
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel solid-state synthesis route for high voltage Na3V2(PO4)2F3−2yO2y cathode materials for Na-ion batteries†
Mainul
Akhtar
ab,
Hafssa
Arraghraghi
ab,
Sylvia
Kunz
ab,
Qingsong
Wang
 ab and
Matteo
Bianchini
*ab
ab and
Matteo
Bianchini
*ab
aDepartment of Chemistry, Universität Bayreuth, Universitätsstrasse 30, 95447 Bayreuth, Germany
bBavarian Center for Battery Technology (BayBatt), Universität Bayreuth, Weiherstrasse 26, 95448 Bayreuth, Germany. E-mail: matteo.bianchini@uni-bayreuth.de
First published on 15th November 2023
Abstract
Polyanionic compounds, particularly Na3V2(PO4)2F3−2yO2y (NVPF3−2yO2y, 0 ≤ y ≤ 1), have emerged as promising cathode materials for Na-ion batteries since they demonstrate electrochemical performances comparable or even superior to most layered transition metal oxides. However, numerous discrepancies are reported in the literature regarding the solid-state synthesis of NVPF3−2yO2y compounds (especially Na3V2(PO4)2F2O and Na3V2(PO4)2FO2). Herein, we report the successful solid-state synthesis of phase pure NVPF3−2yO2y (y = 0, 0.5, 1) compounds from a new reaction pathway using the pyrophosphate (VO)2P2O7 as a precursor. We demonstrate that the novel reaction route offers a more direct pathway to phase pure compounds by combining in situ X-ray diffraction (XRD) during calcination and theoretical calculations (Density Functional Theory). The latter shows that the new pyrophosphate route is thermodynamically favourable as compared to the previously reported ones involving VOPO4. High temperature in situ X-ray diffraction (XRD) was used to observe the structural evolution during synthesis of NVPF3−2yO2y compounds from either VOPO4 or (VO)2P2O7 sources, clearly showing a direct pathway to the final products, with crystallization starting at temperatures as low as 350–450 °C. The desired quality of the products has been verified by XRD, combined with X-ray absorption and X-ray photoelectron spectroscopy, which confirmed the expected V oxidation state. Finally, we have verified the excellent energy storage performances of the NVPF3−2yO2y compounds in Na half cells with realistic areal mass loading of the electrodes and despite the absence of conducting coating on the surface. The intermediate material with y = 0.5 appears particularly promising both in terms of rate capability and capacity retention, thanks to the fully single-phase reaction mechanism during charge/discharge. Our work expands the range of available synthesis reaction routes for NVPF3−2yO2y materials, offering the possibility to better control the oxidation state of V, the stoichiometry in terms of O/F content and the crystal structure, and ultimately the resulting electrochemical performance.
Introduction
Li-ion batteries are already commercialized in portable electronic devices, in the automotive sector and in stationary applications. However, the rapidly rising demand for electric vehicles and for grid storage applications have raised the concern that lithium would not be able to meet the future demand due to the limited and uneven distribution of lithium ores in the Earth's crust and the subsequent high price and volatility.1,2 Na-ion batteries, as an alternative to the Li-ion technology, have emerged as a more sustainable and cost-effective choice because they rely on more abundant and low-cost materials. In addition, the Na-ion battery technology resembles closely the already commercialized Li-ion batteries in terms of working principles and processing, hence they can share the same manufacturing lines and equipment.3 However, Na-ion batteries currently have lower energy densities compared to Li-ion batteries, which limits their widespread applications.4 Thus, ongoing research is focused on improving the performance of Na-ion batteries, including the development of better cathode and anode materials. Hard carbons have received the most attention as anodes, whereas two main classes of active materials such as layered transition metal oxides (NaxTMO2) and polyanionic compounds have been identified as promising materials classes for cathodes.5 The advantages of layered transition metal oxides over the other chemistries arise from their high theoretical specific and volumetric capacities. Moreover, the Na-deficient P-type (P2 or P3) compounds were shown to outperform O3 ones in terms of cycling stability and rate performance due to the faster migration of sodium ions in the structure.6 Nonetheless, layered transitions metal oxides suffer from low average working voltage and irreversible phase changes during the sodiation and de(sodiation) process, that often results in a loss of structural stability and capacity fade.7Alternatively, polyanionic compounds are established as promising cathode materials for Li-ion and Na-ion batteries due to their high operating voltage, stable crystal structure, and high ionic conductivity.8 The vanadium-based oxy-fluorophosphates Na3V2(PO4)2F3−2yO2y (0 ≤ y ≤ 1, in short NVPF3−2yO2y in the following) are of particular interest.9 It has been experimentally demonstrated that the reversible extraction of two Na+ ions from these cathode materials Na3V2(PO4)2F3−2yO2y to form NaV2(PO4)2F3−2yO2y allows for a significant amount of energy to be stored (about 500 W h kg−1).10 NVPF (the y = 0 end member) has been investigated most heavily, and even benchmarked versus several layered oxides in full Na-ion cells, clearly demonstrating its excellent behavior.11,12 In addition, the O/F ratio in these compounds can be adjusted (by regulating the y value and controlling the synthesis conditions to achieve the desired V oxidation state) to tune the electrochemical properties of the material. By doing so, the working voltage, structural stability at high voltage, and high-rate capability of the material can be optimized. Specifically, increasing the O/F ratio results in a lower working voltage, but improved structural stability at high voltage, as well as high-rate capability.13,14
In terms of the synthesis, in 1999 Le Meins et al. first prepared the Na3V2(PO4)2F3 compound by heating a stoichiometric mixture of NaF and VPO4 under argon atmosphere.15 Barker et al. prepared Na3V2(PO4)2F3 in 2006 further describing a two-step process: VPO4 was prepared using a carbothermal reduction process from V2O5 and (NH4)2HPO4 precursors, and then the stoichiometric ratio of VPO4 and NaF were used to prepare Na3V2(PO4)2F3,16 where the residual carbon from the carbothermal reduction also acted to improve the material's electronic conductivity. Following these, several research groups have reported solid-state synthesis of the NaV2(PO4)2F3−2yO2y materials using VPO4 and VOPO4 as precursors.14,17–21 However, the solid-state synthesis of oxy-fluorophosphate compounds requires high temperature, at which fluorophosphates are known to be unstable by easily losing fluorine during the calcination process. This can lead to the formation of impurities such as Na3V2(PO4)3 (NVP) or the excessive substitution of oxygen for fluorine, which can result in the formation of mixed valence compounds of uncontrolled composition.22,23 Broux et al. suggested significant discrepancies in the unit cell volume and phase diagram observed upon cycling of NVPF reported in several articles.14 The subtle differences in structural and electrochemical features of the as-prepared compositions were assigned to the possible partial substitution of oxygen for fluorine and thus to a mixed valence state for vanadium (V3+, V4+) induced by synthesis conditions. Specifically, the phase pure Na3V2(PO4)2F2O (y = 0.5, NVPF2O) compound of the NVPF3−2yO2y (0 ≤ y ≤ 1) series has been found difficult to prepare. For example, Park et al. successfully synthesized the family of Na3V2(PO4)2F3−2yO2y (0 ≤ y ≤ 1) compounds and subsequently neutron diffraction (ND) and X-ray diffraction (XRD) analyses have shown that only the y = 0.5 composition, among all, exhibited an impurity phase Na3V2(PO4)3.24 Furthermore, another issue with the classical solid-state synthesis of Na3V2(PO4)2FO2 (NVPFO2) from VOPO4 and NaF compounds, as described in the literature by Park et al. and Bianchini et al. is the massive volume expansion that occurs during the solid-state synthesis process, most likely due to the gases released.10,24 This uncontrolled volume expansion can prevent the bulk synthesis of NVPFO2 for industrial purposes. Finally, the correct reactions describing the preparation of the family of NVPF3−2yO2y compounds need to be better understood. To correct the reaction proposed by Broux et al.,14 Nguyen et al. investigated the reaction by using thermogravimetric analysis-mass spectrometry (TGA-MS) and found various discrepancies between the expected synthesis reactions (see eqn (4) and (5) below) and the evolved gases. To propose a corrected reaction, the authors needed to assume the presence of significant amount of water in the precursors, the source of which would be unclear in typical anhydrous (or pre-dried) precursors.25
In this work, we report the successful solid-state synthesis of phase pure vanadium-based oxy-fluorophosphate NVPF3−2yO2y (0 ≤ y ≤ 1) compounds from a new route which resolves all the issues mentioned in the preceding section. A two-step route is presented, where VPO4 and (VO)2P2O7 are used as intermediate compounds (see eqn (1)–(3) below), to prepare phase pure products without any observed expansion of the reactants volume. The V valence state on both sides of the equations is also precisely balanced in these reactions, so that no redox activity of V is required during the calcination process, allowing for a more precise control of the O/F ratio. Furthermore, we demonstrated that each compound of the NVPF3−2yO2y (0 ≤ y ≤ 1) family can serve as a stable high voltage cathode for Na-ion batteries. As expected, despite being the members of same crystal framework, the electrochemical properties, in particular the voltage and rate performances, can be controlled based on the composition (O/F ratio) of these compounds. Using a combined theoretical and experimental approach, we demonstrated the advantages of the present synthesis reaction for the NVPF3−2yO2y compounds and revealed their structural and electrochemical properties.
