
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Eco-efficient pickering foams: leveraging sugarcane waste-derived cellulose nanofibres
Pouria
Amani
 ac,
Nasim
Amiralian
*b,
Sandya S. A.
Athukoralalage
b and
Mahshid
Firouzi
*ac
ac,
Nasim
Amiralian
*b,
Sandya S. A.
Athukoralalage
b and
Mahshid
Firouzi
*ac
aARC Centre of Excellence for Enabling Ecoefficient Beneficiation of Minerals, The University of Newcastle, Callaghan 2308, NSW, Australia. E-mail: Mahshid.Firouzi@newcastle.edu.au
bAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, Queensland 4072, Australia. E-mail: n.amiralian@uq.edu.au
cCollege of Engineering Science and Environment, The University of Newcastle, Callaghan, 2308, Australia
First published on 3rd November 2023
Abstract
Foams, known for their diverse material properties, are extensively utilised in fields ranging from food and cosmetics to environmental remediation and mineral processing. Stable foam formation typically requires surface-active substances, such as surfactants, to lower the surface free energy at the air–liquid interface. However, many current foam stabilisers pose environmental risks due to toxicity and non-biodegradability. This study presents cellulose nanofibers (CNF) derived from agricultural waste, as an eco-friendly foam stabiliser. The less dense structure of non-wood biomass yields high aspect ratio and flexible nanofibers, positioning agricultural waste-derived nanofibers as a potential foam stabilising agent. To achieve varying foam stabilities for different applications, the foam generation and stability of 0.1 wt% CNF with different amounts of octylamine (OA) are examined. Insights into the stabilising effect of CNF and OA are drawn from the interfacial tension and dilational rheology of OA and CNF adsorbed layers. Results showed that foam stability peaked with OA concentration at 630 ppm; beyond this, stability decreased because of nanofiber aggregation. The enhanced foam stability is attributed to the increased surface hydrophobicity with OA concentration, which promotes nanofiber adsorption at the air–water interface and improves the interfacial tension and dilational viscoelasticity. Overall, this study provides valuable insights into using agricultural waste-derived nanocellulose as an efficient, eco-friendly, and economical stabilising agent for wet foams.
1. Introduction
Composed simply of bubbles dispersed in water, aqueous foams exhibit a diverse range of material properties, making them integral to an array of applications. This seemingly simple structure belies its significant role in various industries.1 Foams for example, play a pivotal role in environmental remediation, particularly in the treatment of contaminants like per- and poly-fluoroalkyl substances (PFAS), a group of persistent compounds used widely in consumer products, leading to contamination of groundwater and surface water systems.2–4 In the resources sector foams are particularly utilised under challenging conditions, such as high temperatures and elevated salt concentrations for oil and gas recovery.5,6 Additionally, the mineral processing industry utilises foams in several separation techniques, including foam fractionation, ion flotation, and froth flotation.7,8 These techniques rely on the creation of bubbles, which provide a large surface area (air–liquid interface) for separating different substances by adsorbing them to the air–liquid interfaces with the assistance of substances like foamers or collectors.Stable foam formation necessitates the presence of surface-active substances such as surfactants, which act to reduce the surface free energy at the air–liquid interface. Beyond surfactants, particles have also demonstrated their capacity to stabilise foams. This has led to a growing interest in particle-stabilised, or Pickering foams, encouraging numerous studies on their formation and unique characteristics. The multifaceted nature of Pickering foams is shaped by a variety of factors, including particle size, charge, hydrophobicity, and concentration, as well as the charge and concentration of the system's surfactants and salts. A recent comprehensive review by Amani et al.9 delves into the specifics of Pickering foams.
Despite the widespread use of foams in various applications, there are environmental concerns associated with the use of commonly used stabilising substances in applications such as energy, minerals processing. Many commercially available reagents could have adverse environmental or hazardous effects. Additionally, many of these chemical reagents are not biodegradable, further contributing to their potential negative impact on the environment.10,11 For instance, in the mineral processing industry, xanthates and methyl isobutyl carbinol (MIBC) are commonly used as collectors and frothers in the flotation of valuable minerals. However, they are hazardous (irritating to the skin, eyes, and respiratory system) and flammable decomposition products.12,13
This paper aims to investigate a green foam stabiliser, comprised of a plant building block, nanocellulose (NC), extracted from agricultural wastes to generate a stable foam which can be potentially employed in various applications, replacing hazardous reagents. Agricultural waste/residues present a significant environmental challenge when burned because the combustion process releases harmful pollutants and greenhouse gases into the atmosphere. However, advancements in nanotechnology have opened up new possibilities for utilising such waste. One promising nanomaterial derived from biomass is nanocellulose. Nanocellulose (NC) refers to cellulose fibres (CNF)14 or crystals (CNC) that have been broken down to the nanoscale,15 which possesses exceptional mechanical properties, high surface area, high aspect ratio, and biodegradability. The unique properties of nanocellulose make it highly versatile and suitable for numerous applications.16,17 One notable application of nanocellulose is its use as a stabilising agent or Pickering agent, particularly within the pulp and paper industry, replacing petrochemical-derived foaming agents that have been employed to generate and stabilise foams.18–20 Using NC as a stabilising agent in a technique similar to Pickering emulsions, where the nonpolar phase is air instead of oil, results in the formation of low-density cellulose foams or aerogels with good mechanical properties. While most research on nanocellulose-based foams has primarily focused on dry foam and aerogels, several studies have delved into the surface modification of cellulose nanofibers or cellulose nanocrystals. Such modifications control their wettability, which in turn facilitates the formation of nanocellulose-stabilised wet foams.15 High aspect ratio and anisotropic NC have been shown to stabilise wet foams more efficiently than spherical particles, due to their higher surface coverage and the possibility of forming intertwined networks of high mechanical stability to prohibit the bubbles coalescence.21,22 Partially hydrophobic NC can improve the stabilisation of surfactant and NC at the air–liquid interface by the formation of a thick nanofibre network layer on the air bubbles.20,23 Another important factor that needs to be considered for the stability of foam when nanocellulose is used as a stabilising agent, is the electrostatic charge density of NC to prevent flocculation and induce a repulsive disjointing pressure.23 Therefore, the particle size, hydrophobicity, and surface charge of NC play a vital role in the foam stability.
