
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigating structural properties and reaction mechanism of non-stoichiometric spinel LNMO via solid state NMR†
Nahom Enkubahri
Asres
 ab,
Naiara
Etxebarria
a,
Iciar
Monterrubio
ab,
Damien
Saurel
a,
Christian Fink
Elkjær
c,
Montse
Casas-Cabanas
ad,
Marine
Reynaud
a,
Marcus
Fehse
*a and
Juan Miguel
López del Amo
*a
ab,
Naiara
Etxebarria
a,
Iciar
Monterrubio
ab,
Damien
Saurel
a,
Christian Fink
Elkjær
c,
Montse
Casas-Cabanas
ad,
Marine
Reynaud
a,
Marcus
Fehse
*a and
Juan Miguel
López del Amo
*a
aCenter for Cooperative Research on Alternative Energies (CIC energiGUNE), Basque Research and Technology Alliance (BRTA), Alava Technology Park, Albert Einstein 48, 01510, Vitoria-Gasteiz, Spain. E-mail: marcus.fehse@umontpellier.fr; jmlopez@cicenergigune.com
bDepartment of Organic and Inorganic Chemistry, Faculty of Science and Technology, University of the Basque Country, UPV/EHU, B° Sarriena s/n, 48940 Leioa, Spain
cHaldor A/S, Haldor Topsøes Allé 1, 2800 Kgs. Lyngby, Denmark
dIkerbasque – Basque Foundation for Science, Maria Diaz de Haro 3, 48013 Bilbao, Spain
First published on 14th November 2023
Abstract
In this study, 6,7Li solid state nuclear magnetic resonance (ssNMR) has been used to characterize local disorder in LiNi0.5Mn1.5O4 (LNMO) spinel cathodes and to understand their implications for the phase transformation mechanism during electrochemical cycling vs. lithium. By comparing NMR spectra of LNMO samples with slightly varying transition metal stoichiometries, we demonstrate the high sensitivity of ssNMR in the indirect detection of transition metal order and its ability to identify disorder induced by stoichiometric variations. Secondly, we investigate the lithiation reaction mechanism of the non-stoichiometric transition metal-ordered LNMO spinel cathode by ex situ7Li NMR and highlight that its reaction process involves primarily two successive biphasic reactions. However, our results suggest that stoichiometrically induced transition metal disorder promotes a mixed solid solution and biphasic reaction mechanism. Besides an extended solid solution region, we evince that the biphasic reaction involves the presence of intermediate transient species that make it possible to bridge the miscibility gap between the Li1 and Li0.5 phases. These results go beyond our operando XRD results, underlining that local structural analysis by 7Li ssNMR can provide valuable additional insights into the complex reaction mechanism that are difficult to access by other techniques.
1 Introduction
There has been a recent resurgence of interest in the spinel LNMO positive electrode, specifically aiming to establish a systematic comprehension of the relationship between its structural properties and electrochemical performance.1 At the same time, the study of phase transitions during charging and discharging, and the role of structural order and disorder therein, are of great relevance in the field of electrochemical energy storage materials.2These are crucial not only for understanding the prevailing redox mechanism but also for determining the battery performance, including rate capability and cyclability. Despite more than a decade of research on high-voltage spinel, LiNi0.5Mn1.5O4 (LNMO) has yet to unleash its inherent advantages of high energy density (464 W h kg−1), high operating potential of 4.7 V (vs. Li/Li+) and high rate capability in commercial battery applications.3,4
LNMO adopts a spinel arrangement, where Li+ ions are situated at the tetrahedral sites, and the transition metal ions occupy the octahedral sites within an oxygen cubic close-packed lattice. This configuration promotes rapid lithium diffusion in all three dimensions within the framework.5,6 The stoichiometric ideal composition of the material entails Ni and Mn to be in the +2 and +4 oxidation states, respectively. In this ideal stoichiometry the extraction of Li ions is achieved through the oxidation of Ni2+/Ni3+/Ni4+, while Mn4+ ions remain electrochemically inactive. The arrangement of transition metal (TM) cations in the octahedral sites of LiNi0.5Mn1.5O4 can vary depending on the synthesis conditions.7,8 In fact, Ni and Mn can either exhibit ordering in the 4a and 12d sites, respectively within a P4332 space group or be randomly distributed in the 16d sites of a Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m-type unit cell.9,10 The transition metal ordering has been reported to take place when calcining at ≈700 °C.10,11 Previous studies have evinced that elevating the calcination temperatures above the transition temperature results in the partial reduction of Mn(IV) to Mn(III), that can be observed from a progressively growing Mn3+/Mn4+ redox plateau between 4–4.3 V vs. Li+/Li in the electrochemical signature curve.12–16
m-type unit cell.9,10 The transition metal ordering has been reported to take place when calcining at ≈700 °C.10,11 Previous studies have evinced that elevating the calcination temperatures above the transition temperature results in the partial reduction of Mn(IV) to Mn(III), that can be observed from a progressively growing Mn3+/Mn4+ redox plateau between 4–4.3 V vs. Li+/Li in the electrochemical signature curve.12–16
Recent consensus is that the reduction of Mn4+ to Mn3+ in LiNi(II)0.5−yMn(IV)1.5−yMn(III)2yO4 serves as a compensatory mechanism for the formation of a secondary rock salt phase resulting in Ni2+ loss in the spinel, which also explains the release of oxygen, all without necessitating the presence of oxygen defects in the spinel phase.1
Previous studies from our group showed that TM ordering in LNMO leads to the formation of nanosized antiphase domains that are detrimental to Li+ transport, while site mixing in ordered samples leads to performance comparable to that of fully disordered samples.15,16 Recently, Stüble introduced a more accurate categorisation of “ordered” and “disordered” phases in LNMO spinel including both TM order and TM stoichiometry, rectifying the misconception present in some studies that cation order and Mn(III) content are strictly correlated.1
It is general consensus that electrochemical delithiation reaction of LNMO occurs via at least three thermodynamically stable phases, which are the fully lithiated LiNi0.5Mn1.5O4 (Li1, Ni2+), the half-lithiated Li0.5Ni0.5Mn1.5O4 (Li0.5, Ni3+), and the fully delithiated Ni0.5Mn1.5O4 (Li0, Ni4+) compositions. The ordered LixNi0.5Mn1.5O4 undergoes two distinct phase transitions within the range of 1 ≥ x ≥ 0, with their redox potential at around of 4.70 V (Li1/Li0.5) and 4.74 V (Li0.5/Li0) versus Li/Li+, whereas the disordered spinel only undergoes one phase transition from Li0.5 to Li0 and shows solid solution behaviour between Li1 and Li0.5.6,9,17,18 This is believed to be one of the reasons for the superior rate performance and/or cycle life of disordered LNMO in comparison to the ordered LNMO, as reported in previous studies.1,14,16,18,19 By varying the synthetic atmosphere and annealing temperature, it was previously shown that small amount of atomic mixing in a partially ordered spinel, was sufficient to significantly improve electrochemical performance aligning to those of completely disordered spinel.20 Similar observations were also reported by Stüble et al. and DeGennaro et al. that the partially ordered and disordered shows almost similar electrochemical performance.1,21 The reaction mechanism and kinetics of LNMO (de)lithiation are significantly affected by various factors, particularly Mn/Ni ordering and the content of Mn3+, i.e. TM stoichiometry, which are not necessarily coupled as recently shown.1
Neutron powder diffraction (NPD) and Raman spectroscopy are well established techniques to detect TM order in LNMO spinel, nevertheless these techniques are unable to precisely quantify their proliferation within the material. While NPD allows quantitative estimation of TM stoichiometry as well as their position within the lattice, it does not provide information on the disorder of local environments experienced by Li nor on the TM oxidation state. Over the past decade, solid-state NMR spectroscopy has emerged as a valuable technique for studying the chemical environments of lithium (Li+) and sodium (Na+) in cathode materials for batteries.22–27 The sensitivity of 6,7Li magic-angle spinning (MAS) NMR is widely recognized for its ability to detect and characterize the local surroundings of lithium atoms. This technique enables direct assessment of various transition metal (TM) arrangements around lithium, allowing for the quantification and analysis of these distinct environments. In a previous study, Cabana et al. utilized 6Li MAS NMR spectroscopy to examine LiNi0.5Mn1.5O4 samples synthesized at different temperatures, aiming to discern varying levels of ordering. Similarly, Duncan et al. investigated the impact of Ni deficiency in a class of spinel LNMOs LiNixMn2−xO4 (0.3 ≤ x ≤ 0.5) employing 6Li NMR to analyze the effect in local ordering of transition metal ions.10,14 The 6,7Li MAS NMR technique has also been extensively employed to characterise the chemical structure of positive electrode materials in their pristine, partially lithiated, and delithiated states. This technique provides direct insights into the behaviour of mobile Li+ ions, which are directly involved in electrochemical processes. Additionally, this technique is quantitative, enabling the tracking and quantification of the species that are either eliminated or retained in the structure during battery cycling.
In our recent operando X-ray absorption study we have demonstrated that Ni-deficient partially TM ordered and disordered LNMO undergo similar sequences of redox reactions but differences in their local structure evolution were evinced. The aim of this work is to investigate the local and long-range structure of spinel LNMO in order to understand the role of stoichiometric induced TM disorder on the electrochemical reaction (phase transformation) mechanism. With this aim in mind, we have studied a non-stoichiometric (Ni-deficient) “ordered” spinel LNMO alongside with a near stoichiometric “ordered” LNMO sample by ex situ7Li NMR and operando XRD to understand the phase transformation mechanism at different lithiation stages during charging and discharging and to gain insight into the local disorder induced by Ni deficiency. Neutron powder diffraction (NPD) was used to confirm the cation arrangements, while 6,7Li magic angle spinning (MAS) NMR was used to study the local environments.