Experimental
Materials synthesis
Table 1 gathers the precursors used for the synthesis of NVPF3−2yO2y (y = 0, 0.5, 1) materials, referred to as NVPF, NVPF2O and NVPFO2 in the following, respectively. All the samples were prepared by a solid-state method using the following reactions:| For NVPF: 2VPO4(s) + 3NaF(s) = Na3V2(PO4)2F3(s) | (1) |
| V source | Na/F source | Product |
|---|---|---|
| VPO4 (space group: Cmcm) | NaF | Na3V2(PO4)2F3 |
| VPO4, (VO)2P2O7 | NaF + Na2CO3 | Na3V2(PO4)2F2O |
| (VO)2P2O7 (space group: Pca21) | NaF + Na2CO3 | Na3V2(PO4)2FO2 |
For NVPF2O and NVPFO2, using VPO4 and (VO)2P2O7 intermediate compounds:
| VPO4(s) + ½(VO)2P2O7(s) + 2NaF(s) + ½Na2CO3(s) = Na3V2(PO4)2F2O(s) + ½CO2(g) | (2) |
| (VO)2P2O7(s) + NaF(s) + Na2CO3(s) = Na3V2(PO4)2FO2(s) + CO2(g) | (3) |
For NVPF2O and NVPFO2, using VPO4 and VOPO4 as intermediate compounds:
| VPO4(s) + VOPO4(s) + 2NaF(s) + ½Na2CO3(s) = Na3V2(PO4)2F2O(s) + ½CO2(g) + ¼O2(g) | (4) |
| 2VOPO4(s) + NaF(s) + Na2CO3(s) = Na3V2(PO4)2FO2(s) + CO2(g) + ½O2(g) | (5) |
It can be noticed that the main difference between eqn (2) and (3) with respect to eqn (4) and (5) is the required presence of O2 in the products of the latter, leading to the above-mentioned issues. The intermediate compounds VPO4, VOPO4 and (VO)2P2O7 were synthesized via the solid-state reaction of V2O5 and NH4H2PO4 according to the following reactions:
½V2O5(s) + NH4H2PO4(s) = VPO4(s) + NH3(g) + ![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) H2O(g) + ½O2(g) H2O(g) + ½O2(g) | (6) |
| 2VPO4(s) + O2(g) = 2VOPO4(s) | (7) |
| 2VOPO4(s) = (VO)2P2O7(s) + ½O2(s) | (8) |
First, V2O5 (ThermoFisher Scientific; >99.99%) and NH4H2PO4 (Sigma-Aldrich Co. Ltd; 99.5%) were mixed in a Spex ball mill for 90 minutes using zirconia jars. Then, the mixture was pelletized, placed in an alumina crucible covered in gold foil, and heated a first time at 500 °C for 5 h under Ar/6.5% H2 gas flow. The product was further crushed, pelletized, and reduced under Ar/6.5% H2 flow at 800 °C for 9 h to obtain VPO4. The subsequent oxidation of VPO4 calcined at 700 °C for 5 h in air produced VOPO4. To prepare (VO)2P2O7, VOPO4 was further reduced using a mild Ar atmosphere at 700 °C for 20 h. Finally, for Na3V2(PO4)2F3−yOy (0 ≤ y ≤ 1) compositions the stoichiometric amounts of VPO4, VOPO4, (VO)2P2O7, NaF (abcr GmbH; 99.995%), and Na2CO3 (ThermoFisher Scientific; >99.95%) were mixed in Spex ball mill for 90 minutes. The mixtures were pelletized, wrapped with a gold foil, and annealed to 700 °C with a heating rate of 7 °C min−1 for 1 h under Ar atmosphere. Batches of 2–3 g were obtained with good reproducibility.
Materials characterization
Powder X-ray diffraction (XRD) patterns were recorded using a Stoe StadiP diffractometer (acquisition being performed on powder packed in a 0.5 mm diameter borosilicate glass capillary in the 2θ range of 5–50° with a step size of 0.015°). The diffractometer was equipped with a monochromatic Mo-Kα1 (λ = 0.70932 Å) X-ray source and a Mythen2K detector. Rietveld refinements were performed using the FullProf Suite. In situ temperature-resolved XRD was performed by using the Stoe ST2K furnace mounted on the Stoe StadiP diffractometer. Patterns were recorded in the 2θ range of 5–29° with a step size of 0.015° for 10 minutes at each temperature. Powders were filled in sapphire capillaries of 1 mm diameter and heated under Ar gas flow. The exact temperature ramps used are reported in Fig. 4, S5 and S6.†The operando XRD experiment during the initial electrochemical charge/discharge was conducted on a customized CR2023 coin cell with 6 mm diameter quartz glass windows (100 μm thickness). Galvanostatic charging/discharging was performed at C/20 in the voltage range 2.2–4.3 V with constant voltage step at cut-off voltage (30 minutes) to ensure the successful collection of diffraction at fully desodiated states. XRD patterns were collected every 20 minutes with a fixed Mythen 2K detector covering the angular range 0–36°.
Scanning electron microscopy (SEM) images were obtained with a ThermoFischer Scientific Phenom ProX microscope after a gold deposition (sputtering) on the surface of the polycrystalline samples. The images were captured at an acceleration voltage of 10 kV with a magnification of 35000× using a Backscattering electron detector (BSED). X-ray photoelectron spectroscopy (XPS) spectra were collected from a Versa Probe III XPS Microprobe (Physical Electronics PHI), equipped with a monochromatic Al Kα X-ray (1486.6 eV) source. The XPS data were analysed using XPSPEAK version 4.1 program. Survey measurements and detailed spectra were recorded with a constant pass energy of 224 eV and 55 eV, respectively. The V 2p1/2 and V 2p3/2 spectra were fitted with the Lorentz–Gauss and Shirley background. The binding energy was calibrated by using C 1s (284.8 eV) as a reference. X-ray absorption near-edge structure (XANES) spectra at the V K-edge were collected in transmission mode at the beamline KMC2 at BESSY II Light Source – Helmholtz-Zentrum Berlin (HZB), Germany. Reference V foil was used for energy calibration and data was analyzed using the DEMETER package.26
Electrochemical characterization
Electrodes were prepared by dry tape rolling. First, 80 wt% of active material and 15 wt% of carbon black Super P (Alfa Aesar; 99%) were mixed using a mortar and pestle for 20–25 minutes and then 5 wt% of polytetrafluoroethylene, (PTFE, Sigma Aldrich) was added and further mixed for 10–15 minutes. A steel rolling board was used to roll out the electrodes until a shiny and smooth sheet was produced. A 10 mm steel punch was used to cut the self-standing tapes into round electrodes. The loading of the electrodes was 7–10 mg cm−2. The coin half-cells were assembled in an Ar-filled glove box (200 B, MBraun, Germany) (H2O, O2 levels <0.1 ppm) with metallic sodium as a counter and reference electrode, porous glass fiber (Whatman, GF/D grade) as separator, and 120 μL of electrolyte. The electrolyte is 1 M sodium hexafluorophosphate (NaPF6) in ethylene carbonate/dimethyl carbonate (EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMC = 1:1 wt%) with 5 wt% of fluoroethylene carbonate (FEC) as additive. The galvanostatic charge–discharge measurements were performed on a temperature-controlled battery system (Arbin Instruments, USA). All the electrochemical measurements were carried out at 25 °C. The cells were cycled at various specific current rates (0.05–10C, where 1C = 125 mA g−1) between 2.2 and 4.3 V vs. Na+/Na as described in the following.
:DMC = 1:1 wt%) with 5 wt% of fluoroethylene carbonate (FEC) as additive. The galvanostatic charge–discharge measurements were performed on a temperature-controlled battery system (Arbin Instruments, USA). All the electrochemical measurements were carried out at 25 °C. The cells were cycled at various specific current rates (0.05–10C, where 1C = 125 mA g−1) between 2.2 and 4.3 V vs. Na+/Na as described in the following.