Nanocellulose can be extracted from various sources, including agricultural waste, such as sugarcane bagasse, rice straw, grasses, or wood pulp. The utilisation of agricultural waste-derived nanocellulose offers several advantages when compared to wood as a raw material. Due to their different cell wall compositions, e.g., the type of hemicelluloses and lignin and their content, the production of NC from non-wood resources including agricultural waste does not require intensive chemical and mechanical pulping and fibrillation methods and is more cost-effective.24 In addition, they are abundantly available as waste products from farming and other agro-industrial processes. By valorising these waste materials, we not only reduce their environmental impact but also tap into a cost-effective and readily available resource.
This work particularly demonstrates the stabilising effect of a small amount of nanocellulose derived from sugarcane waste as the foam stabilising agent in the presence of octylamine (OA). Utilising comprehensive dynamic and static foam analyses, the influence of OA-treated cellulose nanofibers on wet foam stability is evaluated. To provide insights into the effect of OA on the dynamic adsorption of NC to the air–water interface, the interfacial tension and dilational rheology of air–water interfaces is examined in the presence of OA and nanocellulose. Considering the different structural composition and crystallinity of nanocelluloses derived from non-wood resources, this work demonstrates how the high aspect ratio and flexible sugarcane-derived nanofibres promote shear thinning and thixotropy. These properties result in a gel formation at a relatively low concentration. Further, these properties make sugarcane-derived nanofibres a promising foam stabilising agent, effectively delaying drainage, coalescence, and coarsening, and allows for a significant reduction in the quantity of foaming agents required. By utilising nanocellulose as a foam stabilising agent, we not only minimise the environmental impact associated with excessive foaming agent usage but also harness the unique properties of nanocellulose to enhance foam stability.
2. Experimental
2.1. Materials
The sugarcane trash used in this study was received from Sunshine Sugar. Chemicals employed in this study, sodium hydroxide 99% (Chem-supply), glacial acetic acid (Merc), sodium chlorite-Technical Grade 80% (Sigma-Aldrich), sodium bromide 99% (Sigma-Aldrich), 2,2,6,6-Tetramethylpiperidine 1-oxyl radical (TEMPO) 98% (Sigma-Aldrich), sodium hypochlorite – Technical Grade 8–14% (Ajax Finechem), hydrochloric acid 32% w/w (Chem-supply), and octylamine (OA) 99% (Sigma Aldrich) were used as received. Milli-Q water was used in all experiments.2.2. Preparation of CNF
For preparing cellulose nanofibres, purified sugarcane pulp was first produced using a method reported before.25 Briefly, washed, air dried at room temperature for three days and finely ground sugarcane trash was first soaked in water overnight and delignified with a 2% (w/v) sodium hydroxide solution at a solvent to solid ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at 80 °C for 2 h followed by rinsing with hot water to completely remove soluble lignin and low molecular weight structural components. Delignified fibres were then bleached twice using an acidic solution of 1% (w/v) sodium chlorite (pH = 4, the pH decreased with glacial acetic acid) at 80 °C for one hour at a 30:1 solvent to pulp ratio. The bleached pulp was washed with hot water before storing it in the refrigerator. The lignocellulose content of sugarcane biomass analysed by NREL standards demonstrates the relative content of cellulose, hemicellulose, and lignin are 60, 24, and 4 wt%, respectively.
:1 at 80 °C for 2 h followed by rinsing with hot water to completely remove soluble lignin and low molecular weight structural components. Delignified fibres were then bleached twice using an acidic solution of 1% (w/v) sodium chlorite (pH = 4, the pH decreased with glacial acetic acid) at 80 °C for one hour at a 30:1 solvent to pulp ratio. The bleached pulp was washed with hot water before storing it in the refrigerator. The lignocellulose content of sugarcane biomass analysed by NREL standards demonstrates the relative content of cellulose, hemicellulose, and lignin are 60, 24, and 4 wt%, respectively.
Chemical pre-treatment of bleached pulp was performed using 2,2,6,6-tetramethylpiperidine-1-oxyl radical (TEMPO) mediated oxidation,26 where 500 ml of 1 wt% pulp dispersion was firstly mixed with TEMPO (0.08 g) and NaBr (0.5 g). The oxidation reaction was initiated by adding 10 mmol g−1 of cellulose NaClO solutions at room temperature, pH = 10 for 4 hours. TEMPO-oxidised cellulose pulp was then rinsed with water to remove unreacted TEMPO, inorganic salts, and acids. To produce TEMPO oxidised cellulose nanofibres (CNF), a 0.6% (w/v) dispersion of TEMPO oxidised cellulose pulp was passed through a high-pressure homogeniser for one pass at 400 bar, one pass at 700 bar and three passes at 1100 bar (GEA Niro-Soavi Panda NS1001L 2K Homogeniser).
