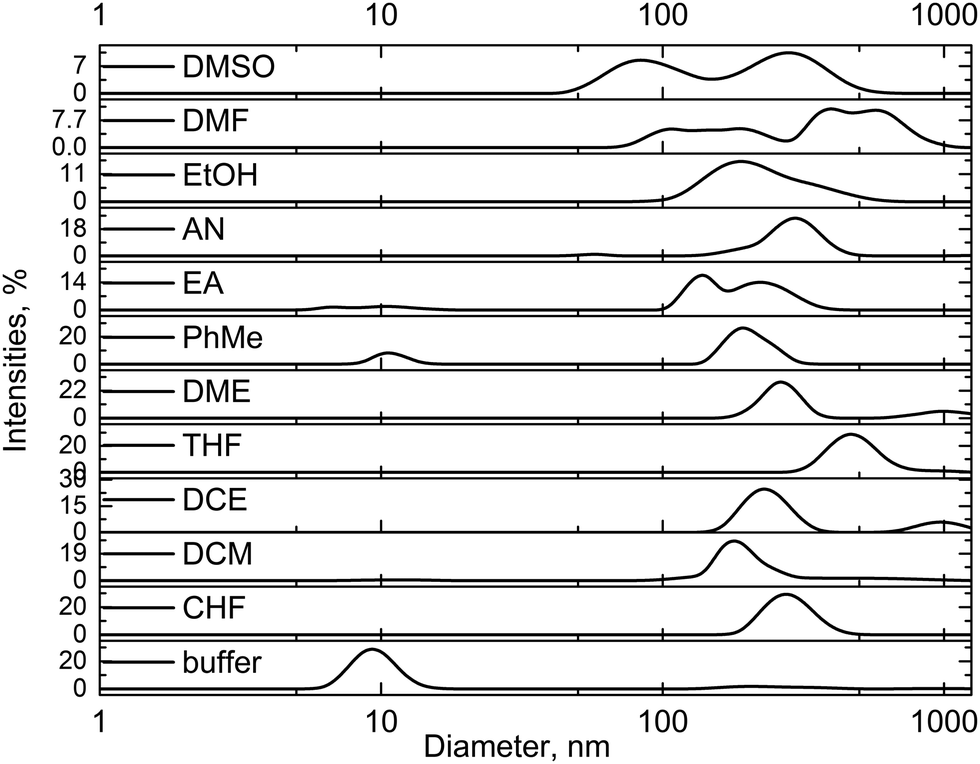 | ||
| Fig. 3 Particle sizes of the enzyme in the buffer solution of pH 7.0 after exposure to the organic solvents, subsequent solvent evaporation and residue dissolution in the buffer. The DMSO and DMF samples were measured without evaporation due to their high boiling points. | ||
Effects of physical properties of the solvent on the GOx catalytic efficiency
At first glance, the general trends between the catalytic efficiencies and the solvent physical properties are not immediately obvious. Our proposed eqn (1) shows a computed correlation between the solvent properties, particle sizes, and experimental catalytic efficiencies (Table S1, ESI†):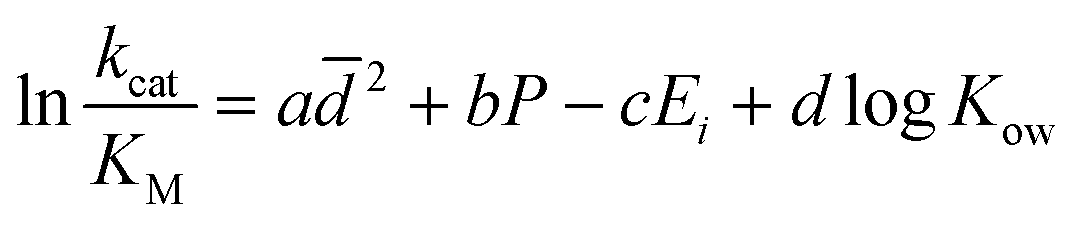 | (1) |
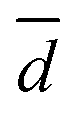 is the average of the particle sizes according to their intensity from the dynamic light scattering experiments, P is the polarity index of the used organic solvent, which is relative to the degree of interaction of the solvent with various polar molecules,32Ei is the energy of intermolecular interaction between the solvent and N-methylacetamide as an amide bond model, which was computed using the MP2/6-311++G(d,p) method, and log
is the average of the particle sizes according to their intensity from the dynamic light scattering experiments, P is the polarity index of the used organic solvent, which is relative to the degree of interaction of the solvent with various polar molecules,32Ei is the energy of intermolecular interaction between the solvent and N-methylacetamide as an amide bond model, which was computed using the MP2/6-311++G(d,p) method, and log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kow is the partition coefficient of the solvent between n-octanol and water.
