
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functionalization of polymeric nanoparticles with targeting VNAR ligands using vinyl sulfone conjugation†
Adam
Leach‡
a,
Marie
Finnegan
b,
Mariana S.
Machado
b,
Laura
Ferguson
c,
John
Steven
c,
Peter
Smyth
a,
Andrew
Porter
cd,
Caroline
Barelle
c,
Efrosyni
Themistou
 *b and
Christopher J.
Scott
*a
*b and
Christopher J.
Scott
*a
aThe Patrick G. Johnston Centre for Cancer Research, School of Medicine, Queen's University Belfast, Belfast, UK. E-mail: c.scott@qub.ac.uk
bSchool of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, UK. E-mail: e.themistou@qub.ac.uk
cElasmogen Ltd, Aberdeen, UK
dThe Institute of Medical Sciences, University of Aberdeen, Aberdeen, UK
First published on 17th April 2023
Abstract
Actively targeted drug loaded nanoparticles represent an exciting new form of therapeutics for cancer and other diseases. These formulations are complex and in order to realize their ultimate potential, optimization of their preparation is required. In this current study, we have examined the conjugation of a model targeting ligand, conjugated in a site-specific manner using a vinyl sulfone coupling approach. A disulfide-functionalized poly(L-lactide)-b-poly(oligo(ethylene glycol) methacrylate)-stat-(bis(2-methacryloyl)oxyethyl disulfide) (PLA-b-P(OEGMA-stat-DSDMA)) diblock copolymer was synthesized by simultaneous ring opening polymerization (ROP) and reversible addition-fragmentation chain transfer (RAFT) polymerization. Subsequently, the disulfide bonds of the polymer were reduced to thiols and divinyl sulfone was attached to the polymer using thiol–ene chemistry to produce the vinyl sulfone (VS)-functionalized PLA-b-P(OEGMA-stat-VSTEMA) amphiphilic block copolymer. Single emulsion – solvent evaporation was employed using a blend of this polymer with poly(D,L-lactide-co-glycolide) (PLGA) to produce VS-functionalized polymeric nanoparticles. The ability of these novel nanoparticles to attach ligands was then exemplified using a single domain variable new antigen receptor (VNAR) with a free carboxyl terminal cysteine residue. The resulting VNAR-functionalized nanoparticles were found to maintain specific affinity to their cognate antigen (DLL4) for at least 72 h at 4 °C. The simplicity of the degradable amphiphilic block copolymer synthesis and the efficiency of VNAR conjugation to the VS-functionalized nanoparticles show the potential of this platform for therapeutic development.
Introduction
The high surface area-to-volume ratio and size (<1 μm) of nanoparticles provide unique biochemical, magnetic, optical and electronic properties.1 As a therapeutic modality, nanoparticles can encapsulate drugs and/or imaging agents, so that nanoparticle properties control the pharmacokinetics and pharmacodynamics of the encapsulated agent,2 rather than the intrinsic properties of the free agent, which can enhance the therapeutic potential of some agents. The biodistribution of nanoparticles can be further enhanced through their vectorization with surface ligands to enhance specific cellular adherence and uptake, for example, at a tumour site.3Although various preclinical studies have demonstrated the therapeutic usefulness of targeted nanotherapeutics, these have not been translated effectively into the clinic to date.4,5 Key parameters needed for the design of these molecules for therapeutic use still require careful examination to derive clinical success and include: optimization of the choice of cancer-associated target, the class of targeting ligand used, and how the ligand is attached to the particle surface.6
A key goal of ligand conjugation to the surface of nanoparticles is to provide controlled coupling to ensure unhindered presentation of the targeting moiety, such as an antibody paratope. This requirement becomes more and more important as the size of the targeting ligand reduces (for example, antibody to antibody fragment to domain binder). To achieve controlled conjugation, some element of selectivity must be present in the nanoconjugation reaction, so that a region distal to the ligand paratope preferentially conjugates to the nanoparticle surface.7 There is increasing interest in this approach, circumventing non-selective coupling chemistries, such as carbodiimide/N-hydroxy succinimide esters, which can lead to random modification of ligand amines, not only preventing paratope presentation in the case of antibodies and other antibody-like proteins, but also reducing the affinity of the ligand.8–11
Previously, we and others have attempted to enhance ligand conjugation methodologies to nanoparticle surfaces. For example, exploitation of disulfide bridges or free cysteines is a more site-specific conjugation approach than relying on amine functionalization by carbodiimide.8–11 Maleimide chemistry facilitates direct conjugation to free or reduced thiols,12 whilst we have also demonstrated the utilization of aminooxy13 and copper free click-chemistry9,11 approaches in the past. In addition, selective carbohydrate oxidation,14 enzymatic amino acid modification8 and noncovalent interactions (e.g. Ni2+-His615) have been exploited by other researchers for site-selective protein conjugation to nanoparticles.
In this current work, we present a novel alternative conjugation approach, exploiting vinyl sulfone (VS) chemistry, which facilitates covalent modification of thiols in free cysteine residues. Herein, a VS-functionalized PLA-b-P(OEGMA-stat-VSTEMA) amphiphilic diblock copolymer was prepared by attachment of the VS group on a thiol-functionalized amphiphilic diblock polymer by thiol–ene chemistry. The latter was prepared from a disulfide-functionalized polymer synthesized by simultaneous ROP and RAFT polymerization using a dual ROP–RAFT agent, after reduction of its disulfide bonds to thiols. Subsequently, this polymer was blended with poly(lactic-co-glycolic acid) (PLGA) and nanoparticles were prepared by a standard facile single emulsion – solvent evaporation methodology16,17 (Scheme 1a). The physicochemical characteristics and cellular cytotoxicity of the resulting nanoparticles were then evaluated. A model targeting ligand, an anti-DLL4 VNAR engineered to display a C-terminal cysteine (Cys) by reduction of disulfide bonds of VNAR dimers, was prepared (Scheme 1b). This was then site-specifically attached to the nanoparticle surface (Scheme 1c) and the specific target binding affinity of the conjugated particles was demonstrated. The VS-functionalized polymer synthesis combined with the high stability of the VS functionality under aqueous conditions18 and the efficiency of VNAR conjugation to subsequent nanoparticles under mild conditions, can be advantageous for future manufacturability of ligand-conjugated VS-functionalized nanoparticles.
