
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the electrochemical performance of covalent organic framework cathodes for Li- and Mg-based batteries: the influence of electrolyte and binder†
Olivera
Lužanin‡
 ab,
Raquel
Dantas‡
c,
Robert
Dominko
abd,
Jan
Bitenc
*ab and
Manuel
Souto
*c
ab,
Raquel
Dantas‡
c,
Robert
Dominko
abd,
Jan
Bitenc
*ab and
Manuel
Souto
*c
aNational Institute of Chemistry, Hajdrihova 19, 1000, Ljubljana, Slovenia. E-mail: jan.bitenc@ki.si
bFaculty of Chemistry and Chemical Technology, University of Ljubljana, Večna pot 113, 1000, Ljubljana, Slovenia
cDepartment of Chemistry, CICECO-Aveiro Institute of Materials, University of Aveiro, Aveiro, 3810-393, Portugal. E-mail: manuel.souto@ua.pt
dAlistore-European Research Institute, CNRS FR 3104, Hub de l’Energie, Rue Baudelocque, 80039, Amiens, France
First published on 3rd October 2023
Abstract
Covalent organic frameworks (COFs) are crystalline porous organic polymers that have recently emerged as promising electrode materials for rechargeable batteries. Herein, we present an approach to improve the electrochemical performance of an anthraquinone-based COF (DAAQ-TFP-COF) cathode material in metal anode (Li, Mg) based batteries through proper selection of the electrolyte and binder. Our results show that the combination of lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) in tetraethylene glycol dimethyl ether (TEGDME) as electrolyte and poly(tetrafluoroethylene) (PTFE) as binder led to the best electrochemical performance with high utilisation efficiency of the redox sites and specific capacities close to the theoretical value. Using such electrolyte and binder, cyclable symmetric cells consisting of two DAAQ-TFP-COF organic electrodes exemplify 79% capacity retention after 2000 cycles at a high current density of 500 mA h g−1. The high reversibility and stability of the COF electrode material upon cycling were confirmed by ex situ IR spectroscopy. In addition, DAAQ-TFP-COF was explored as a cathode in magnesium cells using two different Mg electrolytes; one based on MgCl2 and one containing weakly coordinating anions. Electrochemical characterisation reveals significant differences in the performance of COF in terms of achievable capacities and voltage profiles, pointing towards hindered transport. Our findings demonstrate that the appropriate choice of electrolyte and binder is crucial to maximise the performance of COF-based materials in different post-lithium-ion metal anode batteries.
Introduction
Lithium-ion batteries (LIBs) are among the most promising devices for electrochemical energy storage from renewable sources and for the development of long-range electric vehicles.1,2 However, the increasing demand for some critical raw materials used in LIBs (e.g., lithium, cobalt, nickel) may pose supply risks in the near future3 and raise relevant environmental concerns.4 Electroactive organic materials have attracted much interest as an alternative to inorganic electrodes because they are based on abundant elements (e.g., carbon, oxygen, sulphur, nitrogen), are environmentally friendly, may have high theoretical capacities and their electrochemical performance can be finely modulated by chemical design.4–9 However, organic electrodes based on small molecules generally suffer from high solubility in electrolyte, resulting in fast capacity fading. A successful strategy to overcome the solubility problem is the polymerisation of redox-active building blocks to improve the cycling stability and insolubility in electrolyte.10–12Covalent organic frameworks (COFs) are crystalline porous polymers based on organic building blocks linked by strong covalent bonds.13,14 Electroactive COFs15 have recently emerged as promising electrode materials due to their insolubility in electrolyte, high chemical and structural versatility, tuneable porosity, and the possibility to introduce numerous redox-active centres in a controlled manner.16–21 However, some challenges such as the relatively low electronic conductivity22 or sluggish ion diffusion in bulk COFs23 still need to be addressed to increase their practical capacity. In many cases, COFs have been mixed with conductive carbon substrates (carbon nanotubes,24–28 graphene,29 conductive polymers,30etc.) to increase the electronic conductivity of the composite electrode, but this may reduce the overall energy density of the battery.31 Another recent strategy to increase the practical capacity of the active material is the exfoliation of bulk COFs into few-layer nanosheets to maximise the utilisation efficiency of the redox-active sites.23,32 Nevertheless, the yield for the synthesis of few-layer COFs nanosheets is usually very low, which might be an insurmountable challenge going toward practical applications.16
Much less attention has been invested into studying the influence of the electrolyte and binder on the performance of COF-based batteries and no systematic correlation analysis has been reported to date. It is worth noting that the nature of the electrolyte can have a major impact on the performance of organic batteries.33 On the other hand, although polyvinylidene fluoride (PVdF) is widely used as a binder with inorganic electrode materials, it can have a weaker affinity for organic electrode materials,34,35 and raises serious environmental concerns due to the use of N-methyl-2-pyrrolidinone (NMP) as a solvent.36
Among alternative rechargeable battery technologies, magnesium batteries have attracted a lot of interest because Mg is an earth-abundant element, inexpensive, safe under ambient atmosphere, and has a high volumetric capacity.37 However, Mg2+ insertion in most cathode materials (especially in the inorganic ones) presents several problems because of the strong electrostatic interaction between the divalent cations and the host materials, difficult desolvation and slow Mg2+ diffusion.38,39 Organic materials have been proposed as a versatile and environmentally friendly alternative cathodes that can improve the kinetics and cycling stability in Mg batteries.40,41 In this sense, COFs have been scarcely investigated as electrodes for Mg batteries and, although they are very promising, only a few examples have been reported very recently.42–44
Herein, we report a comprehensive study of the influence of the electrolyte and binder on the performance of COF-based batteries. A redox-active anthraquinone-based COF (DAAQ-TFP-COF) (Scheme 1) was evaluated in four different Li electrolytes with two different binders. The combination of lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) in tetraethylene glycol dimethyl ether (TEGDME) as the electrolyte and poly(tetrafluoroethylene) (PTFE) as the binder resulted in specific capacities close to the theoretical value with high capacity retention. It is worth noting that the bulk COF was not subjected to any processing. Ex situ infrared spectroscopy confirmed the highly reversible reduction of the anthraquinone electroactive moieties. Building on the efficient material utilisation seen in lithium-based electrolytes, DAAQ-TFP COF was also explored as cathode in magnesium batteries. Our results show that in addition to the proper design and processing of the COF as an active material, the choice of the electrolyte and binder can be critical to the performance of COF-based metal anode batteries.
