
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An intrinsically stretchable symmetric organic battery based on plant-derived redox molecules†
Nara
Kim
 *ad,
Samuel
Lienemann
a,
Ziyauddin
Khan
a,
Grzegorz
Greczynski
b,
Aiman
Rahmanudin
a,
Mikhail
Vagin
ad,
Fareed
Ahmed
a,
Ioannis
Petsagkourakis
ac,
Jesper
Edberg
c,
Xavier
Crispin
*ad and
Klas
Tybrandt
*ad
*ad,
Samuel
Lienemann
a,
Ziyauddin
Khan
a,
Grzegorz
Greczynski
b,
Aiman
Rahmanudin
a,
Mikhail
Vagin
ad,
Fareed
Ahmed
a,
Ioannis
Petsagkourakis
ac,
Jesper
Edberg
c,
Xavier
Crispin
*ad and
Klas
Tybrandt
*ad
aLaboratory of Organic Electronics, ITN, Linköping University, 601 74 Norrköping, Sweden. E-mail: nara.kim@liu.se; xavier.crispin@liu.se; klas.tybrandt@liu.se
bThin Film Physics Division, Department of Physics, Chemistry and Biology, Linköping University, 581 83 Linköping, Sweden
cUnit of Bio- and Organic Electronics, Department of Smart Hardware, Digital Systems Division, RISE Research Institutes of Sweden, 602 33 Norrköping, Sweden
dWallenberg Initiative Materials Science for Sustainability, ITN, Linköping University, 601 74 Norrköping, Sweden
First published on 10th November 2023
Abstract
Intrinsically stretchable energy storage devices are essential for the powering of imperceptible wearable electronics. Organic batteries based on plant-derived redox-active molecules can offer critical advantages from a safety, sustainability, and economic perspective, but such batteries are not yet available in soft and stretchable form factors. Here we report an intrinsically stretchable organic battery made of elastomeric composite electrodes formulated with alizarin, a natural dye derived from the plant Rubia tinctorum, whose two quinone motifs enable its uses in both positive and negative electrodes. The quaternary biocomposite electrodes possess excellent electron-ion conduction/coupling and superior stretchability (>300%) owing to self-organized hierarchical morphology. In a full-cell configuration, its energy density of 3.8 mW h cm−3 was preserved at 100% strain, and assembled modules on stretchy textiles and rubber gloves can power integrated LEDs during various deformations. This work paves the way for low-cost, eco-friendly, and deformable batteries for next generation wearable electronics.
Introduction
A strong trend within electronics is towards wearable and implantable electronic devices for new human–machine interfaces, internet-of-things (IoT), healthcare monitoring/therapy, and soft robotics.1,2 In the last decade, there has been remarkable progress in the development of various stretchable electronics devices, including transistors,3 sensors,4 and displays,5 that are mechanically compliant to skin and tissues.6 However, the integration of equivalently stretchable power sources into such wearables has been lagging, as it is more challenging to impart the mechanical characteristics of the human body—dynamic stretchability over 50%, elasticity, and softness—to relatively bulky (often mm-thick) energy storage devices like batteries and supercapacitors. So far, several structural concepts—buckled structures, sponge structures, kirigami/origami designs, rigid island geometries, and fiber shapes—have been successfully applied to demonstrate stretchable batteries and supercapacitors by converting conventional rigid components into stretchable form factors.7,8 Nonetheless, their seamless integration has been hindered due to inevitable drawbacks such as out-of-plane deformations and directionally constrained stretchability. Moreover, such structural strategies typically involve expensive and complicated fabrication processes. Intrinsically stretchable energy storage devices could resolve many of the previously mentioned issues by providing omni-directional stretchability and the potential for high-throughput printing processing, but it requires that all components (encapsulation, current collector, separator, and active electrodes) within the cell are made intrinsically stretchable.An important aspect for wearable applications is that the devices are safe, cheap, biocompatible, and have a low environmental footprint. Li-ion batteries (LIBs) are the most utilized energy storage devices owing to its high energy density and long-term stability.9 Recently, intrinsically stretchable LIBs have been demonstrated by using stretchable organogels as a binder for the electrode materials and/or an electrolyte while employing Au microcracks or metal microparticle/MWCNT composites to create stretchable current collectors.10,11 Despite their high capacity and good stretchability (i.e. 99 mA h g−1 @ 70%), LIBs inherently suffer from high manufacturing costs, the use of finite transition metal raw materials and has safety issues due to the use of flammable organic electrolytes.12 Moreover, the lack of stretchable packaging that protects the reactive constituents from oxygen and water under mechanical deformation hinders their use in realistic settings. Other battery chemistries using more earth-abundant elements, such as Na and Zn ions, as electrode materials have emerged as a relatively cheap alternative.13,14 Nonetheless, the use of toxic metal oxides as cathode materials and the CO2 emissions during mining and battery production still poses environmental concerns.15,16
In contrast, organic batteries are based on the most abundant elements on the surface of the earth; C, H, and O. Already in the 1970s, small molecules and redox polymers have been implemented as electrode materials in Li metal–organic batteries, and lately in alkali ion-organic batteries.17,18 Recently, all-organic batteries have received increasing attention to take full advantages of using organic materials.19,20 Meanwhile, in 2012, lignin, which is the second most abundant biopolymer on earth and constitutes almost 30% of wood, has shown the potential to store electric charges via the quinone/hydroquinone redox couple.21 Inspired by this work, several naturally-derived quinone-rich compounds, including humic acid, juglone, tannin, emodin, and alizarin, have been employed as active materials for energy storage devices.22–26 The use of such biomaterials extracted from plants offers tremendous advantages in terms of cost effectiveness, environmental friendliness, and renewability.23 Moreover, the battery chemistry based on these redox reactions offers high-rate performance and less barrier requirement than e.g. Li-ion batteries. Nonetheless, stretchable organic batteries have been demonstrated only once in the form of ultrathin (1 μm thick) rechargeable device,27 and so far, there is no report on stretchable organic batteries based on plant-derived redox molecules.
Here we report a stretchable symmetric organic battery built with all intrinsically stretchable layers: elastomeric biocomposite electrodes, gold nanowire–elastomer composite current collectors, and a quasi-solid-state hydrogel electrolyte/separator. All layers were prepared via aqueous solution processes (solution mixing, coating, casting, filtering) and the devices were assembled via dry transfer processes owing to the good adhesion between the layers. The biocomposite electrode possesses excellent multifunctionality, including electrical conductivity (70 S cm−1), ionic conductivity (12 mS cm−1), charge storage capacity (6.8 mA h cm−3), and stretchability (>300%). The favorable properties originate from the four constituent organic materials—conducting polymer (CP), ionic liquid (IL), sulfonated alizarin (alizarin red S, ARS), and polyurethane elastomer—and their hierarchically self-organized structure achieved by simple aqueous solution blending and casting. Owing to the existence of both quinone and hydroquinone motifs in ARS, the intrinsically stretchable symmetric full cell was constructed by using the biocomposite electrodes as both positive and negative electrodes, exhibiting an energy density of 3.8 mW h cm−3, good rate capability (maximum power density of 0.9 W cm−3), and excellent stability under stretching (>90% capacity retention at 100% tensile strain loading or after 1000 stretching cycles at 50% strain). The symmetric architecture of our organic battery simplified the design and the fabrication process of a battery module. Based on its skin-like mechanical properties and planar geometry, an wearable organic battery module was seamlessly and conformably integrated onto a stretchy textile and a glove. The module demonstrated stable powering of an integrated LED under folding, twisting, and stretching deformations.
