
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThiophene-fused boron dipyrromethenes as energy efficient near-infrared photocatalysts for radical polymerizations†
Alex
Stafford
a,
Seth R.
Allen
a,
Tod A.
Grusenmeyer
b,
Connor J.
O'Dea
 a,
Laura
Estergreen
a,
Sean T.
Roberts
*a and
Zachariah A.
Page
*a
a,
Laura
Estergreen
a,
Sean T.
Roberts
*a and
Zachariah A.
Page
*a
aDepartment of Chemistry, The University of Texas at Austin, Austin, Texas 78712, USA. E-mail: roberts@cm.utexas.edu; zpage@utexas.edu
bAir Force Research Laboratory, Materials and Manufacturing Directorate, Wright-Patterson Air Force Base, OH 45433, USA
First published on 13th September 2023
Abstract
Rapid near-infrared (NIR) light driven polymerizations have the potential to enable energy-efficient, benign, and multimaterial manufacturing for applications ranging from tissue engineering to soft robotics. However, achieving photopolymerization rates with NIR light that are viable for emergent additive manufacturing technologies (∼0.1–1.0 M s−1 at light intensities <40 mW cm−2) has proven challenging due to its inherently low energy. Herein, we begin to take down these barriers through a systematic investigation of four distinct thiophene-fused boron dipyrromethene (BODIPY) photocatalysts. Through extended π-conjugation, the thiophene-fused BODIPYs effectively absorb NIR (>780 nm) light and drive efficient acrylate polymerizations upon exposure to low-intensity (2.5–40 mW cm−2) NIR light emitting diodes (LEDs). The installation of bromine atoms is shown to further improve NIR light-fueled photopolymerization efficiency by a factor of ∼1.5×. This enhancement was rationalized by the formation of long-lived spin-triplet excited states via intersystem crossing, as observed using ultrafast transient absorption spectroscopy. However, the triplet yields are modest (∼6–14%), suggesting efficient charge transfer from singlet excited states also occurs within the present photosystem. Finally, optimization of resin formulations containing non-halogenated thiophene-fused BODIPY photocatalysts is shown to provide unprecedented polymerization rates (0.33 M s−1) upon exposure to an 850 nm LED at an intensity of 20 mW cm−2. The structure–reactivity relationships identified herein provide key insights that will inform further design and optimization of energy-efficient, NIR light-driven polymerizations with utility in photocurables for coatings, adhesives, and 3D printing.
Introduction
Light as an energy source to drive chemical reactions has seen rapid growth in the past decade, owing in part to the spatiotemporal control it offers and the wide availability of energy-efficient LEDs. A myriad of phototransformations have been developed, ranging from photoredox catalysis1 to photodeprotection,2 and photo-acid and -base generation.3 These transformations have been leveraged in light-driven polymerizations4–7 for adhesives, paints, coatings, lithography, and 3D printing.8–13 However, high-energy ultraviolet (UV) light currently serves as the predominant source to activate these reactions, which has limited the scope of materials that can be produced due to inherent scattering and extraneous absorption and degradation. While significant progress has been made to address this shortcoming by developing rapid and efficient photopolymerizations that are fueled by visible light,14 extending light-driven photopolymerizations into the near-infrared (NIR) spectral region remains challenging. However, addressing this need can enable benign manufacturing for tissue engineering applications and provide a wide spectral window for wavelength-selective multimaterial fabrication of soft robotics owing to the reduced propensity of NIR light to scatter when transmitting through a material.15–17Photosystems for light-driven polymerizations can be generally classified as either Type I or Type II. Type I photosystems consist of a photoinitiator that, upon irradiation, will undergo intramolecular bond homolysis to generate the reactive radical species to initiate polymerization. This process typically results in rapid initiation, yet it is limited in scope due to requiring high-energy UV or violet photons.18 Alternatively, Type II photosystems involve an intermolecular process of either hydrogen bond abstraction, energy transfer, or electron transfer following light absorption. Thus, Type II photosystems operating via a redox mechanism, consist of a chromophore (e.g., photocatalyst) that interacts with either an electron donor or donor/acceptor pair post-irradiation to generate the reactive radical species. This electron cascade enables the use of lower-energy (longer-wavelength) light, but the diffusion-limited bimolecular step(s) typically decrease the overall polymerization rate relative to unimolecular, Type I photosystems.19 The slow photopolymerization rates in turn limit applicability in processes requiring rapid photocuring (i.e., light-driven conversion of liquid to solid on timescales of seconds to minutes), such as stereolithographic 3D printing. Overcoming this shortcoming necessitates further mechanistic insight into redox-based, Type II photopolymerizations.
Traditionally, Type II photosystems have been dominated by xanthenes20 and cyanines21,22 for blue/green and red/NIR light photopolymerizations, respectively. Additionally, Lalevée and coworkers23 have demonstrated NIR-enabled photopolymerizations using squarines, squaryliums, porphyrins, and boron dipyrromethenes (BODIPYs) having peak absorption bands ranging from ∼500 to 650 nm. However, the limited NIR absorption by these compounds (molar absorptivity, ε, at λ ≥ 780 nm is <500 M−1 cm−1) necessitated the use of high-intensity lasers (400 mW cm−2) and incorporation of thermal initiators to induce polymerizations that reached maximum monomer-to-polymer conversion on the minute time-scale (∼1.5–5 min exposure time) (Table S1†). In parallel with our efforts, Allonas and coworkers24 demonstrated how tailoring borate coinitiators (electron donors) together with a penta-methine cyanine dye complex could enable rapid NIR-fueled photopolymerizations. The work we present is complementary as it focuses on tailoring a distinct light-absorbing photoredox catalyst component, and demonstrates the importance of identifying structure–reactivity relationships in photosystems to enable rapid and efficient NIR light-driven polymerizations.
Recently, our group has taken a systematic approach to examining BODIPY as a scaffold for designing visible light-activated photoredox catalysts25 owing to its synthetic modularity that provides tunable solubility, strong absorption, and photostability26 (Fig. 1). Using BODIPYs excited by low-intensity green (530 nm) and far-red (740 nm) LEDs, we have demonstrated rapid acrylate polymerizations (rp ≈ 0.95 and 0.66 M s−1, respectively). Key to the success of this endeavor was the realization of BODIPY halogenation (i.e., heavy-atom effect27–29) as a general strategy to access long-lived spin-triplet excited states. This facilitated diffusion-limited photoredox catalysis, enhancing the photopolymerization efficiency (Φp = photons absorbed/rate of polymerization).
 | ||
| Fig. 1 Overview of the present work that enables low-intensity NIR polymerizations using thiophene-fused BODIPY catalysts, while providing key insight into the governing principles behind efficient conversion of low-energy photons to the formation of radicals. | ||
Building upon this understanding of reactivity in the visible and far-red spectral regions (∼530–740 nm), we aimed to develop an efficient photopolymerization system the operates in the NIR range (∼780–1000 nm). To that end, we synthesized a series of π-extended thiophene-fused BODIPYs capable of effectively absorbing NIR light and examined their utility as photoredox catalysts to induce polymerization (Fig. 1). The present photosystem provides high sensitivity to NIR light (rp up to 0.33 M s−1, 850 nm LED, ILED = 20 mW cm−2) while maintaining temporal control (no polymerization in the absence of light). Furthermore, systematic photophysical characterization using a combination of steady-state and ultrafast transient absorption spectroscopies is used to rationalize the observed photopolymerization rate enhancements for halogenated thiophene-fused BODIPYs relative to their non-halogenated counterparts. However, it was also demonstrated that rapid NIR light-driven photopolymerizations could be accomplished via optimization of resin formulations comprising one of the non-halogenated BODIPY derivatives. These unveiled design principles will inform future photoredox catalyst design and optimization, enabling low-energy (i.e., benign) chemical transformations such as rapid and efficient manufacturing.