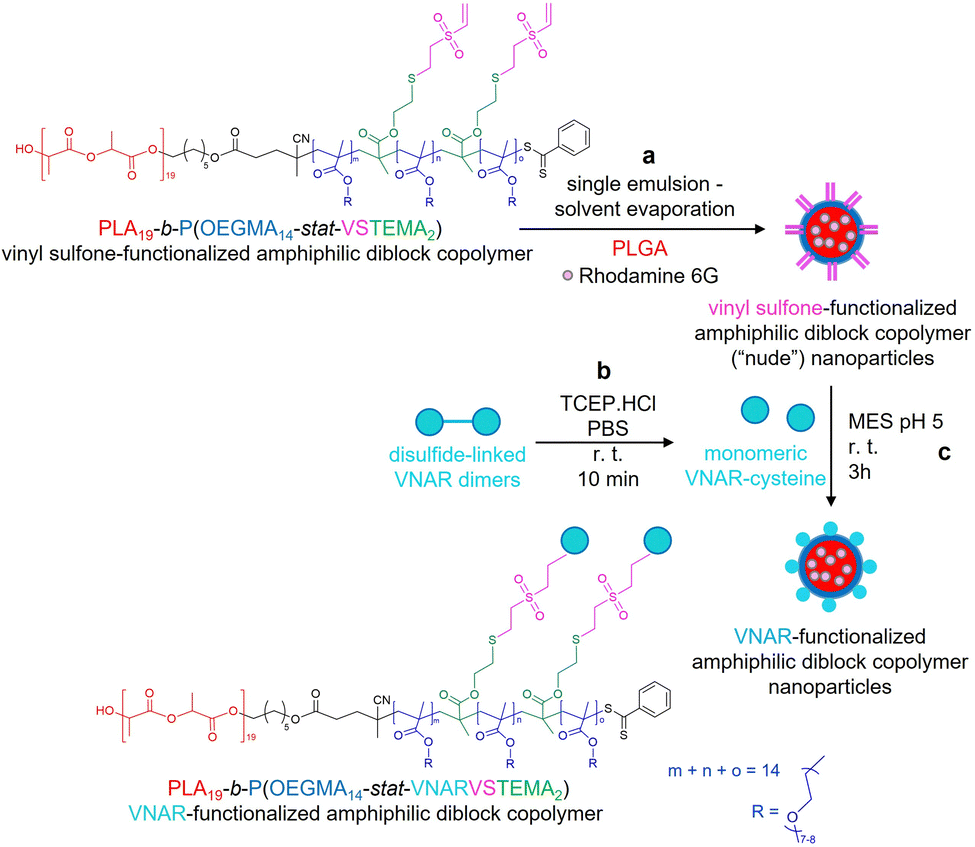 | ||
| Scheme 1 Preparation of rhodamine 6G encapsulated VNAR-functionalized amphiphilic PLA19-b-P(OEGMA14-stat-VSTEMA2) diblock copolymer nanoparticles. (a) Preparation of vinyl sulfone-functionalized amphiphilic diblock copolymer nanoparticles by single emulsion – solvent evaporation from a mixture of the vinyl-sulfone functionalized linear amphiphilic PLA19-b-P(OEGMA14-stat-VSTEMA2) diblock copolymer (10% w/v), PLGA (90% w/v) and rhodamine 6G (0.02% w/v) in DCM using 2.5% w/v PVA in MES buffer (pH 5.0). (b) Reduction of disulfide-linked VNAR dimers (10 μg, 0.83 nmol of monomeric VNAR) to yield monomeric VNARs bearing free C-terminal Cys residues, by incubation with TCEP (8.3 nmol). (c) Conjugation of monomeric free C-terminal Cys bearing VNARs (0.0001% w/v) to “nude” vinyl sulfone-functionalized amphiphilic diblock copolymer nanoparticles to yield VNAR-functionalized amphiphilic diblock copolymer nanoparticles. | ||
Experimental
Materials
(3S)-cis-3,6-dimethyl-1,4-dioxane-2,5-dione (L-lactide, LA, 98%), 4-(dimethylamino)pyridine (DMAP, ≥99%), 1,2-dichloroethane (anhydrous, 99.8%), anhydrous ethanol (≤0.005%), tributylphosphine (Bu3P, mixture of isomers, 97%), divinyl sulfone (DVS, 97%), chloroform-d (CDCl3, 99.8 atom % D), butylated hydroxytoluene (BHT, ≥99%), Resomer® RG 502 H, poly(D,L-lactide-co-glycolide) (PLGA, ≥99%), rhodamine 6G (95%), dichloromethane (DCM, ≥99.0%), 2-(N-morpholino)ethanesulfonic acid (MES) hydrate (≥99.5%), polyvinyl alcohol (PVA, Mw 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–23000, 87–89% hydrolyzed, >95%), Tween-20 (≥97%), acetonitrile (≥99.0%), dimethyl sulfoxide (DMSO, ≥99.5%), bovine serum albumin (BSA, ≥96%), tetrahydrofuran (THF, ≥99.9%), trimethylamine (Et3N, ≥99%), bromophenol blue (90%), 2-mercaptoethanol (BME, ≥99.0%), glycerol (≥99.0%), acrylamide/bis-acrylamide (29:1, 30% solution), sodium dodecyl sulfate (SDS, ≥99.0%), glycine (≥99.0%), Brilliant Blue staining solution and Tris-HCl (≥99.0%) were all obtained from Sigma-Aldrich. PLGA-PEG-NHS (AI064, Lot#: 180521RAI-A, Mn 27677 g mol−1) was obtained from PolySciTech, Akina Inc. Oligo(ethylene glycol) methacrylate (OEGMA, MPEG 350) monomer was kindly donated by GEO Specialty Chemicals (UK). 2,2′-Azobis(2-methylpropionitrile) (AIBN, >98%) was purchased from Fluorochem. Spectra/Por® 6 dialysis membrane (molecular weight cut-off (MWCO) of 1 kD) was purchased from Spectrum Labs (USA). Ethyl acetate (reagent grade, 99%), acetone (laboratory grade), micro-bicinchoninic acid protein assay kit (microBCA) and phosphate buffered saline (PBS) tablets, prepared according to manufacturer's instructions, were obtained from Thermo-Fisher Scientific. DLL4-Fc fusion protein was purchased from Sino Biological, black high-binding plates from Greiner Bio-One, white tissue-culture treated plates from Corning CoStar and CellTiter-Glo reagent from Promega. PANC-1 cells were obtained from ATCC and maintained according to ATCC instructions. VNARs were raised against DLL4 by Elasmogen Ltd using a phage display technique.19
000–23000, 87–89% hydrolyzed, >95%), Tween-20 (≥97%), acetonitrile (≥99.0%), dimethyl sulfoxide (DMSO, ≥99.5%), bovine serum albumin (BSA, ≥96%), tetrahydrofuran (THF, ≥99.9%), trimethylamine (Et3N, ≥99%), bromophenol blue (90%), 2-mercaptoethanol (BME, ≥99.0%), glycerol (≥99.0%), acrylamide/bis-acrylamide (29:1, 30% solution), sodium dodecyl sulfate (SDS, ≥99.0%), glycine (≥99.0%), Brilliant Blue staining solution and Tris-HCl (≥99.0%) were all obtained from Sigma-Aldrich. PLGA-PEG-NHS (AI064, Lot#: 180521RAI-A, Mn 27677 g mol−1) was obtained from PolySciTech, Akina Inc. Oligo(ethylene glycol) methacrylate (OEGMA, MPEG 350) monomer was kindly donated by GEO Specialty Chemicals (UK). 2,2′-Azobis(2-methylpropionitrile) (AIBN, >98%) was purchased from Fluorochem. Spectra/Por® 6 dialysis membrane (molecular weight cut-off (MWCO) of 1 kD) was purchased from Spectrum Labs (USA). Ethyl acetate (reagent grade, 99%), acetone (laboratory grade), micro-bicinchoninic acid protein assay kit (microBCA) and phosphate buffered saline (PBS) tablets, prepared according to manufacturer's instructions, were obtained from Thermo-Fisher Scientific. DLL4-Fc fusion protein was purchased from Sino Biological, black high-binding plates from Greiner Bio-One, white tissue-culture treated plates from Corning CoStar and CellTiter-Glo reagent from Promega. PANC-1 cells were obtained from ATCC and maintained according to ATCC instructions. VNARs were raised against DLL4 by Elasmogen Ltd using a phage display technique.19
The OEGMA monomer was passed through a basic alumina column to remove the inhibitor and the LA monomer was recrystallized from ethyl acetate (4–5 times) prior to its use in the polymerization reaction. The dual ROP–RAFT agent used in the synthesis of the amphiphilic block copolymer was prepared in-house following the procedure reported previously by Themistou et al.20 The disulfide-based dimethacrylate (DSDMA) was synthesized following the method reported by Rosselgong et al.21
Synthesis of VS-functionalized PLA19-b-P(OEGMA14-stat-VSTEMA2) amphiphilic block copolymer
For the synthesis of the VS-functionalized PLA19-b-P(OEGMA14-stat-VSTEMA2) amphiphilic block polymer (Scheme S1, ESI†) used in this work, a similar synthetic procedure to the one described by Themistou et al.20 was used. More specifically, simultaneous ROP and RAFT polymerization was firstly employed for the synthesis of a branched disulfide-functionalized PLA19-b-P(OEGMA14-stat-DSDMA1) amphiphilic block copolymer at 55% w/w solids. The protocol for the synthesis of the disulfide-functionalized polymer was as follows: 0.50 g of LA (3.47 mmol), 0.73 g of OEGMA (1.62 mmol), 0.03 g of DSDMA (0.12 mmol), 0.04 g of ROP–RAFT agent (0.12 mmol), 4.0 mg of AIBN (0.02 mmol), 0.06 g of DMAP (0.46 mmol) and 0.89 mL of 1,2-dichloroethane were added to a 5 mL round-bottom flask equipped with a magnetic stir bar and a rubber septum. The mixture was bubbled with nitrogen gas for 20 min and the flask was placed in a preheated oil bath at 74 °C. The reaction was left to proceed for 24 h. The reaction product was cooled to 20 °C and a sample was withdrawn for proton nuclear magnetic resonance (1H NMR) spectroscopy analysis in CDCl3 to determine the monomer conversions. The polymer was purified by dialysis against acetone (5 times) using a dialysis membrane of 1 kD MWCO. Acetone was removed using a rotary evaporator and the polymer was left under vacuum for 48 h. The obtained dried PLA19-b-P(OEGMA14-stat-DSDMA1) diblock copolymer was characterized by 1H NMR spectroscopy in CDCl3 (Fig. S1a, ESI†) and size exclusion chromatography (SEC) in THF (Fig. S2, ESI†). The SEC analysis of the polymer gave a number average molecular weight (Mn) of 20900 g mol−1, a weight average molecular weight (Mw) of 31000 g mol−1 and dispersity (Đ) of 1.49.