DFT calculations
All DFT calculations were performed using the Vienna Ab initio Software Package (VASP)27,28 within the projector augmented-wave approach using the Perdew–Burke–Ernzerhof (PBE)29 with SCAN (strongly constrained and appropriately normed) functional.30 A plane-wave energy cut-off of 520 eV and 3 × 3 × 3 gamma centered k-point grid were used for all total energies.31 All the structures were obtained from the ICSD database32 (Table S1†) and the primitive unit cells were used. For the NVPFO2 and NVPF, where Na ions in the structure are disordered (in the 8i and 4c/8f Wyckoff sites, respectively), a configuration with triangular arrangement of the Na in the most frequently occupied sites were used. For NVPF2O, where additional disorder exists on the O/F site (8j), we generated 14 configurations using the SUPERCELL program33 and selected the one with lowest total electronic energy for the rest of the work. The 3 structures are shown in Fig. S1.† For the reaction Gibbs free energies, we calculated the energies of the O2 and CO2 gases as isolated dimers in a box using SCAN. The obtained energies (approximated as enthalpies) were then used to estimate Gibbs free energies of reaction ΔrG (T) = ΔrH (0 K) − TΔrS (T) for the various synthesis reactions at different temperatures: 0, 298.15, 400, 500, 700, and 1000 K. We neglected the entropies of the solids but retain the larger entropies of CO2 and O2 gases obtained from the NIST-JANAF database at each considered temperature. To confirm the validity of results, we also compared the standard enthalpies of reaction of VPO4 being oxidized to VOPO4 and (VO)2P2O7 obtained from experimental works34 with those that we calculate using total energies, obtaining satisfactorily matching results.Results and discussion
Structural and morphological characterization
Fig. 1a shows the X-ray diffraction patterns of the NVPF3−2yO2y compounds (y = 0, 0.5, 1) solid-state synthesized according to eqn (1)–(3). The diffraction pattern of NVPF corresponds to the orthorhombic structure with space group Amam. This was first described by Bianchini et al. explaining the subtle distortion of the tetragonal structure (space group P42/mnm) earlier described by Le Mains et al. This distortion is due to a specific distribution of the Na+ ions over three different crystallographic sites within the interstitial sites of the polyanionic framework that lowers the electrostatic interaction between the Na+ ions and is likely to aid Na+ diffusivity.15,35 However, due to the increasing screening of Na+ charges by O2−, the orthorhombic distortion diminishes in the compounds NVPF2O and NVPFO2, which are correctly indexed with the tetragonal space group P42/mnm (see Fig. 1a, insets). Hence, our results support the findings of Broux et al. that the orthorhombic distortion persists only shortly after partial oxygen replacement for fluorine in NVPF3−2yO2y and that the orthorhombic splitting at the 400 and 040 diffraction lines disappears beyond y = 0.3.14 | ||
| Fig. 1 (a) Rietveld refinement of powder XRD patterns of three NVPF3−2yO2y (y = 0, 0.5, 1) samples (λ = 0.70932 Å, 2θ range = 5–50°). NVPF is indexed in the orthorhombic space group Amam, NVPF2O and NVPFO2 in the tetragonal space group P42/mnm. The insets (zoom on angular regions 2θ = 19.2° to 20.4° and 2θ = 35° to 40°) show the orthorhombic splitting and the quality of fit at higher angles. The black lines, red dotted lines, and blue lines correspond to calculated, observed and difference of observed and calculated values, respectively. The vertical green lines correspond to Bragg positions. (b) and (c) show the changes of the c lattice parameter and unit cell volume (V) as function of oxygen substitution for fluorine in NVPF3−2yO2y. The black symbols are from our work, the red ones from Park et al.24 | ||
To confirm the precise structure of the NVPF3−2yO2y compounds, structural models were refined against XRD patterns by Rietveld refinement using the Fullprof program. The unit cell parameters and obtained structural parameters are tabulated in Table 2 as well as in Tables S2–S4.† The structural framework is as expected, built by V2O8+2yF3−2y bi-octahedral units, sharing corners with PO4 tetrahedra. As reported by Park et al. and Nguyen et al., with changing composition in the NVPF3−2yO2y family, a linear evolution of unit cell parameters and unit cell volume should be expected.24,25 In fact, the substitution of O2− for F− leads the vanadium to oxidize (V3+ to V4+) to compensate for the charge. This also results in the creation of short vanadyl bonds (V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) leading to the contraction of the unit cell (particularly along c) in the solid solution between the two end members, NVPF and NVPFO2. Based on the lattice parameters determined from Rietveld refinement (Fig. 1a), the c cell parameter and unit cell volume (V) decrease linearly with decreasing fluorine content (Fig. 1b and c). As expected, the lattice parameters change more significantly along the c axis as compared to a or b (Table 2) due to the greater elongation of vanadium bi-octahedra along the c axis. As explained by Park et al., the lattice parameter c results from the sum of the height of the vanadium bi-octahedra (h, i.e. the collinear sequence of F–V–F–V–F bonds in NVPF, for example) and the distance between terminal anions (d).24 Hence, when y increases from 0 to 1, vanadium oxidizes from V3+ to V4+ and thus the h value decreases due to smaller V4+ ionic radius. On the other hand, due to the stronger repulsion between O2−–O2− compared to F−–F− repulsion, d increases with increasing y value. The increase in h is more pronounced than the decrease in d, so overall the c parameter decreases, as does the volume of the unit cell. As a comparison, we also synthesized NVPF2O by the classical route (eqn (4)). The XRD pattern of NVPF2O synthesised from the VPO4 and VOPO4 precursors is shown in Fig. S2a.† The diffraction pattern of NVPF2O clearly exhibits a few impurity peaks, indexed to the Na3VF6 and rhombohedral NVP phases.24,36 This implies that the obtained composition is likely not NVPF2O precisely. More importantly, as observed by Le Bail and Rietveld refinement, the unit cell volume of the synthesized sample is significantly lower than expected (867.83(1) vs. ∼872 Å3), very close to the one expected for NVPFO2 (866.339(9), Table S4†), indicating that most of the fluorine has likely been lost and replaced by oxygen in the structure. In view of this, we attempted to synthesize the desired compound NVPF2O at various temperatures and time, but we were unable to achieve the required composition by this route with VOPO4 (Fig. S2b†). The exact reason is yet unknown, but it further proves the difficulty in reproducing the reported synthesis reaction, as already reported by Nguyen et al., and the related lack of clarity on the underlying reaction that occurs (eqn (4)).25 On the other hand, we were able to obtain pure NVPFO2 from the synthesis reaction utilizing VOPO4 (eqn (5)); however, we observed this results in a product with a porous appearance which underwent significant volume expansion after outgassing (Fig. S3†). This is a major disadvantage for commercial production of NVPFO2 using VOPO4 as the reactant.
O) leading to the contraction of the unit cell (particularly along c) in the solid solution between the two end members, NVPF and NVPFO2. Based on the lattice parameters determined from Rietveld refinement (Fig. 1a), the c cell parameter and unit cell volume (V) decrease linearly with decreasing fluorine content (Fig. 1b and c). As expected, the lattice parameters change more significantly along the c axis as compared to a or b (Table 2) due to the greater elongation of vanadium bi-octahedra along the c axis. As explained by Park et al., the lattice parameter c results from the sum of the height of the vanadium bi-octahedra (h, i.e. the collinear sequence of F–V–F–V–F bonds in NVPF, for example) and the distance between terminal anions (d).24 Hence, when y increases from 0 to 1, vanadium oxidizes from V3+ to V4+ and thus the h value decreases due to smaller V4+ ionic radius. On the other hand, due to the stronger repulsion between O2−–O2− compared to F−–F− repulsion, d increases with increasing y value. The increase in h is more pronounced than the decrease in d, so overall the c parameter decreases, as does the volume of the unit cell. As a comparison, we also synthesized NVPF2O by the classical route (eqn (4)). The XRD pattern of NVPF2O synthesised from the VPO4 and VOPO4 precursors is shown in Fig. S2a.† The diffraction pattern of NVPF2O clearly exhibits a few impurity peaks, indexed to the Na3VF6 and rhombohedral NVP phases.24,36 This implies that the obtained composition is likely not NVPF2O precisely. More importantly, as observed by Le Bail and Rietveld refinement, the unit cell volume of the synthesized sample is significantly lower than expected (867.83(1) vs. ∼872 Å3), very close to the one expected for NVPFO2 (866.339(9), Table S4†), indicating that most of the fluorine has likely been lost and replaced by oxygen in the structure. In view of this, we attempted to synthesize the desired compound NVPF2O at various temperatures and time, but we were unable to achieve the required composition by this route with VOPO4 (Fig. S2b†). The exact reason is yet unknown, but it further proves the difficulty in reproducing the reported synthesis reaction, as already reported by Nguyen et al., and the related lack of clarity on the underlying reaction that occurs (eqn (4)).25 On the other hand, we were able to obtain pure NVPFO2 from the synthesis reaction utilizing VOPO4 (eqn (5)); however, we observed this results in a product with a porous appearance which underwent significant volume expansion after outgassing (Fig. S3†). This is a major disadvantage for commercial production of NVPFO2 using VOPO4 as the reactant.