 | ||
| Scheme 1 Chemical structure of DAAQ-TFP-COF. | ||
Results and discussion
First, a detailed analysis of the electrolytes, binders and electrode composition used in reported COF cathode materials for LIBs was carried out (Table S1†).23,24,26–31,43,45–57 In most cases, LiPF6 in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1/1) or LiTFSI in 1,3-dioxolane (DOL)/1,2-dimethoxyethane (DME) were used as electrolyte, while PVdF is the most commonly used binder. We have chosen DAAQ-TFP-COF as a model COF active material for this comprehensive investigation because of: (i) its relatively high theoretical capacity; (ii) the high chemical stability of the β-ketoenamine linkages; (iii) it can be easily obtained in large quantities; (iv) interestingly, two independent research groups reported very different capacities (54 and 110 mA h g−1) when the bulk COF was used as a cathode in lithium half-cells using different electrolytes.23,30As previously reported,58DAAQ-TFP-COF was synthesised by condensation of 2,6-diaminoanthraquinone (DAAQ) and 1,3,5-triformylphloroglucinol (TFP) (Scheme S1†) and characterised by several techniques to confirm its chemical composition, crystallinity, redox activity and high porosity (Fig. S1–S6, ESI†). Electrodes were prepared by mixing the active material (DAAQ-TFP-COF), carbon black and the binder (PVdF or PTFE) in a weight ratio of 6/3/1. Electrochemical experiments were first carried out in a two-electrode Swagelok setup using Li metal as the anode (see ESI† for Experimental details). Four different electrolytes were selected for the study. The first two are ether-based 1 M LiTFSI solutions in DOL/DME and TEGDME. These solvent/salt combinations have demonstrated favourable electrochemical performance with COFs in previous studies, as shown in Table S1.† The third electrolyte is a commercial 1 M LiPF6 in EC/DEC, which is commonly used for electrochemical characterisation of inorganic materials. The fourth electrolyte is 1 M LiClO4 in cyclic carbonate (GBL), known for its high anodic and thermal stability.59
We first compared the cycling stability of DAAQ-TFP-COF electrodes at the current density of 150 mA g−1 with the electrodes containing PVdF binder (Fig. 1a and S8, ESI†). Using LiPF6 in EC/DEC (1/1) and LiClO4 in γ-butyrolactone (GBL) as electrolytes, the worst performances were obtained showing capacity retention of ∼58% and ∼67%, respectively, after 100 cycles (Fig. 1a, S8 and Table S2, ESI†). The capacity fading observed when using LiPF6 in EC/DEC could be explained by undesired side reactions31,60–62 or higher solubility of the redox polymer in carbonate-based electrolytes.63 A comprehensive electrochemical study on (poly)anthraquinone linear polymer reported that oxidation become progressively more hindered as the cycling of the polymer progresses in carbonate-based electrolyte.64 It appears that this is the case for the inferior electrochemical performance of DAAQ-TFP-COF as well (Fig. S9†). Hindered oxidation can lead to incomplete charging of the organic cathode, followed by gradual capacity fade. This hypothesis of incomplete charging also explains coulombic efficiency over 100% observed in 1 M LiPF6 in EC/DEC. In contrast, when LiTFSI in DOL/DME (1/1) was used as electrolyte the initial capacity was 118.0 mA h g−1 and a much better capacity retention (90%) was observed after 100 cycles (Table S2, ESI†). The electrochemical performance was further improved by replacing DOL/DME with TEGDME solvent since the specific capacity was maintained at 118.3 mA h g−1 after 100 cycles. The origin of different behaviour in selected electrolytes might be a consequence of the nature of the solvent/solvent mixture used. Charge distribution on the solvent molecule can also influence the stability of the ionic or radical C–O fragment during the redox process which can affect the stability of the material in the electrolyte in question.64
PTFE is an alternative binder that can be dispersed in water and exhibits high chemical and mechanical stability as well as strong hydrophobicity.36 Up to our knowledge, PTFE has not been explored as a binder in COF-based cathode materials for LIBs (Table S1†) and has only been used in two examples of COF-based anodes without any comparative analysis with PVdF.65,66 PTFE-based DAAQ-TFP-COF electrodes were prepared (see ESI† for experimental details) to compare their performance with those based on PVdF. We first confirmed that the PTFE-based electrodes were insoluble in all the electrolytes used (Fig. S7, ESI†). In general, all the initial capacities and capacity retentions of DAAQ-TFP-COF electrodes increased when PTFE was used as a binder, regardless of the electrolyte (Fig. S10, S11 and Table S2, ESI†). One of the main reasons for the better electrochemical performance of the PTFE-based electrodes is due to its fibre-like structure, which results in a more porous electrode, as can be observed by comparing the morphology of electrodes based on PVdF or PTFE binders (Fig. S12, ESI†). Such porosity may facilitate the ion diffusion and accessibility to the redox-active sites, as well as allow easier swelling of the polymer electrode. The capacity retention tendency was similar to that obtained with PVdF-based electrodes, with LiTFSI in TEGDME and LiPF6 in EC/DEC showing the best and worst electrochemical performance, respectively. It is important to highlight that DAAQ-TFP-COF shows a capacity of 147.5 mA g−1 after 100 cycles (99% capacity retention) at a rate of 150 mA g−1 when using LiTFSI in TEGDME as electrolyte and PTFE as a binder (Fig. 1c). After subtracting the carbon black capacity contribution (Fig. S13 and Table S3, ESI†), the capacity obtained corresponds to the 88% capacity utilisation of the active material at 150 mA g−1 (theoretical capacity = 151 mA h g−1, see ESI†). We note that our results are similar to those of the same active material when exfoliated (DAAQ-ECOF) and using PVdF as a binder (capacity of 145 mA g−1 after 70 cycles at a rate of 20 mA g−1).23 These results suggest that both proper binder selection and the exfoliation of the active material can lead to improved utilisation of redox-active sites. However, the top-down exfoliation of bulk COFs still presents some challenges such as the difficulty in precisely controlling the thickness of the exfoliated material and the relatively low yield for the synthesis of few-layer COF nanosheets, limiting the homogeneity and scalability of the active material.21 The use of PTFE binder is a more straightforward approach allowing significantly higher process yields while being both more cost-effective and simpler.