2 Experimental
For the labelling of the LNMO samples investigated in this study we use a nomenclature in which the description of LNMO is followed by a letter D or O indicating whether the sample is predominantly transition metal disordered or ordered, this is followed by a two digit suffix stating the Ni stoichiometry in the formula unit per 1 Li, according to the classification recently proposed by Stüble et al.1 The description of LNMO-O-45 would hence translate to a predominantly transition metal ordered spinel with LiNi0.45Mn1.55O4. Two high-purity samples of TM ordered LNMO with two different Mn/Ni stoichiometries (LNMO-O-45 and LNMO-O-48) were provided by Topsoe.2.1 Material characterisation
Neutron powder diffraction (NPD) of pristine and ex situ cycled TM ordered (LNMO-O) were carried out at D1B and D2B diffractometer at the Institute Laue-Langevin (ILL), Grenoble France28–30 using a wavelength of 1.288 and 1.595 Å, respectively. The use of different beamlines resulted in different angular resolution. The samples were mounted on a 6 mm diameter vanadium sample holder and placed on a carousel that allows automatic sample change. The neutron diffraction patterns were collected at RT with a high statistic to enhance the data accuracy. Rietveld analysis was carried out using the FullProf suite.31Raman spectra of the material was recorded with a Renishaw spectrometer (Nanonics Multiview 2000) operating with an excitation wave-length of 532 nm. Spectra were acquired with 15 s of exposition time of the laser beam to the sample.
The magnetic properties have been characterised using the ACMS option of a PPMS Evercool II instrument from Quantum Design. Magnetic susceptibility has been performed in AC mode, from 5 to 300 K , with an AC field of 10 Oe (see Fig. S1a (left)†). Magnetisation versus magnetic field has been performed in DC mode at 5 K, scanning DC magnetic field from −4 to 4 T (see Fig. S1b (right)†); the magnetisation at 4 T is considered to be the value at saturation. Please see SI for more detail on the method used for extracting Mn3+ excess from magnetisation at saturation.
Micrographs were taken on a Thermo Fisher Quanta 200 FEG high-resolution Scanning Electron Microscope (SEM). The working voltages of the Quanta 200FEG 20 kV and Everhart–Thornley detector (ETD) was used for imaging. Particle sizes were evaluated with a MasterSizer 3000 (Malvern Panalytical, Netherlands).
The samples were analyzed with inductively coupled plasma – atomic emission spectroscopy (ICP-AES) using a Horiba Ultima 2 (Jobin Yvon, Longjumeau, France) in conjunction with a AS500 autosampler and Activanalyst software (version 5.4). The ICP-AES operating conditions were as follows: 1.0 kW of RF power, 13 l min−1 of a plasma-gas flow rate, 0.2 l min−1 of a sheath-gas flow rate and 0.25 l min−1 of a nebulizer-gas flow rate. Solutions were introduced into the plasma torch using nebulizer and a cyclonic type of spray chamber at a flow rate of 0.87 ml min−1. Calibration solutions were prepared using a commercial calibration standards of Li, Ni and Mn (Scharlab, Barcelona, Spain) at concentration of 1000 mg l−1. Nitric acid 69% analytical grade, Hydrochloric acid 37% Ultratrace from Scharlab (Barcelona, Spain) and Ultrapure Water from Fischer Scientific (Waltham, Massachusetts, USA) were used for dilutions. The most prominent analytical lines of Li 670.784 nm, Ni 216.556 nm and Mn 257.610 nm were selected for measurements. Concentrations of these elements were quantified using four-point external calibration curve within the concentration range of 0.01–100 mg l−1.
6,7Li Magic Angle Spinning solid state Nuclear Magnetic Resonance (MAS NMR) experiments were performed on a Bruker 300WB spectrometer charged to a field of 4.69 T equipped with a standard 1.3 mm MAS probe. Spinning frequencies were set to 50 kHz. A rotor synchronised spin-echo pulse sequence was used with typical 90 and 180° pulses of 1.3 and 2.6 μs, respectively. A recycle delay of 0.7 s and 2 s was used and around 89k and 1k scans where typically acquired in a 6Li and 7Li NMR experiment, respectively. The recycle delays were set to ensure full relaxation of the spins. The spectra were referenced to a 1 M solution of LiCl (set at 0 ppm). The spectra were analysed and deconvoluted using the Dmfit software package.32
6Li exhibits smaller quadrupolar and dipolar couplings to the nuclear and electronic spins compared to7Li due to its smaller quadrupole moment and gyromagnetic ratio. As a result, 6Li typically produces higher-resolution NMR spectra with fewer spinning sidebands, enabling a clearer understanding of the local environment. Therefore, we measured 6Li NMR spectra in our studies of the pristine materials. However, one drawback of 6Li is its significantly lower natural abundance. Typically for the small sample volumes taken from the cycled cells (i.e. ex situ samples), 6Li enrichment is necessary. Furthermore, no distinction was observed in the NMR resonance for the pristine material between 7Li and 6Li, which had no impact on the analysis. Consequently, 7Li NMR was utilized for the ex situ investigation of LNMO-O-45.
2.2 Electrode formulation and cell assembly
For the ex situ7Li NMR and operando XRD experiments, LNMO powder samples were mixed in NMP based slurry together with PVdF binder (Kynar HSV900; Arkema, France) and carbon additive (C65; Imerys, Switzerland) in ratio of 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:4 and deposited on single side carbon coated Al foil (Armor, France) with active material loading around 10.6 mg cm−2. After cutting, pressing and drying, electrodes were mounted in CR2032 coin cells for ex situ NMR samples. All reported C-rates are relative to theoretical capacity of 148 mA h g−1 The cells were precycled for two complete cycles and then stopped at the desired SoC, the corresponding electrochemical cycling profiles of these ex situ samples are depicted in Fig. S6 and S7 (ESI).† The voltage of the cells were held at the desired SoC until they were rapidly disassembled and the recovered electrode material was transferred to the NMR rotor, to minimise relaxation previously reported.33 For the operando XRD measurements, the slurry was prepared as for NMR in ratio of 84:8:8 and coated on 6 μm Al foil current collector with active material loading around 6.1 mg. After cutting and drying, the positive electrode was assembled in specially designed in situ cell for operando XRD measurements, recently described elsewhere.34 The cells were assembled in an argon-filled glove-box (≤0.1 ppm H2O and O2) with a 16 mm diameter LNMO positive electrode, a quartz fiber separator (QM-A; Whatman) and a 16 mm diameter lithium disc counter-electrode, using LP30 electrolyte.
:3:4 and deposited on single side carbon coated Al foil (Armor, France) with active material loading around 10.6 mg cm−2. After cutting, pressing and drying, electrodes were mounted in CR2032 coin cells for ex situ NMR samples. All reported C-rates are relative to theoretical capacity of 148 mA h g−1 The cells were precycled for two complete cycles and then stopped at the desired SoC, the corresponding electrochemical cycling profiles of these ex situ samples are depicted in Fig. S6 and S7 (ESI).† The voltage of the cells were held at the desired SoC until they were rapidly disassembled and the recovered electrode material was transferred to the NMR rotor, to minimise relaxation previously reported.33 For the operando XRD measurements, the slurry was prepared as for NMR in ratio of 84:8:8 and coated on 6 μm Al foil current collector with active material loading around 6.1 mg. After cutting and drying, the positive electrode was assembled in specially designed in situ cell for operando XRD measurements, recently described elsewhere.34 The cells were assembled in an argon-filled glove-box (≤0.1 ppm H2O and O2) with a 16 mm diameter LNMO positive electrode, a quartz fiber separator (QM-A; Whatman) and a 16 mm diameter lithium disc counter-electrode, using LP30 electrolyte.