| Sample | a (Å) | b (Å) | c (Å) | V (Å3) | R WP (%) | R Bragg (%) |
|---|---|---|---|---|---|---|
| Na3V2(PO4)2F3 | 9.0357(1) | 9.0475(1) | 10.74322(8) | 878.27(1) | 12.8 | 4.82 |
|
||||||
| (VO) 2 P 2 O 7 source | ||||||
| Na3V2(PO4)2F2O | 9.04091(6) | 9.04091(6) | 10.6699(1) | 872.13(1) | 13.3 | 5.79 |
| Na3V2(PO4)2FO2 | 9.03231(5) | 9.03231(5) | 10.61917(7) | 866.339(9) | 12.6 | 5.05 |
|
||||||
| VOPO 4 source | ||||||
| Na3V2(PO4)2F2O | 9.03714(9) | 9.03714(9) | 10.62611(3) | 867.83(1) | 23.8 | 13.40 |
| Na3V2(PO4)2FO2 | 9.03170(6) | 9.03170(6) | 10.63043(1) | 867.14(1) | 14.1 | 5.97 |
We further analysed our NVPF3−2yO2y (y = 0, 0.5, 1) samples to prove the vanadium oxidation state by X-ray photoelectron spectroscopy (XPS) and X-ray absorption near edge structure (XANES). XPS analysis has been used to assess the vanadium oxidation state near the surface (Fig. 2a), while XANES could confirm this at the bulk level (Fig. 2b). V 2p XPS spectra of each compound in the NVPF3−2yO2y series exhibit two broad peaks that correspond to the V 2p3/2 and V 2p1/2 energy levels. The NVPF exhibits two broad peaks at binding energies of 516.98 and 524.11 eV, which are in good agreement with the known values of V3+ 2p3/2 and V3+ 2p1/2, respectively.37 This supports that vanadium is present in its trivalent state in NVPF. The binding energies of NVPFO2 are located at 517.72 and 524.57 eV, which are consistent with the values of V4+ environment in this polyanionic compound.38 Finally, the intermediate compound NVPF2O shows binding energies of 517.35 for V 2p3/2 and 524.34 for V 2p1/2, which are in between those of the two end members, confirming the mixed V3+/V4+ valence state (see also Fig. S4†).
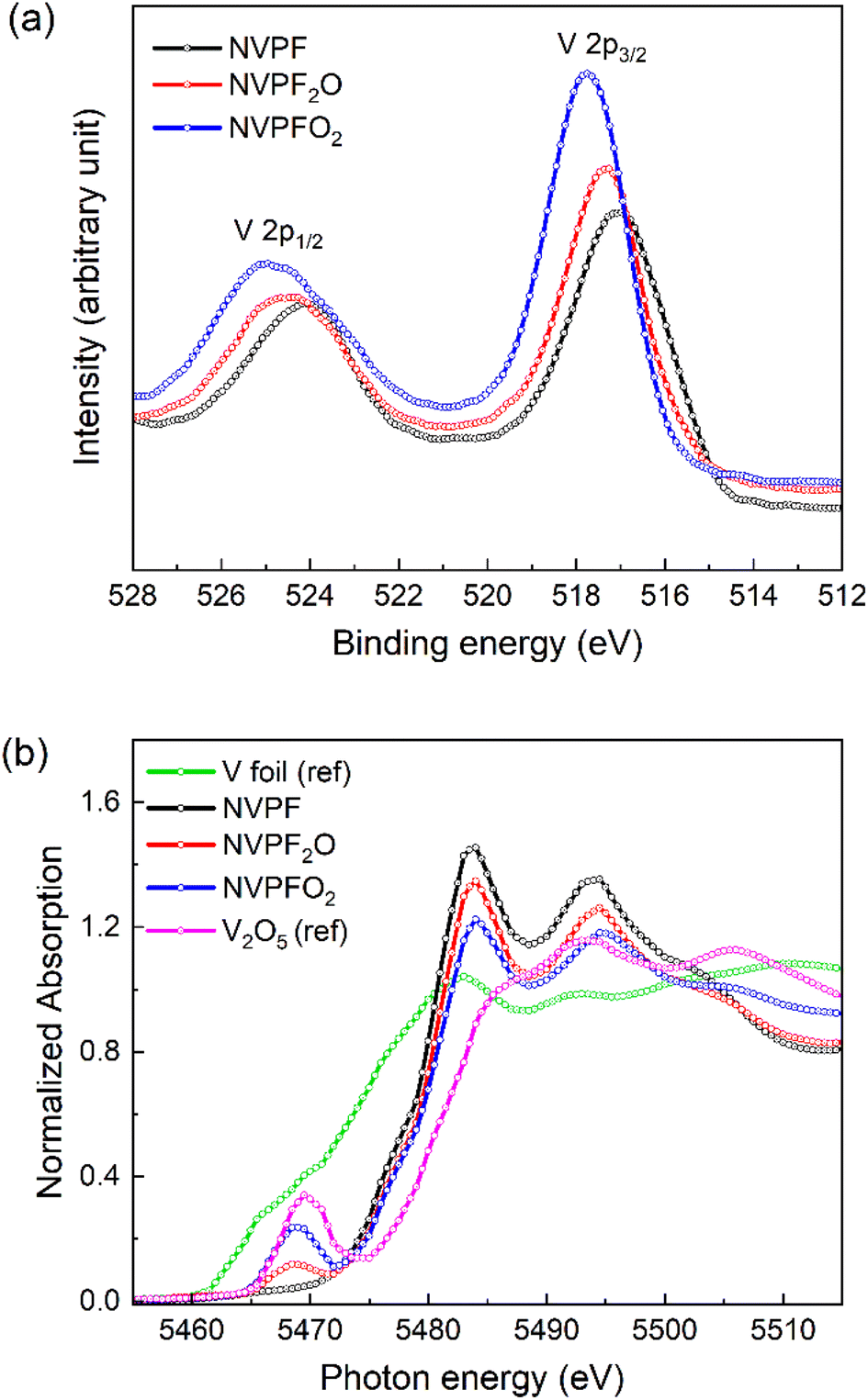 | ||
| Fig. 2 Characterization of the oxidation state of V in NVPF3−2yO2y (y = 0, 0.5, 1) samples. (a) XPS spectra of V 2p energy levels, (b) V K-edge XANES spectra. V foil and V2O5 are reported as reference samples. | ||
XANES was also carried out at the V K-edge to get further insights into the changes on the vanadium bulk oxidation state and the local structure upon partial oxygen substitution for fluorine in the NVPF3−2yO2y (0 ≤ y ≤ 1) compounds (Fig. 2b). As expected, the K edge of the spectrum gradually moves to higher energies as the y value increases, implying the oxidation of the vanadium. The pre-edge signal also exhibits obvious changes as a function of y values, indicating changes in vanadium local structural environment. These signals can be attributed to the 1s to 3d forbidden transitions, which are favored by orbital 3d–p hybridizations in a distorted V4+ octahedra.20 The constant rise in pre-edge signal intensities with increasing oxygen content indicates more distortion of the local symmetry of the vanadium environment or in other words the V2O10F bi-octahedra in NVPFO2 are more distorted than the V2O9F2 in NVPF2O.24
We further analyzed the morphology of the samples obtained by our novel synthesis route. Fig. 3 displays scanning electron microscopy (SEM) images for NVPF, NVPF2O and NVPFO2 powders. It can be observed that the fine particles are largely agglomerated, with irregular shapes in all the samples distributed in a wide size range from 200 nm to 2 μm. Overall, no clear trend in the particles size or shape is apparent with increasing O content in our samples.