In a second reaction step, the disulfide methacrylate units on the polymer were reduced to 2-thioethyl methacrylate (TEMA) using Bu3P as the reducing agent. This reaction was performed to transform the disulfide-functionalized polymer to the thiol-functionalized PLA19-b-P(OEGMA14-stat-TEMA2) polymer, with 1 eq. of DSDMA giving 2 eq. of TEMA monomer units. More specifically, 504 mg of the dried disulfide-functionalized PLA19-b-P(OEGMA14-stat-DSDMA1) polymer (containing 52.3 μmol of disulfide bonds) and 31.7 mL of THF (1.6% w/v) were added to a round-bottomed flask equipped with a magnetic flea and sealed with a rubber septum. The mixture was purged with nitrogen gas for 20 min and placed in an oil bath at 30 °C. A mixture of Bu3P (156.8 μmol, 31.7 mg, 3.0 eq. relative to disulfide bonds of the polymer) and Et3N (109.7 μmol, 11.1 mg, 2.1 eq. relative to disulfide bonds of the polymer) in 5 mL THF was added to the flask via a syringe under nitrogen atmosphere. The final mixture was left for 2 h in an oil bath at 30 °C. To ensure the cleavage of the disulfide bonds in the DSDMA, a sample was extracted for THF SEC analysis (Mn = 17400 g mol−1, Mw = 24300 g mol−1 and Đ = 1.40, Fig. S2, ESI†).
The third reaction step involved the attachment of DVS to the polymer, by reacting the thiol groups of the TEMA monomer units produced in the second reaction step, with one of the double bonds of DVS molecules, converting the TEMA to VSTEMA monomer units. For this to be achieved, a high excess (15 eq. relative to thiol groups of the polymer) of DVS (1.57 mmol, 185.2 mg) dissolved in 5 mL of THF was added to the reaction flask under nitrogen atmosphere. The reaction was allowed to proceed for 15 h at 30 °C. The final product was dialyzed against acetone (MWCO 1 kD, 5 times) and the solvent was removed using a rotary evaporator and by drying under vacuum. The dried VS-functionalized PLA19-b-P(OEGMA14-stat-VSTEMA2) amphiphilic block copolymer was characterized by 1H NMR in CDCl3 (Fig. S1b, ESI†) and THF SEC (Mn = 17600 g mol−1, Mw = 25200 g mol−1 and Đ = 1.43, Fig. S2, ESI†).
Polymer characterization
The molecular weight distributions of all the functionalized amphiphilic block copolymers synthesized in this work were determined using an Agilent 1260 Infinity SEC system equipped with a refractive index (RI) detector. The instrument setup was comprised of a guard column and two Agilent PL gel 5 μm MIXED-C columns in series. The RI detector and the columns were maintained at 30 and 25 °C, respectively. THF (HPLC-grade) containing 2.0% v/v Et3N and 0.05% w/v BHT inhibitor was used as eluent at a flow rate of 1.0 mL min−1. DMSO was used as a flow rate marker. The instrument calibration was performed using a series of near-monodisperse poly(methyl methacrylate) (PMMA) standards from Agilent with MWs of 1010, 1950, 6850, 13900, 31110, 68750, 137800, 320000, 569000 and 1048000 g mol−1. Cirrus software from Agilent was used for the SEC chromatogram analysis. The 1H NMR spectra of all polymer samples were recorded using a Bruker Avance 400 MHz spectrometer.
Polymeric nanoparticle formation
Formation of VS-functionalized nanoparticles was performed using a standard single emulsion – solvent evaporation methodology16,17 (Scheme 1a). A total mass of 20 mg of PLGA 502H and PLA-b-P(OEGMA-stat-VSTEMA) was dissolved in 1 mL of DCM. For N-hydroxy succinimide (NHS)-functionalized nanoparticles, 14 mg PLGA 502H and 6 mg PLGA-PEG-NHS were dissolved in 1 mL of DCM. For the preparation of fluorescent nanoparticles, rhodamine 6G (0.02% w/v) was added to the organic solvent, at this point. The polymer-containing solution was added dropwise via a 25G needle to 7 mL of 2.5% w/v PVA in MES buffer (pH 5.0). Emulsification was performed via pulsed probe sonication on ice (18 cycles, 3 s on, 2 s off, 90 s total, 50% amplitude, FB120 Sonic Dismembrator, Fisher Scientific) before stirring overnight to allow DCM evaporation. The resulting particles were washed three times by centrifugation (16100 × g, 20 min) and were resuspended in MES buffer (pH 5). Prior to nanoparticle conjugation, VNARs were incubated with 10 molar equivalents of TCEP.HCl for 15 min at room temperature to reduce inter-VNAR disulfide bonds. Nanoparticles were resuspended at 2.5 mg mL−1 and stirred with VNAR (0.0001% w/v, 2.08 μM) for at least 3 h. The resulting PLA-b-P(OEGMA-stat-VNARVSTEMA) amphiphilic block copolymer nanoparticles were then subjected to three centrifugal wash steps with PBS (16100 g, 20 min).
Success of nanoparticle functionalization was determined by microBCA protein assay as per manufacturer's instructions. The VNAR that was conjugated to nanoparticles was quantified via comparison to standards comprising known concentrations of VNAR added to nude nanoparticles.
Polymeric nanoparticle characterization
Post-formulation, nanoparticle hydrodynamic diameter and dispersity were determined by dynamic light scattering (DLS) (NanoBrook Omni, Brookhaven Instruments). Particles were suspended at 0.1 mg mL−1 in deionized water and were placed in 2.5 mL polystyrene cuvettes (1 cm path length). Samples were measured at 25 °C using a red (633 nm) laser diode and a detection angle of 90°. Measurements were performed in triplicate for each independent experiment and averaged. For stability assessments, nanoparticles were stored in aqueous PBS buffer at 4 °C and assessed as above at predetermined intervals. Additionally, nanoparticle surface charge was measured by phase analysis light scattering (PALS) using the same instrument, fitted with an electrode submerged in the particle suspension. Measurements were performed in triplicate and averaged. Nanoparticle tracking analysis (NTA) was also performed to determine particle size. Particles were suspended in deionized water (0.05 mg mL−1). 3 × 30 s video captures were taken and averaged (Malvern Panalytical NanoSight NS300).Modified enzyme-linked immunosorbent assay (modified ELISA)
A solution of recombinant DLL4-Fc fusion protein antigen in PBS was added to a black high-binding plate (0.5 μg mL−1, 100 μL per well) and incubated at 4 °C overnight. Excess protein was discarded and wells were washed 4 times with 0.1% v/v Tween-20 in PBS buffer. The remaining non-specific binding sites were blocked with a solution of 1% w/v BSA in PBS (150 μL per well) for 1 h and were washed again 4 times with PBS-Tween. Rhodamine-encapsulated nanoparticles suspended in PBS were added (100 μL per well, 0.5 mg mL−1 polymer) and the plate was incubated at room temperature for 2 h before washing (8 times) with PBS-Tween. A 1:1 mixture of acetonitrile and DMSO (50 μL per well) was then added and the fluorescence was read (525ex/555em nm, Synergy HT, BioTek). For stability binding assessments, nanoparticles were stored in aqueous PBS buffer at 4 °C and assessed as above at predetermined intervals (24 and 48 h post-formulation).