2.3 Operando XRD
Operando XRD measurements were performed on a lab-scale Bragg–Brentano diffractometer (Bruker D8 advance, with Cu Kα1,2 tube source and LYNXEYE 1D detector), using a custom-made in situ electrochemical cell equipped with Beryllium Windows, previously described elsewhere.34 Galvanostatic cycling with potential limitation was performed using a Bio-Logic ST-150 potentiostat at a C/n rate (expressed as 1 mol of Li reacted in n hours per mole of LNMO). The electrochemical cycling during operando measurements were carried out at C/20 rate within voltage window of 3.5 to 5 V vs. Li+/Li in a 2θ range of 8–22° and 35–52°. 20 min patterns were collected sequentially with an angular step size of 0.1°. The electrochemical tests were carried out at room temperature.3 Results
In the first part, see Section 3.1, two pristine LNMO samples are compared by different material characterisation techniques (NPD, ICP, SEM, Raman, magnetic measurements and 6Li NMR) to highlight the influence of slight deviations of the TM stoichiometry on the TM order and its effect on the electrochemical properties. In the second part we characterize the lithiation reaction mechanism of one of the previously examined samples from local and crystal structure perspective by employing ex situ7Li NMR and operando diffraction analysis, see Section 3.2. These findings are further discussed in Section 4.3.1 Comparison of two transition metal ordered pristine LNMO
In a first step, two predominantly TM ordered LNMO samples were analyzed by NPD. It is a highly informative technique since it allows not only to determine phase purity and crystal phases but, more importantly, to elucidate the presence of TM ordering and estimate TM stoichiometries, thanks to the sufficiently different neutron cross section of Ni and Mn. In Fig. 1(a) the NPD patterns and Rietveld refinements of the two pristine materials are compared. Selected fitting parameters are presented in Table 1. Single phase TM ordered space group P4332 with antiphase model as previously proposed16 was used for the refinement of both samples. The good agreement of the calculated with the experimental pattern underlines the phase purity and the predominantly TM-ordered nature of both LNMO samples. Nevertheless, faint features at 35.0 (d = 2.14) and 45.2° (d = 2.07) which are not comprised in the spinel phase refinement can be made out in the bottom pattern (indicated by arrow markers) which could hint presence of a rocksalt impurity. For both samples, a Ni-deficient stoichiometry was determined from these NPD refinements, resulting in Mn/Ni ratios higher than stoichiometrically expected value of 3/1. In accordance with the nomenclature proposed by Stüble et al.,1 the Ni-deficient LNMO samples with Mn/Ni ratios of 3.41 and 3.14 will be referred to as LNMO-O-45 and LNMO-O-48, respectively. The refinement also reveals larger crystal domains and larger antiphase domains for the LNMO-O-48 sample, which is consistent with the fact that a higher degree of order is achieved in a more stoichiometric compositions. The full set of refinement parameters are reported in Table S2.† | ||
| Fig. 1 (a) Rietveld refinement of Neutron diffraction pattern for LNMO-O-45 (top) and LNMO-O-48 (bottom), respectively. Arrow markers in the bottom plot indicate features not comprised in the spinel phase refinement. (b) Raman spectra of pristine LNMO-O-45 and LNMO-O-48. (c) Comparison of Mn3+ content based on magnetic saturation (grey bars) and Curie temperature (red markers) of LNMO-O-45 and LNMO-O-48. | ||
ICP analysis was used to support stoichiometry determination. The analysis revealed that the ratio of sum of both transition metals to Li is close to the expected value of 2/1 for both samples, which underlines the assumption that the Ni deficiency is compensated by a Mn excess to maintain charge neutrality. Furthermore, ICP analysis yields to Mn/Ni ratios of 3.4(2) and 3.0(2), for LNMO-O-45 and LNMO-O-48, respectively. This former value corresponds to an excess of Mn of y = 0.05 in LiNi(II)0.5−yMn(IV)1.5−yMn(III)2yO4 for sample LNMO-O-45, in agreement with NPD data. For sample LNMO-O-48, no Ni deficiency was evinced in the overall sample, which could be due to the presence of minor Ni rich rocksalt segregation besides the LNMO-O-48 spinel phase, as hinted by the faint additional NPD feature, vide supra.
Scanning Electron Microscopy (SEM) has been carried out to compare particle morphology and size of the two pristine LNMO materials. The micrographs depicted in Fig. S2† illustrate that both materials have similar primary and secondary particle size and shape, consisting of spherical particles with an average diameter of 8 μm which consist of smaller polygonal shaped particles with average size of 1 μm.
Raman spectra of the two pristine TM ordered LNMO samples are depicted in Fig. 1(b) showing that both spectra largely superimpose in peak position and shape. This structural fingerprint indicates that both LNMO samples have same local structure as well as similar degree of structural order. Moreover, both spectra reveal the unique features of TM ordered LNMO at 223, 242 as well as the 593 cm−1.35 In the case of the LNMO-O-48 these features stand out more clearly than in the LNMO-O-45. Another noteworthy difference between the two spectra is the more pronounced shoulder peak at ≈665 cm−1, which has been attributed to bending vibrations of the TM in the 16d sites of the disordered Fdm phase.33 The discrepancy between NPD and Raman are a result of the different length scales probed, moreover it should be noted that Raman spectroscopy does not provide quantitative information on TM order.
To maintain the charge neutrality, the Ni-deficient stoichiometry entails an excess of Mn in combination with the reduction of Mn4+ to Mn3+, resulting in the following formula; LiNi(II)0.5−yMn(IV)1.5−yMn(III)2yO4. For each deficient Ni2+, two Mn3+ are formed. The evaluation of the Mn3+ content is hence an indirect way of probing the TM composition. Due to its difference in magnetic moment, the Mn3+ can be probed via its magnetisation saturation. We found ≈0.01 Mn3+ content in the LNMO-O-48 sample and 0.19 of Mn3+ for the LNMO-O-45, see Fig. 1(c). This difference is in qualitative agreement with the NPD and ICP results, which point towards stronger Ni deficiency and hence more elevated Mn3+ content in the LNMO-O-45 compared to LNMO-O-48. While for the LNMO-O-48 the determined Mn excess (0.01) lies within stoichiometry determined by ICP and NPD, the Mn excess determined based on Mn saturation of LNMO-O-45 exceeds those indicated by ICP and NPD (y = 0.05 yielding to expected Mn3+ content of 0.1). This deviation could be due to the fact that the orbital moment is not entirely quenched or to the presence of amorphous Mn(III)-oxide segregation.36 Interestingly, these magnetic measurements also reveal an elevated Curie temperature TC for the LNMO-O-48 which suggests a higher degree of TM ordering, according to previous study.37
The impact of TM stoichiometry and ordering can also be observed in the electrochemical signature of the LNMO sample. The electrochemical cycling curve of the LNMO-O-45 along with the corresponding derivative (dQ/dV) are shown in Fig. 2. Two processes can be easily observed: a shorter one centered at 4.1 V, referred to as the 4 V capacity, contributing approximately 15 mA h g−1, and a longer process at ≈4.75 V, which accounts for the majority of the capacity.
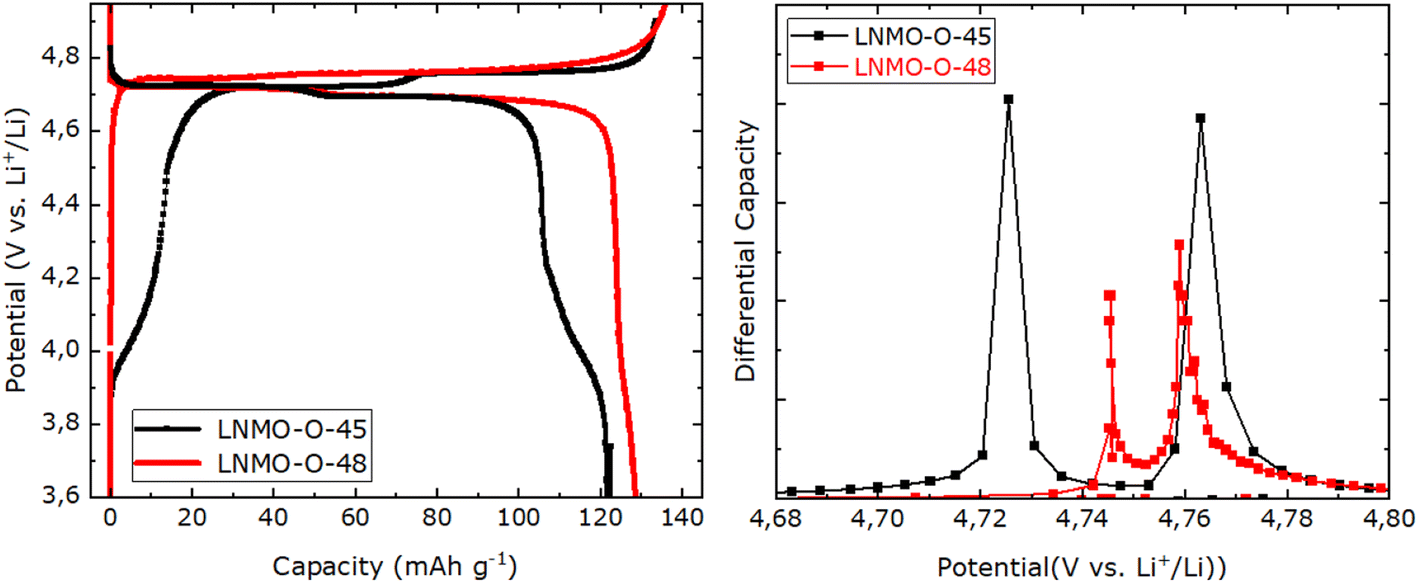 | ||
| Fig. 2 (Left) Electrochemical cycling curve of LNMO-O-45 (black) and LNMO-O-48 (red) at C/10 vs. Li metal in the third cycle. (Right) Corresponding derivative of the Ni charge redox reaction. | ||
According to previous works, the first minor region at around 4.0 to 4.3 V can be attributed to the Mn3+/Mn4+ redox couple.18,21 This can be used as an indication of Mn3+ presence in the spinel structure. This is in agreement with the absence and presence of this plateau in this voltage range for the LNMO-O-45 and LNMO-O-48 samples, respectively (see Fig. 2). Continuing the charge reaction above 4.3 V for LNMO-O-45, a slope leads up to an extended plateau, which can be subdivided into two subsequent plateaus which are highlighted in the derivative curve, see Fig. 2 (right). These two plateaus are expressed by two consecutive peaks at 4.72 and 4.76 V vs. Li+/Li and which are attributed to the redox couples Ni2+/Ni3+ and Ni3+/Ni4+, respectively. In contrast, in the charge/discharge curves of near stoichiometric LNMO-O-48, the Ni plateaus merge into a single long flat plateau at 4.7 V reflected by a much narrower voltage step between the Ni2+/Ni3+ and Ni3+/Ni4+ redox couples.