Kow is the partition coefficient of the solvent between n-octanol and water.
The 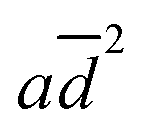 , bP, c Ei, and d
, bP, c Ei, and d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) logKow terms in eqn (1) correspond to the effects of aggregation of the enzyme, the polarity of the solvent, the energies of intermolecular interaction between the solvent and the peptide residues, and the tendency of the organic solvent to adsorb to lipids, respectively. The corresponding descriptors of eqn (1) were calculated to be a = 4.4 × 10−6 nm−2, b = 0.25, c = −0.017 mol kJ−1, and d = 0.25, and the coefficient of determination, R2, of the computed and experimental kcat/KM was 0.901. Fig. 4a shows that the experimental and calculated catalytic efficiencies well correlate with each other. It is worth noting that if the
logKow terms in eqn (1) correspond to the effects of aggregation of the enzyme, the polarity of the solvent, the energies of intermolecular interaction between the solvent and the peptide residues, and the tendency of the organic solvent to adsorb to lipids, respectively. The corresponding descriptors of eqn (1) were calculated to be a = 4.4 × 10−6 nm−2, b = 0.25, c = −0.017 mol kJ−1, and d = 0.25, and the coefficient of determination, R2, of the computed and experimental kcat/KM was 0.901. Fig. 4a shows that the experimental and calculated catalytic efficiencies well correlate with each other. It is worth noting that if the 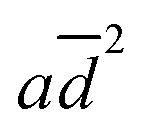 term of eqn (1) is replaced by a
term of eqn (1) is replaced by a![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) , R2 decreases to 0.858. Therefore, these catalytic efficiencies are proportional to the surface area of the particles. Moreover, eqn (1) allows estimation of the influence of each effect on the activity of the enzyme after its exposure to the solvents. Fig. 4b shows that the polarity index of the solvent has a major impact on the activity of all solvents. Gupta and colleagues studied the influence of the polarity index of organic solvents on the activities of polyphenol oxidase, peroxidase, acid phosphatase, and trypsin after their exposure to organic solvents. They noticed that solvents with polarity indexes of 5.8 and above are “good” solvents because these solvents did not irreversibly denature the enzymes, whereas solvents with polarity indexes less than 5.0 denatured the enzymes completely.33 However, the authors did not investigate water-immiscible organic solvents such as CHF, DCM, EA and PhMe. Our study expanded the importance of the polarity index for additional solvents that have not been investigated by Gupta et al. Additionally, we observed that the activity of GOx depends on the particle size of the aggregated enzyme due to the frustrated diffusion of the substrate towards the active site. When the enzyme aggregates into larger structures, its surface area per cofactor unit drastically decreases. For water-soluble apolar solvents such as THF and DME, aggregation of GOx is an important factor for its activity. In the case of water-insoluble apolar solvents, such as CHF, DCM, DCE and PhMe, the logKow of these solvents has a tangible effect on the GOx activity.