2.3. Foaming procedure
TEMPO-oxidised cellulose nanofibres-stabilised foams were prepared by adding octylamine (OA) to 0.1 wt% CNF dispersions. First, 50 ml mixtures of OA and CNF were stirred at 600 rpm for two minutes. Then, the mixture was placed in a glass column with an inner diameter of 40 mm and a height of 250 mm. For foam generation, a fixed gas (air) flow rate of 0.2 L min−1 was injected into the 50 ml of dispersion through a frit plate with a porosity of G3 (16–40 μm pore sizes) placed inside the glass column. The glass column's height allowed a maximum of 20 cm of foam generation. Therefore, the gas was sparged until the foam height reached the 20 cm height limit. In cases where the maximum foamability of the solution was less than 20 cm of the foam height, the gas was sparged for 2 minutes, ensuring that the maximum foam height is reached. The foam height was recorded over time as the gas was sparged, to evaluate the foamability of the CNF and OA mixtures.2.4. Foam stability tests
The foam stability of CNF and OA mixtures was assessed by stopping the gas injection and monitoring the foam structure including its height, bubble size and distribution for up to 60 minutes in a 20 cm glass column. A side-mounted prism on the glass column and a high-resolution camera facilitated clear observation of bubble dynamics over time. The foam height decay rate and the evolution of the bubble size over time were analysed using image analysis to assess the foam stability of each dispersion. All experiments were conducted at a fixed room temperature of 19 °C.2.5. Sample characterisation
 | (1) |
 | (2) |
 | (3) |
| E = |E|cosφ + i|E|sinφ = E′ + iE′′ | (4) |
3. Results and discussion
3.1. CNF surface characteristics
TEMPO-oxidation treatment selectively oxidises the primary hydroxyl groups while leaving unaffected the secondary hydroxyl groups. Here, 1 mol of the C6 primary hydroxyl group is converted to 1 mol of the C6 carboxylate group via C6 aldehyde by consuming 2 mol of NaClO.26 The degree of oxidation (DO) of cellulose determined from the conductometric titration is 27%, and the carboxylate content calculated was 1.7 mmol g−1 of cellulose. The XRD pattern of CNF in Fig. 1b represents the Cellulose I structure with the peaks at 2θ = 18.5° and 22.4° and the crystallinity index value of 75%. Various amounts of OA were added to the CNF to achieve different levels of surface modification. The objective of this modification was to investigate its impact on the stability of the resulting foam. Since the interaction between OA and nanofibres occurs via electrostatic adsorption, it is anticipated that the addition of OA would not alter the crystalline structure of CNF. | ||
| Fig. 1 (a) FTIR spectra of CNF and CNF mixed with different concentrations of OA ranging from 100–3200 ppm, (b) XRD graph of CNF and bleached cellulose, (c) contact angle value of CNF and CNF with various OA concentrations, and TEM images of: (d) CNF, (e) CNF mixed with 100 ppm OA, (f) CNF mixed with 250 ppm OA, (g) CNF mixed with 630 ppm OA, (h) CNF mixed with 1000 ppm OA (the scale bar is 500 nm). | ||
Fig. 1a features an intensive band around 1610 cm,1 which is attributed to the carboxylate group that has been introduced through TEMPO oxidation.31 The peaks at approximately 2905 cm−1 and 3300 cm−1 are assigned to the sp3-hybridised CH stretching and OH stretching, respectively.32 The observed peaks between 1000–1060 cm−1 correspond to C–O–C stretching vibrations, while the peak at 1201 cm−1 is associated with –OH in-plane bending. Additionally, the peaks at 1112 cm−1 and 1165 cm−1 correspond to C–H deformation stretching vibration, and asymmetry stretch vibration, respectively. All these peaks are characteristics of the cellulosic polymer.33 Octylamine attaches to the CNF through electrostatic adsorption. The appearance of a peak at 2855 cm−1 and a slight shift of peak from 2897 to 2922 cm−1 with increasing octylamine content and an increase of the intensity in this peak confirm the presence of octylamine groups on the surface of nanofibres.
Fig. 1c shows the contact angle value measured for the CNF and CNF mixed with different OA contents. The contact angle of CNF is 23.2°, demonstrating the hydrophilic nature of CNF due to its COOH and OH surface functionalities. As the concentration of OA in the mixture increased from 30 ppm to 1000 ppm, the contact angle rose by 22%. For the films prepared with 30 ppm OA + 0.1 wt% CNF, the contact angle is 55.8, and it continued to increase to 67° for the 1000 ppm OA + 0.1 wt% CNF. A higher concentration of OA (e.g. 1400 ppm and above) leads to the agglomeration of fibres, and the formation of bundles, which could affect the roughness of paper films ultimately the CA value.
The increase in the hydrophobicity is attributed to the coverage of hydroxyl groups by OA containing long carbon chains, as well as partial dissociation of the charges on CNF and octylamine. This observation is supported by the zeta potential measurement of CNF (Fig. 2), where the charge density of nanofibres decreases from −59 mV to −45 mV with increasing the OA concentration from 0 ppm to 1000 ppm.
 | ||
| Fig. 2 Zeta potential data as a function of OA concentration in 0.1 wt% CNF dispersions at their associated natural pH. Due to the formation of large aggregates at OA concentrations above 1400 ppm, the surface charge is measured only at lower OA contents. | ||
Fig. 1d–h shows the high aspect ratio and flexible nanofibres obtained from sugarcane waste. The residual hemicellulose in pulp, protects the fibres' structure during the TEMPO treatment, retaining the fibres' flexibility and length. The addition of low concentrations of OA does not affect the morphology of the nanofibres. However, at the OA concentration of 1000 ppm, significant agglomeration of the nanofibres occurs, leading to the formation of bundles (Fig. 1h). It was not feasible to capture TEM images of nanofibres at higher OA concentrations, as the nanofibres aggregates were too big to fit onto the TEM grid.