Results and discussion
Elastomeric biocomposite electrodes
Fig. 1a shows the chemical structures of the four constituent materials: poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS), 1-ethyl-3-methylimidazolium:tricyanomethanide (EMIM:TCM), 1,2-dihydroxyanthraquinone-3-sulfonic acid (ARS), and water-borne polyurethane (WPU). The WPU, which constitutes >60 wt% of the composite, forms an elastomeric matrix that imparts the superb mechanical properties—stretchability, elasticity, and softness—to the composite. The conducting polymer PEDOT:PSS, which is a complex of positively doped PEDOT and charge-balancing PSS polyanion, creates the electrical pathways within the elastomeric matrix. Achieving high electrical conductivity in an elastomeric composite system requires not only a highly conductive filler material but also a well-interconnected percolation network. The addition of an IL such as EMIM:TCM has been shown to significantly increase the electrical conductivity of PEDOT:PSS, from approximately 1 S cm−1 to above 103 S cm−1, by changing the morphology of PEDOT:PSS from amorphous granules to semi-crystalline nanofibrils.28 In composites, a fibrous nanostructure of the conductive filler is beneficial from a percolation network perspective due to the low percolation threshold for 1D-shaped fillers.29 Equally important, ILs act as plasticizers for PEDOT:PSS, which enables the conductive network to deform along with the elastomer matrix and thereby drastically improves the mechanical and electromechanical properties of a composite system.30 Moreover, since ILs are molten salts, they can induce high ionic conductivity within the material, which recently was demonstrated for an IL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :WPU composite with an approximate conductivity of 10 mS cm−1.31 One should note that transport of both electrons and ions is essential for the functionality of electrodes in supercapacitors and batteries, where the electrochemical charging occurs throughout the whole electrode material. Lastly, to maximize the charge storage capacity of the composite electrodes, alizarin, a common red dye that can be derived from the roots of the plant Rubia tinctorum (Fig. 1b), was introduced in its sulfonated form to the composite electrodes. Among the various redox-active molecules found in nature, alizarin was chosen because: (i) it has both two hydroxyl and two carbonyl groups in the aromatic backbone, which leads to either a reduction at negative potential with 4 hydroxyl groups; or an oxidation at positive potential with 4 carbonyl group; each of those reactions in aqueous medium proceed often as a single step two-electron/two-proton reaction (see redox molecule in Fig. 1a).24 (ii) Its sulfonated sodium salt (ARS) is highly soluble in water. (iii) The strong interaction between the sulfonate groups in ARS and positively charged PEDOT chains facilitates the formation of ARS–PEDOT interfaces for efficient electronic charge transfer during redox reactions.32 Note that alizarin, like other redox-active biomolecules, is electrically insulating; thus, the combination with electrical conductors is essential for electrochemical energy storage through electronic charge transport to and from the redox sites in alizarin.
:WPU composite with an approximate conductivity of 10 mS cm−1.31 One should note that transport of both electrons and ions is essential for the functionality of electrodes in supercapacitors and batteries, where the electrochemical charging occurs throughout the whole electrode material. Lastly, to maximize the charge storage capacity of the composite electrodes, alizarin, a common red dye that can be derived from the roots of the plant Rubia tinctorum (Fig. 1b), was introduced in its sulfonated form to the composite electrodes. Among the various redox-active molecules found in nature, alizarin was chosen because: (i) it has both two hydroxyl and two carbonyl groups in the aromatic backbone, which leads to either a reduction at negative potential with 4 hydroxyl groups; or an oxidation at positive potential with 4 carbonyl group; each of those reactions in aqueous medium proceed often as a single step two-electron/two-proton reaction (see redox molecule in Fig. 1a).24 (ii) Its sulfonated sodium salt (ARS) is highly soluble in water. (iii) The strong interaction between the sulfonate groups in ARS and positively charged PEDOT chains facilitates the formation of ARS–PEDOT interfaces for efficient electronic charge transfer during redox reactions.32 Note that alizarin, like other redox-active biomolecules, is electrically insulating; thus, the combination with electrical conductors is essential for electrochemical energy storage through electronic charge transport to and from the redox sites in alizarin.
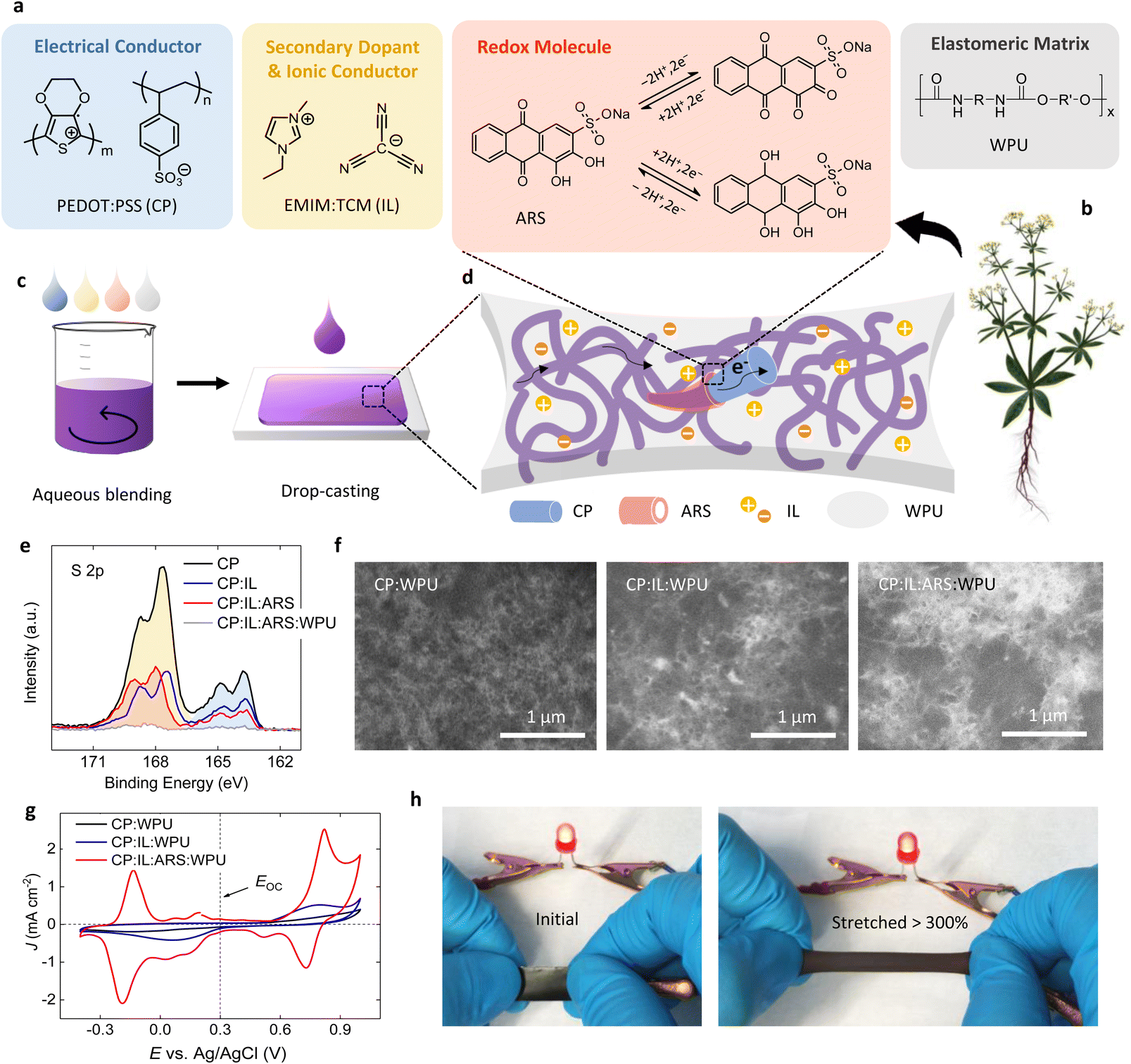 | ||
| Fig. 1 Elastomeric biocomposite electrode. (a) Chemical structures of the four organic components: conducting polymer (CP) PEDOT:PSS, ionic liquid (IL) EMIM:TCM, redox-active molecule ARS, and elastomer WPU. (b) Alizarin can be extracted from the root of the plant Rubia tinctorum. Reproduced with permission from ref. 26. (c) All component materials are soluble or dispersible in water, thereby enabling aqueous blending. (d) The biocomposite film obtained by drop-casting the composite ink possesses a self-organized hierarchical structure: an electrically conductive CP fibrillar network coated with redox-active ARS molecules, which is embedded in an ionically conductive and elastic IL:WPU matrix. (e) High resolution XPS spectra of the S 2p region for CP and the composites when each component is added. S 2p doublet peaks come from the sulfonate moieties in PSS or ARS (171–167 eV, light yellow or orange color) and from the thiophene rings in PEDOT (167–163 eV, light blue color). (f) SEM images of the composites containing various components, showing a morphological evolution from granules (CP) to dense fibrils (CP:IL:ARS) embedded in WPU. (g) Cyclic voltammogram (CV) of the composites containing various components in a three-electrode measurement (0.7 M H3PO4 electrolyte). Intense oxidation and reduction peaks appear after the addition of ARS. The dashed line indicates open circuit voltage (EOC ≈ 0.3 V vs. Ag/AgCl). CV scan rate, 2 mV s−1. (h) Photographs of the CP:IL:ARS:WPU biocomposite film that can be stretched over 300% while maintaining the electrical conduction needed to light up an LED. | ||
All constituents are soluble/dispersible in water and can thereby be blended into an aqueous composite solution (Fig. 1c and S1†). To effectively combine and utilize the desired properties of each material, it is of uttermost importance to create a favorable morphology within the composite. In our carefully designed composite system, a hierarchical structure depicted in Fig. 1d—an electrically conductive fibrillar network of CP with redox-active ARS molecules attached to the surface, embedded in an ionically conductive elastomeric matrix of IL:WPU—was spontaneously organized through electrostatic and van der Waals interactions between constituent molecules. The formation of such a hierarchical morphology was revealed by X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM), and X-ray diffraction (XRD) studies.