Results and discussion
Inspired by prior reports of NIR light-absorbing thiophene-fused BODIPYs used to generate singlet oxygen for photodynamic therapy by accessing their triplet excited-state manifold,30–32 we synthesized a series of derivatives containing a meso-trifluoromethyl bridgehead and para-methoxyphenyl (R-group) substituents (Fig. 2A). The initial thiophenepyrroles (1) were synthesized via a Knoevenagel–Hemetsberger33 reaction with ethyl azidoacetate and thiophene-carboxaldehyde. Saponification of the ester (1) to the carboxylic acid followed by a coupling reaction with trifluoroacetic anhydride yielded the meso-trifluoromethyl dipyrrole (2). Lastly, reaction with boron-trifluoride diethyl etherate gave the non-halogenated BODIPY derivatives (BODIPY–R′–H), which can be followed by a reaction with bromine to yield the corresponding halogenated versions (BODIPY–R′–Br). Ultimately, four derivates were synthesized and characterized, where R′ is either a hydrogen (–H–), bromine (–Br–), or para-methoxyphenyl (–Ph–). | ||
| Fig. 2 (A) Synthesis of thiophene-fused BODIPYs with images of the four derivatives dissolved in CH3CN (10 μM). Reagents and yields: (i) NaOEt/EtOH, then reflux in toluene (54%); (ii) Br2 in CH2Cl2 (99%); (iii) 4-methoxyphenylboronic acid and Pd(PPh3)4 in toluene/2 M Na2CO3 (94%); (iv) NaOH in EtOH/H2O (94%); (v) TFA/TFAA, then NEt3, BF3OEt2 in toluene (12%); (vi) Br2 in DCM (83%). (B) Normalized BODIPY absorbance and NIR LED emission profiles. Symbols are indexed for clarity. | ||
Upon synthesizing the above thiophene-fused BODIPY derivatives, we next opted to examine their steady-state photophysical properties to gain insight into their potential reactivity as NIR-activated photoredox catalysts (Fig. 2B and Table 1). Incorporation of the electron rich para-methoxyphenyl substituents in combination with the electron deficient meso-CF3 bridgehead provided a so-called “push–pull” system with absorbance maxima of 730, 765, 757, and 748 nm for BODIPY–H–H, –Br–Br, –Ph–H, and –Ph–Br, respectively as 3–6 μM solutions in CH3CN (Fig. 2B and Table 1). Similar absorbance profiles were also found for solutions in isobornyl acrylate monomer (4.74 mM, reaction conditions, vide infra) and in CH2Cl2 and toluene (Table S2 and Fig. S1–S4†). Notably, bromination of BODIPY–H–H to –Br–Br resulted in a ∼35 nm red-shift, while bromination of BODIPY–Ph–H to –Ph–Br resulted in a ∼15 nm blue-shift. Furthermore, maximum molar absorptivity (εmax) values in dilute solution (3–6 μM, CH2Cl2) varied considerably for the four derivatives from 161![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ± 3000, 40000 ± 2000, 85000 ± 3000, and 46000 ± 1000 M−1 cm−1 for BODIPY–H–H, –Br–Br, –Ph–H, and –Ph–Br. Similar trends were observed in isobornyl acrylate, 4.74 mM (Fig. S5–S8†). Variable-temperature UV-vis absorption spectroscopy of BODIPY–Br–Br in 1,2-dichlorobenzene (7 μM and 138 μM) from 0 to 100 °C resulted in no significant change in the spectral profile, suggesting the reduced ε is intrinsic and not a result of aggregation (Fig. S9†). These results signify the importance of carefully quantifying spectral profiles for dyes used to drive photochemical reactions to determine structure–reactivity relationships that inform design principles. Overlaying the absorbance profiles with two LEDs having peak emission wavelengths (λemmax) of ∼780 and ∼850 nm shows excellent overlap for the ∼780 nm LED and modest overlap for the ∼850 nm LED (Fig. 2B), suggesting NIR light excitation could be used effectively to induce polymerization.
000 ± 3000, 40000 ± 2000, 85000 ± 3000, and 46000 ± 1000 M−1 cm−1 for BODIPY–H–H, –Br–Br, –Ph–H, and –Ph–Br. Similar trends were observed in isobornyl acrylate, 4.74 mM (Fig. S5–S8†). Variable-temperature UV-vis absorption spectroscopy of BODIPY–Br–Br in 1,2-dichlorobenzene (7 μM and 138 μM) from 0 to 100 °C resulted in no significant change in the spectral profile, suggesting the reduced ε is intrinsic and not a result of aggregation (Fig. S9†). These results signify the importance of carefully quantifying spectral profiles for dyes used to drive photochemical reactions to determine structure–reactivity relationships that inform design principles. Overlaying the absorbance profiles with two LEDs having peak emission wavelengths (λemmax) of ∼780 and ∼850 nm shows excellent overlap for the ∼780 nm LED and modest overlap for the ∼850 nm LED (Fig. 2B), suggesting NIR light excitation could be used effectively to induce polymerization.