3.2. Foam characteristics
The foamability and foam stability of OA and CNF mixtures were investigated to evaluate the potential synergistic behaviour between OA and CNF for obtaining a stable wet foam. These experiments were conducted for 0.1 wt% CNF dispersions containing different concentrations of OA ranging from 30 ppm up to 3200 ppm. Surface tension measurements and dilational interfacial rheology for OA and CNF mixtures were also conducted to provide better insights into the surface activity of OA treated CNF particles and their dynamic adsorption behaviour.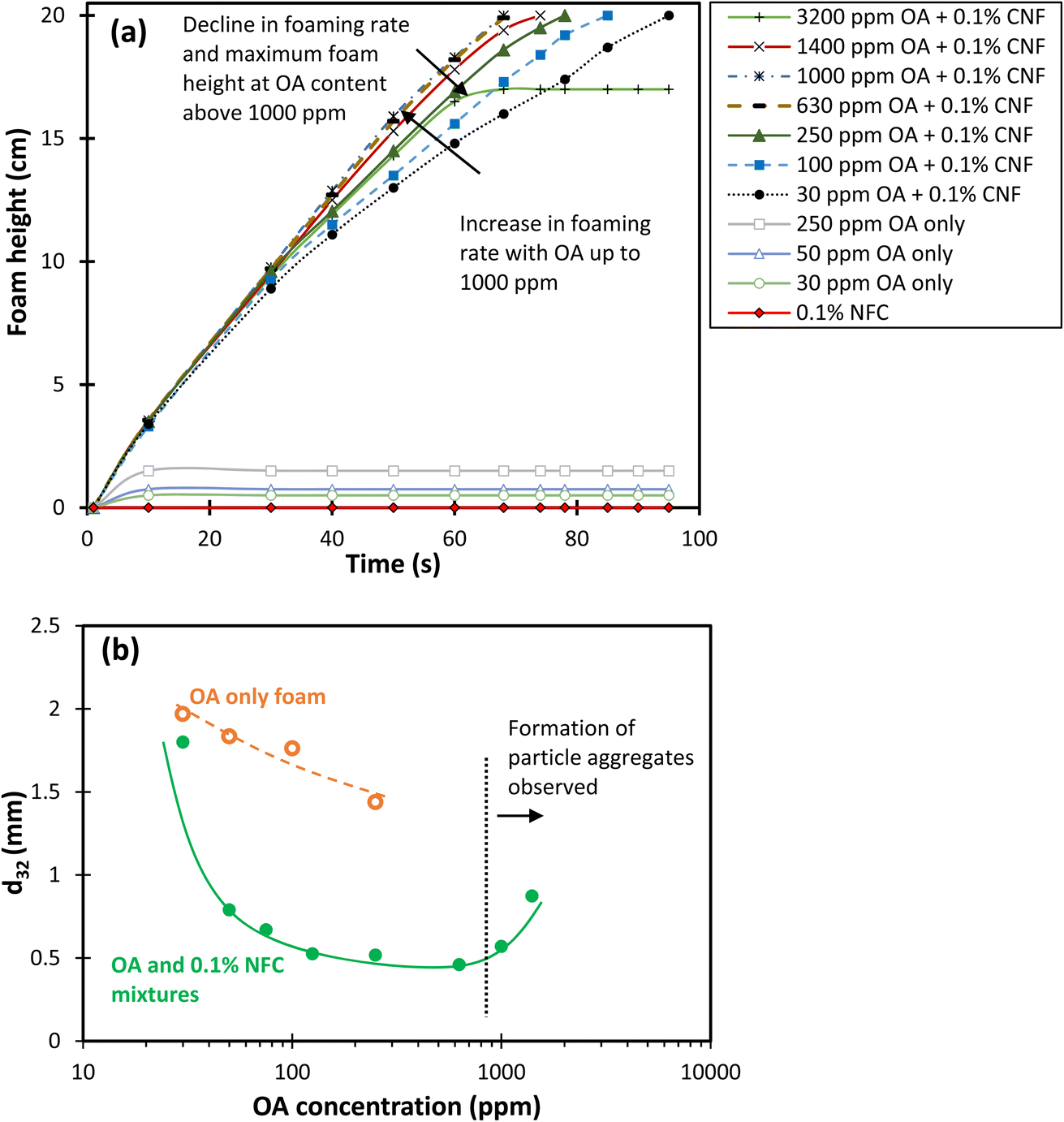 | ||
| Fig. 3 (a) Evolution of foam height during gas injection and (b) the Sauter mean bubble diameter of foams generated from CNF and OA solutions as a function of OA concentration at the maximum foamability, just before stopping the gas injection. | ||
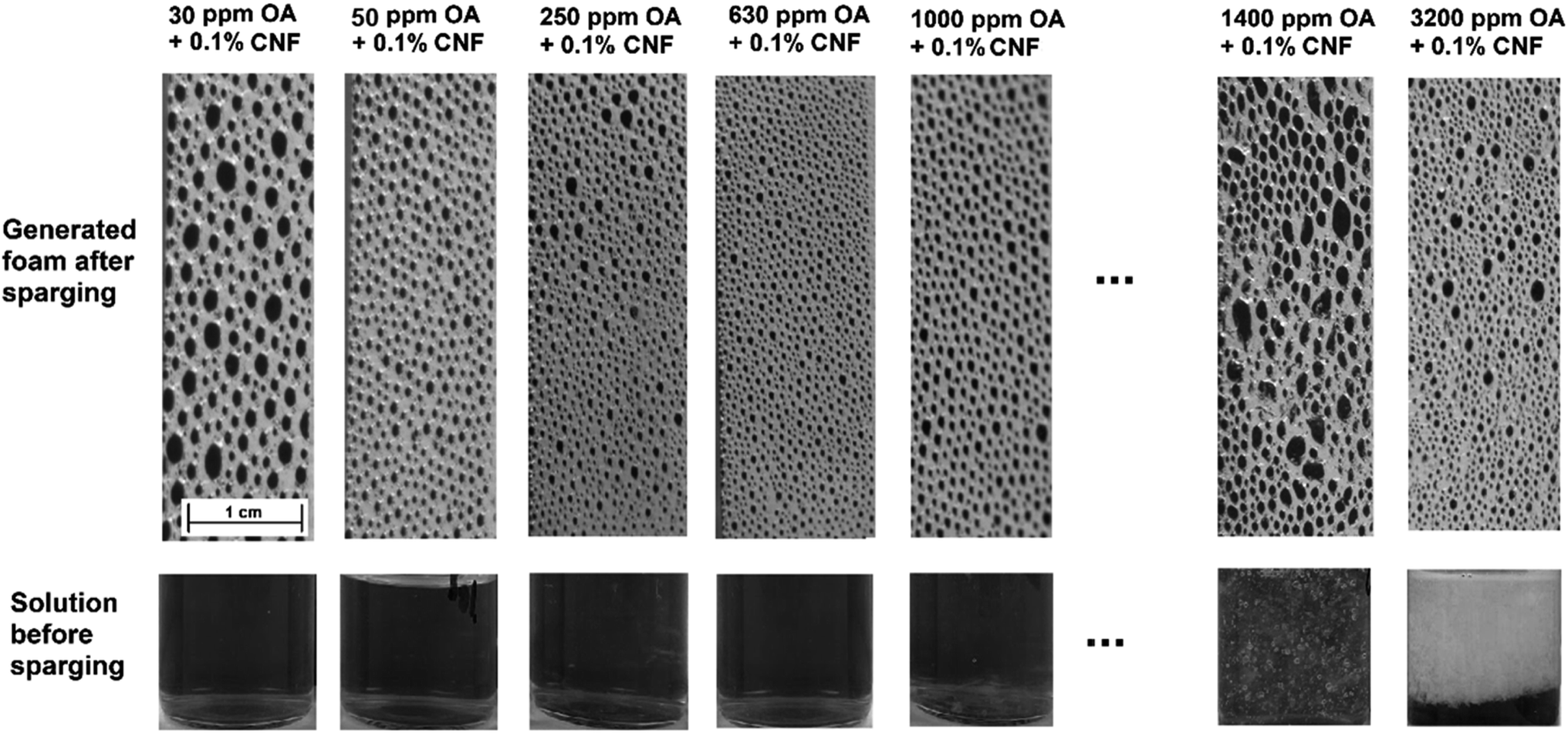 | ||
| Fig. 4 Images of 0.1 wt% CNF dispersions containing 30–3200 ppm OA before and after the foam generation through gas sparging. At 1000 ppm OA, the formation of larger aggregates was visually observed and by increasing the OA concentration to 1400 ppm and 3200 ppm, the majority of the nanofibers were precipitated, clearly evident at 3200 ppm OA. This resulted in less amount of CNF being available to stabilise the foam, resulting in a reduction in the foam stability at higher OA concentrations. | ||
In addition to the foam generation rate, the Sauter mean bubble size of the combined CNF and OA foams network at the maximum foamability was measured and is presented in Fig. 3b to provide information on the dynamic foam structure and available surface area. The data presented in Fig. 3b demonstrates that as the OA content increases from 30 ppm to 400 ppm, there is a consistent reduction in the Sauter mean bubble diameter from 1.8 mm to 0.45 mm. With further increasing the OA content from 400 to 630 ppm, the Sauter mean bubble diameter appears to stabilise, remaining relatively constant. However, upon increasing the OA content above 630 ppm, the average bubble size exhibits an increase, which could be assigned to the formation of CNF aggregates resulting from the enhanced hydrophobicity of nanofibers and reduced electrostatic repulsion between CNF. Fig. 4 shows the behaviour of 0.1 wt% CNF dispersions containing 30–3200 ppm OA before and after foam generation. It clearly illustrates the escalation in bubble size within the foam network and the precipitation of CNF at elevated OA concentrations, more than 630 ppm OA.
3.3. Foam stability
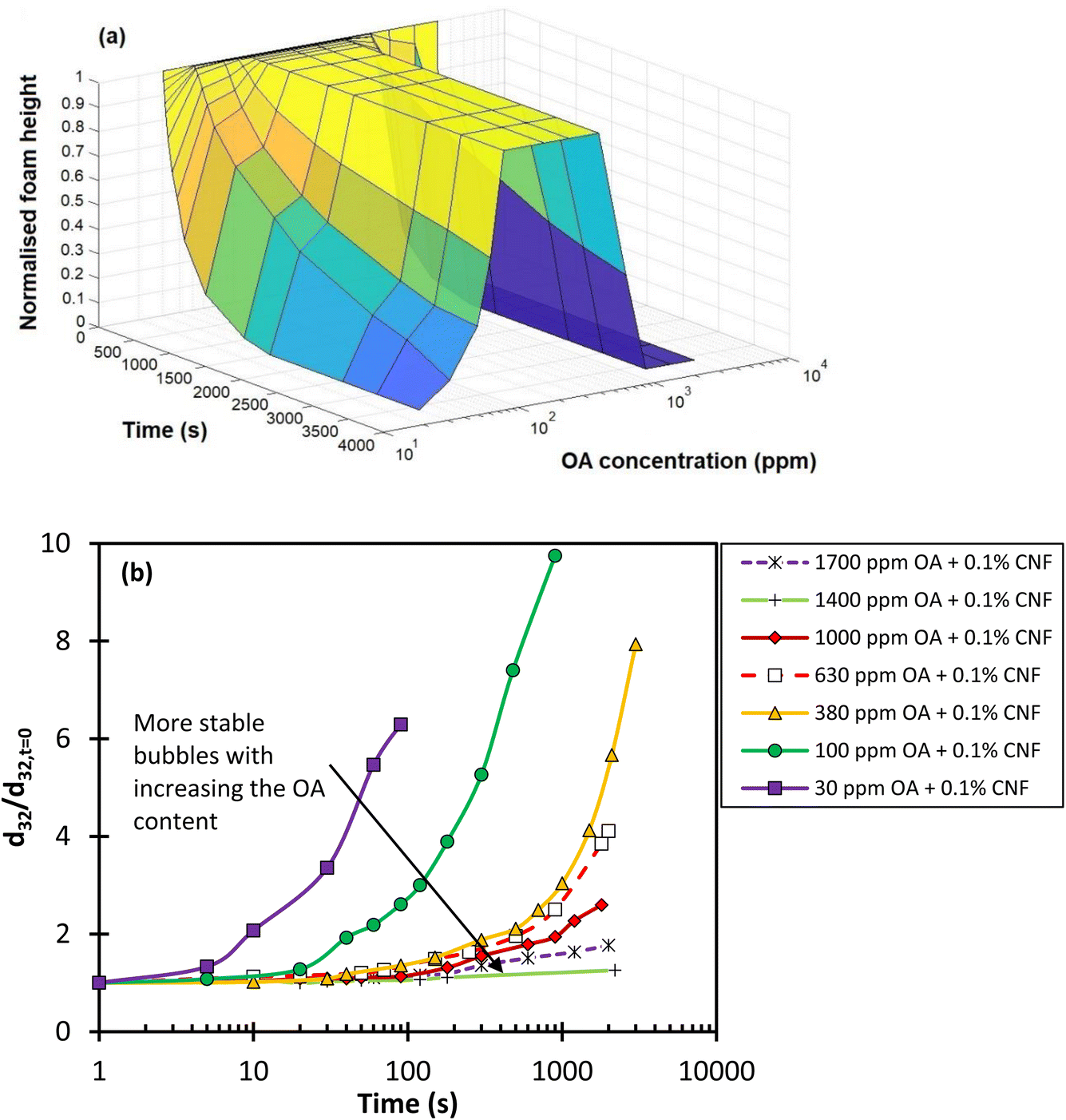