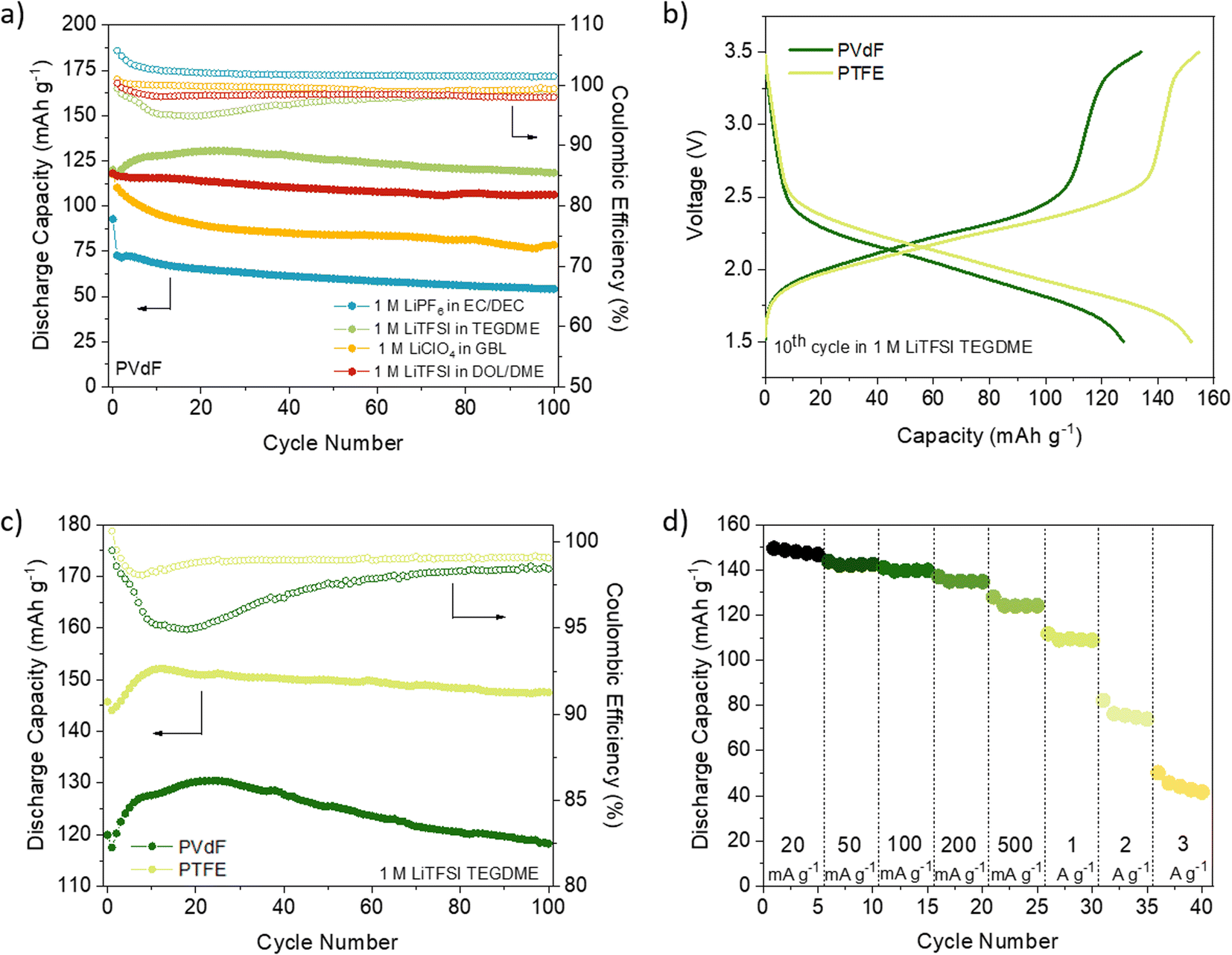 | ||
Fig. 1 (a) DAAQ-TFP-COF electrode performance with PVdF as a binder at 150 mA g−1 (1C) in four different electrolytes: 1 M LiTFSI DOL/DME (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, vol%) (red), 1 M LiTFSI in TEGDME (green), 1 M LiPF6 in EC/DEC (1:1, vol%) (blue), and 1 M LiClO4 in GBL (orange). (b) Comparison between PVdF (dark green) and PTFE (light green) as binders in 1 M LiTFSI TEGDME electrolyte. (c) Stability and coulombic efficiency of DAAQ-TFP-COF cathode in 1 M LiTFSI TEGDME at 150 mA g−1 using PVdF or PTFE as binder. (d) Rate capability test in 1 M LiTFSI TEGDME. :1, vol%) (red), 1 M LiTFSI in TEGDME (green), 1 M LiPF6 in EC/DEC (1:1, vol%) (blue), and 1 M LiClO4 in GBL (orange). (b) Comparison between PVdF (dark green) and PTFE (light green) as binders in 1 M LiTFSI TEGDME electrolyte. (c) Stability and coulombic efficiency of DAAQ-TFP-COF cathode in 1 M LiTFSI TEGDME at 150 mA g−1 using PVdF or PTFE as binder. (d) Rate capability test in 1 M LiTFSI TEGDME. | ||
Despite achieving a high level of capacity utilisation, the overpotential observed in the 1 M LiTFSI TEGDME electrolyte for DAAQ-TFP-COF is unusually high for anthraquinone-based materials, measuring around 210 mV (Fig. S14, ESI†). In contrast, when the cell is cycled in the DOL/DME solvent combination, the overpotential is 85 mV lower under identical conditions. In order to differentiate between the anode and cathode contributions to the overpotential, an electrochemical test using a cyclable symmetric cell was carried out (see ESI† for Experimental details).67 In this controlled environment, where the performance of the cell is determined solely by the electrochemical performance of DAAQ-TFP-COF electrode, the overpotential decreases to 62 mV, aligning closer to the value obtained in the DOL/DME half-cell. The differences observed between the half cell and the symmetric cell suggest that the major overpotential contribution mainly originates from the Li metal anode. The influence of the Li metal anode is also evident in the rate capability test performed in the half-cell configuration (Fig. 1d and S15†). The rapid capacity degradation at higher currents was again attributed to processes occurring at the Li metal anode. In contrast, DAAQ-TFP-COF demonstrated good cycling stability in the symmetric cell, retaining 79% of its initial capacity at a current density of 500 mA g−1 after 2000 cycles (Fig. 2c). It is worth noting that the TEGDME solvent has notably higher viscosity when compared with the DOL/DME mixture, likely leading to impeded transport of active species within the electrolyte. This can, in turn, affect the overpotential of the organic cathode and the initial activation behaviour observed during the first few cycles. Therefore, in addition to selecting an appropriate binder and electrolyte that maximise capacity utilisation of the active material, it is crucial to recognise the contribution of the metal anode in the 2-electrode setup and to ensure that experimental setups allow for a thorough evaluation of the COF cathode material alone as exemplified by the symmetric cell test.67