In the case of the non-stoichiometric TM ordered LNMO (LNMO-O-45), based on capacity values obtained in Fig. 2 (left), the 4 V capacity in the third cycle at 0.1C is around 12% of the total capacity following eqn (1). This is in general agreement with 5% Ni deficiency evinced by NPD and ICP which entail a 10% Mn3+ content for the LNMO-O-45.
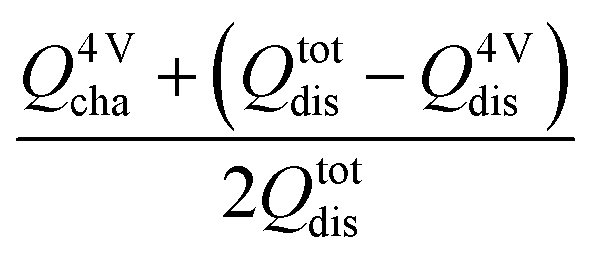 | (1) |
As reported by Song et al., TM cation ordering influences the voltage of the redox plateaus of nickel, or, more precisely, on the voltage of the Ni2+/3+ plateau.12 This effect is difficult to spot in the voltage vs. capacity profile but is clear in the dQ/dV profile shown in Fig. 2 (right). According to Song and co-workers, the presence of TM order is reflected by the difference in potential between the two Ni redox plateaus. An increase in TM order leads to elevation of the Ni2+/Ni3+ redox plateau and hence results in a comparatively smaller voltage gap to the Ni3+/Ni4+ plateau which remains unaltered regardless of the TM stoichiometry, as recently demonstrated by Stüble et al. by comparing LNMO samples of varied TM order and stoichiometry.1 For the LNMO-O-45, we observe a gap of 38 mV between the Ni2+/3+ and the Ni3+/4+ plateaus in the charge curve. Within the compositions screened by Stüble, this value is in best agreement with the one of their partially ordered LNMO (LNMO-43). Our observed value falls in between their reported values of 20 and 60 mV for the ordered and disordered LMNO, respectively. This is an indication for the coexistence of different TM ordering in the LNMO-O-45. The degree of TM ordering in spinel LNMO can affect the electronic band structure of the Ni ions, which in turn influences their redox behaviour as observed by different Ni redox plateaus in the voltage curve. A potential reason for this could be the differences in orbital overlap of the Ni ions as function of TM ordering. In this regard a more distinct electronic states of Ni2+ and Ni3+ within TM ordered LNMO would result in a higher driving force (voltage) needed for the delithiation compared to TM disordered structure in which a larger orbital overlap of Ni is prevalent.
Moreover, their reported 4 V capacity contribution attributed to Mn3+ of around 13.7% is in good agreement with our findings for LNMO-O-45 sample underlining similar TM stoichiometry. Similarly, Duncan et al. showed the effect of Mn3+ presence due to Ni deficient TM stoichiometry on the TM ordering by comparing voltage gap of samples with varying degree of Ni deficiency.14 Based on their voltage gap classification, the expected Ni stoichiometry in our LNMO-O-45 sample falls within the range of 0.4 to 0.45, which aligns well with our results obtained from ICP and NPD analyses.
In Fig. 3 the 6Li NMR spectra of the pristine non-stoichiometric LNMO-O-45 and the near-stoichiometric LNMO-O-48 are shown for comparison. In both spectra, a set of signals are observed between 700 and 1100 ppm. These shifts are clearly out of the range of the shifts typically observed in the 6Li solid-state NMR spectra of diamagnetic compounds (10 to −10 ppm). The large shifts observed in the spectra of LNMO are typically observed in paramagnetic solids and are due to the Fermi contact interactions between Li ions and the unpaired electrons in the t2g and eg orbitals of the transition-metal ions in the local coordination shell.10 The magnitude and sign of the induced paramagnetic shifts of the NMR signals are determined by the bond distances and angles between the metals and Li centres, as well as oxidation states and types of orbitals overlapping along the pathway. In this 3d-transition-metal system, we assume the hyperfine shifts are additive, a consequence of minimal spin–orbit coupling; as such, one can decompose the shift into a set of pathways between Li and nearby transition metal ions.38 The spectra of LNMO-O-48 and LNMO-O-45 shown in Fig. 3 are both characterised by a dominant signal observed at 1017 and 1032 ppm for LNMO-O-45 and LNMO-O-48, respectively, in agreement with previous 6Li solid state NMR characterisations of ordered LNMO samples.10,14,39,40 These signals are assigned to lithium environments with Mn/Ni ratios in the first coordination shell of 3/1. The slightly lower chemical shift of this dominant feature in the LNMO-O-45 sample has its origin in the less oxidised Mn, i.e. higher Mn3+ content.14 Perfectly TM ordered spinel LNMO would only contain a single lithium environment, hence its NMR spectra should consist of a single signal. Indeed, a dominant single peak at 1032 ppm is observed here for the LNMO-O-48 sample shown in Fig. 3 (right), indicating the presence of a nearly ideal TM order and stoichiometric structure (9Mn:3Ni). Additional signals at lower ppm values are also observed in both spectra. The chemical shifts and relative intensities of all the signals obtained by deconvolutional fit are summarised in Table 2.
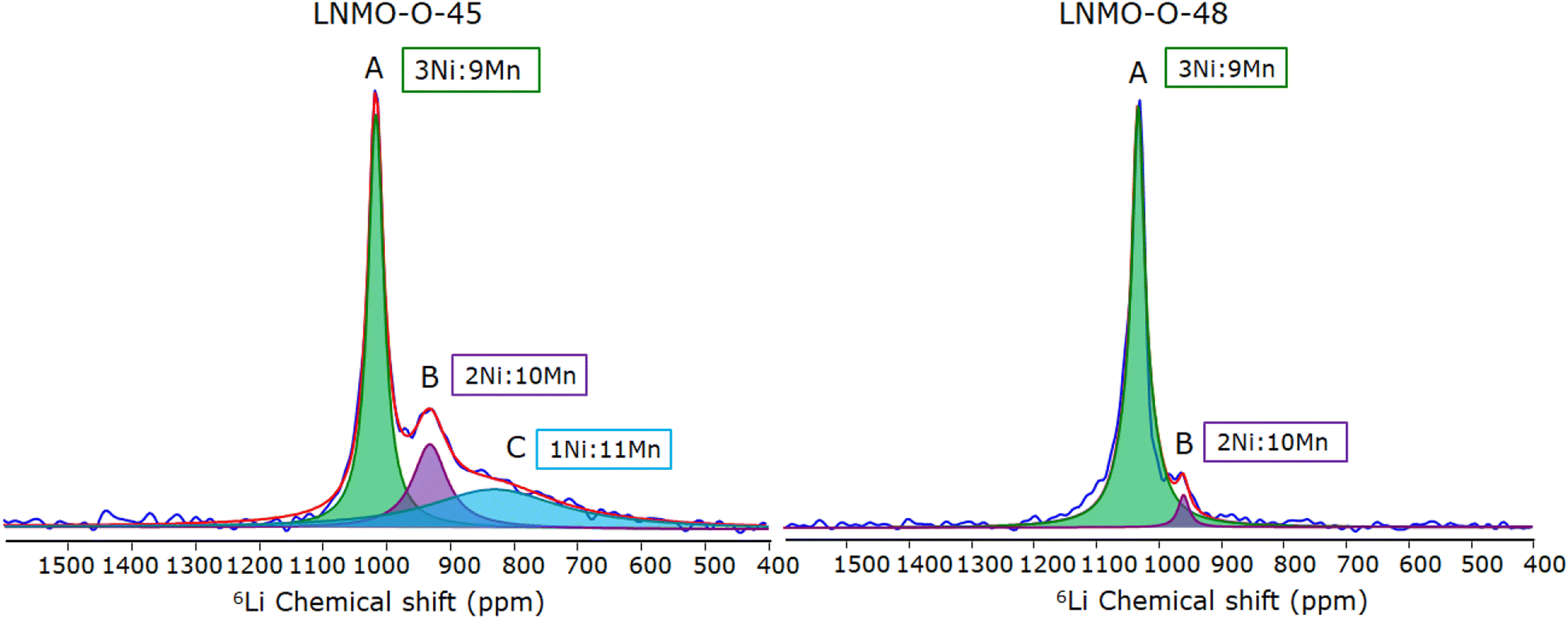 | ||
| Fig. 3 6Li NMR spectra of pristine TM ordered LNMO samples: non-stoichiometric LNMO-O-45 (left) and near-stoichiometric LNMO-O-48 (right). | ||
| TM numbera | LNMO-O-45 | LNMO-O-48 | |||||
|---|---|---|---|---|---|---|---|
| #Mn | #Ni | 6Li shift [ppm] | Int.b [%] | LW [ppm] | 6Li shift [ppm] | Int.b [%] | LW [ppm] |
| a Refers to the number of TM atoms in the first shell around Li. b Values are not T2-weighted, which could influence absolute integration values. | |||||||
| 9 | 3 | 1017 | 48 | 29 | 1032 | 95 | 29 |
| 10 | 2 | 932 | 20 | 73 | 960 | 5 | 20 |
| 11 | 1 | 830 | 32 | 256 | — | — | |
The additional minor signal observed around 962 ppm in LNMO-O-48, indicates that not all the Li in the spinel are surrounded by 3 Ni2+ and 9 Mn4+. This minor resonance feature can hence be attributed to a Mn-rich Li environment (10Mn:2Ni; i.e. presence of Mn3+) in the first TM coordination shell of Li as indicated in the Table 2. This local Mn excess evinced by Li NMR is congruent to the global Mn excess observed by NPD and magnetic measurements.