| BODIPY–R′–X | λ max (nm) | ε max (M−1 cm−1) | E S1 (eV) |
|
|
ΔGoxd (eV) | ΔGredd (eV) |
|---|---|---|---|---|---|---|---|
| a Measured using UV-vis absorption spectroscopy in CH2Cl2 (3–6 μM). Averages provided from 3 samples with ±1 standard deviation from the mean. b Identified as the absorption and fluorescence (emission) crossover point. c Calculated from ES1 and ground state redox potentials (see Cyclic Voltammetry Data in the ESI). d Calculated using the Rehm–Weller equation (see Rehm–Weller Analysis section in the ESI for details). | |||||||
| –H–H | 730 | 161000 ± 3000 |
1.67 | −0.98 | 1.02 | 0.08 | −0.72 |
| –Br–Br | 765 | 40000 ± 2000 |
1.53 | −0.46 | 1.25 | 0.24 | −0.64 |
| –Ph–H | 763 | 85000 ± 2100 |
1.57 | −0.82 | 0.95 | 0.32 | −0.47 |
| –Ph–Br | 748 | 46000 ± 1000 |
1.57 | −0.74 | 0.77 | 0.61 | −0.94 |
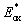

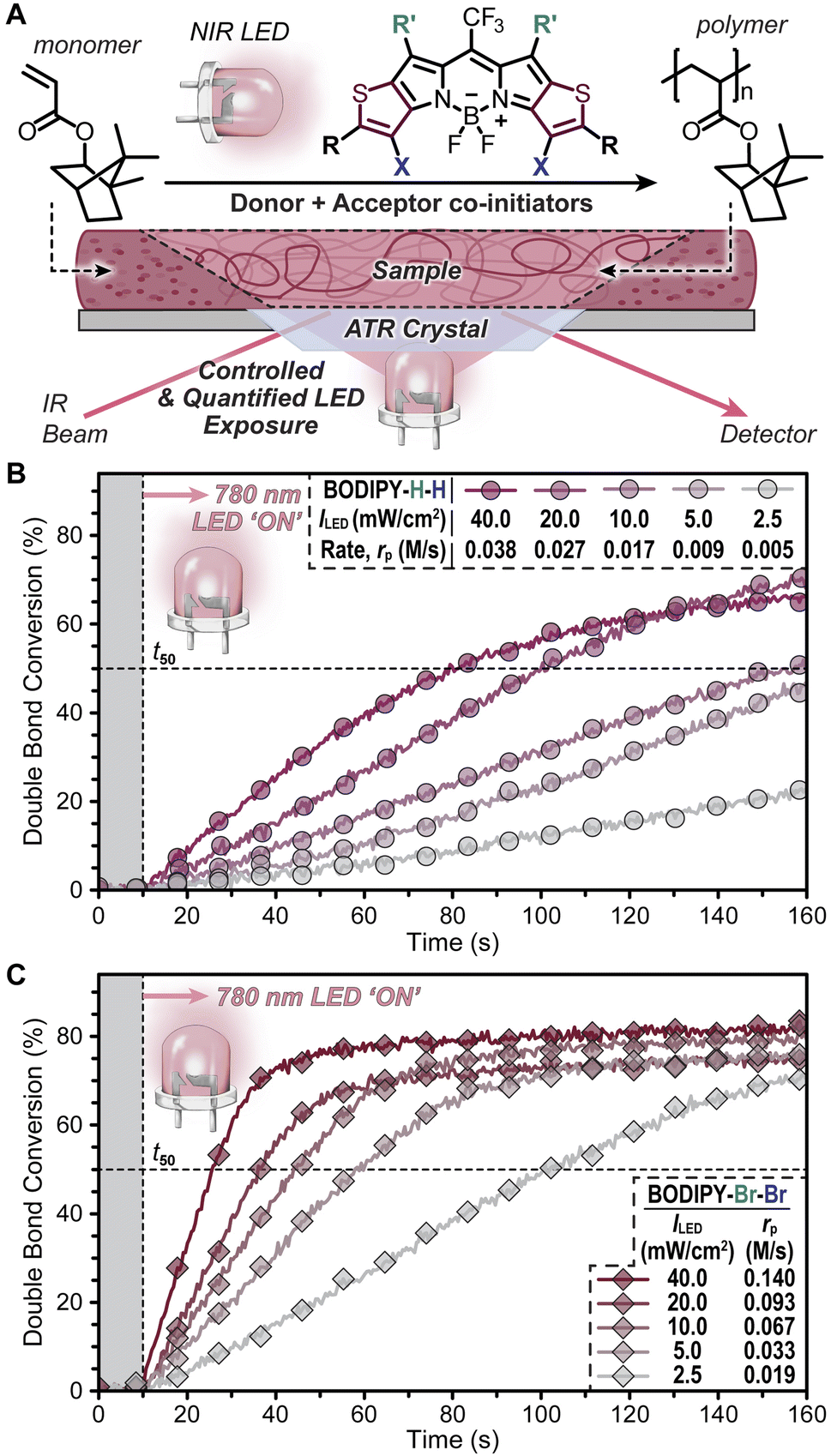
The thiophene-BODIPY derivatives were next examined as NIR photoredox catalysts (Fig. 3). The other Type II photosystem components present in the resin were a borate salt (Borate V, 0.1 mol%) as the electron donor and a diphenyliodonium salt (HNu 254, 1.0 mol%) as the electron acceptor (Fig. S10†), which were selected as effective co-initiators based on our prior reports.14,25 A Rehm–Weller analysis34 was performed to identify the most likely mechanism for photoredox catalysis with the present 3-component system. Excited-state oxidation ( ) and reduction (
) and reduction (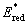 ) potentials for the thiophene-fused BODIPYs were estimated using the singlet excited state energy, ES1, calculated from the crossover in absorption and fluorescence (emission) profiles, in combination with ground state redox potentials obtained from cyclic voltammetry (Table 1 and Fig. S1–S4†), with the latter being used to determine electrochemical oxidation of Borate V (0.43 V) and reduction of HNu 254 (−1.19 V)25 (Fig. S11–S14 and Table S3†). Using these results, a negative change in free energy (ΔG) was calculated for reductive quenching (ΔGred) in all four cases, while the free energy change for oxidative quenching (ΔGox) was positive. This result indicates that electron donation from Borate V to photoexcited BODIPY is the most likely initial mechanistic step in photoredox catalysis.
) potentials for the thiophene-fused BODIPYs were estimated using the singlet excited state energy, ES1, calculated from the crossover in absorption and fluorescence (emission) profiles, in combination with ground state redox potentials obtained from cyclic voltammetry (Table 1 and Fig. S1–S4†), with the latter being used to determine electrochemical oxidation of Borate V (0.43 V) and reduction of HNu 254 (−1.19 V)25 (Fig. S11–S14 and Table S3†). Using these results, a negative change in free energy (ΔG) was calculated for reductive quenching (ΔGred) in all four cases, while the free energy change for oxidative quenching (ΔGox) was positive. This result indicates that electron donation from Borate V to photoexcited BODIPY is the most likely initial mechanistic step in photoredox catalysis.
 | ||
| Fig. 3 (A) Illustration of real-time Fourier transform infrared (RT-FTIR) spectroscopy using attenuated total reflectance (ATR) and irradiating with NIR light from the bottom-up to convert isobornyl acrylate monomers to polymer. Photopolymerization with a 780 nm LED at variable intensity (ILED) for (B) BODIPY–H–H and (C) BODIPY–Br–Br. Grey bar represents the initial dark period prior to turning the light ‘ON’ and the point at which each trace crosses the dashed line represents the time to reach 50% monomer conversion (t50). Symbols are indexed for clarity. | ||
For the polymerizations, isobornyl acrylate (IBOA, 4.74 M) was selected as a representative monomer owing to its biorenewable sourcing and low volatility (Fig. 3A). Additional monomers were also evaluated to demonstrate the versatility of the present NIR light-reactive photopolymerization systems (Fig. S15–S17†). The rate of polymerization (rp) was monitored using a unique real-time Fourier transform infrared (RT-FTIR) spectroscopy setup in an attenuated total reflectance (ATR) configuration, where light exposure occurs from the same face of the ATR crystal as the incident IR beam (Fig. 3A, S18 and S19†). This setup facilitates tunable and quantifiable light dosage from the NIR LEDs by mitigating non-uniform attenuation that would arise from a more traditional top-down irradiation approach.