Gel electrophoresis
10 μL of a 0.2 mg mL−1 (16.67 mM) VNAR solution in PBS was mixed with 1 μL of 1000 mg mL−1 (357 mM) PLA-b-P(OEGMA-stat-VSTEMA) solution in DMSO. The resulting mixture was incubated at room temperature for 1 h. A volume of 2.75 μL 5× loading buffer (0.25% bromophenol blue, 50% glycerol, 10% SDS, 500 mM BME, 250 mM pH 6.8 Tris-HCl) was added and heated at 95 °C for 10 min. Denatured samples were electrophorized in 15% SDS-containing polyacrylamide gels for 2 h at 125 V. The resolving gel was boiled in Brilliant Blue stain before incubating at room temperature for 1 h, destaining overnight and imaged (SynGene G:BOX).Cell line viability
Pancreatic carcinoma PANC-1 cells were seeded in white 96-well plates (2.5 × 103 per well) and allowed to adhere overnight. Growth media was then replaced, and the cells were treated with 20 μL of nanoparticle suspension in PBS to a specified final polymer concentration between 12.5–200 μg mL−1. After 24, 48 or 72 h post-treatment, cell viability was assessed using Cell-Titer Glo reagent (as per manufacturer's instruction).Results
Preparation of the PLA19-b-P(OEGMA14-stat-VSTEMA2) amphiphilic block copolymer
In this work, a VS-functionalized PLA-based PLA17-b-P(OEGMA14-stat-VSTEMA2) amphiphilic block copolymer was prepared (Scheme S1, ESI†). For the preparation of this polymer, a synthetic methodology developed by Themistou et al. was used to initiate both ROP and RAFT polymerizations.20 Combination of these two polymerization techniques allows the synthesis of functionalized block copolymers that can be used in various biomedical applications.22,23 Here, a one-step ROP–RAFT polymerization reaction was employed using a ROP–RAFT dual agent20 (Scheme S1a, ESI†). This reagent has the ability to initiate both the ROP of LA through its hydroxyl group and the RAFT polymerization of a methacrylic OEGMA/DSDMA mixture through its dithiobenzoate functionality, resulting in the formation of the branched PLA-b-P(OEGMA-stat-DSDMA) amphiphilic block copolymer. For this reaction, LA:OEGMA:DSDMA:ROP–RAFT agent:AIBN:DMAP molar ratios of 30:14:1:1:0.2:4 were used in dichloroethane as a solvent (55% w/w, 74 °C, 24 h). 1H NMR (CDCl3) spectroscopy was used to determine the monomer conversions. These were calculated by taking into account the resonance intensities of the unreacted LA (5.03–5.10 ppm) and OEGMA (5.52 ppm and 6.08 ppm) monomers in the 1H NMR (CDCl3) spectrum of the polymerization reaction product. The calculated monomer conversions obtained were 95% for LA monomer and 99% for OEGMA monomer. Based on the 1H NMR spectrum of the purified (after dialysis against acetone), dried PLA-b-P(OEGMA-st-DSDMA) block copolymer, and by comparing the peak at 3.50 ppm corresponding to POEGMA with the PLA peak at 5.10 ppm, the degree of polymerization (DP) of LA was found to be 19. Therefore, the polymer observed after dialysis was the PLA19-b-P(OEGMA14-stat-DSDMA1). The molecular weight distribution (MWD) of this disulfide-containing block copolymer is shown in the SEC data provided in Fig. S2 (ESI†). A number average molecular weight (Mn) value of 20 900 g mol−1 and a dispersity (Đ) value of 1.49 were obtained. This relatively narrow (for a branched polymer) Đ value indicates formation of mostly intramolecular rather than intermolecular bonds,24,25 resulting in less branching. In a second step, the synthesized branched PLA19-b-P(OEGMA14-stat-DSDMA1) amphiphilic block copolymer was treated with an excess of Bu3P (Scheme S1b, ESI†), causing cleavage of the disulfide bonds. The cleavage of one DSDMA unit resulted in the formation of two TEMA units. The SEC chromatogram of the resulting thiol-functionalized linear PLA19-b-P(OEGMA14-stat-TEMA2) amphiphilic block copolymer appeared to have a narrower MWD (Fig. S2, ESI†), with a Đ value of 1.40. The Mn value of this linear polymer was found to be 17400 g mol−1. Following its synthesis, the PLA19-b-P(OEGMA14-stat-TEMA2) block copolymer was allowed to react with DVS (Scheme S1c, ESI†). In order to avoid any (inter- or intra-molecular) cross-linking, a large excess of DVS of 15 eq. in respect to TEMA thiol group, was used. Using excess of DVS ensured that the DVS molecule reacts only from one side (only one of its double bonds reacts with a thiol group on the polymer chain). This reaction converted the TEMA units of this polymer to VSTEMA units, and therefore, a vinyl sulfone-functionalized PLA19-b-P(OEGMA14-stat-VSTEMA2) amphiphilic diblock copolymer was formed with a Mn of 17600 g mol−1 and a Đ value of 1.43.
Preparation of VS-functionalized nanoparticles
Themistou et al. have previously used VS chemistry to produce functionalized electrospun fibers22 and optical fiber overlayers.26 In this work, this chemistry is used for the preparation of nanoparticles. More specifically, the synthesized VS-functionalized PLA19-b-P(OEGMA14-stat-VSTEMA2) amphiphilic block copolymer was blended with carboxylate-terminated PLGA 502H prior to dissolution and particle formation. PLGA 502H was chosen as it has been shown previously to form monodisperse, stable particles of a size ∼200 nm using a similar formulation methodology to the one adopted here.16,17 An initial study was conducted wherein the VS-functionalized polymer was blended with PLGA 502H (to a final mass of 20 mg) at varying mass percentages (10, 50 and 97.5% w/w) before dissolution in DCM and particle formation. The resulting particle size was determined by DLS. The formulations with 10% w/w and 50% w/w VS-functionalized polymer content were noted to have significantly lower hydrodynamic diameters (Fig. 1A and C) and polydispersity values (Fig. 1B and C) than the 97.5% w/w formulations (194.9 and 174.4 nm, respectively, versus 304.8 nm). It was, therefore, decided to proceed with the 10% w/w VS-functionalized polymer formulation for further experimentation.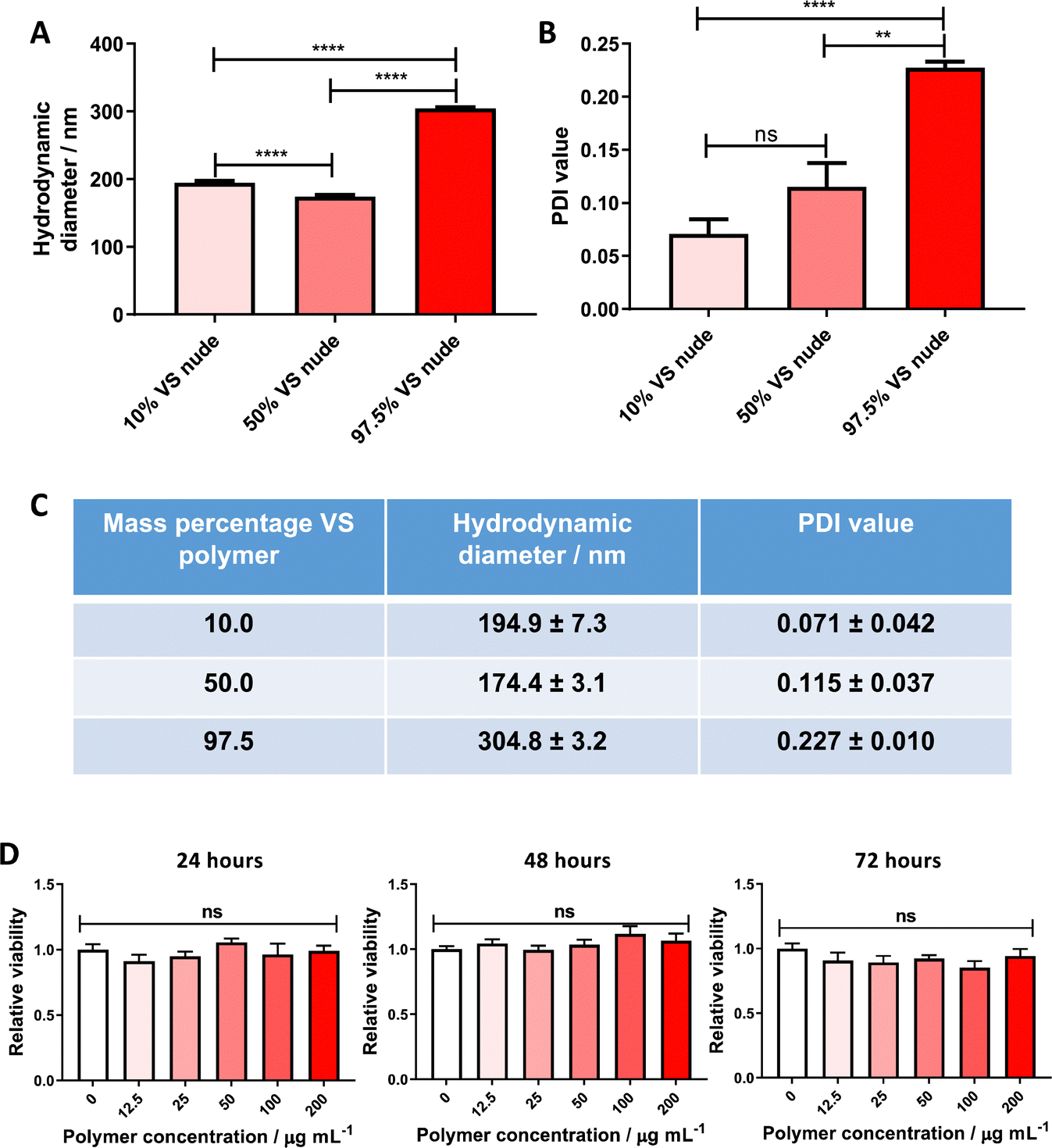 | ||