Compared to the LNMO-O-48, the spectrum of the LNMO-O-45 shown in Fig. 3 (left) has a lower relative intensity for the signal ascribed to the ideal structure (9 Mn4+ and 3 Ni2+ in the first sphere of coordination) at 1017 ppm, as well as at least two strong additional signals are observed. The presence of multiple strong resonances is a clear indication of TM disorder, as previously demonstrated.15 As a higher concentration of Mn is present in LNMO-O-45, these additional features observed at lower ppms are assigned to Li ions in a Mn-rich environment. In particular, the signal at 932 ppm is in agreement with Li ions coordinated to 10Mn:2Ni and the broad signal at 830 ppm to a 11Mn:1Ni environment.10 The fact that the latter is not visible in the spectrum of LNMO-O-48 indicates that a higher Mn excess stoichiometry is required to form such Mn rich local environments. However, it should be noted that the extreme case of pure Mn environment (12Mn:0Ni) which would correspond to the Li environment in spinel LiMn2O4 located at ≈512 ppm is not observed in either of the two samples.39
An increase in linewidth (LW) of the peaks centered at 932 and 830 ppm of the LNMO-O-45 with increasing Mn share is observed. This can be explained by either the higher number of unpaired electrons in Mn3+ compared to Ni2+ or Mn4+, and/or the local heterogeneity (i.e. TM disorder) of Li environments at these positions.
3.2 Reaction mechanism of non-stoichiometric TM ordered LNMO
Fig. 4, left shows the operando XRD profiles of the LNMO electrode during charge from the fully discharged state and the following full discharge, both at 0.05C rate. A reversible capacity of ≈129 mA h g−1 was obtained. In the XRD profiles, three regions of change can be identified attributed to the Li0, Li0.5, and Li1 phases respectively, following previous reports.41,42 It should be noted that the two peaks observed for the (311) reflection are stemming from the bichromatic Cu Kα1,2 radiation. Continuous peak shifts for the Li1 phase at the beginning of the charge and the end of the discharge are observed, which point towards a predominantly solid solution driven reaction mechanism at high lithiation degree. In the region between the 50 and 80 mA h g−1 (≈0.6 ≤ x ≤ 0.4) the decrease of the Li1 and the rise of Li0.5 phase are observed which indicates a biphasic transition. A closeup of this region is depicted in Fig. S5, see ESI.† This underlines that in the Ni2+to Ni3+ transition (Li1 to Li0.5 phase) two different reaction mechanism occur successively, firstly a solid solution reaction (blue) followed by primarily biphasic region (red). Upon further delithiation only the decrease of Li0.5 peak intensity and rise of the completely delithiated Li0 phase intensity are observed which illustrates that the Ni3+ to Ni4+ reaction is a purely biphasic transition (red). Interestingly at very high SOC a peak shift of the solely existing Li0 phase is observed. Lattice contraction of the Li0 phase has been observed previously, but its origins remain unclear.14,18,41,43 Upon discharge the above described changes are reversed, resulting in pattern congruent to pristine. This accentuates the good reversibility of the structural changes of the LNMO spinel phase, in accordance with findings from ex situ NPD, see Fig. S4.†
 | ||
| Fig. 4 (Left) Operando XRD profiles for diffraction reflection (311) of 125 patterns (Cu Kα1,2) acquired during one complete charge and discharge at rate of 0.05C, for a LNMO-O-45 electrode. Prevalent phase reaction mechanism are indicated by blue (solid solution, single phase) and red (biphasic reaction). (Right) Corresponding electrochemical cycling curve with approximate lithiation degree. | ||
 | ||
| Fig. 5 7Li NMR spectra of ex situ LNMO samples at selected stages during (top) charge (delithiation) and (bottom) discharge (lithiation) reaction along with corresponding cycling curve in which points at which spectra were acquired are indicated by colour coded markers.* indicates side band. | ||
This first delithiation step corresponds to the plateau observed between 4.0–4.3 V in the capacity vs. voltage profile of Fig. 5(top). The 7Li NMR spectrum of a sample extracted from a cell charged to 4.05 V (i.e., x ≈ 0.95 in LixNMO, corresponding to the removal of 0.05 Li+ ions per formula unit), which corresponds to 7 mA h g−1 shows a similar pattern to the one obtained at 3.5 V but with a small shift of peak position of 6 ppm towards higher frequency as well as a decay in intensity of the signals at 937 and 837 ppm (see Fig. 5(top)). This small shift on the centre of the spectrum and the decay in the two broad signals can be ascribed to changes in the first and second coordination shell of the Li ion as a result of partial oxidation of Mn3+ to Mn4+. This is in agreement with the observed chemical shift difference between two pristine LNMO samples (i.e. LNMO-O-45 and LNMO-O-48), vide supra. As stated in Table 2, the latter two last signals are assigned to Li environments with increased Mn content (i.e. presence of Mn3+) in the first Li coordination shell of TM ions. At 4.05 V, where extraction of 0.05 Li is achieved, an oxidation of about half of Mn3+ to Mn4+ as the total amount of Mn3+ present in the material was estimated at ≈12% from electrochemistry (and magnetic measurement) vide supra.
Upon further charging to 4.3 V (i.e. x ≈ 0.9, corresponding to the extraction of approximately 0.1 Li+ ion) also equivalently to 13 mA h g−1, both signals at 937 and 837 ppm are no longer observable in agreement with the total oxidation of Mn3+ to Mn4+. In this spectrum a single resonance at 1017 ppm is observed, resembling that of an ideally TM ordered 9Mn:3Ni Li environment., see Table 2). This confirms Cabana's model for a spinel sample with complete ordering of TM ions, where the ratio of Mn4+ to Ni2+ is in accordance with the overall spinel stoichiometry LiNi0.5Mn1.5O4.10 This model suggests that LiNi0.5Mn1.5O4 would exhibit a sole lithium environment, as also observed by Duncan in fully stoichiometric LNMO (y = 0.0 in LiNi(II)0.5−yMn(IV)1.5−yMn(III)2yO4).14 It is noticeable that this signal at 1017 ppm is broader and has shifted back to the lower frequency by 14 ppm compared to the one observed at 4.05 V, This is likely due to a slight relaxation of the system i.e. rapid exchange of Li+ ions between the TM ordered (9Mn:3Ni) and Mn rich sites.
Beyond the Mn3+/4+ redox plateau (above 4.6 V, x > 0.9), five ex situ7Li NMR measurement points were taken to shed light on the Ni redox activity providing insights on its reaction mechanism. The electrodes studied at the first Ni plateau (i.e. Ni2+/3+) are equivalent to the extraction of x = 0.15 to 0.5 Li. In this range of lithiation, the resonance around 1019 ppm attributed to the Li rich (Li1) phase is solely present. The absence of any other NMR signal suggests no additional phase present at this SoC hinting a single phase solid solution reaction driven mechanism. This is in agreement with operando XRD of LNMO-O-45, see Fig. 4, showing a single phase behaviour between x ≈ 1 and 0.7 which also points towards a solid solution mechanism.
At x ≈ 0.6 the appearance of a new NMR signal can be observed centred at around 730 ppm. This new resonance can be assigned to the Li0.5 phase, in agreement with Shimoda.44 The existence of two separate signals attributed to Li1 and Li0.5 phases confirms a two-phase reaction during the first charge plateau corresponding to Ni2+ to Ni3+ redox reaction. Notably, the integrated intensity of the peak assigned for Li0.5 is comparable to that of the Li1 phase. This observation suggests that the biphasic phase transformation from Li1 to Li0.5 occurs within a narrow lithiation region, with 0.7 > x > 0.4. Furthermore, a closer look at the NMR spectra in this lithiation region (see Fig. 6(b) hints that additional species with chemical shift between the two thermodynamically stable main phases (i.e. Li1 and Li0.5) are present. The implications of such intermediate phases are further discussed in Section 4.
 | ||
| Fig. 6 7Li NMR spectra at x ≈ 0.6 (discharge) corresponding to phase transition region between Li1 and Li0.5 phases. (Left) Fitted using solely signals for the Li1 and Li0.5 phases. (Right) Fitted with two additional intermediate components. Black: experimental, light blue: sum of calculated, dark blue: Li0.5 component, olive: Li1 component and red and light green: for intermediate components. | ||
The appearance of a new 7Li NMR resonance at 730 ppm at lower frequency, can be explained by the oxidation of the Ni2+ to Ni3+, which leads to the formation of the new Li0.5 phase clearly distinct from Li1 phase that contains Ni only in a +2 state. In this 3d-transition-metal systems, hyperfine shifts mainly depend on the pathways between Li and nearby transition metal ions. Consequently, during delithiation when the phase transforms from Li1 to Li0.5, this transformation also involves a valence change from Ni2+ to Ni3+. Therefore, in the Li1 phase, Ni exists solely in the +2 form, whereas in the new Li0.5 phase, Ni is in the +3 form. This change could lead to a reduced spin density transfer between the orbital pathways of the transition metal ion and Li due to the absence of one unpaired electron in Ni3+ in the Li0.5 phase resulting in a lower frequency shift for Li0.5 phase compared to Li1 phase. At the end of the first Ni plateau (x ≈ 0.4), it can be observed that the new Li0.5 phase is prevalent with a small remnant of Li1 phase, in agreement with the almost complete transformation of Li1 to Li0.5 phase. Upon further delithiation (x ≤ 0.4) the latter is the only Li containing phase observed by NMR, the Li0 phase being undetectable in Li NMR experiments. However, the coexistence of Li0.5 and Li0 was evinced by operando XRD, vide supra.