 | ||
| Fig. 3 Scanning electron microscopy images of (a) NVPF, (b) NVPF2O, and (c) NVPFO2 samples. | ||
In situ synthesis investigation
To further observe the structural evolution during synthesis of all the tested compositions, high temperature in situ X-ray diffraction was carried out from room temperature to 700 °C (Fig. 4a–c, S5 and S6†). The characteristic high intensity peaks of the reactants in the precursor mixture for all the samples are clearly visible at the beginning of the experiment. To ensure reliability, we refined the diffractogram of the final product after the in situ measurement and verified it matches the expected structure. From Fig. 4, we first observe that the phase formation temperature of NVPF3−2yO2y compounds from either (VO)2P2O7 or VOPO4 (Fig. S5†) sources is low, ranging from 350 to 450 °C, as compared to the calcination temperature commonly employed for synthesizing these compositions (700 °C). For example, the appearance of the 111 characteristic reflection suggesting the phase formation of NVPF3−2yO2y compounds was observed at around 390, 440 and 430 °C for NVPF, NVPF2O and NVPFO2 respectively, when prepared from (VO)2P2O7 source, and 350 and 340 °C for NVPF2O and NVPFO2 respectively, when prepared from VOPO4 source. From this one may conclude that: (i) the phase formation temperature is lower for NVPF than for NVPF2O and NVPFO2 made by the pyrophosphate (ii) the phase formation temperature is higher for NVPF than for NVPF2O and NVPFO2 made by VOPO4, and (iii) in both cases the phase formation temperatures of NVPF2O and NVPFO2 are similar. However, care must be taken. A clear difference in the onset temperature (390 vs. 460 °C) for the phase formation of two NVPF samples (referred as NVPF_p1 and NVPF_p2) was observed when two different precursor sources were used (for V2O5: ThermoFisher Scientific; >99.99%, and Sigma-Aldrich Co. Ltd; 99.95%; for NaF: abcr GmbH; 99.995%, and Grüssing GmbH; >99.0% (Fig. S6†)). | ||
| Fig. 4 In situ high-temperature X-ray diffraction patterns (in 2D contour plots) during the solid-state synthesis of (a) NVPF from VPO4 and NaF (eqn (1)), (b) NVPF2O from VPO4, (VO)2P2O7, NaF, and Na2CO3 (eqn (2)), (c) NVPFO2 from (VO)2P2O7, NaF, and Na2CO3 (eqn (3)). (Left) Temperature ramp used over time. The temperature range was from 25 to 700 °C. | ||
As can be noticed from the SEM images (Fig. S7a–d†), such precursors had different particles size by a factor of 2–3. Precursor reagents with different particle sizes can significantly alter the phase formation kinetics for NVPF3−2yO2y compounds. In a previous report, Semykina et al. also demonstrated that two (3NaF + 2VPO4) reactant mixtures prepared using mortar and pestle or high energy ball milling displayed a discernible difference in the phase composition and the onset temperature for phase formation (for example, 650 and 560 °C for the first and second instances).39 Therefore, while care must be taken in evaluating the relative crystallization temperature of the different NVPF3−2yO2y products, as this can be influenced by a myriad of factors such as precursors choice, how they are mixed and synthesized (including how (VO)2P2O7 and VOPO4 are prepared), etc., we clearly show that thermodynamically and kinetically this occurs already at low temperatures well below 500 °C. Here, it needs to be mentioned that further investigations using TGA-MS analysis combined with detailed refinement of in situ data is needed to properly explain the discrepancies especially in the formation of NVPF2O phase from VOPO4 source, which will be object of future work. A second observation that can be done from Fig. 4, S5 and S6† is that most synthesis reactions appear to go directly from the precursors to the final product without any intermediate step. An exception can be noticed in Fig. 4a (around 2θ = 10° and 14.3°) and Fig. S5a† (around 2θ = 8.25°, 10.1°, 14.3°, and 15°), where some faint reflections can be noticed during the synthesis of NVPF (y = 0) from VPO4 and NaF, and NVPF2O (y = 0.5) from VPO4, VOPO4, NaF, and Na2CO3. On the other hand, no intermediate steps appear when using the pyrophosphate precursors.
Theoretical analysis of the synthesis process
To substantiate our experimental results, we performed DFT calculations to determine the Gibbs free energy of reaction for the synthesis of NVPF3−2yO2y using either (VO)2P2O7 or VOPO4 sources as a function of absolute temperatures (Fig. 5a and b). As observed from the thermodynamic calculations, all the synthesis reactions of NVPF3−2yO2y either with (VO)2P2O7 or VOPO4 sources are feasible (negative ΔrG), except the synthesis reaction of NVPFO2 from VOPO4 source at a very low temperature below 150 K.40 However, as the temperature rises, this reaction is the one that is most favored since it has the steepest negative slope. This is because of the large amount of CO2 and O2 gases that are produced by this reaction (eqn (5)), which also results in practice in a porous and highly expanded reaction product. On the other hand, NVPF has a constant ΔrG since according to eqn (1) there is no gas uptake or release. Overall, Fig. 5 shows how the synthesis of both NVPF2O and NVPFO2 is more favorable (more negative ΔrG) across all temperatures up to 1000 K when using a pyrophosphate precursor (red lines) rather than VOPO4 (blue lines). This is mostly the result of the more negative reaction enthalpy (value at 0 K in Fig. 5) when using (VO)2P2O7, despite the fact that using VOPO4 one would release additional O2 as product (see eqn (4) and (5)) and get a higher entropic gain at increasing temperatures (notice the higher slope of the blue curves in Fig. 5). Yet our calculations show that in the temperature range of interest during the synthesis, i.e., below 1000 K, reactions from VOPO4 never achieve a more negative Gibbs energy of reaction than reactions from (VO)2P2O7.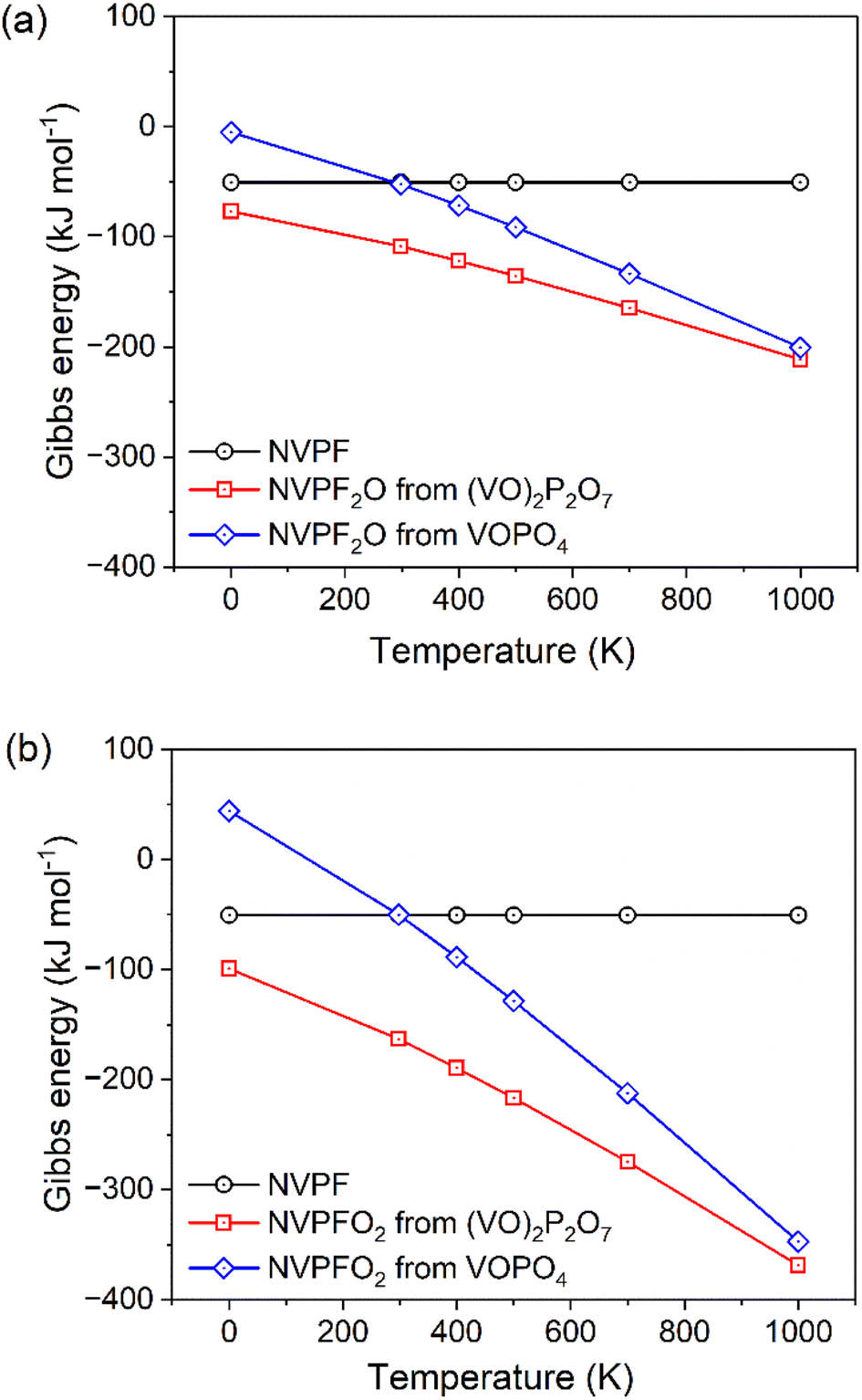 | ||
| Fig. 5 Computed Gibbs free energies for the different reactions as a function of absolute temperature. (a) NVPF from VPO4 and NaF (eqn (1)), NVPF2O from VPO4, (VO)2P2O7, NaF, and Na2CO3(eqn (2)), and NVPF2O from VPO4, VOPO4, NaF and Na2CO3 (eqn (4)). (b) NVPF from VPO4 and NaF (eqn (1)), NVPFO2 from (VO)2P2O7, NaF, and Na2CO3 (eqn (3)), and NVPFO2 from VOPO4, NaF, and Na2CO3 (eqn (5)). | ||
Thanks to this consideration, we can rationalize the fact that the synthesis of NVPF2O is successful via the reaction of eqn (2) while it is harder to achieve for the one of eqn (4). Moreover, it can be noted that the absolute values of ΔrG at the highest temperatures or 700 and 1000 K are significantly more negative (by a factor of 2) when making NVPFO2 rather than NVPF2O, also rationalizing the fact that NVPFO2 is easier to obtain phase-pure and it can be obtained regardless of the precursor choice. However, from a practical perspective, the synthesis from (VO)2P2O7 is still favorable also for NVPFO2 because it leads to less gas releases and hence lower expansion of the reactants during the calcination.