, R2 decreases to 0.858. Therefore, these catalytic efficiencies are proportional to the surface area of the particles. Moreover, eqn (1) allows estimation of the influence of each effect on the activity of the enzyme after its exposure to the solvents. Fig. 4b shows that the polarity index of the solvent has a major impact on the activity of all solvents. Gupta and colleagues studied the influence of the polarity index of organic solvents on the activities of polyphenol oxidase, peroxidase, acid phosphatase, and trypsin after their exposure to organic solvents. They noticed that solvents with polarity indexes of 5.8 and above are “good” solvents because these solvents did not irreversibly denature the enzymes, whereas solvents with polarity indexes less than 5.0 denatured the enzymes completely.33 However, the authors did not investigate water-immiscible organic solvents such as CHF, DCM, EA and PhMe. Our study expanded the importance of the polarity index for additional solvents that have not been investigated by Gupta et al. Additionally, we observed that the activity of GOx depends on the particle size of the aggregated enzyme due to the frustrated diffusion of the substrate towards the active site. When the enzyme aggregates into larger structures, its surface area per cofactor unit drastically decreases. For water-soluble apolar solvents such as THF and DME, aggregation of GOx is an important factor for its activity. In the case of water-insoluble apolar solvents, such as CHF, DCM, DCE and PhMe, the logKow of these solvents has a tangible effect on the GOx activity.
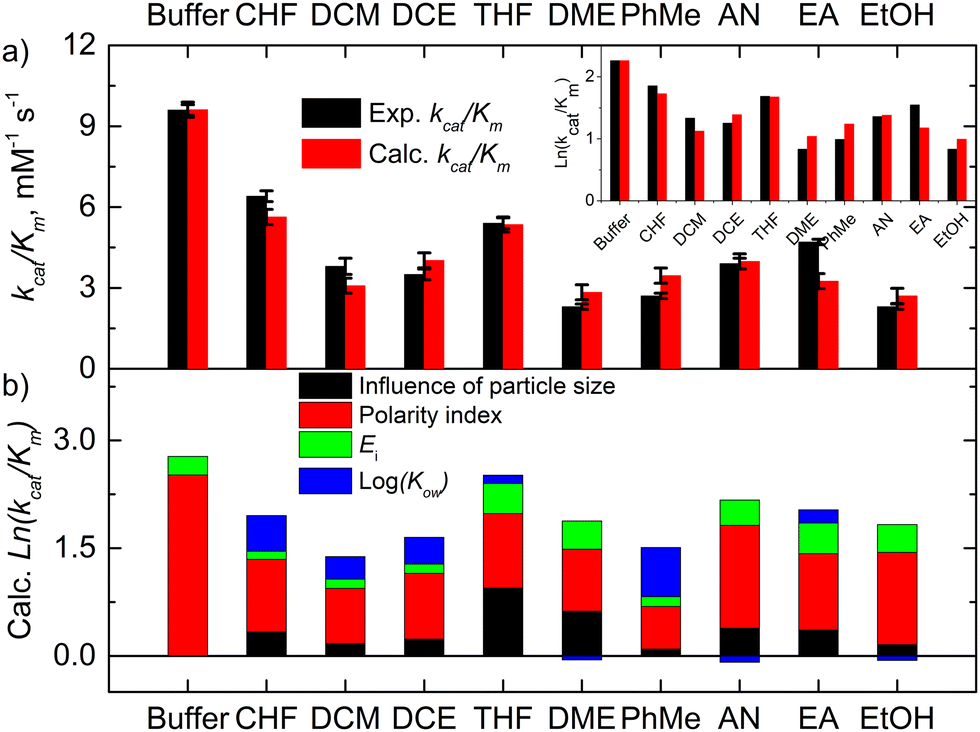 | ||
| Fig. 4 The experimental and calculated kinetic data of GOx and their correlation: (a) the experimental and calculated catalytic efficiencies after GOx exposure to the solvent and (b) the effects of the particle size, the polarity index, and logKow on these catalytic efficiencies. | ||
To estimate the ability of these solvents to form intermolecular solvent–protein H-bonds in the binding pocket of GOx, H-bond energies of these solvents with the amide moiety of amino acid residues were computed using the Møller–Plesset (MP2) perturbation theory (Fig. S1 and S2, ESI†). The H-bonded complexes between N-methylacetamide as the amide bond model and the corresponding organic solvent were optimized. The energies of amide–water and amide–amide H-bonds were estimated to be −15.5 kJ mol−1 and −26.5 kJ mol−1, respectively. Dixon et al. have found similar results.34 Also, the amide–amide energy in peptides can be in the range from −10.5 kJ mol−1 to −40.0 kJ mol−1 depending on the conformation of the peptide and exposure to water.35 Some of the amide–solvent interaction energies listed in Table S1 (ESI†) therefore should be able to break the abovementioned H-bonds if they have lower (more negative) values. However, this process is