Fig. 1e shows the evolution of high resolution XPS S 2p spectra of the composite as the various constituents are added. XPS survey spectra and high-resolution spectra of C 1s, N 1s, and O 1s core levels are shown in Fig. S2.† S 2p spectrum from the PEDOT:PSS exhibits two distinct spin-split doublets, which originate from the thiophene rings in PEDOT (167–163 eV) and from the sulfonate moieties in PSS (171–167 eV). In discord with the existing molar ratio (RM) of PEDOT to PSS in pristine PEDOT:PSS (RM = 1/1.9), the intensity ratio between S 2p peaks from PEDOT and PSS is approximately 1/2.8, indicating an excessive content of PSS on the surface, which is attributed to the well-known PEDOT-rich core/PSS-rich shell structure.33 However, upon the addition of IL, not only strong N(1s) peaks from C–N and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds in the IL appear at 401.6 eV and 398.5 eV, but the PEDOT/PSS S 2p intensity ratio is also increased to 1/1.9, which is the same value as the original RM in PEDOT:PSS. This implies that the core/shell structure of the PEDOT:PSS complex is modified by the interaction with the IL, which agrees with the previously observed morphological transformation towards nanofibrillar structures.28 These observations are fully confirmed by the corresponding O 1s spectra, which reveal that the intensity ratio of the peak at 532.9 eV from PEDOT to the peak at 531.3 eV from PSS increases upon the addition of IL. When ARS is added to the CP:IL composite, a strong Na 1s peak from ARS appears, and the intensity ratio between S 2p doublets from thiophene rings and sulfonate moieties decreases to approximately 1/3.0 due to the contribution from the sulfonate groups in ARS. In addition, the S 2p3/2 peak from sulfonate groups shifts from 167.5 eV to 168.0 eV upon the addition of ARS. Considering that the S 2p signal of sulfonate groups in ARS appears at a higher binding energy than that in PSS (Fig. S3†), this peak shift can be ascribed to the dominant contribution of ARS despite a lesser amount of ARS than PSS (RM of ARS to PSS is 1/1.6). Together with the fact that the Na content (3.5 at%) estimated from the XPS spectrum of the CP:IL:ARS composite is much higher than the 0.9 at% expected from the mixed composition, this result implies an enrichment of ARS on the surface of the composite film. Finally, when WPU is added, Na 1s and S 2p peaks from the CP:IL:ARS composite almost disappears and only the peaks from WPU are detected in the XPS spectra of the final CP:IL:ARS:WPU composite, meaning that the CP:IL:ARS composite phase is completely embedded in the WPU matrix. This is further supported by the disappearance of the O 1s peak from PSS.
N bonds in the IL appear at 401.6 eV and 398.5 eV, but the PEDOT/PSS S 2p intensity ratio is also increased to 1/1.9, which is the same value as the original RM in PEDOT:PSS. This implies that the core/shell structure of the PEDOT:PSS complex is modified by the interaction with the IL, which agrees with the previously observed morphological transformation towards nanofibrillar structures.28 These observations are fully confirmed by the corresponding O 1s spectra, which reveal that the intensity ratio of the peak at 532.9 eV from PEDOT to the peak at 531.3 eV from PSS increases upon the addition of IL. When ARS is added to the CP:IL composite, a strong Na 1s peak from ARS appears, and the intensity ratio between S 2p doublets from thiophene rings and sulfonate moieties decreases to approximately 1/3.0 due to the contribution from the sulfonate groups in ARS. In addition, the S 2p3/2 peak from sulfonate groups shifts from 167.5 eV to 168.0 eV upon the addition of ARS. Considering that the S 2p signal of sulfonate groups in ARS appears at a higher binding energy than that in PSS (Fig. S3†), this peak shift can be ascribed to the dominant contribution of ARS despite a lesser amount of ARS than PSS (RM of ARS to PSS is 1/1.6). Together with the fact that the Na content (3.5 at%) estimated from the XPS spectrum of the CP:IL:ARS composite is much higher than the 0.9 at% expected from the mixed composition, this result implies an enrichment of ARS on the surface of the composite film. Finally, when WPU is added, Na 1s and S 2p peaks from the CP:IL:ARS composite almost disappears and only the peaks from WPU are detected in the XPS spectra of the final CP:IL:ARS:WPU composite, meaning that the CP:IL:ARS composite phase is completely embedded in the WPU matrix. This is further supported by the disappearance of the O 1s peak from PSS.
Fig. 1f presents the morphological evolution of the composite as the components are introduced to the WPU matrix. When the aqueous mixture of CP and WPU dispersions was drop-cast and dried to form a solid film, the composite has the morphology of conductive CP granules (bright contrast) embedded in insulating WPU matrix (dark contrast). When IL is introduced into the CP dispersion before the addition of the WPU, the conductive CP phase, embedded within the WPU matrix, exhibits an interconnected nanofibrous structure of bright contrast. The preservation of granular CP and fibrillar CP:IL morphologies in the mixtures with WPU may result from insignificant interaction between the CP and WPU dispersion particles.30 When ARS is added to the CP:IL mixture solution before the addition of WPU, the composite film still possesses a fibrillar network but with thicker and denser fibrils. Since CP, IL, and ARS are all charged molecules, complicated electrostatic interactions between different cations and anions will occur in the mixture state. Nonetheless, based on the facts that (i) the CP nanofibrils, once induced by the interaction with the IL, retain their morphological features and high electrical conduction even after washing out the IL with water,34 and that (ii) ARS is abundantly distributed on the surface of the CP:IL:ARS composite film as revealed in the XPS analysis, we believe that the thick fibrous structures resulting from the addition of ARS were formed by the attachment of ARS molecules onto the surface of the existing CP nanofibrils. The XRD analysis reveals that the highly ordered π–π stacking peak of PEDOT observed for the CP:IL composite,30 which leads to the formation of CP nanofibrils,28 remains to a similar extent after the addition of ARS (Fig. S4†). Therefore, owing to the preserved fibrillar network of the conductive PEDOT phase and the favorable PEDOT–ARS interface formed on the surface of this conductive pathway, we could simultaneously achieve multiple functionalities—high electrical and ionic conductivities, excellent redox activity, and rubber-like elasticity—when the conductive network was embedded in the elastomeric WPU matrix.
Fig. 1g shows cyclic voltammograms (CVs) of the different composite films used as working electrodes in a three-electrode setup. In all cases, the open circuit potential (EOC) with respect to an Ag/AgCl reference electrode measured in 0.7 M H3PO4 electrolyte was approximately 0.3 V. The CV of the CP:WPU film has a blunt rectangular shape which is typical of capacitor with high resistance. With the addition of IL, very broad and low intensity peaks appear irreversibly in both anodic and cathodic traces. When ARS is introduced to the composite system, the redox-active features of ARS become dominant in the CV; i.e., both prominent oxidation and reduction peaks as well as their reverse peaks, which originate from benzoquinone/hydroquinone conversion reactions of two hydroxyl and two carbonyl groups in ARS, appear at around 0.8 V and −0.2 V vs. Ag/AgCl reference electrode, respectively. These two sets of reversible redox peaks present at both positive and negative potentials with respect to EOC justify the use of our electrode containing ARS as both positive and negative electrodes in the energy storage device.35 Moreover, the potentials of two redox processes are located within the thermodynamic stability window of water (from −0.26 V to 0.97 V vs. Ag/AgCl in 0.7 M H3PO4 electrolyte), which enables the use of aqueous electrolytes in the devices. Fig. 1h shows that the CP:IL:ARS:WPU composite film possesses high stretchability (>300%), while showing a good electrical conduction during a tensile stretching.