Electrochemical characterization
The electrochemical performance of the NVPF3−2yO2y (y = 0, 0.5, 1) compounds was tested by galvanostatic charge–discharge measurements in a coin-type half-cell configuration with sodium metal anode. The electrodes were prepared with a high mass loading (7–10 mg cm−2, >1 mA h cm−2) to attain high areal capacity in order to realize performances more closely related to practical technologies. It is to be noted that no carbonaceous coating was used on the surface of the samples during synthesis, which may a priori limit the electronic conductivity and hence rate performances of the materials. Fig. 6a and b demonstrate the charge–discharge voltage profiles and the associated dQ/dV plots for the NVPF3−2yO2y electrodes during the 10th cycle measured at C/20 rate in the voltage window of 2.2–4.3 V (vs. Na+/Na). During the initial cycling in half cells, unexpected polarization steps in the discharge profile arise due to the overpotential generated in the sodium counter electrode from the parasitic reactions induced by the electrolyte degradation and the formation of a high resistance film on its surface.14,41 For example, the 1st cycle discharge profiles for all the tested electrodes exhibit polarization steps around 3.4 V, as shown in Fig. S8a.† In addition, the extent of this effect with cycling is shown in Fig. S8b,† where the voltage step is clearly visible on 1, 3 and 5th cycles before completely disappearing on the 10th cycle. This results in a lack of precision in the quantitative assessment of the electrodes during the initial cycling for the half cells. Therefore, we have taken into consideration the 10th cycle for the comparison. The three compositions yield discharge capacities of about 125 mA h g−1 at a current rate of C/20, as shown in Fig. 6a, implying the participation of two Na+ ions per formula unit in the charge–discharge reaction (theoretical capacity 125 mA h g−1). However, significant differences are observed in their average charge–discharge voltages, and in the shape of the voltage profiles. The NVPF and NVPFO2 electrodes clearly display flat plateaus signifying the reversible insertion/deinsertion of Na+ ions through (multiple) biphasic processes.42 On the other hand, the electrochemical curve of NVPF2O features two sloping plateaus indicating most likely the existence of a unique (or at most two) solid-solution composition domain(s) upon Na+ extraction/insertion. In addition, the average potential of the cell progressively decreases from 3.89 to 3.78 V as the y value increases from 0 to 1 in NVPF3−2yO2y (i.e., as fluorine is replaced by oxygen). As suggested by Broux et al., this voltage decrease is caused by the highly covalent vanadyl bond (VO) that forms when the y value increases at the expense of a more ionic V–F bond.14 More precisely, a more covalent environment for the transition metal ion causes its antibonding levels to have energies that are closer to the Fermi energy of sodium, which results in voltage drop for the involved redox couple vs. Na+/Na. On the other hand, as shown in Fig. S9a and b,† charge–discharge voltage profiles and related dQ/dV plots for NVPF2O and NVPFO2 prepared from VOPO4 source reveal that, while the NVPFO2 behaviour is the same regardless of the V source, a clear difference is seen for NVPF2O. The NVPF2O cathode made from VOPO4 delivers a discharge capacity of 116 mA h g−1 at a current rate of C/20. In this case, smaller discharge capacity may be attributed to the impurity phases in the sample. In addition, the voltage profile and dQ/dV plots of this NVPF2O resemble those of pure NVPFO2, which further corroborates the XRD results discussed above (see Fig. S2†), proving that the synthesis of NVPF2O from VOPO4 does not indeed yield the intended product, but rather NVPFO2 plus impurities.
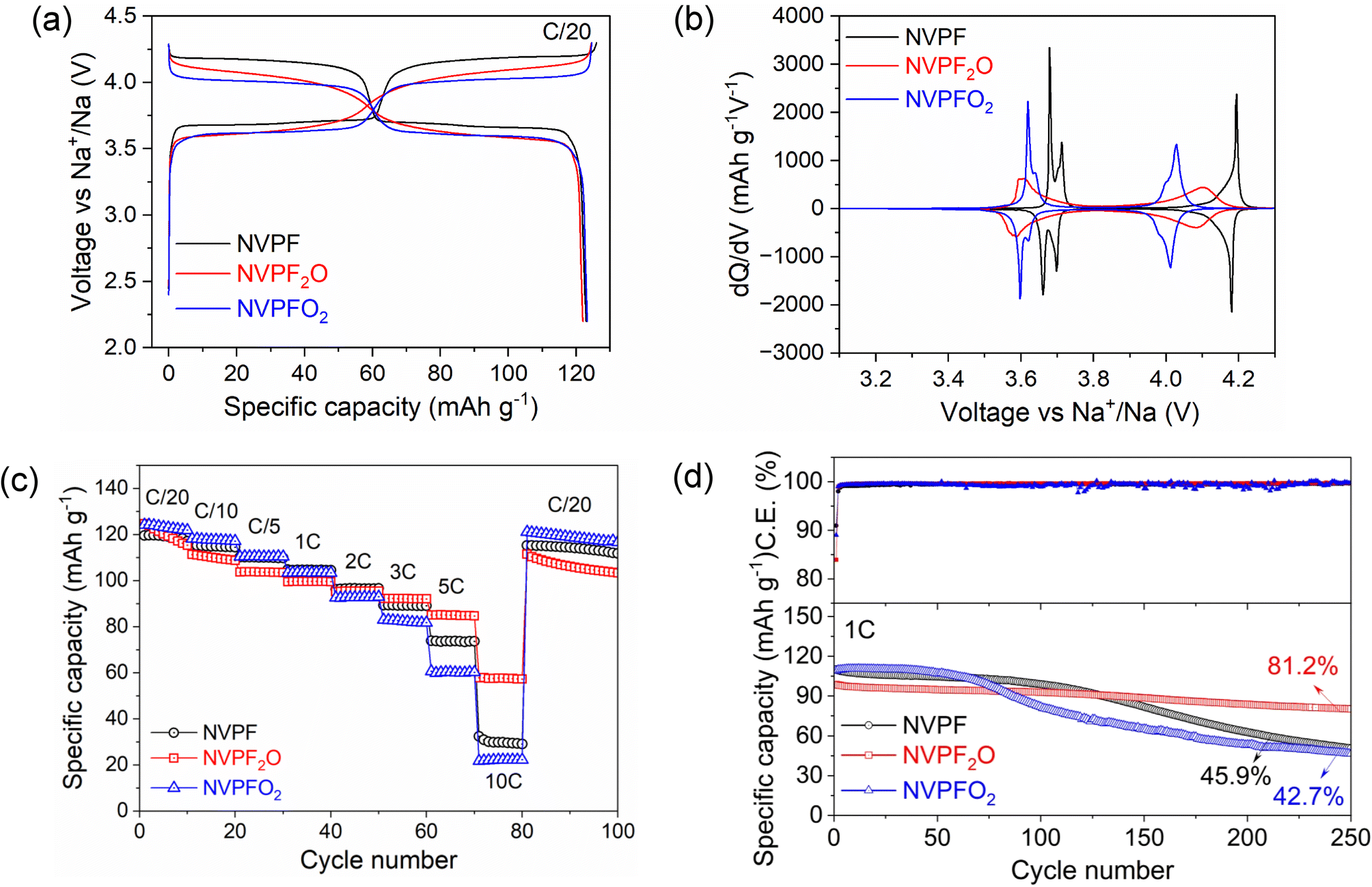 | ||
| Fig. 6 The electrochemical performances of NVPF3−2yO2y (y = 0, 0.5, 1) cathodes in 2.2–4.3 V vs. Na+/Na. (a) Potential profiles for the 10th cycles at C/20, (b) corresponding derivative curves for the 10th cycles, (c) specific discharge capacity during symmetric rate performance test, and (d) cycling performance at 1C rate, including coulombic efficiency (C. E., top) and discharge capacity evolution over cycling (bottom). | ||
To observe the electrochemical performances at high current rates, all the samples were tested for 10 consecutive charge/discharge cycles at each current rate ranging from C/20 to 10C (Fig. 6c). It can be noticed that NVPF2O exhibits the best specific discharge capacities especially at higher current rates. For example, NVPF2O can achieve discharge capacities of 92.23, 85.12, and 57.85 mA h g−1 at a current rate of 3, 5, and 10C, respectively, whereas corresponding discharge capacities are 89.35, 73.89, and 32.39 mA h g−1 for NVPF, and 82.99, 60.66, and 21.72 mA h g−1 for NVPFO2. Nonetheless, NVPFO2 regains its initial capacity the best among the three samples when cycled again at a lower current rate of C/20, perhaps also because of the lower operating voltage. The superior rate performance of NVPF2O can be attributed to the enhanced electronic properties, which are favoured by the mixed V3+/V4+ valence states in the structure.14 Furthermore, the solid solution reaction in NVPF2O might be beneficial for the faster transport of Na+ ions due to fewer number of phase boundaries to overcome,43 as we also verify in the following (see Fig. 7).