Initial photopolymerizations were undertaken with BODIPY–H–H and –Br–Br (0.1 mol%) using the 780 nm NIR LED at intensities ranging from 2.5 to 40 mW cm−2 (Fig. 3B and C, and Table S1†). This LED intensity (ILED) range was selected given its relevance to emergent light-based 3D printing technologies – as digital light processing (DLP) systems typically provide <40 mW cm−2 and liquid crystal display (LCD) systems <10 mW cm−2.35 Also relevant to photocurables is the time it takes to convert from a liquid resin to a solid network (i.e., gel point), which can be correlated to double bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) conversion for a particular resin. Thus, we chose to characterize and report rp in M s−1 along with the time to reach 50% CC conversion post-light exposure (t50), which provides metrics for comparison across different photopolymerization conditions (e.g., intensity) and photosystems. Note that monofunctional acrylates were selected here to avoid gelation for ease of cleaning the ATR crystal, yet the monomers will react in an analogous fashion to multifunctional acrylics for photocurables.
C) conversion for a particular resin. Thus, we chose to characterize and report rp in M s−1 along with the time to reach 50% CC conversion post-light exposure (t50), which provides metrics for comparison across different photopolymerization conditions (e.g., intensity) and photosystems. Note that monofunctional acrylates were selected here to avoid gelation for ease of cleaning the ATR crystal, yet the monomers will react in an analogous fashion to multifunctional acrylics for photocurables.
Starting with BODIPY–H–H, samples were prepared by dissolving all photosystem components in IBOA, degassing the mixture with inert gas to remove oxygen that inhibits polymerization, and placing the mixture on the ATR crystal (Fig. S20†). Excellent temporal control of photopolymerization was observed, with no polymerization occurring during the first 10 seconds of the RT-FTIR spectroscopy measurement in the absence of light, followed by immediate polymerization upon irradiation with a 780 nm LED (Fig. 3B). Increasing ILED from 2.5 to 40 mW cm−2 resulted in an ∼8× increase in rp. Specifically, rp values were 0.005 ± 0.001, 0.009 ± 0.001, 0.017 ± 0.001, 0.027 ± 0.002, and 0.038 ± 0.003 M s−1 for 2.5, 5, 10, 20, and 40 mW cm−2, respectively (Fig. 3B inset). Furthermore, a t50 value of ∼70 seconds was measured for the highest ILED tested (40 mW cm−2).
Performing the same characterization with BODIPY–Br–Br under otherwise identical conditions resulted in a ≥4× increase in rp relative to resins with BODIPY–H–H (Fig. 3C). Specifically, rp values were 0.019 ± 0.006, 0.033 ± 0.006, 0.067 ± 0.003, 0.093 ± 0.009, and 0.14 ± 0.001 M s−1 for 2.5, 5, 10, 20, and 40 mW cm−2, respectively (Fig. 3C inset). Additionally, a t50 value of ∼15 seconds was measured when using an ILED = 40 mW cm−2, while also being <60 seconds at much lower intensities; t50 = 48 seconds using an ILED = 5 mW cm−2.
Resins containing BODIPY–Br–Br were irradiated using an 850 nm LED equipped with a 775 nm longpass filter (ILED = 40 mW cm−2) to mitigate excitation by non-NIR photons (Fig. S21†), while monitoring with RT-FTIR spectroscopy. This provided an rp = 0.070 ± 0.007 M s−1 (Fig. S22†). However, resins with BODIPY–H–H showed little-to-no polymerization under these conditions (Fig. S22†). At this stage, the faster polymerization kinetics measured for BODIPY–Br–Br relative to –H–H were attributed at least in part to its stronger NIR light absorption. Additionally, it was hypothesized that the halogens may facilitate intersystem crossing (ISC) to long-lived triplet excited states, which could further enhance rp (vide infra).
Using analogous photopolymerization conditions, resins comprising BODIPY–Ph–H or –Ph–Br were characterized using RT-FTIR spectroscopy (Fig. 4 and Table S1†). Owing to their similar and appreciable absorption overlap with the 850 nm LED output profile, it was decided to systematically compare polymerizations at this lower-energy exposure (again equipped with a 775 nm longpass filter) as opposed to the 780 nm LED. Both derivatives demonstrated good temporal control with rapid responses to 850 nm NIR light. For BODIPY–Ph–H, rp values as a function of ILED were 0.008 ± 0.002, 0.015 ± 0.002, 0.028 ± <0.001, 0.062 ± 0.004, and 0.091 ± 0.003 for 2.5, 5, 10, 20, and 40 mW cm−2, respectively (Fig. 4A inset). Additionally, a t50 value of 27 seconds was measured at 40 mW cm−2. In comparison to BODIPY–Ph–H, the –Ph–Br analogue had an rp that was ∼1.2× larger, with a maximum value of 0.11 ± 0.02 M s−1 and a t50 of 17 seconds at an ILED of 40 mW cm−2 (Fig. 4B). Notably, the slight blue shift in absorption and moderate decrease in molar absorptivity of the brominated (–Ph–Br) derivative resulted in ∼1.5× fewer 850 nm LED photons absorbed relative to the non-halogenated one (–Ph–H) at an equimolar concentration. This suggested that halogenation results in a >1.2× increase in photopolymerization efficiency.
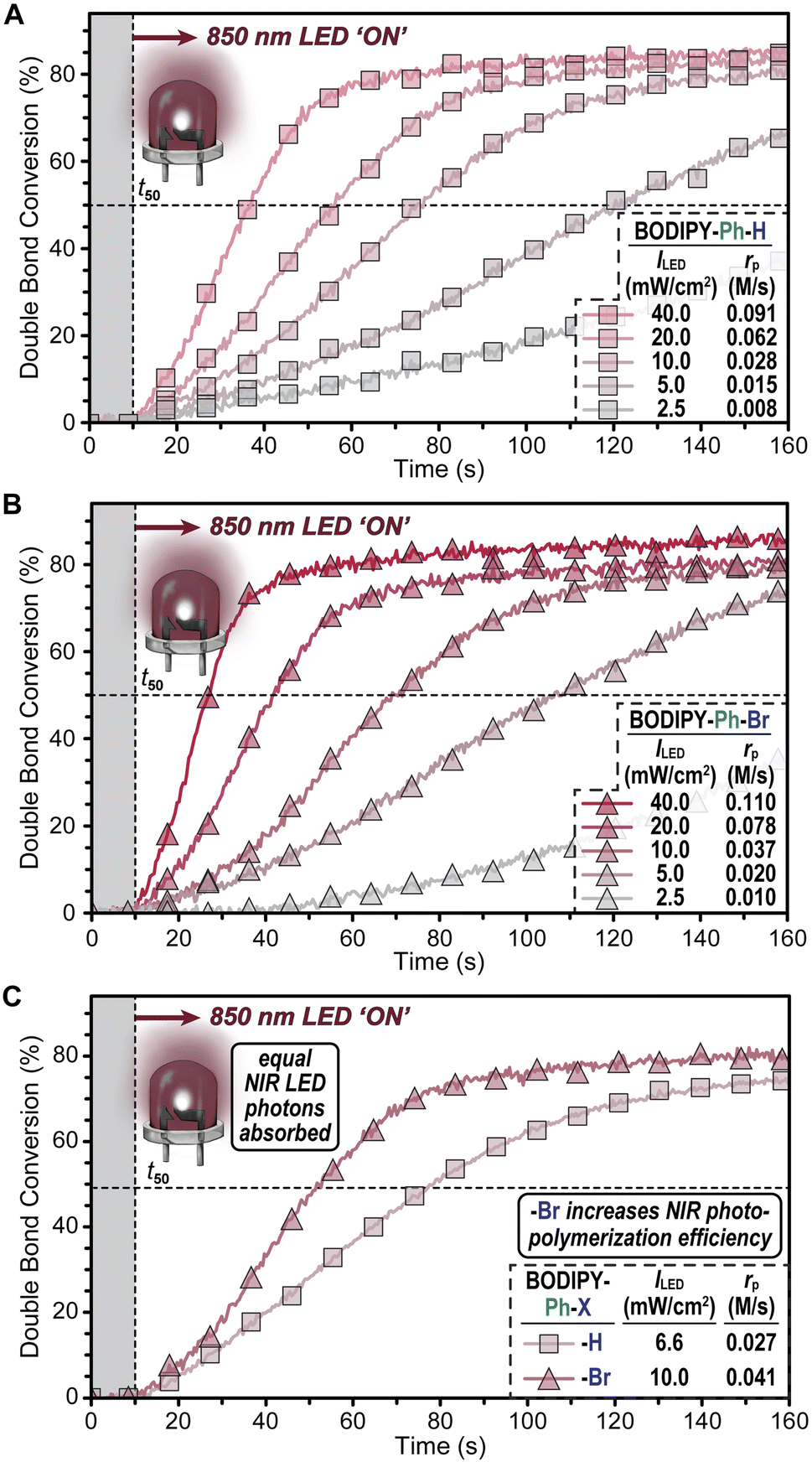 | ||
| Fig. 4 Photopolymerization with an 850 nm LED at variable intensity (ILED) for (A) BODIPY–Ph–H and (B) BODIPY–Ph–Br. (C) Photo-polymerization comparison between BODIPY–Ph–H and –Ph–Br at an ILED that results in an equivalent photon absorption rate by each. Grey bar represents the initial dark period prior to turning the light ‘ON’ and the point at which each trace crosses the dashed line represents the time to reach 50% monomer conversion (t50). Symbols are indexed for clarity. | ||
To better compare the intrinsic photopolymerization efficiencies of resins comprising these two photoredox catalysts, the number of photons absorbed by each equimolar photosystem was equated through controlling ILED (Fig. 4C, and Tables S4 and S5†). Based on the overlap of the absorption cross-section by each BODIPY with the 850 nm LED emission profile, an ILED of 10 mW cm−2 for BODIPY–Ph–Br was found to provide an equivalent photon absorption rate as an LED set to an intensity of ∼6.6 mW cm−2 for BODIPY–Ph–H. Under these conditions, resins with BODIPY–Ph–Br polymerized ∼1.5× faster relative to those containing BODIPY–Ph–H. Specifically, rp values of 0.041 ± <0.001 and 0.027 ± 0.007 M s−1 were measured for the halogenated (–Ph–Br) and non-halogenated (–Ph–H) derivatives, respectively. Thus, the incorporation of heavy atoms was shown to result in a statistically significant increase in NIR photopolymerization efficiency.
Our data highlights that the brominated thiophene-fused BODIPYs BODIPY–Br–Br and BODIPY–Ph–Br exhibit enhanced photopolymerization rates relative to their non-brominated analogues, BODIPY–H–H and BODIPY–Ph–H respectively. These performance gains are observed even after accounting for differences in the spectral overlap of each BODIPY with the output of the 780 and 850 nm LEDs used as light sources for photopolymerization, suggesting that the performance gains of BODIPY–Br–Br and BODIPY–Ph–Br reflect changes in their excited state dynamics induced by the presence of bromine atoms. To ascertain the origin of these changes, we used ultrafast transient absorption spectroscopy (TA) to interrogate the excited state dynamics of the 4 thiophene-fused BODIPYs (Fig. 5).
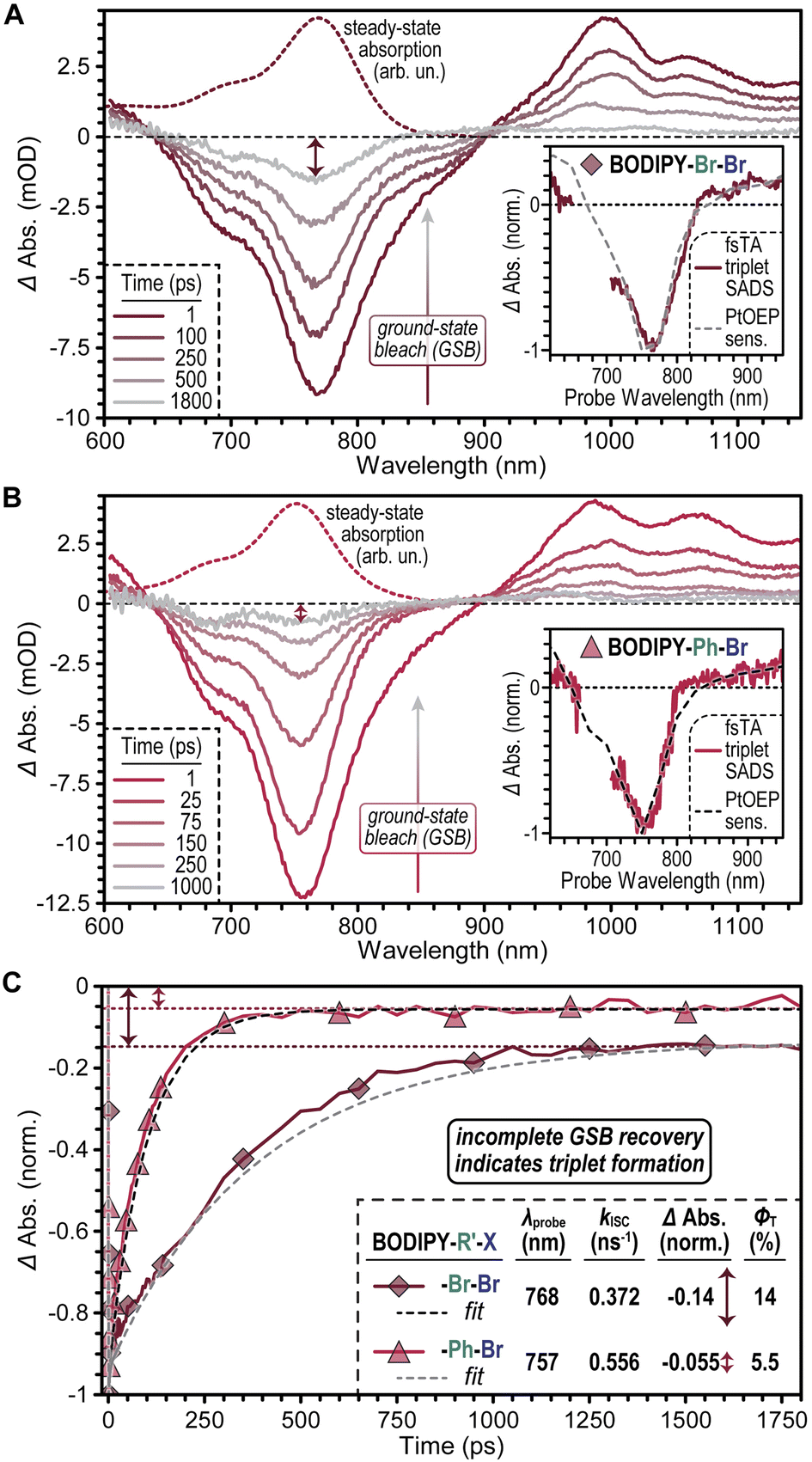 | ||
| Fig. 5 TA spectra of (A) BODIPY–Br–Br and (B) BODIPY–Ph–Br. Dashed lines denote the ground state absorption spectra of each molecule for comparison with the ground state bleach. The insets in each figure compare transient spectra of the T1 state of each molecule measured via triplet sensitization to species-associated decay spectra (SADS) that highlight the state formed by each molecule over ∼1 ns. (C) Dynamics of the ground state bleach (GSB) recovery of BODIPY–Br–Br and BODIPY–Ph–Br. The residual population seen at long delay times (dashed lines) represents the fraction of photoexcited molecules that undergo ISC to access the T1 state used to estimate triplet yield, ΦT. Symbols are indexed for clarity. | ||
TA spectra of BODIPY–Br–Br recorded 1 ps following photoexcitation (Fig. 5A) show a broad negative photobleach peak at 765 nm that is attributed to depletion of its ground state given that it mirrors BODIPY–Br–Br's steady-state absorption spectrum. At longer wavelengths, a small negative shoulder was observed at ∼860 nm, as well as positive bands peaked at 995 and 1060 nm. These features are respectively assigned to stimulated emission and induced absorption bands associated with BODIPY–Br–Br's S1 state. As time progresses, the stimulated emission and S1 induced absorption bands decay in concert and, in their place, a broad induced absorption appears from ∼800 nm to the limit of the NIR window probed (∼1120 nm). Triplet sensitization experiments (Fig. 5A inset) indicated that these features arise due to formation of BODIPY–Br–Br's T1 state. Importantly, TA spectra of BODIPY–H–H (Fig. S25†) show no evidence of ISC occurring upon photoexcitation. Rather, photoexcited BODIPY–H–H molecules return to the ground state (S0) with a timescale of ∼3 ns, which matches the S1 excited state lifetime captured in time-resolved emission experiments (Fig. S1†).
Notably, as BODIPY–Br–Br's T1 state was formed, its ground state bleach underwent a concomitant decay to less than 20% of its initial amplitude (Fig. 5C). One potential explanation for this decay is that induced absorption bands associated with BODIPY–Br–Br's T1 state spectrally overlap with its ground state bleach, which would lead to a partial cancellation of these features as the T1 state is formed. Alternatively, ISC to form the T1 state could compete with direct relaxation to the ground state, S0, from S1. We favor the latter explanation for several reasons. First, prior reports of halogenated BODIPYs have suggested that minimal spectral overlap occurs between their ground state absorption and triplet induced absorption spectra.25,36 These reports appear to be supported by our data as BODIPY–Br–Br's ground state bleach shows minimal spectral evolution as it relaxes (Fig. S27†). If an induced absorption band from BODIPY–Br–Br's T1 state grew in that partially cancelled its ground state bleach, this band would therefore need to directly match the lineshape of BODIPY–Br–Br's ground state bleach to preclude a spectral perturbation in the bleach lineshape, which is unlikely.
Fitting our TA data using a global target analysis model, we found BODIPY–Br–Br's intersystem crossing rate to be 0.372 ns−1 while the rate of S1 relaxation to S0 was 2.29 ns−1, which together gave a triplet yield of 14% (see “Transient Absorption (TA) Characterization” section, page S36 in the ESI† file for details). However, once formed, the triplet exhibited a lifetime of 1.2 μs. Thus, while only 14% of photoexcited BODIPY–Br–Br molecules form their T1 state, this fraction persists for a lifetime that is approximately 400× longer than that of photoexcited BODIPY–H–H molecules (Table S6†). We hypothesize that this long-lived fraction of photoexcited BODIPY–Br–Br molecules contributes to the enhanced photocatalytic performance of BODIPY–Br–Br over BODIPY–H–H.
Comparing the TA spectra of BODIPY–Ph–Br (Fig. 5B) and BODIPY–Ph–H (Fig. S25†) leads to a similar set of conclusions regarding the impact of bromination on the photoexcited dynamics of thiophene-fused BODIPYs. Halogenation to BODIPY–Ph–Br enhances its ability to undergo ISC relative to BODIPY–Ph–H. Notably, we found that while BODIPY–Ph–Br exhibits a slightly faster ISC rate relative to BODIPY–Br–Br (0.556 ns−1vs. 0.372 ns−1, respectively), BODIPY–Ph–Br shows a reduced triplet yield (ΦT) of 5.5% (Fig. 5C), due to an acceleration of its S1 → S0 decay rate (9.54 ns−1) relative to BODIPY–Br–Br (2.29 ns−1). We hypothesize that the faster S1 decay of BODIPY–Ph–Br is tied to the additional nuclear degrees of freedom introduced by its pendant phenyl groups. Rotation of such groups appended to other BODIPY derivatives has been suggested to facilitate nonradiative decay from S1 to S0.37 The lower triplet yield found for BODIPY–Ph–Br relative to BODIPY–Br–Br also explains the more moderate gain in rp observed for BODIPY–Ph–Br over BODIPY–Ph–H (1.5×) relative to those measured for BODIPY–Br–Br over BODIPY–H–H (4×).
Given the less than unity ΦT values of BODIPY–Ph–Br, and BODIPY–Br–Br, we decided to systematically examine if co-initiator (Borate V) and photoredox catalyst concentration could be adjusted to improve rp by inducing a higher rate of reactive collisions involving catalyst molecules in their S1 state (Fig. 6 and Table S1†). To this end, resins containing BODIPY–Ph–H were optimized given the lack of long-lived triplet states, relatively small decrease in rp, and ease of synthesis compared to BODIPY–Ph–Br. As a baseline, resins comprising BODIPY–Ph–H had a high rp from which to build; rp = 0.062 ± 0.004 M s−1 using an ILED = 20 mW cm−2 and a [Borate V] and [BODIPY–Ph–H] = 0.1 mol%. Incrementally increasing the [Borate V] up to 10× resulted in a corresponding increase in rp of up to ∼3× (Fig. 6, S34, and Table S7†). Specifically, rp values of 0.092 ± 0.02, 0.11 ± 0.006, 0.14 ± 0.02, and 0.19 ± 0.01 M s−1 for a [Borate V] of 0.1, 0.2, 0.4, 0.6, and 1.0 mol% were measured, respectively. With these rates, the t50 value was decreased from ∼31 to ∼12 seconds in going from 0.1 to 1.0 mol% Borate V. From there, increasing [BODIPY–Ph–H] incrementally by 10× using 1.0 mol% [Borate V] resulted in an additional ∼2× increase in rp (Fig. 6, S35 and Table S7†). Specifically, rp values of 0.19 ± 0.03, 0.24 ± 0.02, 0.26 ± 0.03, and 0.33 ± 0.06 M s−1 for a [BODIPY–Ph–H] of 0.2, 0.4, 0.6, and 1.0 mol% were measured, respectively. This lead to a further decrease of t50 from ∼12 seconds to only ∼5 seconds. This result is competitive with the best reported NIR photopolymerization system to-date, which has a rp of 1.68 M s−1, albeit a maximum double bond conversion of <40%, using an 850 nm LED at ILED = 40 mW cm−2.24 Notably, unlike the prior system, no additional solvent was required to dissolve the photoredox catalyst herein. Thus, NIR photopolymerizations operating with BODIPY-based photoredox catalysts can be effectively optimized by tailoring the concentration of each individual redox-active component, which will facilitate applications in rapid, low-energy photocurable technology (e.g., coatings, adhesives, and 3D printing).
 | ||
| Fig. 6 Effect of electron donor (borate) and photoredox catalyst (BODIPY–Ph–H) concentration on the rate of polymerization using an 850 nm LED with a 775 nm longpass filter set to an incident intensity of 20 mW cm−2. Grey bar represents the initial dark period prior to turning the light ‘ON’ and the point at which each trace crosses the dashed line represents the time to reach 50% monomer conversion (t50). Symbols are indexed for clarity. | ||
Conclusions
Thiophene-fused BODIPYs were demonstrated to be effective photoredox catalysts for efficient NIR light-driven polymerizations. Four derivatives were examined having either two (BODIPY–X–X) or four (BODIPY–Ph–X) para-methoxyphenyl (= Ph) substituents and four or two bromine atoms (X = Br), respectively. Characterization of optoelectronic properties and associated Rehm–Weller analysis for the present photosystem indicated that reductive quenching was a likely electron transfer mechanism to form initiating radicals for polymerization. Photopolymerization kinetics were monitored using RT-FTIR spectroscopy with simultaneous irradiation from NIR LEDs having peak emission wavelengths of 780 or 850 nm and exciting at different intensities from 2.5–40 mW cm−2. Systematic photopolymerization comparisons using RT-FTIR revealed that incorporation of bromine atoms resulted in a ∼1.2–3.6× increase in rp, reaching values in excess of 0.1 M s−1. Furthermore, ultrafast TA measurements of the thiophene-fused BODIPYs unveiled several key parameters, including that their singlet excited states were short-lived (<3 ns), and BODIPY halogenation facilitated ISC to long-lived (>1 μs) triplet excited states. However, due to the competitive S1 → S0 decay rates, triplet yields of the halogenated BODIPYs were modest (∼6–14%). Taken together with the high rp values achieved using non-halogenated BODIPY photocatalysts, these results suggest that electron transfer from S1 is relatively efficient in this photosystem, despite its short lifetime. In turn, increasing the concentration of photosystem components to address potential diffusion limitations for resins containing the non-halogenated BODIPY–Ph–H derivative resulted in a ∼6× increase in rp (0.06 to 0.33 M s−1) and ∼6× decrease in t50 (31 to 5 seconds) using a low-intensity (20 mW cm−2) 850 nm LED. Going forward, combining photoredox catalyst optimization with chemical modifications to co-initiators to, for example, lower the oxidation potential of the electron donor is anticipated to result in further improvements in NIR light-driven photopolymerization efficiency. Such advancements will enable the integration of NIR light photocurable resins into emergent manufacturing technologies, such as 3D bioprinting and multimaterial-printing, for the energy efficient fabrication of next generation materials.Author contributions
Conceptualization (AS, ZAP); methodology (AS, SRA, TAG, CJO, LE, STR, ZAP); investigation (AS, SRA, TAG, LE); visualization (AS, SRA, TAG, CJO, STR, ZAP); funding acquisition (TAG, STR, ZAP); project administration (STR, ZAP); supervision (STR, ZAP); writing – original draft (AS, STR, ZAP); writing – review & editing (AS, SRA, TAG, CJO, STR, ZAP).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge primary support from the Department of Defense under Grant No. W911NF2210115 (AS, CJO, ZAP; synthesis, steady-state characterization, photopolymerizations, and materials and supplies). Partial support was provided by the National Science Foundation under Grant No. CAT-2155017 (SRA, LE, STR, ZAP; TA spectroscopy) and CMMI-2053567 (SRA, STR), and the Robert A. Welch Foundation under Grant No. F-2007 (AS, ZAP) and F-1885 (STR) and Research Corporation for Science Advancement under Award #28184 (CJO, ZAP). AFRL authors acknowledge funding support from Air Force Research Laboratory contract FA8650-22-D-5408 (TAG).Notes and references
- M. Shaw, J. Twilton and D. J. MacMillan, Org. Chem., 2016, 81, 6898 CrossRef CAS PubMed.
- M. Hansen, W. Velema, M. Lerch, W. Szymanski and B. Feringa, Chem. Soc. Rev., 2015, 44, 3358 RSC.
- N. Zivic, P. Kuroishi, F. Dumur, D. Gigmes, A. Dove and H. Sardon, Angew. Chem., Int. Ed., 2019, 58, 10410 CrossRef CAS PubMed.
- J. Trotta and B. Fors, Synlett, 2016, 27, 702 CrossRef CAS.
- Y. Ma, V. Kottisch, E. McLoughlin, Z. Rouse, M. Supej, S. Baker and B. Fors, J. Am. Chem. Soc., 2021, 143(50), 21200 CrossRef CAS PubMed.
- A. Doerr, J. Burroughs, S. Gitter, X. Yang, A. Boydston and B. Long, ACS Catal., 2020, 10(24), 14457 CrossRef CAS.
- X. Zhang, L. Cox, Z. Wen, W. Xi, Y. Ding and C. Bowman, Polymer, 2018, 156, 162 CrossRef CAS PubMed.
- Y. Bao, Macromol. Rapid Commun., 2022, 2200202 CrossRef CAS PubMed.
- M. Sandmeier, N. Paunović, R. Conti, L. Hofmann, J. Wang, Z. Luo, K. Masania, N. Wu, N. Kleger, F. Coulter, A. Studart, H. Grützmacher, J. Leroux and Y. Bao, Macromolecules, 2021, 54(17), 7830 CrossRef CAS.
- D. Estabrook and E. Sletten, ACS Cent. Sci., 2020, 6(9), 1482 CrossRef CAS PubMed.
- Q. Ma, S. Liu, M. Le Dot, H. Mokbel, Y. Zhang, B. Graff and J. Lalevée, Polym. Chem., 2021, 12, 6386 RSC.
- A. Camposeo, L. Persano, M. Farsari and D. Pisignano, Adv. Opt. Mater., 2019, 7(1), 1800419 CrossRef PubMed.
- K. Jung, N. Corrigan, M. Ciftci, J. Xu, S. E. Seo, C. J. Hawker and C. Boyer, Adv. Mater., 2020, 32, 1903850 CrossRef CAS PubMed.
- D. Ahn, L. Stevens, K. Zhou and Z. Page, ACS Cent. Sci., 2020, 6(9), 1555 CrossRef CAS PubMed.
- P. Lu, D. Ahn, R. Yunis, L. Delafresnaye, N. Corrigan, C. Boyer, C. Barner-Kowollik and Z. A. Page, Matter, 2021, 4, 2172–2229 CrossRef CAS.
- J. L. Pelloth, P. A. Tran, A. Walther, A. S. Goldmann, H. Frisch, V. X. Truong and C. Barner-Kowollik, Adv. Mater., 2021, 33, 2102184 CrossRef CAS PubMed.
- S. Xu, J. Yeow and C. Boyer, ACS Macro Lett., 2018, 7, 1376–1382 CrossRef CAS PubMed.
- K. Lee, N. Corrigan and C. Boyer, Angew. Chem., Int. Ed., 2021, 60, 8839 CrossRef CAS PubMed.
- S. Dadashi-Silab, S. Doran and Y. Yagci, Chem. Rev., 2016, 116, 10212 CrossRef CAS PubMed.
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165 RSC.
- Y. Pang, S. Fan, Q. Wang, D. Oprych, A. Feilen, K. Reiner, D. Keil, Y. Slominksy, S. Popov, Y. Zou and B. Strehmel, Angew. Chem., Int. Ed., 2020, 59, 11440 CrossRef CAS PubMed.
- B. Strehmel, C. Schmitz, C. Kütahya, Y. Pang, A. Drewitz and H. Mustroph, Beilstein J. Org. Chem., 2020, 16, 415 CrossRef CAS PubMed.
- A. Bonardi, F. Bondardi, G. Noirbent, F. Dumur, C. Dietlin, D. Gigmes, J. Fouassier and J. Lalevée, Polym. Chem., 2019, 10, 6505 RSC.
- J. Zhou, L. Pitzer, C. Ley, T. Rölle and X. Allonas, Polym. Chem., 2022, 13(47), 6475–6483 RSC.
- A. Stafford, D. Ahn, E. Raulerson, K. Chung, K. Sun, D. Cadena, E. Forrister, S. Yost, S. Roberts and Z. Page, J. Am. Chem. Soc., 2020, 142(34), 14733 CrossRef CAS PubMed.
- P. Lu, K. Chung, A. Stafford, M. Kiker, K. Kafle and Z. Page, Polym. Chem., 2021, 12, 327 RSC.
- X. Zhang, H. Hu, W. Liu, Y. Wang, J. Liu and P. Wu, J. Phys. Chem. Lett., 2021, 12(38), 9205–9212 CrossRef CAS PubMed.
- J. Koziar and D. Cowan, Acc. Chem. Res., 1978, 11(9), 334–341 CrossRef CAS.
- M. Monka, I. Serdiuk, K. Kozakiewicz, E. Hoffman, J. Szumilas, A. Kubicki, S. Park and P. Bojarski, J. Mater. Chem. C, 2022, 10, 7925–7934 RSC.
- J. Wang, J. Li, N. Chen, Y. Wu, E. Hao, Y. Wei, X. Mu and L. Jiao, New J. Chem., 2016, 40, 5966 RSC.
- S. G. Awuah, S. K. Das, F. D'Souza and Y. You, Chem.–Asian J., 2013, 8, 3123 CrossRef CAS PubMed.
- S. G. Awuah, J. Polreis, V. Biradar and Y. You, Org. Lett., 2011, 13(15), 3884 CrossRef CAS PubMed.
- W. Heaner, C. Gelbaum, L. Gelbaum, P. Pollet, K. Richman, W. DuBay, J. Butler, G. Wells and C. Liotta, RSC Adv., 2013, 3, 13232–13242 RSC.
- M. Narayanan, G. Kodali, V. Singh, V. Velvadupa and R. Stanley, J. Photochem. Photobiol., A, 2012, 249, 9–14 CrossRef CAS.
- D. Ahn, L. M. Stevens, K. Zhou and Z. A. Page, Adv. Mater., 2021, 33, 2104906 CrossRef CAS PubMed.
- X. Miao, W. Hu, T. He, H. Tao, Q. Wang, R. Chen, L. Jin, H. Zhao, X. Lu, Q. Fan and W. Huang, Chem. Sci., 2019, 10, 3096–3102 RSC.
- S. Radunz, W. Kraus, F. Bischoff, F. Emmerling, H. Tschiche and U. Genger, J. Phys. Chem. A, 2020, 124(9), 1787–1797 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, equipment and instrumentation, absorption and fluorescence spectroscopy, Rehm–Weller analysis, material characterization. See DOI: https://doi.org/10.1039/d3ta04462a |
| This journal is © The Royal Society of Chemistry 2023 |