Optimization of composition for multi-functionality
In a composite system, there is typically a trade-off between different properties that can be adjusted by changing the component ratio. Thus, optimization of the composition is critical to attain the desired multi-functionality in a composite material. In our previous report, we found that the optimal composition was CP:IL:WPU = 3:5:17 w/w with respect to high electrical conductivity (≈100 S cm−1) and high stretchability (>500%).30 Without IL, the CP:WPU composite exhibits a very low conductivity (≈10−4 S cm−1) and a poor stretchability (<15%) due to rigid granular non-plasticized CP domains that barely form a percolative network but act as fracture points (Fig. 2b and c).30 Using the previously optimized CP:IL:WPU composite as a starting point, here we focused on investigating the effect of added ARS on the electrochemical, ionic, electrical, mechanical and electromechanical properties. For the studied CP:IL:ARS:WPU composites, mA/mp denotes a mass ratio of ARS to PEDOT.
 | ||
| Fig. 2 Effect of the amount of ARS on various properties. (a–d) Areal discharge capacity (a), ionic conductivity (σion, (b)), electrical conductivity (σdc, (c)), and stress–strain curve (d) of the CP:IL:ARS:WPU composite films with varying amount of ARS. mA/mP denotes a mass ratio of ARS to PEDOT. The mass ratio between other components was fixed at CP:IL:WPU = 3:5:17. Error bars represent the standard deviation (N = 3 for B and C). (e) Resistance change (R/R0) upon tensile stretching. (f) Optical microscopy images taken under 50% strain show that the separation between a conductive CP-rich phase (dark area) and an insulating CP-deficient phase consisting of WPU matrix (bright area due to the transmission of a backlight) occurs at mA/mp = 4. | ||
Fig. 2a shows how the areal discharge capacity of the composite film increases with the added amount of ARS. Note that all samples compared in Fig. 2 were prepared to have the same amount of CP:WPU in a given area of the film, with a thickness of ca. 40–60 μm. The almost linear increase of areal discharge capacity from 3.4 μAh cm−2 without ARS to 44 μAh cm−2 with mA/mp = 4 implies that most ARS molecules participate in the charge storage reactions, which indicates a good ARS–PEDOT interface. The inclusion of ionic species such as IL and ARS in the composites is also beneficial for ionic conduction. The ionic conductivity of the CP:WPU composite was enhanced from 0.85 mS cm−1 to 3.7 mS cm−1 by adding IL (Fig. 2b). With the addition of ARS, the ionic conductivity increases further to 12 mS cm−1 for mA/mp = 3. Although increasing the ARS content resulted in improved charge storage capacity and ionic conductivity, it caused deterioration of other composite properties. Fig. 2c shows how the electrical conductivity of the composite film decreases with increasing amount of ARS. Remarkably, there is a minor decrease in conductivity from 87 S cm−1 without ARS to 70 S cm−1 with mA/mp = 3, presumably due to the retention of the conductive fibrillar network. For mA/mp = 4, the electrical conductivity drops down to 45 S cm−1, indicating a partial disruption of the conductive network. Interestingly, the effect of increasing the ARS content on the maximum stretchability does not show a monotonous trend (Fig. 2d). The fracture strain increases from 550% without ARS to 660% with a small amount of ARS (mA/mp = 1). Then, the fracture strain gradually decreases as more ARS is added but remains above 300% for mA/mp = 3. Surprisingly, the composite becomes stretchable up to 810% after further addition of ARS (mA/mp = 4). Despite the fluctuations in mechanical properties, the electromechanical properties decline in a similar manner as the electrical conductivity, exhibiting slight decreases with increasing ARS content up to mA/mp = 3 followed by an abrupt deterioration at mA/mp = 4 (Fig. 2e). Optical microscopic images (Fig. 2f) show that the increasing content of ARS induces quite different morphological features in the composite at 50% strain. For ARS content up to mA/mp = 2, the stretched composite film looks uniform and continuous. As the content increases to mA/mp = 3, the film becomes rougher, but still does not show any major fractures at 50% strain. When the amount of ARS reaches mA/mp = 4, two distinct phases are observed: the dark area is a conductive CP-rich phase, and the bright area is an insulating CP-deficient phase, while both phases are supposed to be embedded in WPU matrix. The observed phase separation for mA/mp = 4 explains the abrupt increase in resistance of the composite film during stretching. Meanwhile, such phase separation seems to generate better connectivity between the WPU domains, as this composite has the highest fracture strain. To maintain adequate electrical, mechanical, and electromechanical properties while maximizing the charge storage capacity, the optimum ARS content was set to mA/mp = 2.5–3. The optimized CP:IL:ARS:WPU composite shows a 68% elastic recovery with viscoelastic hysteresis under cyclic strain loading and a Young's modulus of 12 MPa, similarly to that of the CP:IL:WPU composite (Fig. S5†).30
Intrinsically stretchable symmetric organic battery
The optimized elastomeric biocomposite electrodes constitute a critical building block for the development of intrinsically stretchable symmetric organic batteries, as the electrodes can function as both positive and negative electrodes. However, to achieve the desirable mechanical properties of the whole device, all layers of the batteries—electrolyte, separator, current collectors, substrate, encapsulation, and electrodes—must possess intrinsic stretchability and elasticity while maintaining their functionality under mechanical deformation. Moreover, there should be a good adhesion between the layers to avoid delamination. Fig. 3a illustrates the architecture of the developed biocomposite-based symmetric organic battery that consists of all intrinsically stretchable component layers. The device structure is built up around styrene–ethylene/butylene–styrene (SEBS) substrates, current collectors based on Au-coated TiO2 nanowires (AuNWs), and a relatively dry PVA:H3PO4 hydrogel acting as both a quasi-solid-state electrolyte and a separator. The stress–strain curves of these components are shown in Fig. S6.†
 | ||
| Fig. 3 Architecture of the full cell. (a) Exploded view of the organic battery comprising all intrinsically stretchable layers: AuNW current collectors embedded in SEBS substrates, redox-active biocomposite electrodes, and PVA:H3PO4 hydrogel electrolyte. (b) Cross-sectional SEM image and false color EDX elemental mapping. (c) R/R0vs. strain of a biocomposite film, an AuNW layer embedded in SEBS, and a biocomposite/AuNW bilayer on SEBS. Note that the sheet resistance of the biocomposite film (Rsheet ≈ 3.5 Ω sq−1) is an order of magnitude higher than that of the AuNW current collector (Rsheet ≈ 0.5 Ω sq−1). (d) Schematic illustration of the complete device. The device was characterized by connecting the edge of each current collector to a potentiostat while clamped on a linear stretching stage (Fig. S11†). (e) Photographs of the device stretched up to 120% strain. | ||
PVA-based hydrogel electrolytes (Fig. 3a, top left) have been commonly used to build flexible and stretchable electrochemical energy storage devices, often by soaking the electrodes in hydrogel solution.7,36 Here, to improve the mechanical integrity of the electrolyte/separator layer, an elastomer-like solid hydrogel film was separately prepared and sandwiched between two biocomposite electrodes. Prior to assembly, the dried hydrogel film was rehydrated to approximately 45 wt% water content by dropping a small amount of aqueous ARS solution onto it. The ARS was introduced to the electrolyte layer to suppress leakage of ARS from the electrodes by concentration matching.26
The current collectors based on AuNWs (Fig. 3a, bottom left) have several advantages, including excellent electrochemical stability, biocompatibility, high electrical conductivity, and good electromechanical characteristics.37 We found a strong synergistic effect in the bilayer structure of the AuNWs and the composite electrode, with a remarkable improvement in electromechanical properties of the bilayer in comparison to that of each individual layer (Fig. 3c). A possible explanation of this effect is that the electrode composite is bridging cracks in the AuNW network under tensile strain. Moreover, the exposed NWs can penetrate the composite electrode when assembled, thereby providing an excellent low-resistance electrical contact to the embedded conductive network. Owing to the low sheet resistance (<0.5 Ω sq−1) of our AuNW current collectors that cover nearly the entire area of the composite electrodes, our device demonstrates a noticeably low equivalent series resistance (ESR ≈ 3 Ω).