 | ||
| Fig. 7 Operando X-ray diffraction of NVPF2O during the first complete cycle. The cell was cycled at C/20 rate between 2.2 and 4.3 V, while acquiring XRD patterns every 20 minutes. A clear single-phase solid solution behaviour is observed throughout the full cycle. | ||
To better observe the kinetic properties of the electrodes, we performed cyclic voltammetry as gathered in Fig. S10.† The linear relationship between ip and ν1/2 confirms diffusion-controlled behaviour in all the electrodes, with the apparent diffusion coefficients obtained from the Rendles–Sevcik equation reported in Table S5.† From these results it appears that the diffusion coefficients for the three materials are comparable, spanning about a factor of 3 between best and worst material. We also find that NVPFO2 seems the material with best kinetic properties, while NVPF appears to be the worst. Interestingly, this matched our observation in Fig. 6c up to the rate of 1C. At even higher rates up to 10C, we had instead observed NVPF2O to yield the best performances. The results obtained from the Rendles–Sevcik equation should nonetheless be taken with caution, as there may be differences in the active surface area of the materials due to the different synthesis which are not taken into account here, as well as the fact that the equation assumes solid solution behavior. This is certainly not true for NVPF and NVPFO2,10,42 while it is accurate for NVPF2O as shown later in Fig. 7. What we can nonetheless conclude is that NVPF2O and NVPFO2 have the advantage over NVPF of a lower voltage of the second charge plateau, which allows higher capacity to be more easily achieved within a fixed voltage window despite the increasing polarization at high rates (note that both charge and discharge are conducted at high rates in our experiments). Moreover, NVPF2O has the additional advantage of having a voltage curve with higher slope, which further helps in this respect (see Fig. 6b or S10a,† where the second plateau of NVPF is quickly lost). Therefore, a larger voltage window or slower rate during charge would be needed to allow high-rate discharge capabilities in NVPF, although this may be detrimental for electrolyte stability. It should be noted here that these results hold for non-coated samples, while it has been demonstrated that carbon-coated NVPF can achieve outstanding rate capabilities at rates beyond 10C.44
Long term cycling stability in half cells has also been tested at different rates. Fig. S11† compares the cycling performance of NVPF3−2yO2y (y = 0, 0.5, and 1) compounds at current rate of C/10 for 50 cycles. All the samples initially offer a discharge capacity of about 120 mA h g−1; in particular, 120.08 mA h g−1 for NVPF, 121.63 mA h g−1 for NVPF2O, and 124.04 mA h g−1 for NVPFO2. However, the samples show different capacity retention capabilities. Among the compositions, NVPFO2 shows the highest capacity retention (96.5%) at low rate after 50 cycles as compared to NVPF (89.1%) and NVPF2O (84.0%). Fig. 6d reports instead the cycling stability at 1C rate over 250 cycles. There, the initial capacities are lower due to the higher rates, especially for NVPF2O (≈100 mA h g−1, in good agreement with the tests shown in Fig. 6c). However, over 250 cycles, NVPF2O clearly performs best with a capacity retention of 81.2%. Taken together, these results demonstrate that NVPF3−2yO2y materials with y > 0 can offer very stable cycling due to their robust structural network. Overall, as previously suggested,45 NVPF2O might perform particularly well within this class of compounds. However, it is important to mention that the actual stability can only be assessed in full cells, which will be object of future work.
As for the samples prepared with the classical synthesis route, which can be seen in Fig. S9d,† among the two comparative samples synthesized from VOPO4 source, NVPF2O exhibits slightly better cycling performance retaining the discharge capacities of 111.7 mA h g−1 (97.2% retention) as compared to the discharge capacity of 118.8 mA h g−1 (96.1% retention) for NVPFO2. Interestingly, NVPF2O made from VOPO4 (which is nearly NVPFO2 in composition as determined by XRD and by its voltage profile) restores its initial capacity after cycling at higher rate (Fig. S9c†). This suggests that a slight change in fluorine to oxygen could have a noticeable impact on the electrochemical performance of NVPF3−2yO2y compounds.
Finally, as mentioned above, we further verified that phase-pure NVPF2O reacts as a single phase during (de)sodiation. Fig. 7 gathers the result of our operando XRD experiment. As can be clearly observed, even for the peaks at high angles, there is no trace of discontinuity in the Bragg reflections. Instead, these shift as befits a solid solution mechanism. The sign of the angular shift depends on the hkl values, as during desodiation in this family of materials, the a–b unit cell parameters decrease, while c increases.42,46 This yields the peculiar behavior observed in Fig. 7, where peaks cross during charge. Overall, our results clearly support that NVPF2O, differently than NVPF or NVPFO2, reacts via a unique single-phase mechanism. This fact appears to be beneficial, as observed above, because the lower voltage, the slopy voltage curve, the high Na+ diffusion coefficient and the lack of phase transitions are all factors allowing high-rate capability and long-term structural stability during cycling in this material.
Conclusion
In the present work, we have reported the successful solid-state synthesis of phase pure vanadium-based oxy-fluorophosphate Na3V2(PO4)2F3−2yO2y (y = 0, 0.5, 1) compounds from a novel route by using (VO)2P2O7 as an intermediate precursor. The present solid-state reaction route offers a new approach to produce phase pure NVPF3−2yO2y compounds while resolving most of the difficulties previously reported by various research groups. Herein, the intermediate compounds VPO4 and (VO)2P2O7 (instead of VOPO4) were used to obtain the phase pure NVPF2O compound. By preparing and exploiting these intermediates, the oxidation state is tailored to the one needed in the product. By decoupling the oxidation state from the overall composition, we demonstrate it is easier to tune the O/F composition as desired. In addition, the use of (VO)2P2O7 also eliminated the issue of obtaining a porous product (no volume expansion) when synthesizing NVPFO2. Using DFT calculations, we have proven that the syntheses of NVPF3−2yO2y using (VO)2P2O7 are more thermodynamically favourable as compared to the reactions employing a VOPO4 precursor. From high temperature in situ X-ray diffraction, we have for the first time demonstrated that the phase formation temperature of NVPF3−2yO2y compounds from either (VO)2P2O7 or VOPO4 sources is low, ranging between 350 and 450 °C, as opposed to the calcination temperature typically used for synthesizing those compositions (700 °C). Using precursors from different sources, we have also shown how the precursor reagents with various particle sizes can significantly change the phase formation kinetics for NVPF3−2yO2y compounds. Finally, we have reported that the NVPF, NVPF2O and NVPFO2 synthesized by the novel route can serve as stable high voltage cathodes for sodium ion batteries. All the samples yielded discharge capacities of about 125 mA h g−1 at a current rate of C/20. However, the average discharge voltage, cyclability, and rate performance differs as a function of oxygen substitution for fluorine in NVPF3−2yO2y. For example, from NVPF to NVPFO2, the average discharge voltage drops by over 100 mV. The best specific capacities, particularly at higher current rates, are demonstrated by NVPF2O (92.23, 85.12, and 57.85 mA h g−1 at current rates of 3, 5, and 10C, respectively, compared to 89.35, 73.89, and 32.39 mA h g−1 for NVPF, and 82.99, 60.66, and 21.72 mA h g−1 for NVPFO2). Among the three samples, NVPF2O also shows the most stable cycling behavior in half cells over 250 cycles at 1C rate, yet this has to be verified in full cells. Nonetheless, by operando XRD, we showed that a specific advantage of this materials over the other compositions, that could rationalize its outstanding behavior, is the unique solid solution reaction mechanism during charge and discharge and the ensuing slopy voltage curve.In summary, we have demonstrated the advantages of the pyrophosphate-based synthesis for NVPF3−2yO2y compounds using a combined theoretical and experimental approach and revealed the excellent energy storage performance of these materials under realistic conditions of high areal mass loading of the electrode and absence of conducting coating on the surface. Our works expand therefore the range of suitable routes to achieve NVPF3−2yO2y materials, being at present the most promising material class as cathode for Na-ion batteries.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge HZB/BESSY for the allocation of synchrotron XAS measurements at beamline KMC2 (Proposal No. 222-11655-ST) and beamline scientist Dr Götz Schuck for the assistance with XAS experiments.References
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 3, 18013 CrossRef.