At fully charged state x = 0 (i.e. 4.95 V), as depicted in the Fig. 5(top), no 7Li NMR resonances of neither the Li1 nor Li0.5 phases are present, suggesting that the transformation to fully oxidised spinel phase (Li0) devoid of lithium is achieved.
During subsequent lithiation to x ≈ 0.2, the reappearance of the Li0.5 phase at 780 ppm is found. Upon further lithiation at x ≈ 0.6, Li1 phase reemerged and coexisted momentarily with the Li0.5 phase suggesting a biphasic reaction, as depicted in Fig. 5(bottom) Between Li content of 0.4 and 0.7, the Li0.5 phase gradually vanishes, as observed during charging in a similar lithiation range. Simultaneously, the Li1 phase becomes the predominant phase, as evidenced in the x ≈ 0.7 sample, indicating that at higher Li content the mechanism is essentially a solid solution, in agreement with operando XRD findings. Upon reaching full lithiation in the range of 0.9 < x < 1, the NMR features linked to the presence of the ordered (9Mn:3Ni) at 1013 ppm as well as Mn rich local Li environments (932 ppm and 830 ppm) were observed. This observation reflects the reversible reduction of Ni4+ to Ni2+ and Mn4+ to Mn3+, respectively. Furthermore, a shift in the center of gravity toward lower frequency was observed yielding to congruent NMR spectrum to that of the pristine state. This concordance serves as compelling evidence for the reversible relithiation of the sites within the LNMO spinel structure.
4 Discussion
Monitoring local Li environments at different stages of SoC upon delithiation and lithiation of the non-stoichiometric partially ordered LNMO-O-45 spinel via7Li NMR and contrasting them with operando XRD resulted in plentiful insights whose implications will be further discussed in the following.At the beginning of the charge reaction in the 4 V plateau region, we observe the preferential delithiation of the Mn-rich (Mn > 9) local Li environments. This is reflected by the disappearance of the two NMR signals (at 932 ppm and 830 ppm) assigned to these Li environments when reaching 4.3 V. This implies that the Mn oxidation and Li vacancy formation upon electrochemical charging occur in close vicinity. Albeit appearing logical, this spatial concurrence has not been shown in any previous LNMO studies to the best of the authors knowledge. Moreover, the chemical shift of the main feature observed in this SoC region hints that a fast Li exchange occurs between the TM ordered stoichiometric (9Mn:3Ni) and Mn-rich sites.
A second noteworthy finding is the extended solid solution region evinced for this TM ordered LNMO sample by both ex situ7Li NMR and operando XRD. This finding is reflected by the absence of the Li0.5 phase approximately until about 1/3 of total charge is reached, well beyond Mn3+ related 4 V plateau. Only upon further delithiation, the biphasic reaction mechanism expected for the TM ordered LNMO in the Ni2+/3+ plateau is observed. These findings underline the importance of both structural parameters, TM stoichiometry and TM order to adequately describe LNMO characteristics, as the former greatly affects the latter. Although TM order features are salient for the LNMO-O-45 sample in both Raman spectroscopy and Neutron diffraction, its electrochemical mechanism strongly deviates from the expected predominantly biphasic reaction mechanism of TM ordered LNMO.17,18,44,45 Indeed our findings show that already small Mn-excess stoichiometry induces TM disorder (by partially populating Ni 4b sites) which result in an extended solid solution reaction region in the phase transformation from Li1 to Li0.5 phase as well as the presence of intermediate species.
The existence of additional spectral features besides those of the expected Li1 and Li0.5 phase towards the end of the Ni2+ to Ni3+ transition (Fig. 6) is another interesting observation revealed by the ex situ7Li NMR. The fact that such intermediate environments were not observed in the ideal stoichiometric and fully TM ordered LNMO investigated by Shimoda et al.44 hints that their presence could be related to the stoichiometric induced TM disorder in the LNMO-O-45. In Fig. 6 the ex situ NMR spectrum at SoC x ≈ 0.6 is depicted. It is evident that the experimental spectrum cannot be matched by solely using two signals for the expected Li1 and Li0.5 phases, see Fig. 6 (left). Our Gaussian and Lorentzian fit analysis suggest that there are at least two additional broad features present at this SoC, see Fig. 6 (right). These additional broad features, exhibiting a paramagnetic shift between those of the Li1 and Li0.5 phases, suggest the existence of intermediate Li environments. However, these intermediate species do not result in distinct resonances, as observed for the Li1 and Li0.5 phases; instead, they represent a continuous distribution of environments between the two equilibrium phases. We believe this intermediate region exhibits characteristics of a mix of both phases could suggest a narrow miscibility gap between the two phases with a benefit of facilitating a facile Li hopping between the Li1 and Li0.5 phase, possibly correlated with a blend of Ni2+ and Ni3+ states as well as various lithiation states as suggested by ex situ NMR having a chemical shift between the Li1 and Li0.5 phases. The presence of these intermediate environments may contribute to narrowing the lattice mismatch or miscibility gap between the fully lithiated (Li1) and half lithiated (Li0.5) phases. This phenomenon has been reported mainly for LFP,46,47 but a clear correlation between cation ordering, reaction mechanism, and rate has yet to be fully established for spinel LNMO. Battery performance is not solely constrained by Li transport in the electrolyte and electron movement within the electrodes but also by the phase transformations occurring during lithiation and delithiation. Therefore, the absence of a phase transformation or the presence of an intermediate phase that narrows the miscibility gap is generally considered a key factor in achieving high rates and better cyclability. In this regard Omenya et al. showed that by partially substituting Fe with V the miscibility gap in LFP can be significantly decreased. Moreover, this leads to a reduction of the solid solution formation temperature in the two-phase region and enhances Li solubility. In our spinel LNMO case, the local environment change stems from the Ni-deficient stoichiometry resulting in partial “substitution” of Ni2+ sites by Mn (Mn4+ and Mn3+). Such stoichiometrically induced partial disorder can hence be a viable approach to close the miscibility gap and enable better cycling performance. If these intermediate species exist only on a local structural level or also exist as long range intermediate crystal phases cannot be answered with certainty at this point. The analysis of our operando XRD data in this region of SoC see Fig. S5 (see ESI†) doesn't provide conclusive evidence, if besides the evident features of biphasic transition additional phases are present. Nevertheless the existence of several intermediate species with different lithiation stages besides the thermodynamically stable phases Li1 and Li0.5 was previously suggested based on operando XRD by Komatsu and coworkers.41 Furthermore, it should be noted that the co-existence of multiple local environments at about half lithiation was already hinted in our previous operando X-ray absorption study by a maxima in local disorder of the Ni–O bonds (Debye–Waller factor) for the TM ordered LNMO.15
In Fig. S8 (in ESI†), the peak intensity ratios and chemical shift of the four spectral features present during delithiation and lithiation are depicted in single graph to highlight their evolution as function of SoC. The full data set of deconvoluted spectra is provided in Table S1 in the ESI† along with a discussion of the trends observed.
On a macroscopic level the narrow co-existence range of thermodynamically stable phases Li1 and Li0.5 (0.6 ≤ x ≤ 0.4) evinced by both ex situ NMR and operando XRD points towards the prevalence of a concurrent reaction propagation model rather than particle by particle reaction propagation.41,48 In such concurrent model a quasi-simultaneous transformation occurs in all particles. A scheme of this proposed concurrent model reaction front propagation is illustrated in Fig. 7 along with corresponding NMR spectra. Varying charge states within the same particle, so called intra-particle state of charge heterogeneity, has been observed in several cathode active materials.49,50 This heterogeneity in phases likely also exist in partially delithiated LNMO, as observed by Kuppan et al.48
 | ||
| Fig. 7 Scheme of concurrent reaction propagation model showing the narrow lithiation range of coexistence (0.6 ≤ x ≤ 0.4) of both thermodynamically stable phases (Li1 and Li0.5) as well as intermediates species based on corresponding ex situ7Li NMR spectra. | ||
Another remarkable finding of our ex situ7Li NMR study is the complete delithiation of the spinel structure at the end of the charging process, as evidenced by the absence of signals in the 7Li NMR spectra. It should be considered that NMR is an element-selective technique and only detects the nuclei under investigation. Hence in this case, the Li0 phase present at the lower lithiation state cannot be observed by Li NMR. Our results indicate a complete phase transformation to the Li0 phase (Ni0.5Mn1.5O4), which is confirmed by our diffraction results (NPD and XRD) and is in agreement with the results of other authors.51,52 However, it may contradict studies which claim that incomplete delithiation (only up to 90%) can be obtained with a full charge.43 The complete delithiation of the spinel structure evinced by ex situ7Li here is a strong asset of the LNMO spinel which results in better use of lithium compared to other cathode materials such as NMC.