Each component layer was prepared via various solution processes (i.e., drop-casting, spin-coating, and filtration) and the solid layers were assembled via dry transfer and room temperature lamination. A cross-sectional SEM image of the completed device shows that each layer is well-defined and there are no voids or cracks at the interfaces, owing to the good adhesion between the SEBS, WPU, and PVA-based hydrogel (Fig. 3b). The total thickness of the device is ≈240 μm [≈520 μm] when the SEBS substrates are excluded [included] (Fig. S7†). Note that the thickness of each layer was not optimized, and the total device thickness can be further reduced by decreasing the thicknesses of electrolyte (≈130 μm) and substrates (≈140 μm for each side) to improve the conformability of the device on arbitrary deforming surfaces such as textiles and the human body. Energy dispersive X-ray (EDX) elemental mapping of the device cross-section shows distinct atomic compositions of each layer. Interestingly, phosphorus atoms were detected throughout the full thickness of the electrode layer, indicating that phosphate ions from electrolyte layer can permeate into the bulk of the composite electrode based on good ionic conduction. The full device geometry is illustrated in Fig. 3d. The elongated current collectors were designed to fit the clamps of our linear stretching stage (Fig. S12†) and the devices were characterized under strain with a connected potentiostat. The devices were subject to uniaxial tensile strains up to 120% during the various device characterizations (Fig. 3e).
Fig. 4a shows CVs of full cell devices with and without ARS in the electrode and electrolyte layers. Whereas the device without ARS has a thin rectangular shaped CV of typical supercapacitors, the device with ARS shows strong peaks in the CV during the charging and discharging processes, attributed to the redox reactions of ARS occurring in both the positive and negative electrodes. The observed intense single peak at 1.0 V in the forward scan direction is the result of superimposition of the oxidation and reduction peaks generated at the positive and negative electrodes, respectively. Because both the oxidation and reduction peak potentials are positioned equidistantly (±0.5 V) from EOC, as shown in the half-cell CV (Fig. 1g), the CV of the full device shows only one peak. However, in the reverse scan direction of the CV, which corresponds to discharging of the electrodes, two peaks appear, one stronger at 0.9 V and one weaker at 0.53 V. As the amount of ARS in the electrode increases, more peak splitting was observed during the reverse redox reactions in the half-cell CV at the positive potential side (Fig. S8†). The reason behind this is not clearly understood, but we suppose that more reaction steps can be involved with more stacks of ARS at the PEDOT–ARS interfaces. As shown in the electrochemical impedance spectroscopy (EIS) spectra (Fig. 4a, inset), both devices with and without ARS have similar ESR (Z′-intercept in Nyquist plot), while only the device with ARS show a semicircle due to the charge transfer resistance associated with the redox reactions of ARS.38 The redox peaks in the CV translate into voltage plateaus in the galvanostatic charge/discharge (GCD) curve for the device with ARS (Fig. 4b). In contrast, the GCD curve of the device without ARS shows only linear capacitive behavior. The power law dependance of the peak current (IP = aνb) on the potential scan rate (ν) clearly distinguishes the different charge storage kinetics between the devices with and without ARS: diffusion-controlled, faradaic process for the device with ARS (b ≈ 0.60); and capacitive process for the device without ARS (b ≈ 0.96) (Fig. S9†).38 Therefore, we can classify the device with ARS as a battery, while the device without ARS is categorized as an electrochemical double-layer capacitor or simply a supercapacitor. By introducing ARS into the system, we achieve a 17-fold increase in the areal energy density, from 2.2 μWh cm−2 without ARS to 38 μWh cm−2 with ARS (discharged at 0.5 mA cm−2). Considering the total electrode thickness of 100 μm, the volumetric energy density for the organic battery with ARS is 3.8 mW h cm−3. Meanwhile, the power density of the organic battery scales similarly to that of the pure CP supercapacitor without ARS as shown in the Ragone plot (Fig. 4c), proving the fast surface redox charge storage in our battery system. The excellent rate capability of the organic battery results in a 63% energy density retention for a 14-fold increase in the power density output (8 mA cm−2vs. 0.5 mA cm−2, GCD curves at different current densities are shown in Fig. S10†).
 | ||
| Fig. 4 Performance of the full cell. (a and b) CV (a) and galvanostatic charge/discharge (GCD, (b)) characteristics of the devices with and without ARS (mA/mp = 2.5). The device with ARS exhibits strong redox peaks in CV and voltage plateaus in GCD, whereas the device without ARS shows only capacitive behavior (rectangular-shaped CV and linear dependence in GCD). The inset of (a) shows Nyquist plots of the devices. (c) Ragone plots of the devices with and without ARS. (d and e) CV (d) (Nyquist plots in the inset) and GCD (e) characteristics of the device with ARS under different tensile strains and when released after 120% strain loading (0%, dashed lines in CV and GCD, open ◇ in Nyquist plot). (f) Capacity retention and equivalent series resistance (ESR) after tensile stretching/releasing cycles at 50% strain. CV scan rate, 10 mV s−1. GCD current density, 1 mA cm−2 for (b) and 4 mA cm−2 for (e). | ||
Under tensile strain loading, the CV shape of the intrinsically stretchable organic battery remains nearly unchanged up to 80% strain. As the applied strain increases further, the redox peaks gradually decrease in intensity and the charging and discharging peak separation grows (Fig. 4d). Correspondingly, a gradual increase in the ESR as well as an increase in the charge transfer resistance (diameter of the semicircle) is observed in the EIS spectrum (Fig. 4d, inset. enlarged graph is shown in Fig. S11†). By comparing the change in ESR with the separately measured strain dependence of the lateral current collector/electrode bilayer resistance, we conclude that the change in ESR mainly originates from the resistance change in the current collector/electrode bilayers (Fig. S12†). Despite the 4.3-fold [6.7-fold] increase in the ESR under 100% [120%] strain, the change in the GCD curve upon stretching is quite small, with 91% [85%] of the initial discharge capacity being maintained at 100% [120%] strain. When released after 120% strain loading, the CV, the GCD curve and the EIS spectrum closely return to their original shapes, indicating an excellent elastic recovery in electrochemical performances. The stress–strain curves upon cyclic strain loading at various levels (20–120%) show the dimensional stability of the device with a 91% elastic recovery, which is dominated by the mechanical property of SEBS substrates (Fig. S13†). The stretchability and volumetric energy density of our organic stretchable battery is outstanding compared with those of other intrinsically stretchable organic electrochemical energy storage devices (Fig. S14†). Our device shows excellent stability for 1000 stretching cycles at 50% strain, with nearly unaffected ESR and 90% charge storage capacity retention. The GCD cycling stability of the device was further tested at 4 mA cm−2 after the aforementioned measurements (Fig. S15†). An initial capacity loss of 19% occurs during the first 100 cycles, after which the performance stabilizes with 78% retention of the initial capacity during 200 and 500 GCD cycles. The coulombic efficiency remains above 98% throughout the whole 500 GCD cycles, indicating superior reversibility and cyclic stability.