reversible. According to our theoretical results, water molecules of the system can be replaced with the DME, THF, EA, EtOH and AN molecules. However, the polar DMSO and DMF solvents can break the intermolecular amide–amide bonds. These non-covalent bonds are similar to or stronger than the amide–amide interactions (Fig. S2, ESI†). Our correlation confirms that this effect of the interaction energies is significant only for these solvents (Fig. 4b).
The validity of our developed solvent property–efficiency relationship model (eqn (1)) was tested by predicting the GOx catalytic efficiency after exposure to diethyl ether (Et2O) as an apolar solvent. The properties of Et2O are provided in Table S1 (ESI†). The theoretical and experimental catalytic efficiencies were calculated to be 1.9 ± 0.3 mM−1 s−1 and 2.2 ± 0.1 mM−1 s−1, respectively (Table S1, entry 13, ESI†). Therefore, our model allows us to reliably predict the enzyme catalytic efficiency after exposure to apolar organic solvents. These results agree well within the error limits. Furthermore, the DMF and DMSO solvents can strongly coordinate with amino acids via H-bonds and prevent binding of a substrate. Also, these polar solvents can change the conformation of the enzyme due to the disconnection of the amide–amide H-bonds. Therefore, the enzyme is inactive in these two solvents. Unfortunately, the theoretical catalytic efficiencies after GOx exposure to DMSO and DMF were predicted to be 3.6 ± 0.3 mM−1 s−1 and 6.0 ± 0.3 mM−1 s−1 using eqn (1) and their physical properties. Therefore, the aggregation, polarity and other effects do not provide an answer to the vanishingly low GOx activity in the polar DMSO and DMF solvents.
Molecular dynamics simulations of GOx in the solvents
The behaviour of GOx in organic solvents was further explored using 200 ns molecular dynamics (MD) simulations of the enzyme monomer in water (buffer), DCE, THF, PhMe, EA, AN, DMSO and DMF. To imitate the hydrated GOx as the starting macromolecule for the experiments, water molecules that bound via H-bonds to the protein were retained during the simulation (see the ESI†). Thus, a layer of water molecules was left on the surface of the enzyme. During these MD simulations of GOx, we observed a propensity towards salt bridge formation in the nonaqueous solvents between the positively charged amino acids Arg, His, and Lys and the negatively charged Glu and Asp (Table 1). Yenenler et al. theoretically showed that the salt bridges can impair the catalytic function by reducing the structural flexibility of enzymes during apolar solvent contact.36 Moreover, charged groups on the protein surface increase their solubility.37 Therefore, the formation of the salt bridges could partially neutralize and stabilize charged side chains sticking into the solution and facilitate aggregation of GOx observed in the experiment. Besides, organic