5 Conclusion
In this study, we have shown that NMR is highly sensitive to key structural parameter of TM ordering of the LNMO spinel cathode material. Its ability to evince and quantify TM order degree but also distinguish between thermodynamic and stoichiometry induced disorder is a unique asset. In this regard we demonstrated that even minor deviations from ideal TM stoichiometry (0.02 ≤ y ≤ 0.05 in LiNi(II)0.5−yMn(IV)1.5−yMn(III)2yO4) result in strong changes of the NMR spectra and greatly affect the electrochemical mechanism. By coupling local and long range sensitive techniques we have gained a holistic picture of the delithiation/lithiation process, revealing that Ni deficient LNMO-O-45 does not follow a simple sequence of a two biphasic reactions as previously claimed for such predominantly TM ordered LNMO spinels. Instead our ex situ NMR and operando XRD suggest that delithiation linked to Ni2+/3+ redox reaction occurs firstly via an extended solid solution mechanism followed by the expected two phase reaction mechanism. However, we evince for the first time the presence of reaction intermediates during this later stage which suggest that the formation from Li1 to Li0.5 phase occurs on a local structural level via intermediate steps. We attribute their existence to the stoichiometric induced TM disorder and rationalise their potential for material performance enhancement by closing the miscibility gap. During the Ni3+/4+ redox reaction a two phase reaction mechanism (Li0.5 to Li0) is prevalent resulting in the complete delithiation of the spinel structure at the end of the charge process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors are thankful for beamtime at ILL Neutron facility (Proposals: CRG-2789-2020, 5-23-751, EASY1082) and the support of Oscar Fabelo and Emannuelle Suard. The authors would like to thank Silvia Fuentes Martin for preparing the LNMO laminates. Haldor Topsoe is acknowledged for providing LNMO active material samples. Furthermore the authors are grateful to the European Commission for the support of the work, performed within the EU H2020 project 3beLiEVe (grant agreement 875033). The Spanish MCIN/AEI/10.13039/50110001103 and the Basque Government are also acknowledged for their support through the project ION-SELF ref. PID2019-106519RB-I00 and the PhD grant ref. PRE-2021-2-011, respectively.References
- P. Stüble, V. Mereacre, H. Geßwein and J. R. Binder, On the Composition of LiNi0.5Mn1.5O4 Cathode Active Materials, Adv. Energy Mater., 2023, 13(10), 2203778 CrossRef.
- M. Reynaud, J. Serrano-Sevillano and M. Casas-Cabanas, Imperfect Battery Materials: A Closer Look at the Role of Defects in Electrochemical Performance, Chem. Mater., 2022, 35, 3363 Search PubMed.
- K. Amine, H. Tukamoto, H. Yasuda and Y. Fujita, Preparation and electrochemical investigation of LiMn2xMexO4 (Me: Ni, Fe, and x = 0.5, 1) cathode materials for secondary lithium batteries, J. Power Sources, 1997, 68, 604–608 CrossRef CAS.
- Q. Zhong, A. Bonakdarpour, M. Zhang, Y. Gao and J. R. Dahn, Synthesis and Electrochemistry of LiNi x Mn2 - x O 4, J. Electrochem. Soc., 1997, 144, 205–213 CrossRef CAS.
- R. Amin and I. Belharouak, Part-II: Exchange current density and ionic diffusivity studies on the ordered and disordered spinel LiNi0.5Mn1.5O4 cathode, J. Power Sources, 2017, 348, 318–325 CrossRef CAS.
- J. Xiao, X. Chen, P. V. Sushko, M. L. Sushko, L. Kovarik, J. Feng, Z. Deng, J. Zheng, G. L. Graff, Z. Nie, D. Choi, J. Liu, J. G. Zhang and M. S. Whittingham, High-Performance LiNi0.5Mn1.5O4 Spinel Controlled by Mn3+ Concentration and Site Disorder, Adv. Mater., 2012, 24, 2109–2116 CrossRef CAS PubMed.
- B. Wood, R. Kirkpatrick and B. Montez, Order-disorder phenomena in MgAl2O4 spinel, Am. Mineral., 1986, 71, 999–1006 CAS.
- M. C. Kemei, S. L. Moffitt, L. E. Darago, R. Seshadri, M. R. Suchomel, D. P. Shoemaker, K. Page and J. Siewenie, Structural ground states of (A,A) Cr2 O4 (A = Mg, Zn; A = Co, Cu) spinel solid solutions: Spin-Jahn-Teller and Jahn-Teller effects, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 174410 CrossRef.
- J. H. Kim, S. T. Myung, C. S. Yoon, S. G. Kang and Y. K. Sun, Comparative Study of LiNi0.5Mn1.5O4-δ and LiNi0.5Mn1.5O4 Cathodes Having Two Crystallographic Structures: Fd3m and P4332, Chem. Mater., 2004, 16, 906–914 CrossRef CAS.
- J. Cabana, M. Casas-Cabanas, F. O. Omenya, N. A. Chernova, D. Zeng, M. S. Whittingham and C. P. Grey, Composition-Structure Relationships in the Li-Ion Battery Electrode Material LiNi 0.5 Mn 1.5 O 4, Chem. Mater., 2012, 24, 2952–2964 CrossRef CAS PubMed.
- B. Aktekin, F. Massel, M. Ahmadi, M. Valvo, M. Hahlin, W. Zipprich, F. Marzano, L. Duda, R. Younesi, K. Edström and D. Brandell, How Mn/Ni Ordering Controls Electrochemical Performance in High-Voltage Spinel LiNi0.44Mn1.56O4 with Fixed Oxygen Content, ACS Appl. Energy Mater., 2020, 3, 6001–6013 CrossRef CAS.
- J. Song, D. W. Shin, Y. Lu, C. D. Amos, A. Manthiram and J. B. Goodenough, Role of oxygen vacancies on the performance of Li[Ni 0.5-xMn 1.5+ x]O 4 (x = 0, 0.05, and 0.08) spinel cathodes for lithium-ion batteries, Chem. Mater., 2012, 24, 3101–3109 CrossRef CAS.
- J. H. Kim, A. Huq, M. Chi, N. P. Pieczonka, E. Lee, C. A. Bridges, M. M. Tessema, A. Manthiram, K. A. Persson and B. R. Powell, Integrated nano-domains of disordered and ordered spinel phases in LiNi0.5Mn1.5O4 for li-ion batteries, Chem. Mater., 2014, 26, 4377–4386 CrossRef CAS.
- H. Duncan, B. Hai, M. Leskes, C. P. Grey and G. Chen, Relationships between Mn3+ Content, Structural Ordering, Phase Transformation, and Kinetic Properties in LiNixMn2–xO4 Cathode Materials, Chem. Mater., 2014, 26, 5374–5382 CrossRef CAS.
- M. Fehse, N. Etxebarria, L. Otaegui, M. Cabello, S. Martín-Fuentes, M. A. Cabañero, I. Monterrubio, C. F. Elkjær, O. Fabelo, N. A. Enkubari, J. M. López del Amo, M. Casas-Cabanas and M. Reynaud, Influence of Transition-Metal Order on the Reaction Mechanism of LNMO Cathode Spinel: An Operando X-ray Absorption Spectroscopy Study, Chem. Mater., 2022, 34, 6529–6540 CrossRef CAS PubMed.
- M. Casas-Cabanas, C. Kim, J. Rodríguez-Carvajal and J. Cabana, Atomic defects during ordering transitions in LiNi 0.5 Mn 1.5 O 4 and their relationship with electrochemical properties, J. Mater. Chem. A, 2016, 4, 8255–8262 RSC.
- E. Lee and K. A. Persson, Solid-solution Li intercalation as a function of cation order/disorder in the high-voltage LixNi0.5Mn1.5O4 spinel, Chem. Mater., 2013, 25, 2885–2889 CrossRef CAS.
- L. Wang, H. Li, X. Huang and E. Baudrin, A comparative study of Fd-3m and P4332 “LiNi0.5Mn1.5O4”, Solid State Ionics, 2011, 193, 32–38 CrossRef CAS.
- M. Kunduraci, J. F. Al-Sharab and G. G. Amatucci, High-power nanostructured LiMn 2-xNi xO 4 high-voltage lithium-ion battery electrode materials: Electrochemical impact of electronic conductivity and morphology, Chem. Mater., 2006, 18, 3585–3592 CrossRef CAS.
- M. Casas-Cabanas, C. Kim, J. Rodríguez-Carvajal and J. Cabana, Atomic defects during ordering transitions in LiNi0.5Mn1.5O4 and their relationship with electrochemical properties, J. Mater. Chem. A, 2016, 4, 8255–8262 RSC.
- M. DeGennaro, B. Ganev, M. Jahn, M. Reynaud, M. Fehse, L. Otaegui, M. Cabello, S. Mannori and O. Rahbari, 3beLiEVe: Towards Delivering the Next Generation of LMNO Li-Ion Battery Cells and Packs Fit for Electric Vehicle Applications of 2025 and Beyond, SAE WCX Digital Summit, 2021, pp. 2688–3627, ISSN: 0148-7191 Search PubMed.