Wearable battery module
To magnify the discharge voltage of a single battery (<1 V), we designed and fabricated a module where four cells are connected in series and used it to power an LED in a fully stretchable circuit. Fig. 5a and b show the exploded view and the assembled architecture of a module, respectively. One electrode of each cell lies on the same plane and the cells are connected to each other through the AuNW current collectors. Owing to the symmetric electrode configuration within the cells, only one electrode material needs to be patterned per plane and all interconnects between the cells can be made without bridging the planes, which greatly simplifies the assembly and integration of the device. Additionally, the stretchable circuit comprises a surface-mounted device (SMD) red LED (operating voltage max. 2.4 V), a current limiting SMD resistor (200 Ω), a simplistic switch, and AuNW interconnects between the components. The equivalent circuit diagram is shown in Fig. 5c. The module was first charged at 1 mA cm−2 with respect to the area of a single cell while the switch was open. Next, the switch was closed by the addition of a stretchable and adhesive piece of CP:IL:WPU composite, which started the powering of the integrated LED. Both CV and GCD curves reveal that the discharge voltage is magnified to nearly 4 V for the module, which corresponds to the quadruple of the single cell voltage (Fig. 5d). The discharge time of the module is reduced in comparison to a single cell, probably due to variations in cell performance, which strongly effect the module performance in a serial configuration. Finally, we demonstrated how the stretchable circuit, including the module, can be integrated onto textile or the human body. The thin and flat geometry in combination with the intrinsic stretchability of the entire module, allows for seamless integration onto a textile (Fig. 5e). As expected from the previous characterizations, the stretchable circuit remains functional and powers the LED during various mechanical deformations, including folding, twisting, and stretching (Fig. 5e). Fig. 5f shows how the module, when mounted onto a rubber glove, can conformally follow the deformations of the hand while remaining functional.
 | ||
| Fig. 5 Wearable battery module. (a) Exploded view of a stretchable circuit comprising a module with 4 cells connected in series, a resistor, a switch, and a red LED. (b) Schematic illustration of the complete module in operation. A piece of CP:IL:WPU composite was used as a stretchable conductive glue to close the switch. (c) Equivalent circuit diagram of the stretchable circuit. Rload, Rinterconnect, and Rinternal denote resistances of a current limiting resistor, AuNW interconnects, and the internal resistance of a module, respectively. (d) Comparison of GCD curves (CV curves in the inset) between a single device and a module. The current density is 1 mA cm−2 with respect to a single cell of the module. The CV scan rate is 10 mV s−1 for the single cell and 40 mV s−1 for the module. (e) Photographs of the module attached to a stretchy fabric. The module remains functional (powers the LED) at various deformation conditions. (f) Photographs of the module attached to a human hand wearing a transparent poly glove. | ||
Conclusions
We have developed an intrinsically stretchable organic battery where the charge storage occurs via the quinone/hydroquinone redox couples in the plant-derived biomolecule alizarin. Formulating a quaternary biocomposite for intrinsically stretchable electrodes that possess excellent multifunctionality was the key success factor. Our biocomposite is made up of a unique combination of four organic materials: PEDOT:PSS, IL, ARS, and WPU, and obtained simply via blending of aqueous component solutions/dispersions and drop-casting of the mixture. The final biocomposite shows a self-organized hierarchical morphology comprising: (i) a well-connected conductive network of fibrillated PEDOT:PSS induced by the interaction with IL, which imparts high electrical conductivity (>70 S cm−1) to the composite, (ii) the redox-active biomolecule ARS incorporated onto the conductive network, which provides efficient charge transfer at the favorable ARS–PEDOT interface and charge storage capacity of 6.8 mA h cm−3 to the composite, and (iii) an elastomeric matrix consisting of hygroscopic WPU and ionically conductive IL, which ensures high stretchability (>300%) and good ionic conduction (12 mS cm−1). Interestingly, when the biocomposite electrodes were transferred onto the AuNW/SEBS current collectors, there was a significant improvement in the electromechanical properties of the AuNW current collectors, which might be the result of a conductive bridging effect. Owing to two quinone motifs in ARS (i.e., two hydroxyl and two carbonyl groups) that undergo oxidation at positive potential and reduction at negative potential, respectively, the intrinsically stretchable symmetric organic battery was constructed by sandwiching a hydrogel electrolyte between two identical electrode/current collector bilayers. It is noteworthy that 85% of charge storage capacity was retained upon 120% tensile stretching, while the ESR of the device increased 6.7-fold. The device exhibited elastic recovery of all electrochemical characteristics (i.e., CV, GCD, EIS) when released from 120% strain and stable performance under 1000 times stretching cycles at 50% strain. The battery module, which was attached on a stretchy textile or a rubber glove, stably powered the integrated LED under various mechanical deformation modes (i.e., folding, twisting, stretching), proving its potential as wearable power sources.For commercial applications, however, the energy density of our device (3.8 mW h cm−3) needs to be enhanced further. In the current work, the energy density was suppressed by a large volume of inactive elastomer (WPU > 60 wt%), as well as a limited amount of redox-active materials (PEDOT + ARS < 13 wt%) to ensure good mechanical and electromechanical properties of the electrodes. Although the use of a symmetric electrode configuration and an aqueous electrolyte in our battery is advantageous in terms of simplicity and safety, respectively, the electrode and electrolyte materials incurred low operating voltage (<1 V). Therefore, a wide range of materials and micro-/nano-structural engineering need to be explored to simultaneously achieve high performance, good mechanical conformability, economic and environmental sustainability, and safety in wearable and implantable batteries. We believe that our pioneering work on an intrinsically stretchable plant-based organic battery will stimulate the development of the next-generation wearable and sustainable energy storage devices.
Experimental
Material
PEDOT:PSS (Clevios™ PH1000, 1.1–1.3 wt% aqueous dispersion), EMIM TCM (purity >98%), H3PO4 (85 wt% in H2O) and NH3 (28% in H2O) were purchased from Heraeus, Iolitec, and BASF, respectively. WPU (ALBERDINGK®U4101, 39–41 wt% aqueous anionic dispersion of aliphatic polyether-polyurethane) and SEBS (H1052) was obtained from Alberdingk Boley and Asahi Kasei, respectively. Alizarin Red S, poly(styrene sulfonic acid) (PSSH, average Mw 75 k), Nafion™ perfluorinated resin solution (5 wt%), Nafion™ 115 perfluorinated membrane, and PVA (average Mw 130 k, 99+% hydrolyzed) were purchased from Sigma-Aldrich. All chemicals were used as received without further purification.Composite and hydrogel preparation
CP:IL:ARS:WPU composites were prepared by adding constituent aqueous solutions/dispersions dropwise into CP (PEDOT:PSS) dispersions in the following order. During blending, the CP dispersions were kept stirred (400 rpm) at room temperature by using a magnetic stirrer. IL was dissolved in deionized water (DIW) to a concentration of 30 g L−1, added to the CP dispersion, and stirred overnight (>12 h). ARS was dissolved in DIW to a concentration of 3.43 g L−1 and added to the CP:IL dispersion, and then the mixture was stirred for >1 h. WPU was added to the CP:IL:ARS dispersion and the mixture was stirred for >2 h. Prior to adding WPU, 0.15 wt% of NH3 solution was added with respect to the PEDOT:PSS dispersion to prevent acid-induced aggregation of the anionic WPU. The composition was CP:IL:WPU = 3:5:17 w/w with varying amount of ARS. Composites without some constituents (i.e., CP:IL, CP:IL:ARS, CP:WPU, CP:IL:WPU) were prepared in the same manner, but excluding some addition steps. The dispersions were drop-cast onto cleaned glass substrates and then dried in air or in a vacuum desiccator at room temperature. The dry films were cut and peeled off, and then the free-standing films were transferred on measurement set-ups or AuNW current collectors for the characterization and the fabrication of devices. For XRD samples, drop-cast films were prepared on p-Si substrates. For XPS samples, the solutions/dispersions were spin-coated on p-Si substrates or Au-deposited glass substrates. PVA:H3PO4 hydrogel was prepared as follows. PVA pellets were dissolved in DIW to a concentration of 100 g L−1 under vigorous magnetic stirring in a hot water bath (85 °C) overnight (>12 h). When the PVA solution cooled down to room temperature, H3PO4 solution was added (PVA:H3PO4 = 1:1 w/w), and then the mixture was stirred in a hot water bath (85 °C) for >4 h. The viscous PVA:H3PO4 solution was spin-coated on a glass substrate silanized with trichloro(1H,1H,2H,2H-perfluorooctyl) silane, and then dried in air at room temperature for >3 days. Prior to the device assembly, the dry hydrogel films were cut, transferred on Teflon substrates, and rehydrated with DIW or ARS solution (3.43 g L−1 in DIW).