| Fig. 1 VS-functionalized PLA19-b-P(OEGMA14-stat-VSTEMA2) amphiphilic block copolymer blended with PLGA 502H can be used to form non-cytotoxic nanoparticles. (A) DLS hydrodynamic diameters, (B) DLS PDI values and (C) tabulated DLS data (from (A) and (B)) of unconjugated particle formulations, containing different amounts of VS-functionalized polymer (10%, 50% and 97.5% w/w) to PLGA 502H (total polymer mass remained constant at 20 mg). Measurements performed in triplicate, data pooled from two independent experiments. Statistical significance established by one-way ANOVA (ns p > 0.05, **p ≤ 0.01, ****p ≤ 0.0001) D. Cell viability of PANC-1 cells treated with varying concentrations of 10% VS-functionalized nanoparticles for 24, 48 and 72 h. Viability was measured by CellTiter-Glo assay and normalized to vehicle control, data pooled from two independent experiments. Statistical significance assessed by one-way ANOVA at each timepoint (ns p > 0.05). | ||
In vitro cytotoxicity of VS-functionalized nanoparticles
This nanoparticle formulation was subsequently initially screened for major toxicity towards cell cultures. PANC-1 cells were chosen for this assay, as the most probable downstream application for this targeted system is within an oncology setting. No significant cytotoxicity to PANC-1 confluent monocultures was found, up to a polymer concentration of 0.2 mg mL−1 over 72 hours (Fig. 1D), allowing future experimentation in cell models.Attachment of VNAR to the nanoparticle surface
Upon successful demonstration of the ability to fabricate a monodisperse, non-cytotoxic formulation of the VS-functionalized nanoparticles with hydrodynamic diameter ∼200 nm, they were next tested for their ability to conjugate to engineered VNARs. To show that the VNAR is capable of covalently binding (instead of simply adsorbing) to the VS-functionalized polymer, gel electrophoresis was performed on VNAR samples preincubated either in the presence or absence of VS-functionalized polymer. Non-reducing SDS-PAGE analysis of polymer-incubated VNAR showed a shift to a higher mass, from around 12 to 13 kDa (Fig. 2A), indicating covalent conjugation to the polymer. A band of approximately 24 kDa was observed in the non-reducing SDS-PAGE of the VNAR incubated without polymer (Fig. 2A). It was thought that this species consisted of a VNAR homodimer, linked through the exposed C-terminal Cys residue. It was therefore decided to incubate VNARs with TCEP prior to attempting conjugation to VS-functionalized nanoparticles to increase the availability of the C-terminal cysteine for Michael addition to the VS electrophile (Scheme 1b). Knowing that VS-functionalized polymer mass percentage in the formulation could be varied between 10 and 50%, while maintaining acceptable size and polydispersity, the conjugation of VNAR to particle formulations of 10%, 25% and 50% was tested (Fig. 2B). Efficiency of VNAR conjugation with the 10% VS-functionalized polymer formulations was found to be significantly higher than for the 25% and 50% formulations (4.6 compared to 2.5 and 0.5 μg of conjugated VNAR per mg of nanoparticles, respectively). This unexpected result might be attributed to the increasing density of the surface POEGMA layer as the concentration of the VS-functionalized polymer increases, hindering access of the cysteine nucleophile to the VS functional group. Additionally, conjugation was compared to NHS ester conjugation, a well-validated non-site selective bioconjugation chemistry.27,28 Nanoparticles with theoretically equimolar numbers of surface functional groups, either VS or NHS ester, were prepared. Conjugation to the VS particles was found to be more efficient than to NHS particles (Fig. 2C). The amounts of the functional groups on the polymers were calculated based on the polymer Mn value (17400 and 27677 g mol−1, respectively) and assuming 2 VS or 1 NHS functional groups per polymer chain, therefore, 10% w/v VS and 31% w/v NHS nanoparticles theoretically bear the same number of functional groups.
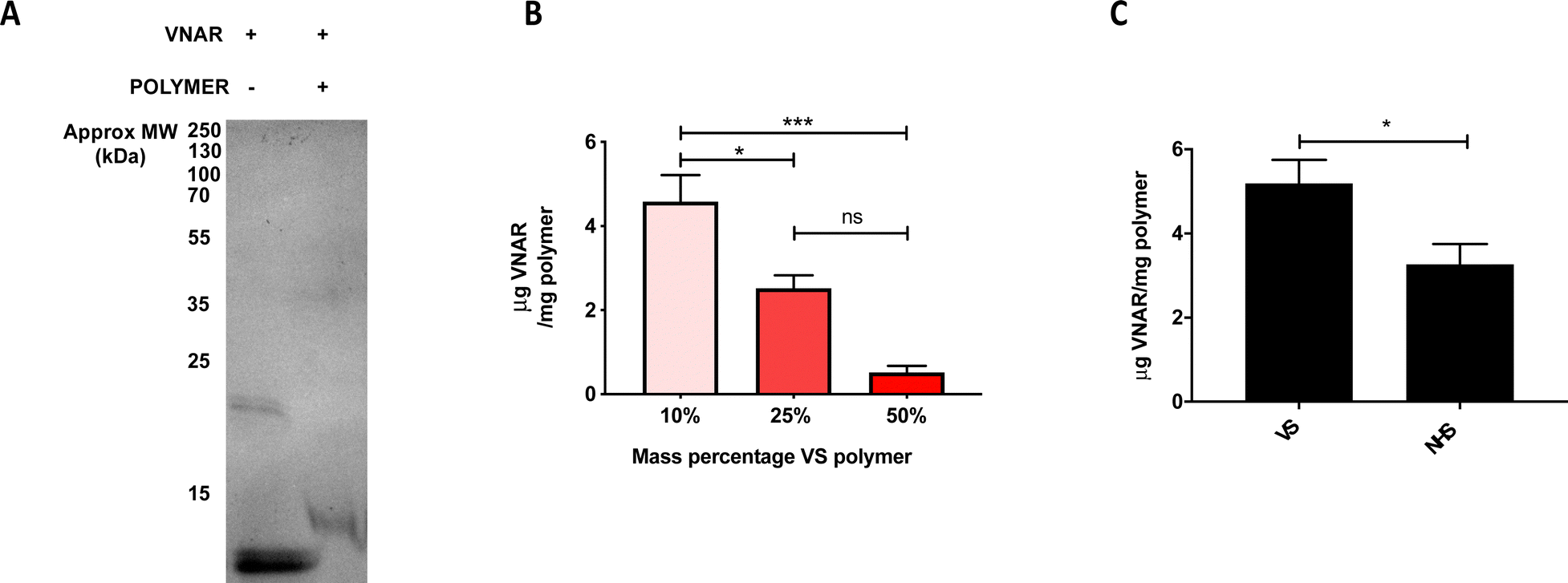 | ||
| Fig. 2 VS-functionalized nanoparticles can conjugate VNARs bearing a single accessible cysteine residue efficiently. (A) SDS-PAGE analysis of VNAR incubated in the presence or absence of VS-functionalized polymer. (B) Quantification of VNAR mass bound to particles by microBCA assay. Three particle formulations with varying VS-functionalized polymer mass percentages (10%, 25% and 50%) were assessed. Measurements were performed in triplicate, data pooled from three independent experiments. Statistical significance established by one-way ANOVA (ns p > 0.05, *p ≤ 0.05, ***p ≤ 0.001). (C) Mass of particle-associated VNAR per mg of particles theoretically bearing equimolar amounts of VS or NHS functional groups. Measured by microBCA assay, measurements performed in triplicate, data from three independent experiments. Statistical significance assessed by unpaired two-tailed t-test, *p < 0.05. | ||
Having established that VNAR conjugation to the VS-functionalized nanoparticles was successful, the physicochemical characteristics of the conjugated and unconjugated nanoparticles were examined to ascertain that they were not altered by the conjugation. No statistically significant difference in the DLS hydrodynamic diameters and PDI values between the unconjugated (195 nm and 0.071, respectively) and conjugated (194 nm and 0.066, respectively) formulations was detected (Fig. 3A, B and D), with a unimodal size distribution observed in both samples (Fig. 3E).