- L. Yang, J. M. L. del Amo, Z. Shadike, S. M. Bak, F. Bonilla, M. Galceran, P. K. Nayak, J. R. Buchheim, X. Q. Yang, T. Rojo and P. Adelhelm, A Co- and Ni-Free P2/O3 Biphasic Lithium Stabilized Layered Oxide for Sodium-Ion Batteries and its Cycling Behavior, Adv. Funct. Mater., 2020, 30, 2003364 CrossRef CAS.
- J. Serrano-Sevillano, D. Carlier, A. Saracibar, J. M. L. D. Amo and M. Casas-Cabanas, DFT-Assisted Solid-State NMR Characterization of Defects in Li2MnO3, Inorg. Chem., 2019, 58, 8347–8356 CrossRef CAS PubMed.
- F. Aguesse, J. M. L. del Amo, L. Otaegui, E. Goikolea, T. Rojo and G. Singh, Structural and electrochemical analysis of Zn doped Na3Ni2SbO6 cathode for Na-ion battery, J. Power Sources, 2016, 336, 186–195 CrossRef CAS.
- G. Singh, J. M. L. D. Amo, M. Galceran, S. Pérez-Villar and T. Rojo, Structural evolution during sodium deintercalation/intercalation in Na2/3[Fe1/2Mn1/2]O2, J. Mater. Chem. A, 2015, 3, 6954–6961 RSC.
- Y. Lee, J. An, S.-A. Park and H. Song, Ex situ 7 Li MAS NMR Study of Olivine Structured Material for Cathode of Lithium Ion Battery, J. Korean Magn. Reson., 2014, 18, 63–68 Search PubMed.
- Y. J. Lee, C. Eng and C. P. Grey, 6Li Magic Angle Spinning NMR Study of the Cathode Material LiNix Mn2x O4:The Effect of Ni Doping on the Local Structure during Charging, J. Electrochem. Soc., 2001, 148, A249 CrossRef CAS.
- M. Reynaud, M. A. Cabanero, M. C. Cabanas, O. R. F. Rosa, M. Fehse and I. Monterrubio, Elucidating the Effect of Stoichiometry, Ni/Mn Ordering and Defects Onthe Electrochemical Properties of High-Voltage Positive Electrode, 2020, DOI:10.5291/ILL-DATA.CRG-2789.
- M. Reynaud, M. Fehse, I. Monterrubio, M. A. Cabanero, M. C. Cabanas and E. SUard, Effect of Stoichiometry, Ordering and Defects on the Electrochemical Properties of High-Voltage Spinel LNMO for Li-Ion Batteries. 2021, DOI:10.5291/ILL-DATA.5-23-751.
- F. Marcus and S. Emmanuelle, Influence of Transition Metal Order and Stoichiometry on Physico-Chemical Properties of High Voltage LNMO Cathode Spinel, Material, 2023 DOI:10.5291/ILL-DATA.EASY-1084.
- J. Rodríguez-Carvajal, Recent advances in magnetic structure determination by neutron powder diffraction, Phys. B, 1993, 192, 55–69 CrossRef.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Modelling one- and two-dimensional solid-state NMR spectra, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- A. Bhatia, Y. Dridi Zrelli, J.-P. Pereira-Ramos and R. Baddour-Hadjean, Detailed redox mechanism and self-discharge diagnostic of 4.9 V LiMn1.5Ni0.5O4 spinel cathode revealed by Raman spectroscopy, J. Mater. Chem. A, 2021, 9, 13496–13505 RSC.
- D. Saurel, A. Pendashteh, M. Jáuregui, M. Reynaud, M. Fehse, M. Galceran and M. Casas-Cabanas, Experimental Considerations for Operando Metal-Ion Battery Monitoring using X-ray Techniques, Chem.: Methods, 2021, 1, 249–260 CAS , https://chemistry-europe.onlinelibrary.wiley.com/doi/pdf/10.1002/cmtd.202100009.
- N. Amdouni, K. Zaghib, F. Gendron, A. Mauger and C. M. Julien, Structure and insertion properties of disordered and ordered LiNi0.5Mn1.5O4 spinels prepared by wet chemistry, Ionics, 2006, 12, 117–126 CrossRef CAS.
- N. Amdouni, K. Zaghib, F. Gendron, A. Mauger and C. Julien, Magnetic properties of LiNi0.5Mn1.5O4 spinels prepared by wet chemical methods, J. Magn. Magn. Mater., 2007, 309, 100–105 CrossRef CAS.
- Z. Moorhead-Rosenberg, K. R. Chemelewski, J. B. Goodenough and A. Manthiram, Magnetic measurements as a viable tool to assess the relative degrees of cation ordering and Mn3+ content in doped LiMn1.5Ni0.5O4 spinel cathodes, J. Mater. Chem. A, 2013, 1, 10745–10752 RSC.
- A. K. Cheetham, C. M. Dobson, C. P. Grey and R. J. Jakeman, Paramagnetic shift probes in high-resolution solid-state NMR, Nature, 1987, 328(6132), 706–707 CrossRef CAS.
- Y. J. Lee, C. Eng and C. P. Grey, [sup 6]Li Magic Angle Spinning NMR Study of the Cathode Material LiNi[sub x]Mn[sub 2-x]O[sub 4]: The Effect of Ni Doping on the Local Structure during Charging, J. Electrochem. Soc., 2001, 148, A249 CrossRef CAS.
- B. Hai, A. K. Shukla, H. Duncan and G. Chen, The effect of particle surface facets on the kinetic properties of LiMn1.5Ni0.5O4 cathode materials, J. Mater. Chem. A, 2013, 1, 759–769 RSC.
- H. Komatsu, H. Arai, Y. Koyama, K. Sato, T. Kato, R. Yoshida, H. Murayama, I. Takahashi, Y. Orikasa, K. Fukuda, T. Hirayama, Y. Ikuhara, Y. Ukyo, Y. Uchimoto and Z. Ogumi, Solid Solution Domains at Phase Transition Front of LixNi0.5Mn1.5O4, Adv. Energy Mater., 2015, 5, 1500638 CrossRef , https://onlinelibrary.wiley.com/doi/pdf/10.1002/aenm.201500638.
- L. Boulet-Roblin, P. Borel, D. Sheptyakov, C. Tessier, P. Novák and C. Villevieille, Operando Neutron Powder Diffraction Using Cylindrical Cell Design: The Case of LiNi0.5Mn1.5O4 vs Graphite, J. Phys. Chem. C, 2016, 120, 17268–17273 CrossRef CAS.
- I. Martens, N. Vostrov, M. Mirolo, M. Colalongo, P. Kúš, M.-I. Richard, L. Wang, X. Zhu, T. U. Schülli and J. Drnec, Revisiting Phase Transformation Mechanisms in LiNi0.5Mn1.5O4 High Voltage Cathodes with Operando Microdiffraction, ACS Materials Lett., 2022, 4(12), 2528–2536 CrossRef CAS.
- K. Shimoda, M. Murakami, H. Komatsu, H. Arai, Y. Uchimoto and Z. Ogumi, Delithiation/Lithiation Behavior of LiNi0.5Mn1.5O4 Studied by In Situ and Ex Situ 6,7Li NMR Spectroscopy, J. Phys. Chem. C, 2015, 119, 13472–13480 CrossRef CAS.
- S. T. Myung, S. Komaba, N. Kumagai, H. Yashiro, H. T. Chung and T. H. Cho, Nano-crystalline LiNi0.5Mn1.5O4 synthesized by emulsion drying method, Electrochim. Acta, 2002, 47, 2543–2549 CrossRef CAS.
- F. Omenya, N. A. Chernova, Q. Wang, R. Zhang and M. S. Whittingham, The structural and electrochemical impact of li and fe site substitution in LiFePO4, Chem. Mater., 2013, 25, 2691–2699 CrossRef CAS.
- H. Liu, F. C. Strobridge, O. J. Borkiewicz, K. M. Wiaderek, K. W. Chapman, P. J. Chupas and C. P. Grey, Capturing metastable structures during high-rate cycling of LiFePO4 nanoparticle electrodes, Science, 2014, 344, 1252817 CrossRef PubMed.
- S. Kuppan, Y. Xu, Y. Liu and G. Chen, Phase transformation mechanism in lithium manganese nickel oxide revealed by single-crystal hard X-ray microscopy, Nat. Commun., 2017, 8, 14309 CrossRef CAS PubMed.
- Z. Xu, et al., Charge distribution guided by grain crystallographic orientations in polycrystalline battery materials, Nat. Commun., 2020, 11, 83 CrossRef CAS PubMed.
- S. L. Spence, A. Hu, M. Jiang, Z. Xu, Z. Yang, M. M. Rahman, L. Li, Y. S. Chu, X. Xiao, X. Huang and F. Lin, Mapping Lattice Distortions in LiNi0.5Mn1.5O4 Cathode Materials, ACS Energy Lett., 2022, 7, 690–695 CrossRef CAS.
- B. Hai, A. K. Shukla, H. Duncan and G. Chen, The effect of particle surface facets on the kinetic properties of LiMn1.5Ni0.5O4 cathode materials, J. Mater. Chem. A, 2013, 1, 759–769 RSC.
- J.-H. Kim, S.-T. Myung, C. S. Yoon, S. G. Kang and Y.-K. Sun, Comparative Study of LiNi0.5Mn1.5O4−δ and LiNi0.5Mn1.5O4 Cathodes Having Two Crystallographic Structures: Fd3m and P 4 3 32, Chem. Mater., 2004, 16, 906–914 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta04649d |
| This journal is © The Royal Society of Chemistry 2023 |