Elemental, morphological, and structural characterization
XPS analyses were conducted in an Axis Ultra DLD spectrometer from Kratos Analytical (UK) with the base pressure during spectra acquisition better than 1.1 × 10−9 Torr (1.5 × 10−7 Pa). The excitation source was monochromatic Al Kα radiation (hν = 1486.6 eV) and the anode power was 150 W. All spectra were collected at normal emission angle. The analyzer pass energy was set to 20 eV, resulting in the spectrometer energy resolution determined from the FE cut-off of Au and Ag samples of 0.38 eV. The calibration of the binding energy scale was confirmed by examining sputter-cleaned Au, Ag, and Cu samples (all in the form of polycrystalline thin films) according to the recommended ISO standards for monochromatic Al Kα sources that place Au 4f7/2, Ag 3d5/2, and Cu 2p3/2 peaks at 83.96, 368.21, and 932.62 eV, respectively. To avoid problems with referencing to the C 1s peak of adventitious carbon, all core level spectra are presented as recorded exploiting the fact that PEDOT:PSS containing materials are known to possess sufficient charge density to ensure Fermi level alignment between specimen and the spectrometer.39 SEM images were acquired with a ZEISS field-emission SEM (Sigma 500 Gemini). Morphological images were acquired at 3 kV accelerating voltage with an InLense detector. XRD patterns were acquired in a θ–2θ geometry using a PANalytical X'Pert PRO diffractometer system equipped with a Cu Kα source operated at 45 kV and 40 mA. Optical microscope images were obtained using a Nikon Optiphot 150 inspection microscope in a bright field mode with a backlight underneath the samples.Ionic, electrical, mechanical, and electromechanical characterization
Through-plane ionic conductivity was measured in a 4-probe configuration using a symmetric H-cell (Scribner Associates Inc., USA) filled with 4 M H3PO4, two counter electrodes (Pt/Nb mesh cylinders), and two references electrodes (two Luggin probes with Ag/AgCl (3 M KCl) as terminals), as shown in Fig. S16.† The free-standing composite film (10 mm × 10 mm) was sandwiched between two Nafion™ 115 membranes to prevent the leakage of IL and ARS from the sample, and then placed between two SEBS gaskets with a hole diameter of 6 mm. Prior to use, the Nafion membranes were activated by sequentially soaking them in 3% H2O2, DIW, and 0.5 M H2SO4 respectively for 1 h at 80 °C, followed by a final rinse in DIW. The pressed gaskets were held in the middle of H-cell using a pinch clamp. Resistance was measured with and without the composite film via linear sweep voltammetry (from −0.1 V to 0.1 V, scan rate of 10 mV s−1) using a Gamry Potentiostat 1010E. In-plane electrical conductivity was measured using the four-lead method with a geometry shown in Fig. S17.† A strip of the free-standing composite film was transferred on a glass substrate where four Au lines were deposited. A current was applied to the outer two Au lines while the voltage drop was measured across the inner two Au lines using a Keithley 2400 Source Meter unit in a 4-wire sensing mode. The film width and thickness were measured by using a surface profilometer (Dektak 3ST, Veeco). Mechanical and electromechanical analyses were conducted by measuring force with a force gauge (M5-2, Mark-10) and measuring resistance with Au-coated 4-point contact pads coupled with a Keithley 2701 Ethernet Multimeter data acquisition system, respectively, while the free-standing film was clamped and stretched on a motorized linear stage (X-LSQ300A-E01, Zaber). The strain rate was 1% s−1 for the resistance-strain measurements and the maximum strain tests, and 2% s−1 for the cycling tests.Half-cell/full cell/module fabrication and characterization
AuNWs were synthesized as described in the previous literature.30 SEBS substrates were prepared by spin-coating a SEBS solution (400 g L−1 in toluene) on cleaned glass wafers and drying on a hot plate at 100 °C. A pattern of AuNWs (18 mm × 5.5 mm) was prepared on a filter membrane by wax-assisted vacuum filtration40 and then transferred by placing a pattern on a SEBS substrate heated on a hot plate at 100 °C and pressing with a rubber hand roller during heating. The AuNW current collector embedded in a SEBS substrate was treated with UV/O2 for 30 min and then spin-coated with a thin layer of WPU in order to improve the adhesion with the composite film while keeping the AuNWs open in the air. The free-standing composite film (6 mm × 6 mm) was transferred onto the WPU-coated AuNW-embedded SEBS substrate. This constitutes a half-cell. Prior to immersing the half-cell in a 0.7 M H3PO4 electrolyte bath, a solution of Nafion (diluted to 2 wt% using isopropanol) was drop-cast on the half-cell and immediately dried on a hot plate at 70 °C. The area of the attached composite film was kept open while the surrounding areas and the AuNW current collector without the composite film were covered with Kapton tape. Half-cell CV and GCD curves were obtained by using a potentiostat (BioLogic SP-200) in a three-electrode setup using an Ag/AgCl reference electrode and a Pt mesh counter electrode. Areal discharge capacity of the half-cell was estimated from a GCD curve in Fig. S8b† (i.e., average of the cathodic discharge capacity from 1.0 V to 0.3 V and the anodic discharge capacity from −0.4 V to 0.3). A full cell device was fabricated by sandwiching PVA:H3PO4 hydrogel between two identical half-cells. Prior to the assembly, some area of the AuNW current collectors without the composite film was covered with Kaptone tape to avoid a direct contact with hydrogel. The device was clamped on a motorized linear stage and connected to a potentiostat to measure EIS (at EDC = 0 V, EAC = 10 mV), CV, and GCD characteristics in the initial, stretched, and released states. A cross section of the device was obtained by freezing and breaking the device in half in a liquid nitrogen bath and then freeze-drying the broken device in a freeze drier (Benchtop Pro, SP Scientific). Cross sectional SEM images and energy dispersive X-ray spectroscopy maps (EDX, BRUKER) were acquired at 2 kV and 6 kV accelerating voltage, respectively. A module with the architecture shown in Fig. 5b was fabricated and characterized in the same method described above. Prior to the assembly, some area of the AuNW current collectors/interconnects without the composite film was covered with SEBS layers to avoid a direct contact with hydrogel. SMD LED and resistor were connected to the stretchable circuit by using silver epoxy (8330S Silver Conductive Epoxy Adhesive, MGchemicals) and protected by a blob of PDMS (Sylgard 184, Dow Corning, 1:10) dropped on top of the LED and resistor and cured at 100 °C for >1 h. EIS, CV, and GCD characteristics of the full cell and module were obtained by using a potentiostat (BioLogic SP-200). Prior to the characterization, the devices were activated by galvanostatic charging up to 1.3 V at 0.5 mA cm−2 and discharging at the same current density once.
Author contributions
N. K., K. T., and X. C. conceived the project. N. K. formulated the composites and conducted the electrochemical, ionic, electrical, mechanical, and electromechanical characterizations. S. L. synthesized AuNWs and conducted the SEM and EDX measurements. Z. K. contributed to the electrochemical characterizations and analyses. G. G. conducted the XPS measurements and contributed to the XPS analysis. A. R., M. V., and F. A. contributed to the characterization and analysis of the ionic transport properties. J. E. contributed to the half-cell tests. N. K. conducted the XRD measurements and fabricated the devices. I. P. contributed to the fabrication of SEBS substrates and the AuNW transfer process. N. K., S. L., and K. T. designed and fabricated the module. K. T. and X. C. supervised the work. N. K. wrote the first draft of the manuscript and all authors contributed to the finalization of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Mohsen Mohammadi, Sangmin Park, and Dr Robert Brooke for assistance with illustrations, Meysam Karami Rad for LabVIEW programming and help with the circuit tests, and Laura Seufert for assistance with the module demonstration. This work was financially supported by the ÅForsk Foundation (19-428), the Swedish Government Strategic Research Area in Materials Science on Advanced Functional Materials at Linköping University (Faculty grant SFO-Mat-LiU no. 2009-00971), the Knut and Alice Wallenberg Foundation (POC “paper batteries” and “high voltage aqueous electrolyte”), and the Swedish Research Council (starting grant no. 2020-05218, no. 2019-04424 and no. 2016-06146). G. G. acknowledges financial support from the Swedish Research Council (no. 2018-03957) and the Swedish Energy Agency grant 51201-1. A. R. acknowledges Marie Skłodowska-Curie Actions Seal of Excellence Fellowship program from the Sweden's Innovation Agency (Vinnova grant no. 2021-01668). This work was partially supported by the Wallenberg Initiative Materials Science for Sustainability (WISE) funded by the Knut and Alice Wallenberg Foundation.References
- T. R. Ray, J. Choi, A. J. Bandodkar, S. Krishnan, P. Gutruf, L. Tian, R. Ghaffari and J. A. Rogers, Chem. Rev., 2019, 119, 5461–5533 CrossRef CAS PubMed.
- J. C. Yang, J. Mun, S. Y. Kwon, S. Park, Z. Bao and S. Park, Adv. Mater., 2019, 31, 1904765 CrossRef CAS PubMed.