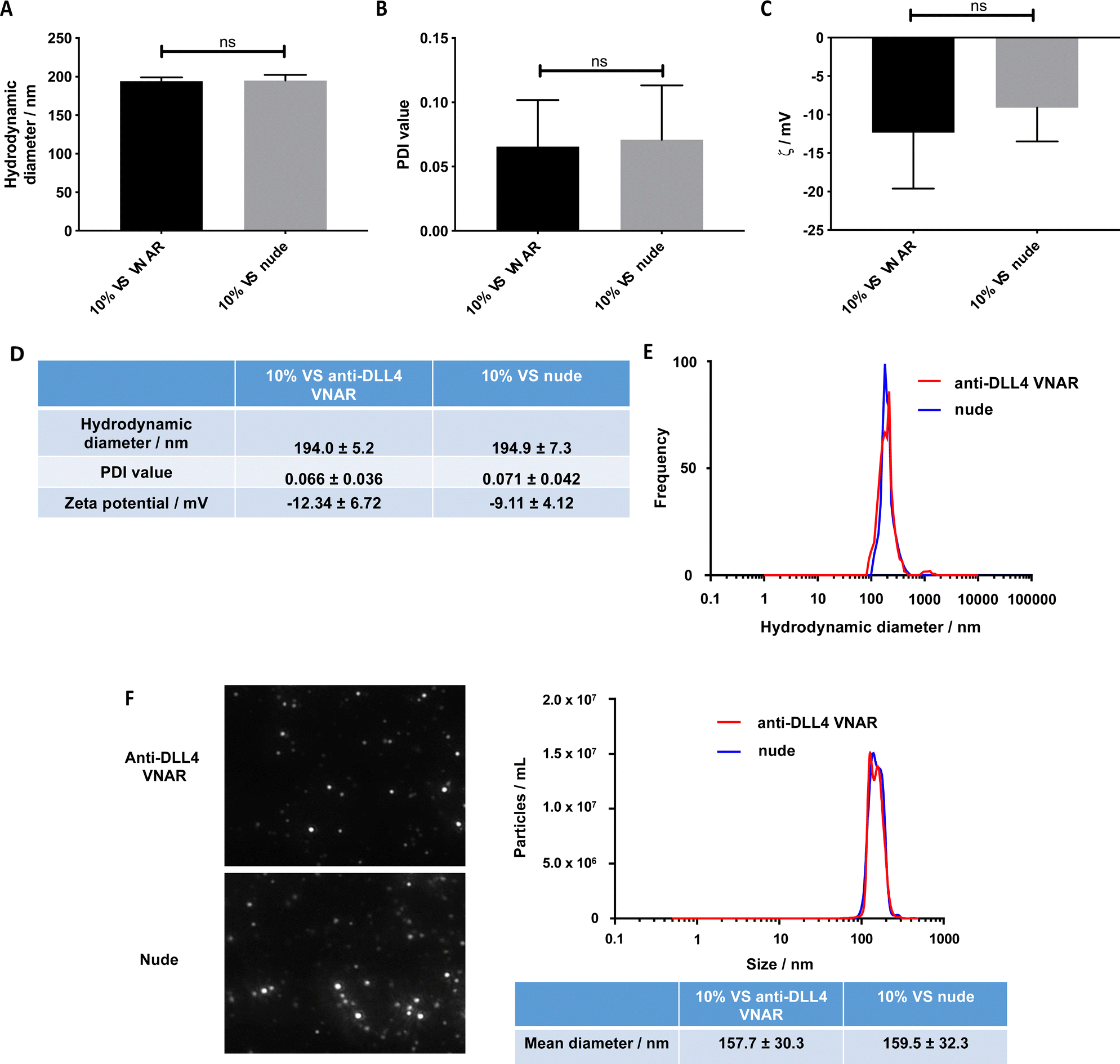 | ||
| Fig. 3 VNAR conjugation to vinyl sulfone particles does not alter their physical characteristics. Conjugated (VNAR) and unconjugated (nude) particle physical properties: (A) DLS zeta average hydrodynamic diameters. (B) DLS PDI values. (C) PALS-measured zeta-potential values. Measurements performed in triplicate, data pooled from three independent experiments. Statistical significance established by unpaired two-tailed t-test (ns p > 0.05). (D) Tabulated data from (A), (B) and (C). (E) Intensity-weighted DLS histograms of 10% VS VNAR conjugated and nude particles. Representative of three independent experiments. (F) NTA screen captures, histograms and tabulated data of 10% VS VNAR conjugated and nude particles. Representative of two independent experiments. | ||
Similarly, no significant difference in their zeta potential was detected (Fig. 3C and D). Complementary characterization by NTA revealed no alteration in either size or spherical morphology and again indicated the presence of a unimodal size distribution (Fig. 3F). Furthermore, the stability of the formulation was assessed over a 72 h time course, over which, nanoparticle size and PDI value remained consistent (Fig. S3A and B, ESI†).
VNAR-functionalized nanoparticle affinity towards their cognate antigen (DLL4)
Finally, an ELISA-like binding assay (Fig. 4A) was used to show that anti-DLL4 VNAR retained target specificity following site-specific conjugation to VS-functionalized nanoparticles (Fig. 4B). Binding was shown to be retained 72 h post formulation, with negligible drop-off in affinity, attesting to the stability of the formulation (Fig. S3 C, ESI†). VS-functionalized polymers, therefore, are a suitable candidate for production of functional VNAR-conjugated therapeutic polymeric nanoparticles.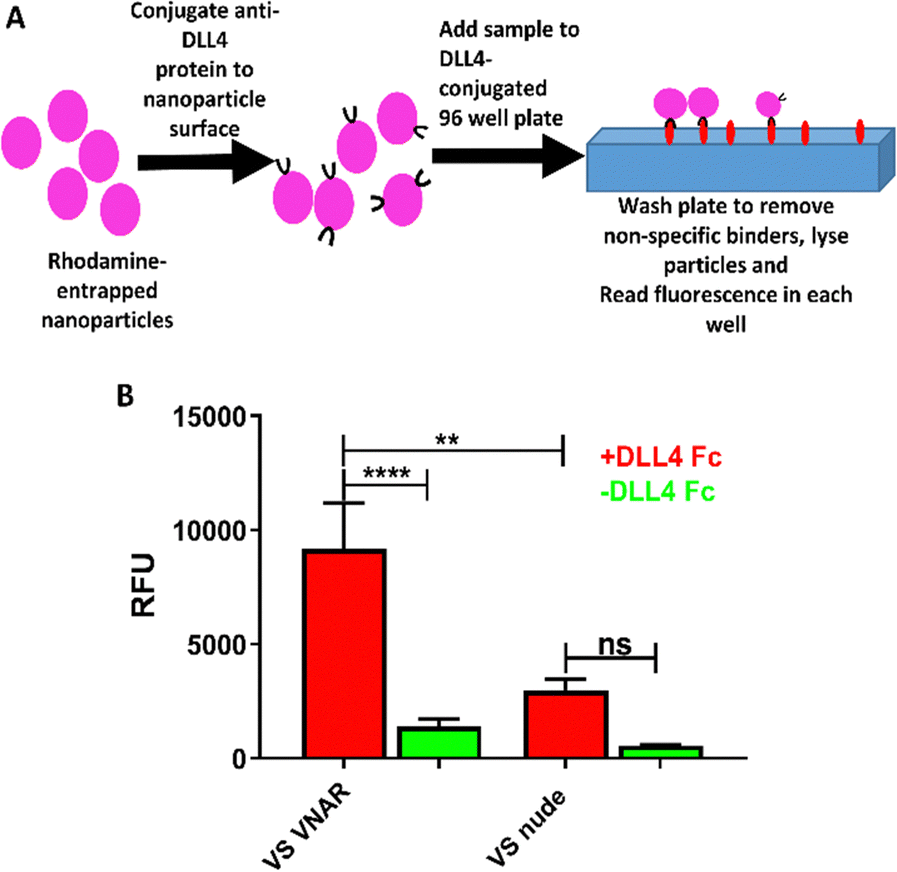 | ||
| Fig. 4 Anti-DLL4 VNAR-conjugated VS-functionalized nanoparticles have specific binding affinity for DLL4. (A) Schematic of the ELISA-like binding assay performed. (B) ELISA-like binding assay of VNAR conjugated and nude 10% VS-functionalized nanoparticles to a DLL4-Fc antigen coated plate. Measurements performed in sextuplicate, data pooled from three independent experiments. Statistical significance established by one-way ANOVA (ns p > 0.05, **p ≤ 0.01, ****p ≤ 0.0001). | ||
Discussion
In this work, a novel method for conjugating VNARs to nanoparticles was developed. More specifically, a VS-functionalized polymer was synthesized and blended with poly(lactic-co-glycolic) acid to form polymeric nanoparticles with favourable characteristics for therapeutic use. Subsequently, it was shown that these particles can conjugate to a model thiol-functionalized targeting ligand, namely an anti-DLL4 VNAR, without alteration of their favourable physical characteristics. Importantly, VNAR conjugation endowed the nanoparticles with specific binding affinity for DLL4.Initially, a VS-functionalized PLA19-b-P(OEGMA14-stat-VSTEMA2) amphiphilic block copolymer was successfully synthesized (Scheme S1, ESI†). A one-step procedure involving the simultaneous ROP of LA and RAFT polymerization of a mixture of OEGMA and DSDMA using a dual ROP–RAFT agent was employed (Scheme S1a, ESI†).20 Subsequently, the disulfide bonds of the resulting disulfide-functionalized PLA19-b-P(OEGMA14-stat-DSDMA1) amphiphilic block copolymer were reduced to thiol groups using Bu3P (Scheme S1b, ESI†). This resulted in the formation of the thiol-functionalized PLA19-b-P(OEGMA14-stat-TEMA2), possessing an average of two thiol groups in each polymer chain. Finally, this polymer reacted with DVS (Scheme S1c, ESI†) to give the VS-functionalized PLA19-b-P(OEGMA14-stat-VSTEMA2) amphiphilic block copolymer. Characterization of the VS-functionalized polymer and its precursors by 1H NMR (Fig. S1, ESI†) and SEC (Fig. S2, ESI†) proved the successful synthesis of the block copolymer and incorporation of VS functional groups on its polymer chain.
The synthesized VS-functionalized PLA19-b-P(OEGMA14-stat-VSTEMA2) amphiphilic block copolymer was then blended with PLGA 502H in different ratios before dissolution and nanoparticle formation via a single emulsion – solvent evaporation approach. The resulting biodegradable monodisperse nanoparticles (of the 10% VS-functionalized polymer formulation) possessed hydrodynamic diameters of around 195 nm (Fig. 1A and C), slightly negative zeta potential (Fig. 3C) and were not cytotoxic (Fig. 1D). Therefore, these nanoparticles do fulfil the generalized rules concerning nanoparticle properties for optimization of in vivo performance.29 These include hydrodynamic diameter around 100–200 nm, sufficiently large to avoid renal/hepatic filtration30 without being so large as to experience extreme protein adsorption and phagocytic uptake.31 A slightly negative (around −10 mV) surface charge also increases circulation time and subsequent tumour deposition.32 Hydrophilic surface layers, such as the POEGMA layer here, are found to sterically oppose adsorption of proteins, such as complement components and apolipoproteins, and reduce macrophage uptake.33–37
Furthermore, by attaching a functional group on a hydrophilic PEG polymer block of the amphiphilic block copolymer, the functional group is preferentially presented to the aqueous environment where it can take part in surface conjugation reactions.3,29 The same phenomenon occurs with POEGMA-containing amphiphilic block copolymers,13 such as the PLA19-b-P(OEGMA14-stat-VSTEMA2) polymer used here. The VS functional group, attached in the hydrophilic part of the polymer, is positioned at the surface of the resulting nanoparticle, permitting thiol-Michael addition reactions to take place there.38