solvents can reduce strong electrostatic forces between oppositely charged residues of amino acids and stabilize the salt bridges on the enzyme surface.38 We think that non-aqueous solvents often have less compact and weaker (easier to destroy) solvation shells and this also helps in aggregation of the enzyme. Almost all salt bridges are formed near the surface, but only one salt bridge as a catalytically important pair, His559–Glu412, was farther away from the surface. Based on our MD simulations, the numbers of the salt bridges increase from around 23 in water to a range of 32–36 in the apolar solvents such as DCE, THF, and EA and to around 38 in polar solvents such as DMSO and DMF (Table 1). Additionally, we found that the aggregated GOx particle sizes linearly correlate with the number of salt bridges on GOx from the MD simulations (Fig. S3, ESI†). As discussed above, the higher density of the salt bridges onto the surface of GOx also suggests the formation of the salt bridges between the enzymes by leading to easier aggregation. Based on the statistical analysis of the high-quality X-ray structures of proteins, Xu et al. consider that salt bridges play an important role in protein–protein binding.39 Moreover, we observed that charged complexes aggregated into nanoparticles because the charged molecules lowered the surface tension of the solution.40A closer look at the catalytic active site during the simulation gives a further possible clue to the passivity of GOx in DMSO and DMF, in contrast with the other organic solvents: the ability of these polar solvents to block the active site. Fig. 5a–d show 200 ns snapshots from the MD simulations. We observed that in DMSO and DMF (Fig. 5a and b), the organic solvent molecules penetrate into the glucose-binding pocket, bordered by Tyr68, Phe414, Trp426, Arg512, Asn514, His516, and His559 (orange in Fig. 5a–d); some of these residues also play an important role in the proton transfer to/from FAD.41 Importantly, DMSO and DMF not only broke through the water layer, but also formed H-bonds with the active site residues or/and the residues at the entrance of the active site, thereby locking the active pocket (Fig. 5a and b). In other solvents, the solvent molecules did not enter the pocket appreciably (Fig. 5d), or they did enter but did not form H-bonds with the protein and therefore did not get locked in the pocket (Fig. 5c). Presumably locking of the glucose pocket may prevent the entering of the substrate into the active site, causing GOx inactivity.
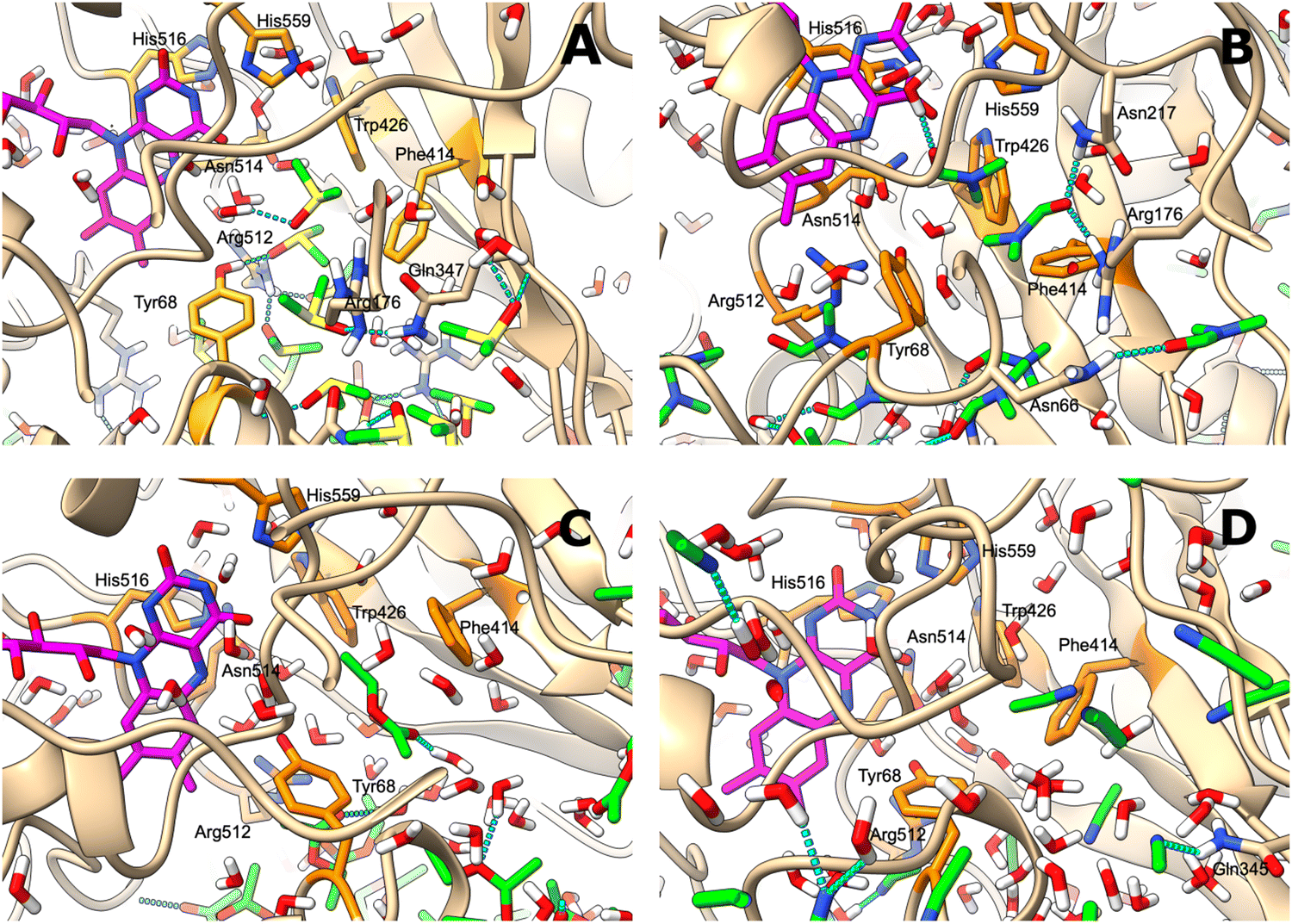 | ||
| Fig. 5 (A–D) Glucose binding pocket, bordered by orange-coloured key side-chains after the 200 ns MD simulation in (A) DMSO; (B) DMF; (C) EA; and (D) AN. FAD is coloured in magenta, and the organic solvent is shown with green-coloured carbons. Hydrogen bonds involving the organic solvent molecules are depicted as broken cyan lines, and the protein side-chains that form hydrogen bonds with the solvent are also shown. | ||