- J. Xu, S. Wang, G.-J. N. Wang, C. Zhu, S. Luo, L. Jin, X. Gu, S. Chen, V. R. Feig, J. W. F. To, S. Rondeau-Gagné, J. Park, B. C. Schroeder, C. Lu, J. Y. Oh, Y. Wang, Y.-H. Kim, H. Yan, R. Sinclair, D. Zhou, G. Xue, B. Murmann, C. Linder, W. Cai, J. B. H. Tok, J. W. Chung and Z. Bao, Science, 2017, 355, 59 CrossRef CAS PubMed.
- S. Wang, J. Xu, W. Wang, G.-J. N. Wang, R. Rastak, F. Molina-Lopez, J. W. Chung, S. Niu, V. R. Feig, J. Lopez, T. Lei, S.-K. Kwon, Y. Kim, A. M. Foudeh, A. Ehrlich, A. Gasperini, Y. Yun, B. Murmann, J. B. H. Tok and Z. Bao, Nature, 2018, 555, 83–88 CrossRef CAS PubMed.
- T. Yokota, P. Zalar, M. Kaltenbrunner, H. Jinno, N. Matsuhisa, H. Kitanosako, Y. Tachibana, W. Yukita, M. Koizumi and T. Someya, Sci. Adv., 2016, 2, e1501856 CrossRef PubMed.
- D. C. Kim, H. J. Shim, W. Lee, J. H. Koo and D.-H. Kim, Adv. Mater., 2020, 32, 1902743 CrossRef CAS PubMed.
- D. G. Mackanic, T.-H. Chang, Z. Huang, Y. Cui and Z. Bao, Chem. Soc. Rev., 2020, 49, 4466–4495 RSC.
- D. G. Mackanic, M. Kao and Z. Bao, Adv. Energy Mater., 2020, 10, 2001424 CrossRef CAS.
- W. Liu, J. Chen, Z. Chen, K. Liu, G. Zhou, Y. Sun, M.-S. Song, Z. Bao and Y. Cui, Adv. Energy Mater., 2017, 7, 1701076 CrossRef.
- D. G. Mackanic, X. Yan, Q. Zhang, N. Matsuhisa, Z. Yu, Y. Jiang, T. Manika, J. Lopez, H. Yan, K. Liu, X. Chen, Y. Cui and Z. Bao, Nat. Commun., 2019, 10, 5384 CrossRef PubMed.
- S. Y. Hong, S. M. Jee, Y. Ko, J. Cho, K. H. Lee, B. Yeom, H. Kim and J. G. Son, ACS Nano, 2022, 16, 2271–2281 CrossRef CAS PubMed.
- K. Liu, Y. Liu, D. Lin, A. Pei and Y. Cui, Sci. Adv., 2018, 4, eaas9820 CrossRef PubMed.
- T. N. Nguyen, B. Iranpour, E. Cheng and J. D. W. Madden, Adv. Energy Mater., 2022, 12, 2103148 CrossRef CAS.
- W.-J. Song, S. Lee, G. Song and S. Park, ACS Energy Lett., 2019, 4, 177–186 CrossRef CAS.
- M. Iturrondobeitia, O. Akizu-Gardoki, O. Amondarain, R. Minguez and E. Lizundia, Adv. Sustainable Syst., 2022, 6, 2100308 CrossRef CAS.
- X. Zhang, L. Yang, Y. Li, H. Li, W. Wang and B. Ye, Environ. Monit. Assess., 2012, 184, 2261–2273 CrossRef CAS PubMed.
- H. Alt, H. Binder, A. Köhling and G. Sandstede, Electrochim. Acta, 1972, 17, 873–887 CrossRef CAS.
- F. Goto, K. Abe, K. Ikabayashi, T. Yoshida and H. Morimoto, J. Power Sources, 1987, 20, 243–248 CrossRef CAS.
- N. Goujon, N. Casado, N. Patil, R. Marcilla and D. Mecerreyes, Prog. Polym. Sci., 2021, 122, 101449 CrossRef CAS.
- J. Kim, Y. Kim, J. Yoo, G. Kwon, Y. Ko and K. Kang, Nat. Rev. Mater., 2023, 8, 54–70 CrossRef.
- G. Milczarek and O. Inganäs, Science, 2012, 335, 1468–1471 CrossRef CAS PubMed.
- F. N. Ajjan, D. Mecerreyes and O. Inganäs, Biotechnol. J., 2019, 14, 1900062 CrossRef CAS PubMed.
- L. Liu, N. Solin and O. Inganäs, Adv. Energy Mater., 2021, 11, 2003713 CrossRef CAS.
- L. Tong, Y. Jing, R. G. Gordon and M. J. Aziz, ACS Appl. Energy Mater., 2019, 2, 4016–4021 CrossRef CAS.
- Y. Liu, S. Lu, S. Chen, H. Wang, J. Zhang and Y. Xiang, ACS Appl. Energy Mater., 2019, 2, 2469–2474 CrossRef CAS.
- J. Edberg, R. Brooke, H. Granberg, I. Engquist and M. Berggren, Adv. Sustainable Syst., 2019, 3, 1900050 CrossRef.
- K. Hatakeyama-Sato, H. Wakamatsu, K. Yamagishi, T. Fujie, S. Takeoka, K. Oyaizu and H. Nishide, Small, 2019, 15, 1805296 CrossRef PubMed.
- S. Kee, N. Kim, B. S. Kim, S. Park, Y. H. Jang, S. H. Lee, J. Kim, J. Kim, S. Kwon and K. Lee, Adv. Mater., 2016, 28, 8625–8631 CrossRef CAS PubMed.
- S. Choi, S. I. Han, D. Kim, T. Hyeon and D.-H. Kim, Chem. Soc. Rev., 2019, 48, 1566–1595 RSC.
- N. Kim, S. Lienemann, I. Petsagkourakis, D. Alemu Mengistie, S. Kee, T. Ederth, V. Gueskine, P. Leclère, R. Lazzaroni, X. Crispin and K. Tybrandt, Nat. Commun., 2020, 11, 1424 CrossRef CAS PubMed.
- Y. Fang, H. Cheng, H. He, S. Wang, J. Li, S. Yue, L. Zhang, Z. Du and J. Ouyang, Adv. Funct. Mater., 2020, 30, 2004699 CrossRef CAS.
- D. Ju, D. Kim, H. Yook, J. W. Han and K. Cho, Adv. Funct. Mater., 2019, 29, 1905590 CrossRef CAS.
- G. Greczynski, T. Kugler, M. Keil, W. Osikowicz, M. Fahlman and W. R. Salaneck, J. Electron Spectrosc. Relat. Phenom., 2001, 121, 1–17 CrossRef CAS.
- Y. Wang, C. Zhu, R. Pfattner, H. Yan, L. Jin, S. Chen, F. Molina-Lopez, F. Lissel, J. Liu, N. I. Rabiah, Z. Chen, J. W. Chung, C. Linder, M. F. Toney, B. Murmann and Z. Bao, Sci. Adv., 2017, 3, e1602076 CrossRef PubMed.
- A. Berndt, D. Pospiech, D. Jehnichen, L. Häußler, B. Voit, M. Al-Hussein, M. Plötner, A. Kumar and W.-J. Fischer, ACS Appl. Mater. Interfaces, 2015, 7, 12339–12347 CrossRef CAS PubMed.
- H. Dong, J. Li, J. Guo, F. Lai, F. Zhao, Y. Jiao, D. J. L. Brett, T. Liu, G. He and I. P. Parkin, Adv. Mater., 2021, 33, 2007548 CrossRef CAS PubMed.
- K. Tybrandt, D. Khodagholy, B. Dielacher, F. Stauffer, A. F. Renz, G. Buzsáki and J. Vörös, Adv. Mater., 2018, 30, 1706520 CrossRef PubMed.
- T. S. Mathis, N. Kurra, X. Wang, D. Pinto, P. Simon and Y. Gogotsi, Adv. Energy Mater., 2019, 9, 1902007 CrossRef CAS.
- G. Greczynski and L. Hultman, Angew. Chem., Int. Ed., 2020, 59, 5002–5006 CrossRef CAS PubMed.
- K. Tybrandt and J. Vörös, Small, 2016, 12, 180–184 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta04153k |
| This journal is © The Royal Society of Chemistry 2023 |