To investigate the stability of the foams formed, two characteristics were used: (I) foam decay rate (the change in the foam height over time), and (II) the evolution of the average bubble size over time. The decay rate better characterises the foam stability in applications where the total volume of the foam is important, while the change in the bubble size represents the evolution of the foam network over time. The latter is particularly important in applications where the mass transfer between two phases or particle attachment onto the surface of bubbles is the main concern.The decay rate and the evolution of the average bubble size of the generated foam from each combined CNF and OA dispersion over 2200 seconds after stopping the gas injection are presented in Fig. 5a. The foam height and bubble size were normalised with respect to the initial foam height and bubble Sauter mean diameter (d32) at the inception point when the gas injection was stopped. As shown in Fig. 5a, the normalised foam height increases with the addition of OA content, peaking at 250–630 ppm. Despite the occurrence of liquid drainage and coarsening within the 250–630 ppm OA content range (as shown in Fig. 6 for 630 ppm of OA over time), the foam network maintained its total height for over 60 minutes, eventually becoming very dry. A further increase in the OA content had the opposite impact and lowered the normalised foam height due to the aggregation of nanofibres and their phase separation in the dispersion. The data in Fig. 5 showed that the foam decayed and completely collapsed within a time range of 50 minutes to 30 seconds when OA concentrations of 1400 ppm and 3200 ppm were used, respectively.
 | ||
| Fig. 5 (a) Normalised foam height, and (b) normalised Sauter mean bubble size over time for different OA concentrations at fixed 0.1% CNF. Foam height and the average bubble size data were normalised with respect to the initial data right after stopping the gas sparging. | ||
 | ||
| Fig. 6 Evolution of foam network and normalised total foam height, liquid drainage, and foam wetness after stopping the gas injection for (a) 630 ppm OA + 0.1 wt% CNF and (b) 1400 ppm OA + 0.1 wt% CNF. At 630 ppm the total foam height remains constant while the liquid drainage is occurring, resulting in the transition to a dry foam with thin liquid films and relatively larger bubbles. At 1400 ppm, the total foam height collapses over time, but the foam wetness tends to remain at ∼0.15. | ||