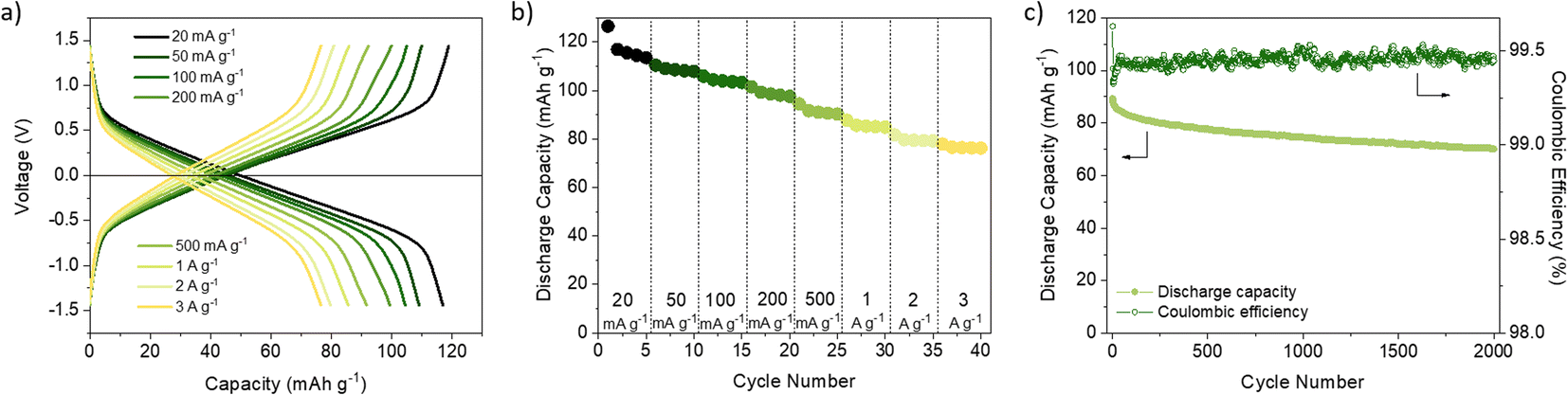 | ||
| Fig. 2 (a) Galvanostatic profiles at different current rates and (b) discharge capacity of (+)DAAQ-TFP-COF‖DAAQ-TFP-COF-M(−) symmetric cells during rate capability test. (c) Cycling stability and coulombic efficiency of (+)DAAQ-TFP-COF‖DAAQ-TFP-COF-M(−) symmetric cells during long-term cycling at a current density of 500 mA g−1. | ||
Ex situ infrared (IR) spectroscopy was performed on cycled electrodes at different charge states to assess the reversibility of the electrochemical performance (Fig. 3). Taking into account the recorded voltage profiles as well as the obtained capacities, it is reasonable to assume that only the anthraquinone moieties participate in the electrochemical reaction. Several characteristic IR bands can be observed in the pristine electrode. The carbonyl stretching of the anthraquinone unit can be found at 1619 and 1673 cm−1, whereas the carbonyl band of the TFP moiety can be observed around 1658 cm−1 in the spectrum of pristine linker (Fig. S2†). However, following the polymerisation process, spectral bands begin to merge, leading us to assume that TFP C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching is part of a broad band appearing at 1544 cm−1, mainly due to CC stretching. The strong band at 1225 cm−1 is related to the C–N stretching.68 Throughout the reduction process, all CO bands diminish in intensity, as shown in Fig. 3c. Upon recharging, carbonyl bands increase in intensity, confirming the good reversibility of the electrochemical process. The strong peak at 1544 cm−1 also decreases in intensity and broadens upon discharge, indicating changes in aromaticity upon reduction. In the half-discharged electrode, a new peak is observed at 1428 cm−1, due to the formation of –C–O− Li+. The band around 1225 cm−1 related to the C–N stretching also showed some changes in the shape and intensity of the peak on discharge, which could be related to the change in conjugation on the anthracene ring. These results are consistent with the good reversibility observed in the galvanostatic cycling.
O stretching is part of a broad band appearing at 1544 cm−1, mainly due to CC stretching. The strong band at 1225 cm−1 is related to the C–N stretching.68 Throughout the reduction process, all CO bands diminish in intensity, as shown in Fig. 3c. Upon recharging, carbonyl bands increase in intensity, confirming the good reversibility of the electrochemical process. The strong peak at 1544 cm−1 also decreases in intensity and broadens upon discharge, indicating changes in aromaticity upon reduction. In the half-discharged electrode, a new peak is observed at 1428 cm−1, due to the formation of –C–O− Li+. The band around 1225 cm−1 related to the C–N stretching also showed some changes in the shape and intensity of the peak on discharge, which could be related to the change in conjugation on the anthracene ring. These results are consistent with the good reversibility observed in the galvanostatic cycling.
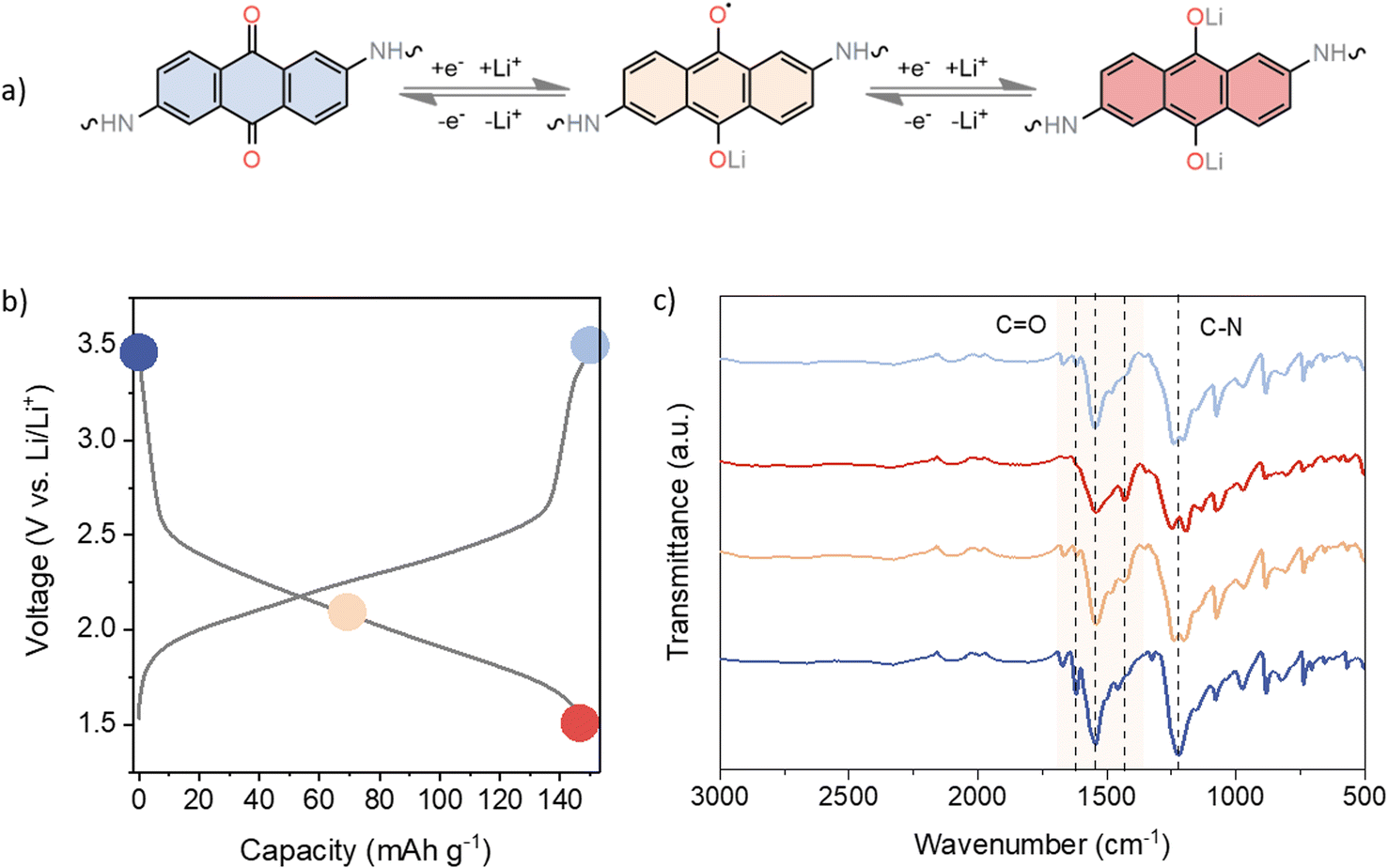 | ||
| Fig. 3 (a) Electrochemical mechanism of DAAQ-TFP-COF. (b) Galvanostatic curve showing the ex situ FTIR sampling points. (c) Ex situ FTIR spectra of DAAQ electrodes in different states of charge: pristine electrode (dark blue), electrode at 0.5 SOC (yellow), electrode at 1.5 V (fully discharged, red), and electrode fully charged back to 3.5 V (light blue). | ||
Encouraged by the good reversibility and high utilisation of non-exfoliated DAAQ-TFP-COF, we decided to extend the study to COF performance in Mg-based batteries. Given the strong tendency of Mg metal to passivate, only handful of salt/solvent combinations are available that enable reversible Mg plating and stripping. In this case, we opted for two electrolytes – one containing chloride species (0.6 M MgTFSI2 – 1.2 M MgCl2 in DME) and another containing weakly coordinating anions (0.2 M Mg(B(hfip)4)2 in DME). It is important to note that in Mg metal half-cells, magnesium metal can significantly affect the overall performance of the cell, especially due to increased cell overpotential stemming from Mg plating/stripping. Both electrolytes chosen for this study enable highly reversible platting and stripping of magnesium metal with reasonable overpotential. Given the high surface area of the chosen COF, we have subjected cathodes to electrochemically assisted swelling, to maximise capacity utilisation.69
Comparison of the galvanostatic curves obtained in two different electrolytes revealed significant differences: COF in chloride-free electrolyte exhibited higher overpotential accompanied by a lower average discharge potential (Fig. 4a and S16, ESI†). Notably, the COF exhibited good stability in both electrolytes, which is a consequence of its robust structure. This stands in stark contrast to linear anthraquinone-based polymers reported in Mg electrolytes.70 However, in the case of the chloride-containing electrolyte, coulombic efficiency surpasses 100% during cycling, likely indicative of side reactions or an inadequate voltage window. The rate capability (Fig. 4c) was performed only in chloride-containing electrolyte, considering the relatively low capacities achieved at low current density in the chloride-free electrolyte. The rate performance was substantially poorer compared to that in lithium-based counterpart. This discrepancy suggests that kinetic limitations become more pronounced when Mg species are involved in the electrochemical reaction.
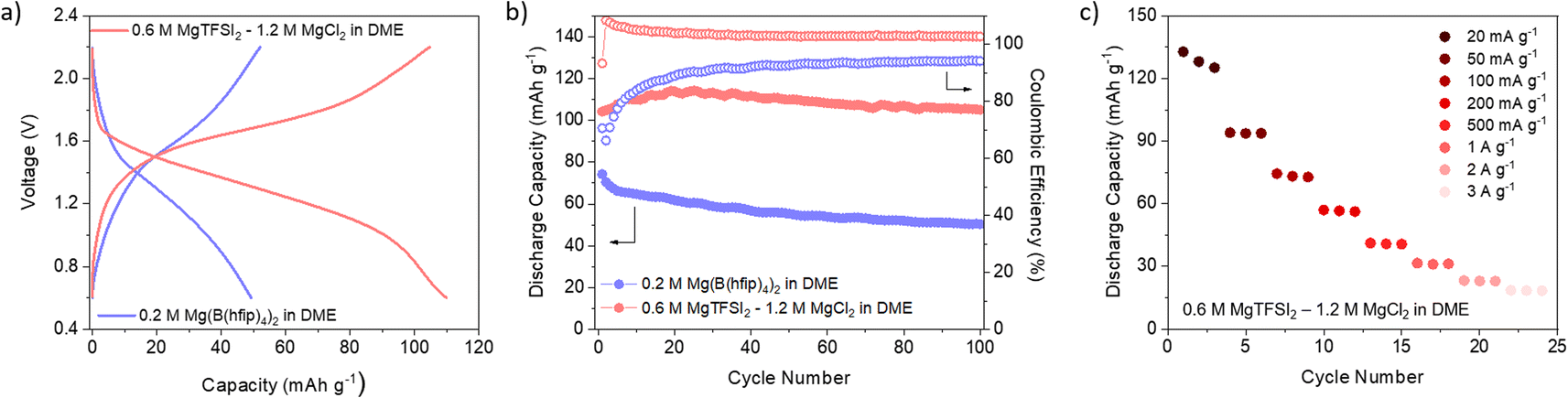 | ||
| Fig. 4 (a) Galvanostatic charge/discharge curves of DAAQ-TFP-COF in 0.6 M MgTFSI2 – 1.2 M MgCl2 DME (red) and 0.2 M Mg(B(hfip)4)2 DME (blue). (b) Evolution of discharge capacity and coulombic efficiency over 100 cycles. (c) Rate capability performance in chloride-containing electrolyte. All the experiments were conducted in Mg metal half-cells. | ||
The selection of the electrolyte in Mg batteries introduces one key distinction compared to Li-based system – the possibility of presence of different ionic species in discharged polymer. In the chloride-containing electrolyte, the coordination of reduced carbonyls was predominantly done with MgCl+ ionic complexes, rather than Mg2+ ions, in agreement with prior reports and confirmed by energy dispersive X-ray spectroscopy (EDX) (Table S4†).71 Conversely, in the chloride-free electrolyte, a large fraction of Mg2+ was present, although ion pairs still partially participate in coordination as evidenced by the increased presence of fluorine in the discharge cathode. This shows that COF performance is governed by the choice of salt/solvent combination, with the effect of electrolyte being more pronounced in Mg batteries than in their lithium counterparts. This raises the challenge of designing COFs that would perform better when coupled with multivalent charge carriers. One of the strategies could be to design COFs with active units in which the position of carbonyl groups is tuned to help accommodate divalent cations.72 Changing the active unit from an anthraquinone-based one to a smaller unit such as benzoquinone could lead to higher potentials and ultimately higher energy densities, as has already been demonstrated in the case of COF cathodes in Li-based batteries.23 An alternative strategy could also be designing hosts with hybrid mechanisms that simultaneously allow for the optimisation of capacity and rate performance of the covalent organic framework.73,74 Potential improvements in COFs for Mg batteries could lead to readily available cathodes for other divalent batteries suffering from similar issues, such as Ca-based ones.
Conclusions
In summary, we have investigated the role of the electrolyte and binder on the performance of a redox-active COF (DAAQ-TFP-COF) electrode material. Our results demonstrate that by identifying the most suitable electrolyte (LiTFSI in TEGDME) and binder (TPFE) for DAAQ-TFP-COF, specific capacities close to the theoretical values (88% real utilisation of the active material) were obtained without the need for extensive COF processing (e.g., exfoliation). The most likely reasons for the improved performance are the better stability of prepared COF-based electrodes in non-carbonate electrolytes and the improved ion diffusion through the PTFE binder. Ex situ IR experiments confirmed the reversibility of the electrochemical process in agreement with the galvanostatic cycling performance. In addition, electrochemical tests were performed using symmetric cells by combining two DAAQ-TFP-COF electrodes to identify the contribution of the metal anode in the 2-electrode setup. To further investigate the electrolyte influence on the cycling of COFs, we have employed DAAQ-TFP-COF in Mg cells using chloride-free and chloride-containing Mg electrolytes We are confident that our results will be very useful in evaluating and comparing the performance of COF-based electrodes in metal-ion batteries as well as stimulating the optimisation of electrolytes and binders for this type of organic batteries. As COFs are interesting host materials for a variety of ions, more efforts should be invested to facilitate the accommodation of multivalent cations, where high performance organic cathode materials are urgently needed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the European Research Council (ERC) under the European Union's Horizon Europe Framework Programme (ERC-2021-Starting Grant, grant agreement no. 101039748-ELECTROCOFS). This work was developed within the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020, UIDP/50011/2020 & LA/P/0006/2020, financed by national funds through the FCT/MEC (PIDDAC). We also thank FCT for funding the PTDC/QUI-ELT/2593/2021 project. The authors would like to acknowledge the financial support from European Union's Horizon 2020 research and innovation program under the Marie Sklodowska Curie framework, grant agreement no. 860403 and Slovenian Research Agency through program P2-0423 and projects N2-0279 and J2-4462.References
- J. B. Goodenough, Nat. Electron., 2018, 1, 204 CrossRef.
- F. Wu, J. Maier and Y. Yu, Chem. Soc. Rev., 2020, 49, 1569–1614 RSC.
- European Commission, Critical Raw Materials Resilience: Charting a Path towards Greater Security and Sustainability, https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:52020DC0474 Search PubMed.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 RSC.
- T. B. Schon, B. T. McAllister, P. F. Li and D. S. Seferos, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC.
- Y. Lu, Q. Zhang, L. Li, Z. Niu and J. Chen, Chem, 2018, 4, 2786–2813 CAS.
- Y. Lu and J. Chen, Nat. Rev. Chem., 2020, 4, 127–142 CrossRef CAS PubMed.
- P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. Liang and Y. Yao, Chem. Rev., 2020, 120, 6490–6557 CrossRef CAS PubMed.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS PubMed.
- B. Esser, F. Dolhem, M. Becuwe, P. Poizot, A. Vlad and D. Brandell, J. Power Sources, 2021, 482, 228814 CrossRef CAS.
- N. Goujon, N. Casado, N. Patil, R. Marcilla and D. Mecerreyes, Prog. Polym. Sci., 2021, 122, 101449 CrossRef CAS.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- K. T. Tan, S. Ghosh, Z. Wang, F. Wen, D. Rodríguez-San-Miguel, J. Feng, N. Huang, W. Wang, F. Zamora, X. Feng, A. Thomas and D. Jiang, Nat. Rev. Methods Prim., 2023, 3, 1 CrossRef CAS.
- M. Souto, K. Strutyński, M. Melle-Franco and J. Rocha, Chem.–Eur. J., 2020, 26, 10912–10935 CrossRef CAS PubMed.
- T. Sun, J. Xie, W. Guo, D. S. Li and Q. Zhang, Adv. Energy Mater., 2020, 10, 1–23 Search PubMed.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC.
- Y. Lu, Y. Cai, Q. Zhang and J. Chen, J. Phys. Chem. Lett., 2021, 12, 8061–8071 CrossRef CAS PubMed.
- D. Zhu, G. Xu, M. Barnes, Y. Li, C. Tseng, Z. Zhang, J. Zhang, Y. Zhu, S. Khalil, M. M. Rahman, R. Verduzco and P. M. Ajayan, Adv. Funct. Mater., 2021, 31, 2100505 CrossRef CAS.
- S. Kandambeth, V. S. Kale, O. Shekhah, H. N. Alshareef and M. Eddaoudi, Adv. Energy Mater., 2022, 12, 2100177 CrossRef CAS.
- S. Haldar, A. Schneemann and S. Kaskel, J. Am. Chem. Soc., 2023, 145, 13494–13513 CrossRef CAS PubMed.
- M. Souto and D. F. Perepichka, J. Mater. Chem. C, 2021, 9, 10668–10676 RSC.
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS PubMed.
- F. Xu, S. Jin, H. Zhong, D. Wu, X. Yang, X. Chen, H. Wei, R. Fu and D. Jiang, Sci. Rep., 2015, 5, 8225 CrossRef CAS PubMed.
- X. Li, H. Wang, Z. Chen, H. Sen Xu, W. Yu, C. Liu, X. Wang, K. Zhang, K. Xie and K. P. Loh, Adv. Mater., 2019, 31, 1–9 Search PubMed.
- S. Xu, G. Wang, B. P. Biswal, M. Addicoat, S. Paasch, W. Sheng, X. Zhuang, E. Brunner, T. Heine, R. Berger and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 849–853 CrossRef CAS PubMed.
- G. Wang, N. Chandrasekhar, B. P. Biswal, D. Becker, S. Paasch, E. Brunner, M. Addicoat, M. Yu, R. Berger and X. Feng, Adv. Mater., 2019, 31, 1–6 Search PubMed.
- C. Yao, Z. Wu, J. Xie, F. Yu, W. Guo, Z. J. Xu, D. Li, S. Zhang and Q. Zhang, ChemSusChem, 2020, 13, 2457–2463 CrossRef CAS PubMed.
- Z. Wang, Y. Li, P. Liu, Q. Qi, F. Zhang, G. Lu, X. Zhao and X. Huang, Nanoscale, 2019, 11, 5330–5335 RSC.
- E. Vitaku, C. N. Gannett, K. L. Carpenter, L. Shen, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2020, 142, 16–20 CrossRef CAS PubMed.
- H. Gao, A. R. Neale, Q. Zhu, M. Bahri, X. Wang, H. Yang, Y. Xu, R. Clowes, N. D. Browning, M. A. Little, L. J. Hardwick and A. I. Cooper, J. Am. Chem. Soc., 2022, 144, 9434–9442 CrossRef CAS PubMed.
- S. Gu, S. Wu, L. Cao, M. Li, N. Qin, J. Zhu, Z. Wang, Y. Li, Z. Li, J. Chen and Z. Lu, J. Am. Chem. Soc., 2019, 141, 9623–9628 CrossRef CAS PubMed.
- M. Li, R. P. Hicks, Z. Chen, C. Luo, J. Guo, C. Wang and Y. Xu, Chem. Rev., 2023, 123, 1712–1773 CrossRef CAS PubMed.
- J. Tong, C. Han, X. Hao, X. Qin and B. Li, ACS Appl. Mater. Interfaces, 2020, 12, 39630–39638 CrossRef CAS PubMed.
- J. Bitenc, U. Košir, A. Vizintin, N. Lindahl, A. Krajnc, K. Pirnat, I. Jerman and R. Dominko, Energy Mater. Adv., 2021, 9793209 CAS.
- D. Bresser, D. Buchholz, A. Moretti, A. Varzi and S. Passerini, Energy Environ. Sci., 2018, 11, 3096–3127 RSC.
- J. Muldoon, C. B. Bucur and T. Gregory, Chem. Rev., 2014, 114, 11683–11720 CrossRef CAS PubMed.
- B. T. McAllister, L. T. Kyne, T. B. Schon and D. S. Seferos, Joule, 2019, 3, 620–624 CrossRef.
- T. Bančič, J. Bitenc, K. Pirnat, A. Kopač Lautar, J. Grdadolnik, A. Randon Vitanova and R. Dominko, J. Power Sources, 2018, 395, 25–30 CrossRef.
- B. Pan, J. Huang, Z. Feng, L. Zeng, M. He, L. Zhang, J. T. Vaughey, M. J. Bedzyk, P. Fenter, Z. Zhang, A. K. Burrell and C. Liao, Adv. Energy Mater., 2016, 6, 1600140 CrossRef.
- Y. Chen, K. Fan, Y. Gao and C. Wang, Adv. Mater., 2022, 34, 2200662 CrossRef CAS PubMed.
- R. Sun, S. Hou, C. Luo, X. Ji, L. Wang, L. Mai and C. Wang, Nano Lett., 2020, 20, 3880–3888 CrossRef CAS PubMed.
- S. Li, Y. Liu, L. Dai, S. Li, B. Wang, J. Xie and P. Li, Energy Storage Mater., 2022, 48, 439–446 CrossRef.
- G. Zou, Z. Tian, V. S. Kale, W. Wang, S. Kandembeth, Z. Cao, J. Guo, J. Czaban-Jóźwiak, L. Cavallo, O. Shekhah, M. Eddaoudi and H. N. Alshareef, Adv. Energy Mater., 2023, 13, 2203193 CrossRef CAS.
- Z. Luo, L. Liu, J. Ning, K. Lei, Y. Lu, F. Li and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 9443–9446 CrossRef CAS PubMed.
- X. Yang, Y. Hu, N. Dunlap, X. Wang, S. Huang, Z. Su, S. Sharma, Y. Jin, F. Huang, X. Wang, S. Lee and W. Zhang, Angew. Chem., Int. Ed., 2020, 59, 20385–20389 CrossRef CAS PubMed.
- Z. Meng, Y. Zhang, M. Dong, Y. Zhang, F. Cui, T.-P. Loh, Y. Jin, W. Zhang, H. Yang and Y. Du, J. Mater. Chem. A, 2021, 9, 10661–10665 RSC.
- J. Sprachmann, T. Wachsmuth, M. Bhosale, D. Burmeister, G. J. Smales, M. Schmidt, Z. Kochovski, N. Grabicki, R. Wessling, E. J. W. List-Kratochvil, B. Esser and O. Dumele, J. Am. Chem. Soc., 2023, 145, 2840–2851 CrossRef CAS PubMed.
- X. Xu, S. Zhang, K. Xu, H. Chen, X. Fan and N. Huang, J. Am. Chem. Soc., 2023, 145, 1022–1030 CrossRef CAS PubMed.
- S. Gu, X. Ma, J. Chen, R. Hao, Z. Wang, N. Qin, W. Zheng, Q. Gan, W. Luo, M. Li, Z. Li, K. Liao, H. Guo, G. Liu, K. Zhang and Z. Lu, J. Energy Chem., 2022, 69, 428–433 CrossRef CAS.
- V. Singh, J. Kim, B. Kang, J. Moon, S. Kim, W. Y. Kim and H. R. Byon, Adv. Energy Mater., 2021, 11, 1–10 Search PubMed.
- S. Jhulki, C. H. Feriante, R. Mysyk, A. M. Evans, A. Magasinski, A. S. Raman, K. Turcheniuk, S. Barlow, W. R. Dichtel, G. Yushin and S. R. Marder, ACS Appl. Energy Mater., 2021, 4, 350–356 CrossRef CAS.
- S. Gu, R. Hao, J. Chen, X. Chen, K. Liu, I. Hussain, G. Liu, Z. Wang, Q. Gan, H. Guo, M. Li, K. Zhang and Z. Lu, Mater. Chem. Front., 2022, 6, 2545–2550 RSC.
- M. Wu, Y. Zhao, R. Zhao, J. Zhu, J. Liu, Y. Zhang, C. Li, Y. Ma, H. Zhang and Y. Chen, Adv. Funct. Mater., 2022, 32, 1–8 Search PubMed.
- G. Zhao, H. Li, Z. Gao, L. Xu, Z. Mei, S. Cai, T. Liu, X. Yang, H. Guo and X. Sun, Adv. Funct. Mater., 2021, 31, 1–9 Search PubMed.
- C. Wu, M. Hu, X. Yan, G. Shan, J. Liu and J. Yang, Energy Storage Mater., 2021, 36, 347–354 CrossRef.
- D. Yang, Z. Yao, D. Wu, Y. Zhang, Z. Zhou and X. Bu, J. Mater. Chem. A, 2016, 4, 18621–18627 RSC.
- C. R. Deblase, K. E. Silberstein, T. T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed.
- Y. Gu, S. Fang, L. Yang and S. Hirano, Electrochim. Acta, 2021, 394, 139120 CrossRef CAS.
- M. Dahbi, F. Ghamouss, F. Tran-Van, D. Lemordant and M. Anouti, J. Power Sources, 2011, 196, 9743–9750 CrossRef CAS.
- Y. Shi, H. Tang, S. Jiang, L. V. Kayser, M. Li, F. Liu, F. Ji, D. J. Lipomi, S. P. Ong and Z. Chen, Chem. Mater., 2018, 30, 3508–3517 CrossRef CAS.
- S. F. Lux, J. Chevalier, I. T. Lucas and R. Kostecki, ECS Electrochem. Lett., 2013, 2, A121–A123 CrossRef CAS.
- V. Perner, D. Diddens, F. Otteny, V. Küpers, P. Bieker, B. Esser, M. Winter and M. Kolek, ACS Appl. Mater. Interfaces, 2021, 13, 12442–12453 CrossRef CAS PubMed.
- S. Phadke, M. Cao and M. Anouti, ChemSusChem, 2018, 11, 965–974 CrossRef CAS PubMed.
- S. Haldar, K. Roy, S. Nandi, D. Chakraborty, D. Puthusseri, Y. Gawli, S. Ogale and R. Vaidhyanathan, Adv. Energy Mater., 2018, 8, 1702170 CrossRef.
- S. Haldar, K. Roy, R. Kushwaha, S. Ogale and R. Vaidhyanathan, Adv. Energy Mater., 2019, 9, 1902428 CrossRef CAS.
- O. Lužanin, J. Moškon, T. Pavčnik, R. Dominko and J. Bitenc, Batteries Supercaps, 2023, 6, e202200437 CrossRef.
- S. Gu, S. Wu, L. Cao, M. Li, N. Qin, J. Zhu, Z. Wang, Y. Li, Z. Li, J. Chen and Z. Lu, J. Am. Chem. Soc., 2019, 141, 9623–9628 CrossRef CAS PubMed.
- X. Wang, H. Dong, A. Eddine Lakraychi, Y. Zhang, X. Yang, H. Zheng, X. Han, X. Shan, C. He and Y. Yao, Mater. Today, 2022, 55, 29–36 CrossRef CAS.
- J. Bitenc, K. Pirnat, T. Bančič, M. Gaberšček, B. Genorio, A. Randon-Vitanova and R. Dominko, ChemSusChem, 2015, 8, 4128–4132 CrossRef CAS PubMed.
- H. Dong, Y. Liang, O. Tutusaus, R. Mohtadi, Y. Zhang, F. Hao and Y. Yao, Joule, 2019, 3, 782–793 CrossRef CAS.
- L. Miao, L. Liu, Z. Shang, Y. Li, Y. Lu, F. Cheng and J. Chen, Phys. Chem. Chem. Phys., 2018, 20, 13478–13484 RSC.
- H. Kong, Y. Guan, J. Wang, W. Sun, L. Chen, J. Ou, L. Xie, F. Fu, H. Zhang and H. Chen, J. Mater. Chem. A, 2022, 10, 20866–20873 RSC.
- W. Sun, C. Zhou, Y. Fan, Y. He, H. Zhang, Z. Quan, H. Kong, F. Fu, J. Qin, Y. Shen and H. Chen, Angew. Chem., Int. Ed., 2023, 62, e202300158 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General methods and materials, synthesis and characterisation of DAAQ-TFP-COF, additional electrochemical experiments, SEM, EDX. See DOI: https://doi.org/10.1039/d3ta05190k |
| ‡ Equally contributed. |
| This journal is © The Royal Society of Chemistry 2023 |