VSs react preferentially with thiols over amine groups,3 and as such would be expected to selectively react with the engineered single cysteine residue at the C-terminus of the anti-DLL4 VNAR rather than the seven lysines found in the anti-DLL4 VNAR sequence. As this is the only non-framework, and therefore, “available” cysteine residue within the VNAR sequence, it represents a unique nucleophilic site. Given the potential for VNARs to spontaneously dimerize, likely through the C-terminal cysteine, it was decided to treat VNARs with a reducing agent to unmask this unique nucleophilic site prior to VS conjugation. Additionally, the VNAR-particle conjugation reaction was performed at pH 5. This is below the pKa of the cysteine sidechains (8.3) and further below that of the lysine sidechains (10).18 Accordingly, it would be expected that the VNAR lysine sidechain amines would be more protonated than the thiol of the C-terminal cysteine. Therefore, it is reasonable to expect that the VNAR site-selectively reacts preferentially with the surface VS electrophiles at the engineered C-terminal cysteine residue at pH 5.39
With this reasoning, the VS-functionalized polymer was investigated as a method of cysteine-selective conjugation to a model targeting ligand, anti-DLL4 VNAR. Covalent conjugation between the VNAR and VS polymer was confirmed through SDS-PAGE (Fig. 2A), however unexpectedly, the conjugation efficiency of VNAR to the nanoparticles increased with decreasing VS polymer mass percentage (Fig. 2B). This may be due to increased steric repulsion within the more dense, bulky POEGMA layer, hindering VNAR conjugation40,41 as VS polymer concentration increased. Conjugation to VS-functionalized particles was then compared to particles functionalized with NHS esters. Particles with theoretically equal numbers of surface functional groups (either VS or NHS) were conjugated to C-terminal cysteine engineered VNAR, with those particles bearing VS functionalities displaying higher conjugation efficiency (Fig. 2C). This confirmed the suitability of this chemistry for efficient conjugation of biomolecules. It should be noted here that there were differences between the polymers other than the functional group, with the VS polymer composed of lactic acid units with pendant PEG moieties and functional groups interspersed among these PEG chains. In contrast, the NHS polymer is composed of lactic and glycolic acid units, a linear PEG block and a terminal functional group. However, despite these differences, the physical properties (hydrodynamic diameter, polydispersity index, zeta potential) of the particles were similar, making the particles reasonably comparable.
Importantly, for therapeutic use, the nanoparticles were found to retain their physical properties upon conjugation with the anti-DLL4 VNAR (Fig. 3). Ensuring this was important, since significant increase of size or neutralization of zeta potential can negatively affect the in vivo properties of the particles, for example, by increasing phagocytic uptake by the reticuloendothelial system.31,42 NTA, which is more sensitive to polydispersity than DLS,43 confirmed that the particles did not aggregate upon VNAR conjugation (Fig. 3F), which was further confirmed via a 72 h DLS stability study (Fig. S3A and B, ESI†).
Finally, conjugation of the anti-DLL4 VNAR (Fig. 4A) was found to give to the VS-functionalized particles specific affinity against DLL4 (Fig. 4B and Fig. S3C, ESI†), a therapeutically relevant target overexpressed in a variety of cancers, supporting the potential of these nanoparticles as candidates for cancer therapy.
Conclusions
The development of novel nanoparticle formulations for targeted cancer therapies has great potential. In this work, the ability of a successfully synthesized VS-functionalized PLA-b-POEGMA amphiphilic block polymer to form VS-functionalized nanoparticles when mixed with PLGA was proven. These nanoparticles appeared to have favourable physicochemical properties for therapeutic use and were able to be efficiently surface functionalized with a VNAR ligand to further enhance their therapeutic utility. This approach has great potential for the development of next-generation targeted cancer therapies, with the simplicity of the functionalized polymer synthesis and conjugation procedure being incredibly attractive. In future work, these VS-functionalized nanoparticles conjugated to targeting ligands, such as VNARs, will be examined in vitro to assess biocompatibility across a broader range of cell types before assessing targeting efficiency and therapeutic payload delivery. Additionally, the VS approach described here can be more generally be applied to facilitate conjugation of various biologically important molecules to a range of organic and inorganic nanosystems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The studentship of A. L. was supported by the John Martin Scholarship at Queen's University Belfast. The authors acknowledge the Engineering and Physical Sciences Research Council (EPSRC) (S3804ASA) for funding the PhD studentship of M. F.References
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373 CrossRef CAS PubMed.