Observations from Fig. 6a for 630 ppm of OA indicate that despite maintaining the foam height, the bubble size significantly increases, and the foam becomes significantly dry (foam wetness reduces). This suggests that monitoring the foam height or even foam half-life, which is commonly used for measuring foam stability, may not be a sufficient indicator of true foam stability. Therefore, it is recommended to consider the mean bubble size in the foam network in conjunction with the foam height when examining foam stability. Fig. 5b illustrates the impact of different OA contents on the evolution of the normalised Sauter mean bubble size diameter over time in a 0.1 wt% CNF solution. The data suggest that in contrast to the foam height, both the mean bubble size and the growth rate of bubble size consistently decrease with an increase in OA concentration. However, it should be noted that at OA concentrations above 630 ppm, as shown in Fig. 5a and illustrated in Fig. 6b for 1400 ppm OA, the foam height collapses and bubbles at the top part of the foam coalesce despite having smaller bubble sizes. This fails the first criterion in analysing foam stability.
Given this, a more effective way to analyse foam stability might be to consider the ratio of the normalised foam height to the normalised mean bubble size (d32) over time, for a wide range of OA concentrations. Fig. 7 provides this analysis. From this figure, it appears that 630 ppm OA offers the highest foam stability, as it maintains the highest ratio of normalised foam height to normalised bubble size.
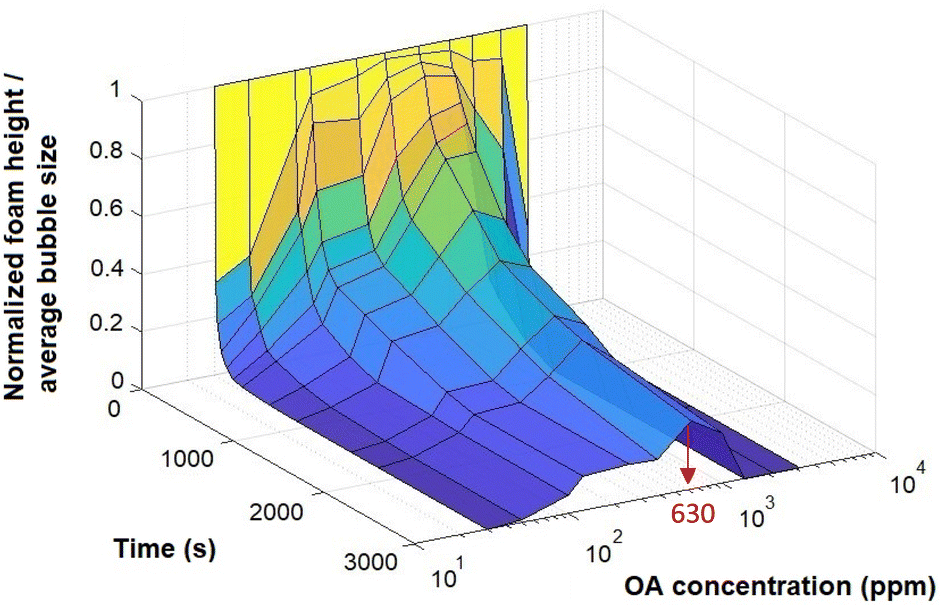 | ||
| Fig. 7 The ratio of the normalised foam height to normalised mean bubble size for 0.1 wt% CNF at different OA concentrations. | ||
3.4. Surface tension and dilational rheology
The stability of foams and emulsions is determined by the equilibrium and dynamic properties of their interfacial layers.35 These properties are directly influenced by the composition and structure of surface-active compounds adsorbed at the interface. Interfacial dilational rheology, which demonstrates dynamic interfacial reactions to changes in the interfacial area, serves as an invaluable tool for exploring the interfacial behaviour within foams and emulsions. To better understand the effect of OA on CNF, the surface activity of OA coated CNF was studied by two complementary methods – surface tension measurements and dilational interfacial rheology.Regarding the surface tension measurements, the CNF alone did not exhibit any surface activity. Fig. 8 presents the equilibrium surface tension data for OA and CNF mixtures. The data indicate that OA concentrations below 250 ppm (the concentration at which OA is soluble in MiliQ water) demonstrate negligible surface activity. However, when CNF is treated with OA, they become more surface active, thereby reducing the equilibrium surface tension. The surface tension dropped to approximately 43.7 mN m−1 when the OA content increases to 125 ppm and remains largely constant with further increases in the OA concentration up to 1000 ppm.