Conclusions
In this work, the mechanism of the inhibition of the activity of glucose oxidase (GOx) from Aspergillus niger by various pure organic solvents as the non-native environment was studied by combining experimental and theoretical methods. Using chiroptical spectroscopy, slight changes in the orientation of FAD to nearby aromatic amino acids in the active centre were observed after exposure of GOx to apolar solvents. The correlation between the measured activities of GOx and the physical properties of the solvents showed that the polarity index of the solvent and aggregation of the enzyme have the greatest influence on the enzyme activity. The activity of the enzyme is inversely proportional to the particle size due to impaired diffusion of the substrate towards the active site and the lower surface area per cofactor unit compared to the monomeric enzyme in the buffer. This process is irreversible, and the nanoparticles remain in aqueous solutions. We explain this phenomenon by the formation of the salt bridges between the acidic and basic moieties of amino acids onto the surface of GOx, which stimulates the enzyme to aggregate towards nanoparticles with a diameter of several hundreds of nanometers. Based on our MD simulations, organic solvent-induced depolarization of the environment leads to the formation of salt bridges on the surface of the enzyme. As a result, the enzymes aggregate by binding through the inter-enzymatic salt bridges. Moreover, we found that DMSO and DMF as polar solvents do not degrade the GOx dimers, but strongly coordinate to the amide bond of amino acid residues in the active centre of GOx and prevent the binding of glucose. According to these results, the enzyme retains its activity after storage in apolar solvents such as chloroform and THF and these solvents can be used for chemical modification of the enzyme. We believe that the given strategy can be transferred to other enzyme classes and can empower researchers to expand a range of biocatalysis applications in various organic solvents.Experimental procedures
Materials, instrumentation, computation details, and the full experimental procedures are provided in the ESI.†Author contributions
V. D. conducted the experiment and wrote the paper. V. K. conducted the MD simulations and wrote the paper. G. B. designed the experiments and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Research Council of Lithuania (grant no. S-MIP-20-45) and by the European Regional Development Fund under the “Promotion of Centers of Excellence and Innovation and Technology Transfer Centers” program No. 01.2.2-CPVA-K-703 (grant No. 01.2.2-CPVA-K-703-03-0010).References
- A. M. Klibanov, Nature, 2001, 409, 241–246 CrossRef CAS PubMed.
- M. N. Gupta, Eur. J. Biochem., 1992, 203, 25–32 CrossRef CAS PubMed.
- R. M. Daniel, J. L. Finney, M. Stoneham, C. Nick Pace, S. Treviño, E. Prabhakaran and J. M. Scholtz, Philos. Trans. R. Soc., B, 2004, 359, 1225–1235 CrossRef PubMed.
- V. Stepankova, S. Bidmanova, T. Koudelakova, Z. Prokop, R. Chaloupkova and J. Damborsky, ACS Catal., 2013, 3, 2823–2836 CrossRef CAS.
- J. A. Bauer, M. Zámocká, J. Majtán and V. Bauerová-Hlinková, Biomolecules, 2022, 12, 472 CrossRef CAS PubMed.
- R. Wilson and A. P. F. Turner, Biosens. Bioelectron., 1992, 7, 165–185 CrossRef CAS.
- Š. Žukauskas, A. Ramanavičius and G. Bagdžiūnas, J. Electrochem. Soc., 2019, 166, B316 CrossRef.
- G. Bagdžiūnas and D. Palinauskas, Biosensors, 2020, 10, 104 CrossRef PubMed.
- G. Bagdžiūnas and A. Ramanavičius, Phys. Chem. Chem. Phys., 2019, 21, 2968–2976 RSC.
- V. Dudkaitė and G. Bagdžiūnas, Biosensors, 2022, 12, 335 CrossRef PubMed.
- E. I. Iwuoha and M. R. Smyth, Anal. Proc., 1994, 31, 19–21 RSC.
- N. Vasileva and T. S. Godjevargova, Mater. Sci. Eng., C, 2005, 25, 17–21 CrossRef.
- N. Balistreri, D. Gaboriau, C. Jolivalt and F. Launay, J. Mol. Catal. B: Enzym., 2016, 127, 26–33 CrossRef CAS.
- M. Karplus and J. A. McCammon, Nat. Struct. Biol., 2002, 9, 646–652 CrossRef CAS PubMed.
- G. Colombo, G. Ottolina and G. Carrea, in Biocatalysis, ed. H. Griengl, Springer, Vienna, 2000, pp. 1–21 Search PubMed.
- N. Milčić, V. Stepanić, I. Crnolatac, Z. Findrik Blažević, Z. Brkljača and M. Majerić Elenkov, Chem. Eur. J., 1923, e20220 Search PubMed.
- M. Mohtashami, J. Fooladi, A. Haddad-Mashadrizeh, M. R. Housaindokht and H. Monhemi, Int. J. Biol. Macromol., 2019, 122, 914–923 CrossRef CAS PubMed.
- L. Yang, J. S. Dordick and S. Garde, Biophys. J., 2004, 87, 812–821 CrossRef CAS PubMed.
- H. Cui, T. H. J. Stadtmüller, Q. Jiang, K.-E. Jaeger, U. Schwaneberg and M. D. Davari, ChemCatChem, 2020, 12, 4073–4083 CrossRef CAS.