- I. F. Uchegbu and A. Siew, J. Pharm. Sci., 2013, 102, 305–310 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- V. J. Venditto and F. C. Szoka, Adv. Drug Delivery Rev., 2013, 65, 80–88 CrossRef CAS PubMed.
- K. Park, ACS Nano, 2013, 7, 7442–7447 CrossRef CAS PubMed.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak, W. C. W. Chan, S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, H. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- D. A. Richards, A. Maruani and V. Chudasama, Chem. Sci., 2016, 1–15 CAS.
- M. Colombo, S. Mazzucchelli, J. M. Montenegro, E. Galbiati, F. Corsi, W. J. Parak and D. Prosperi, Small, 2012, 8, 1492–1497 CrossRef CAS PubMed.
- M. K. Greene, D. A. Richards, J. C. F. Nogueira, K. Campbell, P. Smyth, M. Fernández, C. J. Scott and V. Chudasama, Chem. Sci., 2018, 9, 79–87 RSC.
- J. C. F. Nogueira, M. K. Greene, D. A. Richards, A. O. Furby, J. Steven, A. Porter, C. Barelle, C. J. Scott and V. Chudasama, Chem. Commun., 2019, 55, 7671–7674 RSC.
- M. K. Greene, J. C. F. Nogueira, S. R. Tracey, D. A. Richards, W. J. McDaid, J. F. Burrows, K. Campbell, D. B. Longley, V. Chudasama and C. J. Scott, Nanoscale, 2020, 12, 11647–11658 RSC.
- A. Leach, P. Smyth, L. Ferguson, J. Steven, M. K. Greene, C. M. Branco, A. P. McCann, A. Porter, C. J. Barelle and C. J. Scott, Nanoscale, 2020, 12, 14751–14763 RSC.
- T. J. Gibson, P. Smyth, W. J. McDaid, D. Lavery, J. Thom, G. Cotton, C. J. Scott and E. Themistou, ACS Macro Lett., 2018, 7, 1010–1015 CrossRef CAS PubMed.
- L. García-Fernández, J. Garcia-Pardo, O. Tort, I. Prior, M. Brust, E. Casals, J. Lorenzo and V. F. Puntes, Nanoscale, 2017, 9, 6111–6121 RSC.
- S. Mazzucchelli, S. Sommaruga, M. O’Donnell, P. Galeffi, P. Tortora, D. Prosperi and M. Colombo, Biomater. Sci., 2013, 1, 728–735 RSC.
- D. Schmid, F. Fay, D. M. Small, J. Jaworski, J. S. Riley, D. Tegazzini, C. Fenning, D. S. Jones, P. G. Johnston, D. B. Longley and C. J. Scott, Mol. Ther., 2014, 22, 2083–2092 CrossRef CAS PubMed.
- S. Spence, M. K. Greene, F. Fay, E. Hams, S. P. Saunders, U. Hamid, M. Fitzgerald, J. Beck, B. K. Bains, P. Smyth, E. Themistou, D. M. Small, D. Schmid, C. M. O. Kane, D. C. Fitzgerald, S. M. Abdelghany, J. A. Johnston, P. G. Fallon, J. F. Burrows, D. F. Mcauley, A. Kissenpfennig and C. J. Scott, Sci. Transl. Med., 2015, 7, 1–13 Search PubMed.
- O. Koniev and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495–5551 RSC.
- J. Steven, M. R. Müller, M. F. Carvalho, O. C. Ubah, M. Kovaleva, G. Donohoe, T. Baddeley, D. Cornock, K. Saunders, A. J. Porter and C. J. Barelle, Front. Immunol., 2017, 8, 1361 CrossRef PubMed.
- E. Themistou, G. Battaglia and S. P. Armes, Polym. Chem., 2014, 5, 1405–1417 RSC.
- J. Rosselgong, S. P. Armes, W. Barton and D. Price, Macromolecules, 2009, 42, 5919–5924 CrossRef CAS.
- P. Viswanathan, E. Themistou, K. Ngamkham, G. C. Reilly, S. P. Armes and G. Battaglia, Biomacromolecules, 2015, 16, 66–75 CrossRef CAS PubMed.
- T. J. Gibson, P. Smyth, M. Semsarilar, A. P. Mccann, W. J. Mcdaid, M. C. Johnston, C. J. Scott and E. Themistou, Polym. Chem., 2020, 11, 344–357 RSC.
- J. Rosselgong and S. P. Armes, Macromolecules, 2012, 45, 2731–2737 CrossRef CAS.
- J. Rosselgong, S. P. Armes, W. R. S. Barton and D. Price, Macromolecules, 2010, 43, 2145–2156 CrossRef CAS.
- A. Petropoulou, T. J. Gibson, E. Themistou, S. Pispas and C. Riziotis, Mater. Chem. Phys., 2018, 216, 421–428 CrossRef CAS.
- W. J. McDaid, M. K. Greene, M. C. Johnston, E. Pollheimer, P. Smyth, K. McLaughlin, S. Van Schaeybroeck, R. M. Straubinger, D. B. Longley and C. J. Scott, Nanoscale, 2019, 11, 20261–20273 RSC.
- M. C. Johnston, J. A. Nicoll, K. M. Redmond, P. Smyth, M. K. Greene, W. J. McDaid, D. K. W. Chan, N. Crawford, K. J. Stott, J. P. Fox, N. L. Straubinger, S. Roche, M. Clynes, R. M. Straubinger, D. B. Longley and C. J. Scott, J. Controlled Release, 2020, 324, 610–619 CrossRef CAS PubMed.
- M. J. Ernsting, M. Murakami, A. Roy and S.-D. Li, J. Controlled Release, 2013, 172, 782–794 CrossRef CAS PubMed.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharmacol., 2008, 5, 505–515 CrossRef CAS PubMed.
- C. Fang, B. Shi, Y.-Y. Pei, M.-H. Hong, J. Wu and H.-Z. Chen, Eur. J. Pharm. Sci., 2006, 27, 27–36 CrossRef CAS PubMed.
- J. L. Seok, M. Ankone, E. Pieters, R. M. Schiffelers, W. E. Hennink and J. Feijen, J. Controlled Release, 2011, 155, 282–288 CrossRef PubMed.
- M. T. Peracchia, S. Harnisch, H. Pinto-Alphandary, A. Gulik, J. C. Dedieu, D. Desmaële, J. d’Angelo, R. H. Müller and P. Couvreur, Biomaterials, 1999, 20, 1269–1275 CrossRef CAS PubMed.
- D. Bazile, C. Prud’homme, M. Bassoullet, M. Marlard, G. Spenlehauer and M. Veillard, J. Pharm. Sci., 1995, 84, 493–498 CrossRef CAS PubMed.
- R. Gref, M. Lück, P. Quellec, M. Marchand, E. Dellacherie, S. Harnisch, T. Blunk and R. Müller, Colloids Surf., B, 2000, 18, 301–313 CrossRef CAS PubMed.
- D. E. Owens 3rd and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef PubMed.
- E. Swider, O. Koshkina, J. Tel, L. J. Cruz, I. J. M. de Vries and M. Srinivas, Acta Biomater., 2018, 73, 38–51 CrossRef CAS PubMed.
- S. Chatani, D. P. Nair and C. N. Bowman, Polym. Chem., 2013, 4, 1048–1055 RSC.
- M. Morpurgo, F. M. Veronese, D. Kachensky and J. M. Harris, Bioconjugate Chem., 1996, 7, 363–368 CrossRef CAS PubMed.
- Y. Inoue and K. Ishihara, Colloids Surf., B, 2010, 81, 350–357 CrossRef CAS PubMed.
- Y. Inoue, T. Nakanishi and K. Ishihara, Langmuir, 2013, 29, 10752–10758 CrossRef CAS PubMed.
- K. Xiao, Y. Li, J. Luo, J. S. Lee, W. Xiao, A. M. Gonik, R. G. Agarwal and K. S. Lam, Biomaterials, 2011, 32, 3435–3446 CrossRef CAS PubMed.
- V. Filipe, A. Hawe and W. Jiskoot, Pharm. Res., 2010, 27, 796–810 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb01985j |
| ‡ Current address: Nuntius Therapeutics, London, UK. |
| This journal is © The Royal Society of Chemistry 2023 |