 | ||
| Fig. 8 Equilibrium surface tension of OA solution and mixtures of OA and 0.1 wt% CNF as a function of OA concentration. | ||
Fig. 9 compares the interfacial dilational viscoelastic modulus, represented by the change in the surface tension with respect to the change in the interfacial area at a fixed frequency of 0.01 Hz, for 250 ppm OA only and with 0.1 wt% CNF. The dynamic surface tension data shown in Fig. 10 indicate the higher surface activity of OA + TCNF compared to OA only. The increased surface activity leads to a more viscoelastic interface by improving the surface tension gradient during surface oscillation.35,36 This is evident by higher oscillation amplitude in the surface tension (Fig. 9b), of 250 ppm OA in the presence of 0.1 wt% CNF. The higher oscillation amplitude in the surface tension of 250 ppm OA + 0.1 wt% CNF is attributed to the slow relaxation of the air–water interface after the application of harmonic oscillations due to the slow adsorption of combined OA and CNF, as evidenced in Fig. 10.
 | ||
| Fig. 9 Dynamic interfacial tension and interfacial area as a function of the oscillation time for (a) 250 ppm OA and (b) 250 ppm OA + 0.1 wt% CNF at the oscillation frequency of 0.01 Hz. | ||
 | ||
| Fig. 10 Dynamic surface tension for 250 ppm OA only and 250 ppm OA + 0.1% CNF as a function of time. | ||
4. Conclusions
This study introduced a biodegradable Pickering foam reagent comprised of TEMPO-oxidised cellulose nanofibres (CNF), sourced from Australian sugarcane waste, and treated with octylamine (OA). To evaluate the impact of OA-treated CNF on the stability of wet foams, we conducted both dynamic and static foam analyses in a bubble column equipped with imaging equipment for subsequent image analysis. We proposed a new criterion for assessing foam stability that considers not only the traditional measure of foam height but also the mean bubble size within the foam network. Our findings showed that the foam stability increased with the addition of OA up to an optimum concentration of 630 ppm. Beyond this concentration, however, foam stability declined, a trend we attributed to the aggregation of CNF in the presence of high OA content. This reduced the amount of CNF available in the foam network. The formation of aggregates was visually and microscopically via TEM confirmed.The stabilising effect of OA was attributed to the increased surface hydrophobicity and reduced electrostatic repulsions between CNF, promoting the adsorption of CNF at the air–water interface. Furthermore, we examined the interfacial properties of the adsorbed layers of OA-treated CNF using a bubble/drop profile analysis technique. Even the addition of as little as 0.1 wt% CNF significantly enhanced the surface activity of OA, reducing its surface tension at the air–liquid interface by 19–40%, depending on the OA concentration and enhancing its interfacial dilational viscoelasticity.
Overall, this study advances our understanding of using cellulose nanofibres for the formation and stabilisation of aqueous foams. Promoting the hydrophobic characteristics of nanocellulose facilitates its accumulation at the air–water interface. However, maintaining an appropriate surface charge density and surface hydrophobicity on the nanocellulose are equally important to prevent clumping and promote a repulsive disjoining pressure.
The future work could focus on the development of more environmentally friendly and sustainable materials by using bio-based foaming agents and more environmentally friendly reagents for the surface modification of cellulose nanofibers. Future work will also explore the use of sugarcane-derived CNFs as frothers or collectors in the processing of critical minerals and co-surfactants in the environmental remediation of PFAS, offering a sustainable alternative to conventional petroleum-based reagents.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the financial support from the Australian Research Council (ARC) through the ARC Centre of Excellence for Enabling Eco-efficient Beneficiation of Minerals, grant number CE200100009. The authors also acknowledge the contribution of Dugalunji Aboriginal Corporation on behalf of the Indjalandji-Dhidhanu people using their equipment and in-kind support. N. A. acknowledges the financial support from Advance Queensland Industry Fellowship (AQIRF128-2020) and support received from industry partners Manildra Harwood Sugar trading as Sunshine Sugar.References
- P. Amani, S. I. Karakashev, N. A. Grozev, S. S. Simeonova, R. Miller, V. Rudolph and M. Firouzi, Adv. Colloid Interface Sci., 2021, 295, 102490 CrossRef CAS PubMed.
- T. Buckley, T. Vuong, K. Karanam, P. H. N. Vo, P. Shukla, M. Firouzi and V. Rudolph, Water Res., 2023, 239, 120028 CrossRef CAS PubMed.
- T. Buckley, K. Karanam, X. Xu, P. Shukla, M. Firouzi and V. Rudolph, Sep. Purif. Technol., 2022, 286, 120508 CrossRef CAS.
- T. Buckley, K. Karanam, H. Han, H. N. P. Vo, P. Shukla, M. Firouzi and V. Rudolph, Water Res., 2023, 230, 119532 CrossRef CAS PubMed.