- P. K. Nayak, L. Yang, W. Brehm and P. Adelhelm, Angew. Chem., Int. Ed., 2018, 57, 102–120 CrossRef CAS PubMed.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- I. Hasa, S. Mariyappan, D. Saurel, P. Adelhelm, A. Y. Koposov, C. Masquelier, L. Croguennec and M. Casas-Cabanas, J. Power Sources, 2021, 482, 228872 CrossRef CAS.
- W. Song, X. Ji, Z. Wu, Y. Yang, Z. Zhou, F. Li, Q. Chen and C. E. Banks, J. Power Sources, 2014, 256, 258–263 CrossRef CAS.
- J. Xiao, F. Zhang, K. Tang, X. Li, D. Wang, Y. Wang, H. Liu, M. Wu and G. Wang, ACS Cent. Sci., 2019, 5, 1937–1945 CrossRef CAS PubMed.
- A. R. Armstrong, S. F. Linnell, P. A. Maughan, B. Silván and N. Tapia-Ruiz, in Sodium-Ion Batteries, John Wiley & Sons, Ltd, 2022, pp. 93–127 Search PubMed.
- C. Masquelier and L. Croguennec, Chem. Rev., 2013, 113, 6552–6591 CrossRef CAS PubMed.
- L. H. Nguyen, Crystal chemistry of polyanion vanadium fluorinated oxy-phosphates: from atomic local structure to electrochemical performance in Na-ion batteries, Material Chemistry, Université de Bordeaux, 2019 Search PubMed.
- M. Bianchini, P. Xiao, Y. Wang and G. Ceder, Adv. Energy Mater., 2017, 7, 1700514 CrossRef.
- P. Desai, J. Forero-Saboya, V. Meunier, G. Rousse, M. Deschamps, A. M. Abakumov, J.-M. Tarascon and S. Mariyappan, Energy Storage Mater., 2023, 57, 102–117 CrossRef.
- S. Mariyappan, Q. Wang and J. M. Tarascon, J. Electrochem. Soc., 2018, 165, A3714–A3722 CrossRef CAS.
- R. Fang, J. Olchowka, C. Pablos, R. Bianchini Nuernberg, L. Croguennec and S. Cassaignon, ACS Appl. Energy Mater., 2022, 5, 1065–1075 CrossRef CAS.
- T. Broux, T. Bamine, F. Fauth, L. Simonelli, W. Olszewski, C. Marini, M. Ménétrier, D. Carlier, C. Masquelier and L. Croguennec, Chem. Mater., 2016, 28, 7683–7692 CrossRef CAS.
- J.-M. Le Meins, M.-P. Crosnier-Lopez, A. Hemon-Ribaud and G. Courbion, J. Solid State Chem., 1999, 148, 260–277 CrossRef CAS.
- J. Barker, R. K. B. Gover, P. Burns and A. J. Bryan, J. Electrochem. Soc., 2007, 154, A882–A887 CrossRef CAS.
- R. Gover, A. Bryan, P. Burns and J. Barker, Solid State Ionics, 2006, 177, 1495–1500 CrossRef CAS.
- T. Jiang, G. Chen, A. Li, C. Wang and Y. Wei, J. Alloys Compd., 2009, 478, 604–607 CrossRef CAS.
- Y.-U. Park, D.-H. Seo, B. Kim, K.-P. Hong, H. Kim, S. Lee, R. A. Shakoor, K. Miyasaka, J.-M. Tarascon and K. Kang, Sci. Rep., 2012, 2, 704 CrossRef PubMed.
- P. Serras, V. Palomares, J. Alonso, N. Sharma, J. M. López Del Amo, P. Kubiak, M. L. Fdez-Gubieda and T. Rojo, Chem. Mater., 2013, 25, 4917–4925 CrossRef CAS.
- A. Ponrouch, R. Dedryvère, D. Monti, A. E. Demet, J. M. Ateba Mba, L. Croguennec, C. Masquelier, P. Johansson and M. R. Palacín, Energy Environ. Sci., 2013, 6, 2361 RSC.
- F. Sauvage, E. Quarez, J.-M. Tarascon and E. Baudrin, Solid State Sci., 2006, 8, 1215–1221 CrossRef CAS.
- M. Akhtar, A. Mitra, J.-K. Chang and S. B. Majumder, ACS Appl. Energy Mater., 2022, 5, 4070–4084 CrossRef CAS.
- Y.-U. Park, D.-H. Seo, H. Kim, J. Kim, S. Lee, B. Kim and K. Kang, Adv. Funct. Mater., 2014, 24, 4603–4614 CrossRef CAS.
- L. H. B. Nguyen, T. Broux, P. S. Camacho, D. Denux, L. Bourgeois, S. Belin, A. Iadecola, F. Fauth, D. Carlier, J. Olchowka, C. Masquelier and L. Croguennec, Energy Storage Mater., 2019, 20, 324–334 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- J. Hafner and G. Kresse, in Properties of Complex Inorganic Solids, ed. A. Gonis, A. Meike and P. E. A. Turchi, Springer US, Boston, MA, 1997, pp. 69–82 Search PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. Sun, A. Ruzsinszky and J. P. Perdew, Phys. Rev. Lett., 2015, 115, 036402 CrossRef PubMed.
- I. L. Matts, S. Dacek, T. K. Pietrzak, R. Malik and G. Ceder, Chem. Mater., 2015, 27, 6008–6015 CrossRef CAS.
- M. Hellenbrandt, Crystallogr. Rev., 2004, 10, 17–22 CrossRef CAS.
- K. Okhotnikov, T. Charpentier and S. Cadars, J. Cheminf., 2016, 8, 17 Search PubMed.
- J. W. Buchler, W. Kokisch and P. D. Smith, in Novel Aspects, Springer Berlin Heidelberg, Berlin, Heidelberg, 1978, vol. 34, pp. 79–134 Search PubMed.
- M. Bianchini, N. Brisset, F. Fauth, F. Weill, E. Elkaim, E. Suard, C. Masquelier and L. Croguennec, Chem. Mater., 2014, 26, 4238–4247 CrossRef CAS.
- N. V. Kosova and D. O. Rezepova, J. Power Sources, 2018, 408, 120–127 CrossRef CAS.
- L. Li, X. Liu, L. Tang, H. Liu and Y.-G. Wang, J. Alloys Compd., 2019, 790, 203–211 CrossRef CAS.
- R. Fang, J. Olchowka, C. Pablos, P. S. Camacho, D. Carlier, L. Croguennec and S. Cassaignon, Batteries Supercaps, 2022, 5, e202100179 CrossRef CAS.
- D. O. Semykina, M. R. Sharafutdinov and N. V. Kosova, Inorg. Chem., 2022, 61, 10023–10035 CrossRef CAS PubMed.
- Z. Zhang, J. Sha, Y. Zu, J. Dai and Y. Liu, J. Adv. Ceram., 2019, 8, 527–536 CrossRef CAS.
- A. Rudola, D. Aurbach and P. Balaya, Electrochem. Commun., 2014, 46, 56–59 CrossRef CAS.
- M. Bianchini, F. Fauth, N. Brisset, F. Weill, E. Suard, C. Masquelier and L. Croguennec, Chem. Mater., 2015, 27, 3009–3020 CrossRef CAS.
- J. Zheng, J. Xiao, X. Yu, L. Kovarik, M. Gu, F. Omenya, X. Chen, X.-Q. Yang, J. Liu, G. L. Graff, M. S. Whittingham and J.-G. Zhang, Phys. Chem. Chem. Phys., 2012, 14, 13515 RSC.
- T. Broux, F. Fauth, N. Hall, Y. Chatillon, M. Bianchini, T. Bamine, J. Leriche, E. Suard, D. Carlier, Y. Reynier, L. Simonin, C. Masquelier and L. Croguennec, Small Methods, 2019, 3, 1800215 CrossRef CAS.
- X. Sun, Z. Wang, Q. Hu, X. Mei, Y. Zhao, A. Ndahimana, T. Geng and J. Cui, ACS Appl. Energy Mater., 2022, 5, 15799–15808 CrossRef CAS.
- N. Sharma, P. Serras, V. Palomares, H. E. A. Brand, J. Alonso, P. Kubiak, M. L. Fdez-Gubieda and T. Rojo, Chem. Mater., 2014, 26, 3391–3402 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta04239a |
| This journal is © The Royal Society of Chemistry 2023 |