- H. Cui, L. Eltoukhy, L. Zhang, U. Markel, K.-E. Jaeger, M. D. Davari and U. Schwaneberg, Angew. Chem., Int. Ed., 2021, 60, 11448–11456 CrossRef CAS PubMed.
- K. N. Ingenbosch, J. C. Vieyto-Nuñez, Y. B. Ruiz-Blanco, C. Mayer, K. Hoffmann-Jacobsen and E. Sanchez-Garcia, J. Org. Chem., 2022, 87, 1669–1678 CrossRef CAS PubMed.
- S. S. Tatke, C. K. Loong, N. D’Souza, R. T. Schoephoerster and M. Prabhakaran, Biopolymers, 2008, 89, 582–594 CrossRef CAS PubMed.
- S. Zhang, S. Yu, M. Wang, Z. Cui, B. Chen and T. Tan, Chem. Phys., 2022, 552, 111366 CrossRef CAS.
- T. Tu, Y. Wang, H. Huang, Y. Wang, X. Jiang, Z. Wang, B. Yao and H. Luo, Food Chem., 2019, 281, 163–170 CrossRef CAS PubMed.
- D. Petrović, D. Frank, S. C. L. Kamerlin, K. Hoffmann and B. Strodel, ACS Catal., 2017, 7, 6188–6197 CrossRef PubMed.
- P. K. Robinson, Essays Biochem., 2015, 59, 1–41 CrossRef PubMed.
- A. Guiseppi-Elie, S.-H. Choi and K. E. Geckeler, J. Mol. Catal. B: Enzym., 2009, 58, 118–123 CrossRef CAS.
- M. F. Pignataro, M. G. Herrera and V. I. Dodero, Molecules, 2020, 25, 4854 CrossRef CAS PubMed.
- E. H. Strickland and S. Beychok, CRC Crit. Rev. Biochem., 1974, 2, 113–175 CrossRef CAS PubMed.
- M. K. Sharon and C. P. Nicholas, Curr. Protein Pept. Sci., 2000, 1, 349–384 CrossRef PubMed.
- D. M. Rogers, S. B. Jasim, N. T. Dyer, F. Auvray, M. Réfrégiers and J. D. Hirst, Chem., 2019, 5, 2751–2774 CAS.
- C. Reichardt and T. Welton, Solvents and Solvent Effects in Organic Chemistry, John Wiley & Sons, Ltd, 4th edn, 2010, pp. 425–508 Search PubMed.
- M. N. Gupta, R. Batra, R. Tyagi and A. Sharma, Biotechnol. Prog., 1997, 13, 284–288 CrossRef CAS.
- D. A. Dixon, K. D. Dobbs and J. J. Valentini, J. Phys. Chem., 1994, 98, 13435–13439 CrossRef CAS.
- R. L. Baldwin, J. Biol. Chem., 2003, 278, 17581–17588 CrossRef CAS PubMed.
- A. Yenenler, A. Venturini, H. C. Burduroglu and O. U. Sezerman, J. Mol. Model., 2018, 24, 229 CrossRef PubMed.
- A. J. Doig, FEBS J., 2017, 284, 1296–1305 CrossRef CAS PubMed.
- A. Kütt, S. Selberg, I. Kaljurand, S. Tshepelevitsh, A. Heering, A. Darnell, K. Kaupmees, M. Piirsalu and I. Leito, Tetrahedron Lett., 2018, 59, 3738–3748 CrossRef.
- D. Xu, C. J. Tsai and R. Nussinov, Protein Eng., Des. Sel., 1997, 10, 999–1012 CrossRef CAS PubMed.
- G. Bagdžiūnas, E. Butkus and E. Orentas, Organometallics, 2019, 38, 2647–2653 CrossRef.
- I. Baek, H. Choi, S. Yoon and S. Na, ACS Biomater. Sci. Eng., 2020, 6, 1899–1908 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb02605h |
| This journal is © The Royal Society of Chemistry 2023 |