- P. Amani and M. Firouzi, Gas Sci. Eng., 2023, 114, 204977 CrossRef CAS.
- P. Amani, V. Rudolph, S. Hurter and M. Firouzi, Fuel, 2022, 324, 124675 CrossRef CAS.
- R. Prud'homme, Foams: Theory: Measurements: Applications, Routledge, 2017 Search PubMed.
- G. Bournival, S. Ata and G. J. Jameson, Miner. Process. Extr. Metall. Rev., 2017, 38, 366–387 CrossRef CAS.
- P. Amani, R. Miller, A. Javadi and M. Firouzi, Adv. Colloid Interface Sci., 2022, 301, 102606 CrossRef CAS PubMed.
- G. Z. Kyzas and K. A. Matis, Processes, 2019, 7, 138 CrossRef CAS.
- J. A. Clark and E. E. Santiso, Engineering, 2018, 4, 336–342 CrossRef CAS.
- X.-h. Chen, Y.-h. Hu, H. Peng and X.-f. Cao, J. Cent. South Univ., 2015, 22, 495–501 CrossRef CAS.
- H. Park and L. Wang, Miner. Eng., 2018, 127, 74–80 CrossRef CAS.
- T. Li, C. Chen, A. H. Brozena, J. Y. Zhu, L. Xu, C. Driemeier, J. Dai, O. J. Rojas, A. Isogai, L. Wågberg and L. Hu, Nature, 2021, 590, 47–56 CrossRef CAS PubMed.
- B. Wei, H. Li, Q. Li, Y. Wen, L. Sun, P. Wei, W. Pu and Y. Li, Langmuir, 2017, 33, 5127–5139 CrossRef CAS PubMed.
- Y. Ye, L. Yu, E. Lizundia, Y. Zhu, C. Chen and F. Jiang, Chem. Rev., 2023, 123, 9204–9264 CrossRef CAS PubMed.
- S. J. Eichhorn, A. Etale, J. Wang, L. A. Berglund, Y. Li, Y. Cai, C. Chen, E. D. Cranston, M. A. Johns, Z. Fang, G. Li, L. Hu, M. Khandelwal, K. Y. Lee, K. Oksman, S. Pinitsoontorn, F. Quero, A. Sebastian, M. M. Titirici, Z. Xu, S. Vignolini and B. Frka-Petesic, J. Mater. Sci., 2022, 57, 5697–5767 CrossRef CAS.
- T. Hjelt, J. A. Ketoja, H. Kiiskinen, A. I. Koponen and E. Pääkkönen, J. Dispersion Sci. Technol., 2022, 43, 1462–1497 CrossRef CAS.
- R. Abidnejad, M. Beaumont, B. L. Tardy, B. D. Mattos and O. J. Rojas, ACS Nano, 2021, 15, 19712–19721 CrossRef CAS PubMed.
- N. T. Cervin, L. Andersson, J. B. S. Ng, P. Olin, L. Bergström and L. Wågberg, Biomacromolecules, 2013, 14, 503–511 CrossRef CAS PubMed.
- K. S. Gordeyeva, A. B. Fall, S. Hall, B. Wicklein and L. Bergström, J. Colloid Interface Sci., 2016, 472, 44–51 CrossRef CAS PubMed.
- H. A. Wege, S. Kim, V. N. Paunov, Q. Zhong and O. D. Velev, Langmuir, 2008, 24, 9245–9253 CrossRef CAS PubMed.
- N. T. Cervin, E. Johansson, J.-W. Benjamins and L. Wågberg, Biomacromolecules, 2015, 16, 822–831 CrossRef CAS PubMed.
- N. Amiralian, P. K. Annamalai, P. Memmott, E. Taran, S. Schmidt and D. J. Martin, RSC Adv., 2015, 5, 32124–32132 RSC.
- N. Amiralian, M. Mustapic, M. S. A. Hossain, C. Wang, M. Konarova, J. Tang, J. Na, A. Khan and A. Rowan, J. Hazard. Mater., 2020, 394, 122571 CrossRef CAS PubMed.
- T. Saito, S. Kimura, Y. Nishiyama and A. Isogai, Biomacromolecules, 2007, 8, 2485–2491 CrossRef CAS PubMed.
- D. da Silva Perez, S. Montanari and M. R. Vignon, Biomacromolecules, 2003, 4, 1417–1425 CrossRef CAS PubMed.
- Y. Habibi, H. Chanzy and M. R. Vignon, Cellulose, 2006, 13, 679–687 CrossRef CAS.
- N. Mucic, A. Javadi, N. Kovalchuk, E. Aksenenko and R. Miller, Adv. Colloid Interface Sci., 2011, 168, 167–178 CrossRef CAS PubMed.
- J. Lucassen and M. Van Den Tempel, Chem. Eng. Sci., 1972, 27, 1283–1291 CrossRef CAS.
- J. Levanič, M. Gericke, T. Heinze, I. Poljanšek and P. Oven, Cellulose, 2020, 27, 2053–2068 CrossRef.
- D. J. Mendoza, C. Browne, V. S. Raghuwanshi, G. P. Simon and G. Garnier, Carbohydr. Polym., 2019, 226, 115292 CrossRef CAS PubMed.
- G. Paladini, V. Venuti, V. Crupi, D. Majolino, A. Fiorati and C. Punta, Polymers, 2021, 13(4), 528 CrossRef CAS PubMed.
- N. T. Cervin, L. a. Andersson, J. B. S. Ng, P. Olin, L. Bergström and L. Wagberg, Biomacromolecules, 2013, 14, 503–511 CrossRef CAS PubMed.
- P. Amani, R. Miller, S. Ata, S. Hurter, V. Rudolph and M. Firouzi, J. Ind. Eng. Chem., 2020, 92, 174–183 CrossRef CAS.
- M. Firouzi, V. I. Kovalchuk, G. Loglio and R. Miller, Curr. Opin. Colloid Interface Sci., 2022, 57, 101538 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |