
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling the reaction mechanism of capacity reactivation in silver vanadate cathodes for aqueous zinc-ion batteries†
Ziqing
Wang
 a,
Jiefeng
Diao
ab,
Kenta
Kawashima
a,
Jason A.
Weeks
a,
Rinish Reddy
Vaidyula
a,
Raul A.
Marquez
a,
Nathaniel
Miller
c,
Graeme
Henkelman
ab and
C. Buddie
Mullins
*ade
a,
Jiefeng
Diao
ab,
Kenta
Kawashima
a,
Jason A.
Weeks
a,
Rinish Reddy
Vaidyula
a,
Raul A.
Marquez
a,
Nathaniel
Miller
c,
Graeme
Henkelman
ab and
C. Buddie
Mullins
*ade
aDepartment of Chemistry, The University of Texas at Austin, Austin, Texas 78712, USA. E-mail: mullins@che.utexas.edu
bOden Institute for Computational Engineering and Sciences, The University of Texas at Austin, Austin, Texas 78712, USA
cDepartment of Geosciences, The University of Texas at Austin, Austin, Texas 78712, USA
dTexas Materials Institute, The University of Texas at Austin, Austin, Texas 78712, USA
eMcKetta Department of Chemical Engineering, The University of Texas at Austin, Austin, Texas 78712, USA
First published on 16th August 2023
Abstract
Aqueous zinc-ion batteries are regarded as promising candidates for future energy storage devices because of their high safety. Due to the dissolution in the aqueous electrolytes, most vanadate-based zinc-ion batteries suffer from continuous capacity fading. In some cases, a capacity reactivation process can be observed in vanadate-based cathodes after capacity decay. Herein, we employed electrochemical methods and characterization techniques to study the reaction mechanism of capacity reactivation in Ag0.33V2O5 cathodes. Our preliminary results suggested that the reactivation is due to an in situ crystalline structure evolution in the cathode materials. Under the electrochemical condition, a new phase Zn3(OH)2V2O7·2H2O irreversibly formed on the initial cathodes, which prevented the dissolution of vanadate and further resulted in the capacity increase. Moreover, we confirmed the unique intercalation pseudocapacitive behavior in the reconstructed Zn3(OH)2V2O7·2H2O, which provided fast ionic diffusion to facilitate electrochemical performance. Accordingly, our study offers a new understanding of the capacity change of the vanadate-based cathode materials and provides a more general explanation of the capacity reactivation in aqueous zinc-ion batteries.
1. Introduction
Lithium-ion batteries are currently essential for portable energy storage for digital devices and the transportation sector, however, due to the application of organic electrolytes, there are still many safety concerns limiting their implementation.1–4 In this regard, aqueous zinc-ion batteries (ZIBs) have recently received attention due to their intrinsic advantages, including high safety, low cost, and environmental friendliness.5–10 Different types of cathode materials, including manganese oxides,11–15 vanadium oxides,16–21 Prussian blue analogs,22–24 and even organic cathodes,25–27 have been widely utilized in the ZIBs, among which, vanadate-based cathodes, especially V2O5-originated species, have been extensively studied. Specifically, because of the multivalent property, the open layered structure, and the support from the intercalated cations, V2O5-based materials can store more Zn2+ to provide higher capacity.28–33 Moreover, nanosized morphology efficiently promotes ionic diffusion to boost reaction kinetics.Unfortunately, V2O5-based cathode materials experience obvious dissolution in the aqueous electrolytes, inhibiting the capacity and stability of the batteries. However, it has been reported that a capacity reactivation process occurred during cycling even after capacity fading.34–39 Various possible explanations and hypotheses, including moderate penetration of the electrolyte, gradual utilization of active materials, and slow replacement of host cations between V–O layers, have been proposed to illustrate the increase in capacity.37,40,41 While these factors may contribute to the capacity increase during cycling, they do not fully explain the complex reactions taking place within the cathode materials. Therefore, detailed characterization and electrochemical measurement are necessary to be applied for checking the actual reaction mechanism behind the capacity change.
Recently, it has been reported that vanadate-based cathode materials experienced a structural transition into a new phase during cycling, providing improved electrochemical performance.42,43 The deep reconstruction of the cathode is considered to provide better ion diffusion, higher conductivity, and more ion insertion sites, contributing to the enhanced Zn2+ storage ability and exhibiting higher capacity.44–46 A typical product of this transformation process, Zn3V2O7(OH)2·2H2O, is derived from a rapid reaction between the original vanadium-based cathodes and the electrolytes.47–49 Lu et al. revealed the dissolution mechanism of V2O5 in the aqueous electrolyte by simply submerging a V2O5 cathode into a Zn(CF3SO3)2 electrolyte, demonstrating that a spontaneous reaction between the cathode and electrolyte caused the formation of Zn3V2O7(OH)2·2H2O.47 The nanoflake-like newly transformed phase provided high surface area and electrolyte penetration, leading to better accessibility and faster reaction kinetics. Moreover, Tao et al. proved the enhanced pseudocapacitive behavior in the reconstructed ZnVO materials, significantly improving the rate performance.50 Inspired by the above results, we consider that capacity change is highly related to the in situ reconstruction of cathode materials and the properties of the reconstructed products. Under such circumstances, we employed vanadate-based cathode Ag0.33V2O5 (AgVO), which exhibits capacity reactivation during cycling, to unveil the connection between capacity change and structure evolution to understand the reaction mechanism behind the capacity increase. By applying different characterization techniques, we discovered the in situ transition from the original cathodes into Zn3(OH)2V2O7·2H2O (ZnVO). In the initial cycles, the ZnVO mitigates the dissolution of cathodes via coating on the cathodes as a protective layer. Beyond the protection, we propose that the formation and accumulation of ZnVO result in a capacity increase after degradation. Electrochemical measurement proved the intrinsic intercalation pseudocapacitive behavior of ZnVO, which is confirmed by the density functional theory (DFT) calculations that Zn2+ has fast ionic diffusion in ZnVO. In general, we have connected the capacity drop and capacity reactivation during cycling with the reconstruction of cathode materials, proposing a universal and general explanation for the capacity change in ZIBs.
2. Results and discussion
2.1. Characterization of Ag0.33V2O5 cathode material
The AgVO cathode material was synthesized by a one-step hydrothermal method (synthetic details can be found in the ESI†). The crystalline structure of AgVO was studied by powder X-ray diffraction (XRD), which suggests a high-purity material without detectable crystalline byproducts (Fig. 1a). The absence of the V2O5 peaks illustrates the successful doping of Ag+ into V2O5. The crystal structure sketch image reveals the layered structure consisting of V–O octahedrons with the supporting of Ag+ between the layers. High-resolution transmission electron microscopy (HRTEM) images of AgVO show several lattice fringes belonging to AgVO (Fig. S1†). Scanning electron microscope (SEM) images reveal the nanobelt-like morphology of the AgVO with a width of 200 nm (Fig. 1b). Moreover, the high-angle annular dark-field scanning TEM (HAADF-STEM) elemental mapping image demonstrates the even distribution of Ag, V, and O elements and homogenous composition in AgVO (Fig. S2†). The chemical bonding states of each element were evaluated by X-ray photoelectron spectroscopy (XPS) (Fig. S3 and S4†). The Ag 3d XPS spectrum verifies the existence of Ag+ in the cathode (Fig. S4a†). The 525.2 eV and 517.5 eV peaks in the V 2p spectra correspond to the V 2p1/2 and V 2p3/2 features, respectively, demonstrating clear V4+ and V5+ peaks within both regions (Fig. S4b†). The origin of V4+ is ascribed to a reduction of V2O5 during synthesis, and the introduction of Ag+ provides the charge-compensating ion. The above results indicate that the Ag+ was successfully doped in the V2O5 layers. | ||
| Fig. 1 (a) XRD pattern of AgVO powder. Inset is the crystalline structure of AgVO. (b) SEM images of nanobelt-like morphology of AgVO. (c) Cycling performance of AgVO/Zn full battery under a current density of 0.1 A g−1. (d) XRD pattern of the AgVO cathode after the first cycle at 0.1 A g−1. (e) TEM and HRTEM images of AgVO cathode fully charged to 1.4 V in the first cycle. (f) HAADF-STEM element mapping images of AgVO nanobelt after the first cycle. (g–i) XPS spectra of V 2p3/2, Zn 2p, and Ag 3d, respectively, in the first cycle. | ||
2.2. Formation and characterization of the Zn3(OH)2V2O7·2H2O
The electrochemical performance of the AgVO/Zn batteries is evaluated by employing a 3 mol L−1 (M) Zn(ClO4)2 electrolyte. At a current density of 0.1 A g−1, the battery shows a high initial capacity of approximately 400 mA h g−1 over 100 cycles (Fig. 1c). Moreover, a capacity drop and reactivation process can be observed, which will be discussed in detail later. In order to explore the energy storage mechanism, ex situ XRD, SEM, TEM, and XPS were conducted. Initially, the first-cycle ex situ XRD patterns exhibit stable peaks of AgVO without any shift, indicating no lattice expansion occurred in AgVO when Zn2+ intercalated in the cathode (Fig. S5†). In addition, two new phases, Zn5(OH)8Cl2·H2O and Zn4ClO4(OH)7, experienced a dynamic appearance and disappearance during discharge and charge processes, respectively, which are regarded as byproducts of the simultaneous insertion/extraction of Zn2+ and H+ in the cathode.51–53 In the corresponding ex situ SEM images (Fig. S6†), nanoflower-shaped Zn5(OH)8Cl2·H2O and Zn4ClO4(OH)7 were observed after discharging to 1.0 V, which grew into larger nanoflakes and gradually coated on the cathode surface, however, then started to reduce and finally diminished when charging to 1.2 V, in agreement with the XRD results. Meanwhile, the ex situ XRD of the second cycle exhibits a similar phase transition of zinc hydroxide byproducts, proving the reversible Zn2+ and H+ ion (de)intercalation (Fig. S7†). The simultaneous intercalation of Zn2+ and H+ was confirmed by cyclic voltammetry plots and in situ pH measurements (Fig. S8 and S9†). It is worth noting that after the first cycle, a new peak in the XRD pattern at 30° was observed in Fig. 1d and S5,† representing the formation of Zn3V2O7(OH)2·2H2O (ZnVO). Interestingly, the peaks around 30° were less intense compared to the original AgVO cathode materials. This reduction in peak intensity can be attributed to the decrease of the AgVO phase during cathodic transformation, as well as the overlapping of the peaks of both original AgVO and newly formed ZnVO at 12°, which lowers the relative intensity of the peak at 30°. In addition, the small nanosheet can be observed on the cathode surface in the SEM images (Fig. S6†), uniformly distributed throughout the first cycle. The energy-dispersive X-ray spectroscopy (EDX) results suggest that the main component of nanosheets can be ascribed to the newly formed ZnVO (Fig. S10†). The coverage of ZnVO on the cathode is reinforced by the TEM and HRTEM images, in which a more transparent layer can be observed on the AgVO nanobelt (Fig. 1e). The HRTEM results reveal the main component of ZnVO in the outside layer with a distribution of tiny amount of Ag nanoparticles. The existence of Ag nanoparticles is due to the replacement of Ag+ by intercalated Zn2+, similar to the previous reports related to Ag-doped V2O5 materials.37,54,55 The HAADF-STEM elemental mapping results with the signals from Zn, V, and O in the outside layer confirm the ZnVO layer; while the main Ag signal from the inner bulk material indicates the original AgVO nanobelt within the surrounding of ZnVO layer (Fig. 1f). The HRTEM image of the cathode material after one cycle also exhibits lattice fringes belonging to the Zn5(OH)8Cl2·H2O and Zn4ClO4(OH)7 (Fig. S11†).The phase transition is presented by XPS results as well. As shown in Fig. 1g, the signals for V4+ and V5+ are found in the pristine V 2p XPS spectra, similar to the XPS results of AgVO powder, meaning the electrode preparation does not influence the material properties. However, when the battery was discharged to 0.4 V, the V3+ component emerged via its characteristic peak, illustrating the reduction of vanadium to balance the valence change induced by the insertion of Zn2+ and H+. In the fully charged state, the absence of the V3+ component proves the reversible transformation of vanadium oxide. Interestingly, after one cycle, the distribution of V5+ in the V 2p XPS peak increases, which means AgVO with V4+ and V5+ partially converted into ZnVO with only V5+. Fig. 1h shows the Zn 2p spectra with no Zn signal in the pristine stage, whereas when discharging to 0.4 V, two strong peaks belonging to Zn 2p3/2 and Zn 2p1/2 are seen. Two features at 1024.6 eV (1047.8 eV) and 1022.7 eV (1045.7 eV) can be ascribed to the Zn2+ inserted in the AgVO layered structure and the Zn2+ from ZnVO, respectively.56 A similar feature was also observed in the Zn 2p XPS spectra in the hydrothermally-synthesized ZnVO (Fig. S12a†). In addition, the peaks at 1024.6 eV and 1047.8 eV also originated from the tiny amount of Zn5(OH)8Cl2·H2O and Zn4ClO4(OH)7 byproducts during cycling (Fig. S12b†).57 However, the intensity of peaks belonging to the inserted Zn2+ decreases when fully charged, where most of the Zn2+ was contributed by ZnVO with only a tiny amount of Zn2+ remaining in the V–O layer, indicating the irreversible transition of ZnVO and reversible Zn2+ intercalation. Additionally, the Cl 2p XPS spectra of pre- and post-cycled cathodes reveal the reversible formation of Zn5(OH)8Cl2·H2O and Zn4ClO4(OH)7 (Fig. S13†). There is no Cl 2p signal in the initial cathode, though, when discharged to 0.4 V, two Cl 2p peaks at 208.3 eV and 198.6 eV emerged, corresponding to Cl from ClO4− and Cl−, respectively. These two peaks show significantly lower intensity when fully charged, meaning a low residue of these two compounds on the cathode. Moreover, the state change of Ag is also attractive. As shown in Fig. 1i, only the signal for Ag+ can be found in the pristine cathode, and the component of Ag0 appears when discharging to 0.4 V, verifying the extraction of Ag+ as Ag metals. At the end of the first cycle, the intensity of the two components has barely changed, suggesting an irreversible substitution of Ag+ during the discharge process when Zn2+ intercalated in the cathode. Accordingly, Zn2+ replaced Ag+ to serve as a pillar to support the V–O layered structure when discharge occurs. In the charging process, the intercalated Zn2+ was extracted from the active sites and subsequently formed V2O5 layers. This replacement may be a possible explanation for the formation of ZnVO.
2.3. Enhanced reaction kinetics by Zn3(OH)2V2O7·2H2O
In order to further evaluate the properties of the ZnVO layer on the cathode, ex situ electrochemical impedance spectroscopy (EIS) was employed, and a 3 M ZnSO4 electrolyte was utilized as the comparison. To evaluate the impedance in both electrolytes, the Nyquist plots were fitted by two equivalent circuits (Fig. S14–S16†). As shown in Fig. 2a, the batteries using the 3 M ZnSO4 electrolyte show an obvious increase in Rct during the discharge process, accompanied by a gradual decrease to the original value during charging. This change in Rct is ascribed to the reversible transformation of the Zn4SO4(OH)6·5H2O and Zn4SO4(OH)6·3H2O byproducts on the surface of the cathode, in agreement with the ex situ XRD patterns showing that the peaks of byproducts appeared and disappeared in the first cycle (Fig. S17†). The nanoflake-like byproducts covering the surface provide the barrier for the transport of charge carriers and further decrease the active surface area to increase the internal resistance (Fig. S18†). Furthermore, the ex situ SEM images of the cathodes using 3 M ZnSO4 show a surface morphology change in the first cycle (Fig. S19†). Compared with the batteries using 3 M Zn(ClO4)2 electrolyte (Fig. S6†), the SEM images of the cathodes cycled in the 3 M ZnSO4 exhibit obvious AgVO nanobelts without the coverage of ZnVO nanosheets, indicating that the lack of a protective shield harms the internal resistance. In contrast, other than the increase in the value of Rct at the very beginning, which can be explained by the formation of zinc hydroxides, the value of Rct remains very stable without obvious change in the batteries when using the 3 M Zn(ClO4)2 electrolyte (Fig. 2b). This result is ascribed to the ZnVO nanosheets on the surface, which act as a passive layer to not only prevent the formation of Zn4ClO4(OH)7 but also provide pathways for the Zn2+ diffusion. The enhanced ion diffusion at the interface provided by ZnVO was also confirmed by the contact angle measurement (Fig. 2c and d). On the pristine cathodes, both electrolytes show similar initial contact angles between 110° and 130°, whereas after 5 cycles, the cathode with ZnVO exhibited a significantly lower contact angle of 58.7° compared to the cathode cycled in 3 M ZnSO4 with a slight change of the contact angle (118.6°). This result confirms the improved hydrophilicity endowed by the coating of ZnVO, providing easy access for the transport of ions to aid the electrochemical kinetics.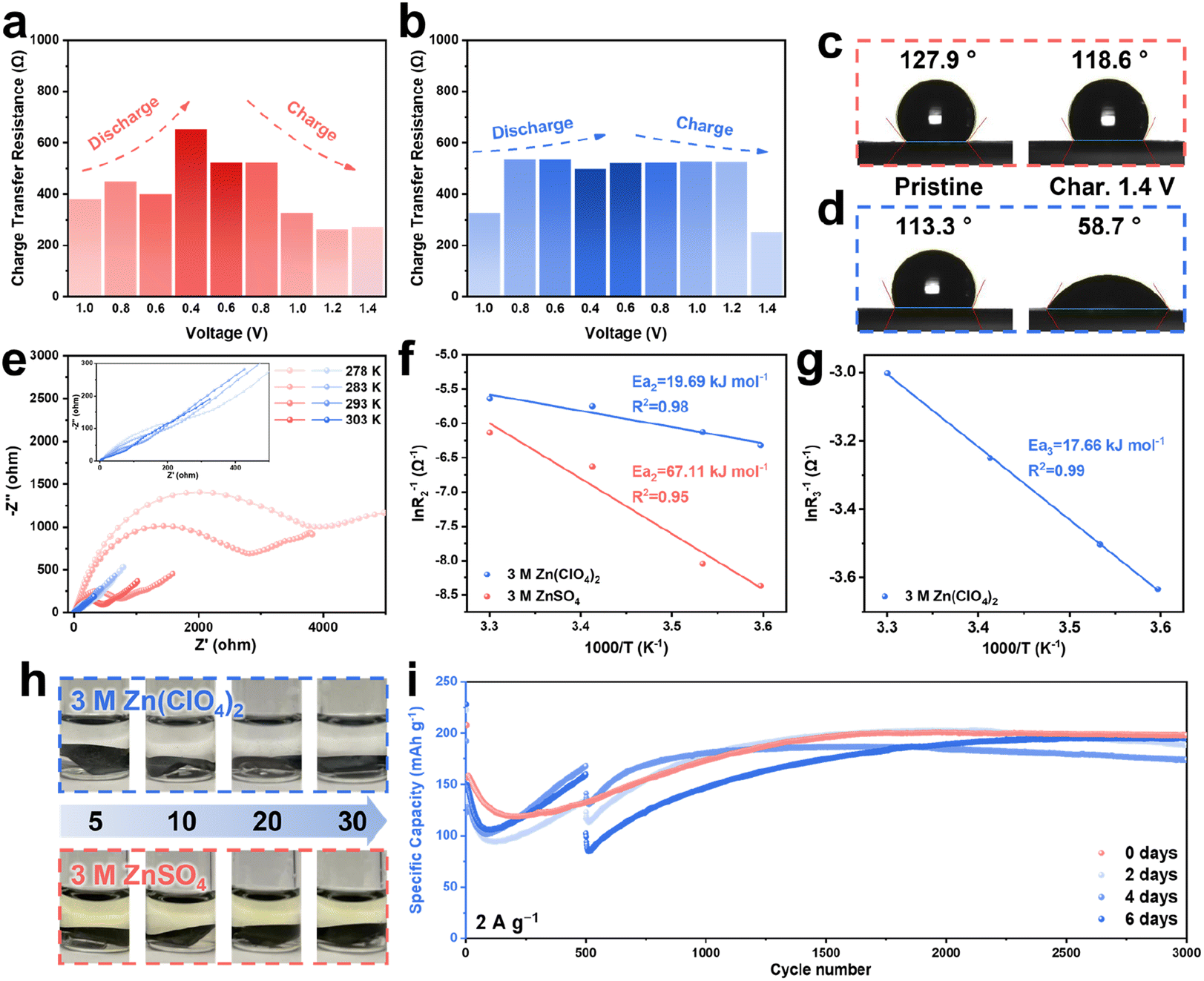 | ||
| Fig. 2 Charge transfer resistance of AgVO/Zn batteries using (a) 3 M ZnSO4 and (b) 3 M Zn(ClO4)2 during the first cycle. Contact angles of pristine and fully charged AgVO cathodes using (c) 3 M ZnSO4 and (d) 3 M Zn(ClO4)2. (e) Nyquist plots at different temperatures of AgVO/Zn batteries using 3 M Zn(ClO4)2 (blue) and 3 M ZnSO4 (red) at a fully charged state after 10 cycles at 0.1 A g−1. The activation energy (f) Ea2 and (g) Ea3 determined by an Arrhenius analysis. (h) Optical images of AgVO cathodes cycled for 5, 10, 20, and 30 cycles in 3 M Zn(ClO4)2 and 3 M ZnSO4 and subsequently immersed in two electrolytes for 30 days, respectively. (i) Cycling performance of the AgVO/Zn batteries at 2 A g−1 with resting for different times. | ||
To fully verify the enhancement of Zn2+ transport due to the resulting ZnVO layer, electrochemical reaction kinetics are explored via CV, EIS, and galvanostatic intermittent titration technique (GITT) tests. First, the CV curves at different scan rates from 0.2 to 1.0 mV s−1 shown in Fig. S20a† present two obvious redox peaks. Fig. S20b† illustrates the theoretically calculated b-values of 0.67 and 0.53 corresponding to the redox peaks 1 and 2, respectively, meaning that these cells show pseudocapacitive storage behavior. The capacitive contribution increases from 16.7 to 32.7% at corresponding scan rates of 0.2 to 1.0 mV s−1 (Fig. S20c†). The excellent rate performance at different current densities from 0.2 to 1.0 A g−1 substantiated the fast reaction kinetics (Fig. S21†). Nyquist plots at different temperatures for the batteries using 3 M Zn(ClO4)2 and 3 M ZnSO4 after ten cycles are presented in Fig. 2e. The plots were fit by two different equivalent circuits (Fig. S14†), and the fitting results of two electrolytes can be found in Fig. S22 and S23.†R1 represents the solution impedance in both electrolytes, R2 corresponds to the electrode/electrolyte impedance of ZnVO/3 M Zn(ClO4)2 and AgVO/3 M ZnSO4, and R3 represents the impedance of the additional interface between AgVO and ZnVO in the 3 M Zn(ClO4)2 electrolyte. It is evident that the impedance of AgVO/ZnVO/3 M Zn(ClO4)2 is dramatically smaller than that of AgVO/3 M ZnSO4 at each temperature. Arrhenius fitting plots of the activation energies Ea2 and Ea3 can be found in Fig. 2f and g, respectively (the detailed calculations can be found in the ESI†). With the presence of the ZnVO layer, the activation energy of AgVO using 3 M Zn(ClO4)2 (19.69 kJ mol−1) is significantly lower than that of the batteries in 3 M ZnSO4 without ZnVO protection (67.11 kJ mol−1). Moreover, the relatively small activation energy Ea3 (17.66 kJ mol−1) between AgVO and ZnVO demonstrates the adequate conductivity and compatibility of the ZnVO layer. The activation energy Ea2 of the battery using 3 M Zn(ClO4)2 without cycling was analyzed as well (Fig. S24 and S25†). Without the ZnVO coating on the surface, the original Ea2 (23.93 kJ mol−1) is higher than that of the cycled batteries (19.69 kJ mol−1), indicating that the ZnVO is beneficial for the reactions occurring at the electrode/electrolyte interface.
Additionally, the GITT results at the 5th cycle illustrate the diffusion coefficient at the interfaces between the cathodes and electrolytes (Fig. S26†). The diffusion coefficient in 3 M Zn(ClO4)2 and 3 M ZnSO4 electrolytes display similar values and trends, indicating that the ZnVO coating does not negatively influence ion diffusion. During the charging process, the cathode with ZnVO in 3 M Zn(ClO4)2 exhibited a higher diffusion coefficient at most states, demonstrating enhanced ion diffusion provided by the ZnVO. To deconvolute the protection and properties of the ZnVO film, density functional theory (DFT) calculations were carried out. The calculated density of states (DOS) of AgVO and ZnVO are shown in Fig. S27a and b,† respectively, indicating AgVO is an electronic conductor while ZnVO is an electronic insulator. The electronic insulator and ionic conductor properties of ZnVO not only optimize the interface but also reduce the activation energy, further hindering the direct contact between active material and electrolyte to suppress the cathodic dissolution ultimately.
As well as accelerating reaction kinetics, the nanosheet-like ZnVO layer may prevent the cathode materials from dissolving. To prove the protective function of ZnVO, the cathodes cycled for different numbers of cycles (i.e., 5, 10, 20, and 30 cycles) were soaked in two electrolytes for 30 days (Fig. 2h). The color of the 3 M ZnSO4 changed dramatically while the 3 M Zn(ClO4)2 electrolytes remained transparent without color change. The inductively coupled plasma mass spectrometry (ICP-MS) results show that the concentrations of vanadium in 3 M ZnSO4 are almost 200–400 times more than those in 3 M Zn(ClO4)2 after different cycles, corresponding to the color difference in both electrolytes (Tables S1 and S2†). The above results demonstrate that the ZnVO film formed in the 3 M Zn(ClO4)2 electrolyte prevented the dissolution of AgVO cathode. Subsequently, the cycling performance of the AgVO/Zn batteries using the 3 M Zn(ClO4)2 electrolyte at 2 A g−1 with rests for two, four, and six days after cycling 500 cycles were evaluated. As shown in Fig. 2i, all three rested cells have a capacity drop after a long-time rest, whereas all of them exhibited a reactivation process and a similar specific capacity of approximately 200 mA h g−1 after reactivation, very close to the specific capacity of the battery without the resting process at 2 A g−1. The reactivation and maintenance of capacity indicate almost no active material loss during the resting period, verifying the effective protection provided by the ZnVO films. In contrast, without the protection of ZnVO layer, cathode materials continuously dissolved into the 3 M ZnSO4 electrolyte, and byproducts formed on the cathode. Consequently, the AgVO/Zn battery cycled in 3 M ZnSO4 electrolyte at 2 A g−1 displays an apparent capacity fading for 1000 cycles with a capacity retention of almost 0% (Fig. S28a†). The corresponding XRD result of cycled AgVO cathode proves that there is no formation of ZnVO and the cathode maintains pristine AgVO very well, confirming the formation of ZnVO protective layer did not occur on the cathode cycled in 3 M ZnSO4 electrolyte (Fig. S28b†).
2.4. Capacity reactivation and reconstruction mechanism of Zn3(OH)2V2O7·2H2O
The cycling performance of the AgVO/Zn batteries in 3 M Zn(ClO4)2 under 0.1, 2, and 10 A g−1 were all similar regarding the capacity change in that the batteries experienced a capacity fading, a subsequent gradual reactivation, and final stable cycling (Fig. 1c, 2i, and 3a). The initial capacity fading can be ascribed to the dissolution of the AgVO cathode in the electrolyte due to the limited protection of the ZnVO films. To prove the dissolution of AgVO in the aqueous electrolyte, the cycling performances of AgVO/Zn batteries using non-aqueous 1 M Zn(ClO4)2/PC and 3 M ZnSO4 aqueous electrolytes were evaluated (Fig. S29†), which show no capacity loss in non-aqueous electrolyte while obvious capacity fading in aqueous electrolyte, indicating the high stability of AgVO in the non-aqueous system and the drastic dissolution in aqueous electrolyte. Looking back to the batteries employing the 3 M Zn(ClO4)2 electrolyte, the reactivation process appeared to deliver an increased capacity after the reduction in capacity. Several explanations and hypotheses, including moderate penetration of the electrolyte, gradual utilization of active material, and slow replacement of host cations between V–O layers, have been proposed to illustrate the increase in capacity.37,40,41 However, we confirm that a reactivation process can be ascribed to the moderate phase transition occurring on the cathode during cycling. | ||
| Fig. 3 (a) Cycling performance at 10 A g−1 of AgVO/Zn full cell using 3 M Zn(ClO4)2. (b) GCD curves at different typical cycles and corresponding (c) differential capacity (dQ/dV) curves. (d) XRD patterns and XPS spectra of (e) V 2p3/2 and (f) Ag 3d at different cycles. (g) Cross-sectional SEM images of the cathodes at different cycles (SSWM is the current collector). | ||
To comprehensively unveil the mechanism behind the capacity change, the cathodes at five different typical points were selected and characterized, which corresponded to the initial drop of capacity (A), the stop of capacity loss (B), the increase in capacity (C), the maximum capacity after reactivation (D), and the stable cycling after reactivation (E), respectively. Galvanostatic charge–discharge (GCD) curves at different states are shown in Fig. 3b, which displays the changes in charge/discharge plateaus, indicating the switch of the energy storage mechanism during cycling. Compared with state A, the GCD curve of state D displays two more obvious plateaus at around 0.6 and 1.0 V during charging, indicating two new redox reactions occurred during charging to provide more energy. Moreover, during discharging, the GCD curve of state D shows two more obvious plateaus at 0.6 and 1.0 V, corresponding to the charging process, which means the redox reactions are reversible. Specifically, differential capacity (dQ/dV) curves derived from these GCD curves were developed to reveal a more detailed mechanism in the capacity drop and increase (Fig. 3c). The first redox peaks on both cathodic and anodic sweeps (0.4–0.8 V) are attributed to the reversible V4+/V3+ redox couples. Moreover, the redox peak at 1.0 V during the anodic sweep was observed in each cycle and experienced a gradual intensity increase with the formation of a new wide peak on the cathodic sweep at around 0.95 V. This redox pair (1.0 V/0.95 V) is induced by the V5+/V4+ transition in ZnVO, which corresponds to the plateau at around 1.0 V in the GCD curves, indicating that the ZnVO moderately replaced AgVO as the active cathode material.47,48,58 A new redox peak pair (1.05 V/0.75 V) emerged during cycling, representing the insertion and extraction of cations in the newly transformed ZnVO. Other than the variety of peak intensities, the redox peak pairs during discharge between 0.6 to 0.8 V moderately shifted toward lower voltages, and the peak at around 0.45 V gradually diminished, demonstrating an energy storage host phase change in the bulk cathode material, which was verified by the XRD patterns at different cycles (Fig. 3d). Initially, from state A to E, the characteristic peaks at around 12.5° have a slight shift toward lower 2θ degrees, which corresponds to the XRD pattern gradually changing from AgVO to ZnVO, illustrating the phase change from AgVO to ZnVO. Since the characteristic peaks at 12.5° of AgVO and ZnVO overlap, in order to clearly distinguish the emergence and the change of ZnVO, the characteristic peak at 30° was focused. The diffraction peak at 30° of ZnVO emerged from state A with a tiny signal, which moderately increased the intensity until AgVO fully transformed into ZnVO from state D. The intensity change of the peak demonstrates the accumulation of ZnVO on the cathode, representing a continuous phase transition from AgVO to ZnVO. Ex situ XRD patterns at state D illustrate the stability of ZnVO without any peak shift and phase change during cycling (Fig. S30†). Meanwhile, the V 2p3/2 XPS results display the trade-off of the vanadium valence in that there was a decrease of V4+ with an increase of V5+, meaning the V5+ in the ZnVO phase gradually became dominant (Fig. 3e). The remaining V4+ after state E may be attributed to a very tiny amount of AgVO residue. Furthermore, the characteristic peak of Ag metal at 38.1° can be observed after the state D and became intense at the state E in the XRD patterns, demonstrating the irreversible replacement of Ag+ by Zn2+ during cycling. More evidentially, the XPS spectra of Ag 3d informed the decrease of Ag+ and the increase of Ag0 component as the cycling progressed, corresponding to the appearance of the Ag metal phase in the XRD results (Fig. 3f). Interestingly, it was reported that the replacement of Ag+ by Zn2+ led to the activation of capacity, where more Zn2+ active sites provided higher capacity.37,54 However, in the battery using the non-aqueous electrolyte, the pristine AgVO phase partially transformed into Zn2(V3O8)2, agreeing with the replacement of Ag+ by Zn2+, while no activation process was observed during cycling (Fig. S31†). Thus, the reactivation cannot be simply explained by the increased Zn2+ intercalation sites originating from the substituted Ag+ positions.
SEM images at different cycles exhibited the coverage of dense ZnVO nanosheets on the cathode surface, preventing the cathode from dissolving (Fig. S32†). More intuitively, cross-sectional SEM images at different cycles emphasize the gradual transition from AgVO to ZnVO (Fig. 3g). The pristine cathode without cycling only has one dense AgVO layer on the current collector. The cross-sectional image at state C exhibits two layers, a ZnVO nanosheets protective layer on top of the untransformed AgVO. Cross-sectional imaging after state E depicts a dominant layer of ZnVO nanosheets on the current collector. The distinct morphological difference during phase transition on the cathode surface may influence the capacity change. Compared with the dense original AgVO cathode, ZnVO layer has a more open topography, larger surface area, and higher thickness. The electrochemically active surface areas (ECSAs) determined by the double layer capacitance (Cdl) results confirm a larger surface area of the active cathode material after state E (231.9 cm2) than that of the pristine one (181.2 cm2), indicating more contact between cathode and electrolyte to facilitate the ion transition to increase the capacity (Fig. S33†). In addition, the higher surface area of ZnVO can improve the contact between ZnVO and conductive materials (SuperP and in situ precipitated Ag nanoparticles) to increase the conductivity and reduce resistance, which may lead to capacity activation.
Regarding the formation mechanism of ZnVO in the vanadate-based cathode materials, it is reported that within a specific pH range of 3.8–7.8, V2O5 can spontaneously transform into ZnVO by reacting with Zn2+-containing electrolytes.47 Even though the original pH of 3 M Zn(ClO4)2 (2.85) is out of that range, the pH increase caused by the H+ insertion probably created a proper pH environment that allows the transition from AgVO to ZnVO to occur with Zn2+ replacing Ag+. In order to reveal the formation mechanism of ZnVO, at first, Na0.33V2O5 (NaVO), which has the same layered structure as Ag0.33V2O5, was electrochemically tested and characterized (Fig. S34†). Even though AgVO and NaVO show the same structure, the NaVO/Zn battery in 3 M Zn(ClO4)2 cycled at 0.1 A g−1 exhibited poor cycling stability without reactivation (Fig. S35†). The XRD pattern of the post-cycled cathode after 150 cycles shows no evidence of ZnVO, and the NaVO structure was maintained without any phase change (Fig. S36†). The lack of structural changes observed in NaVO during cycling can be attributed to the comparatively more difficult replacement between Zn2+ and Na+ in comparison to that between Zn2+ and Ag+. As shown in Fig. S37,† Ag+ and Na+ were replaced by Zn2+, which need to overcome the energy barriers of 0.81 and 2.27 eV, respectively, indicating that replacing Na+ with Zn2+ is hard to occur in NaVO. This result indicates that the reconstruction of NaVO is considerably more challenging than AgVO, thus explaining the observed stability of the NaVO structure after cycling. Moreover, the XRD result in Fig. S38† suggests that the phase transition is not a spontaneous reaction between electrolyte and AgVO. Even after long-time soaking, the XRD pattern still exhibits the main phase of AgVO without the existence of ZnVO (Fig. S38†). In contrast, in the previous reports, V2O5 transferred into ZnVO rapidly after submersion in the electrolyte.47,48,59 The cycling of the AgVO/Zn battery with the soaked cathode shows a similar reduction and reactivation process in the capacity as the pristine cathode without soaking, indicating a similar phase transition only occurred under electrochemical conditions (Fig. S39†). Therefore, the phase transition in the AgVO is related to the substitution of Ag+ by Zn2+ during cycling.
In conclusion, the relationship between capacity change and phase reconstruction is summarized and shown in Fig. S40a.† Initially, without any protection, the pristine cathode dissolved in the aqueous electrolyte, continuously causing capacity decay. After several cycles, although AgVO still suffered from dissolution, a ZnVO protective layer formed on the cathode surface to mitigate the dissolution. Accordingly, the ZnVO layer grew thicker and more robust, allowing more efficient protection of the cathode and eliminating capacity fading. In the subsequent cycles, as well as protection, the well-reconstructed ZnVO replaced AgVO and accumulated to become the main active material, causing an increase in the capacity. On the other hand, the enhanced capacity after reactivation can also be explained by the enlarged surface of ZnVO, which provided abundant Zn2+ storage sites, strong connection with the current collector, open transport tunnels, and enlarged contacting surface with conductive materials and electrolyte, facilitating ion diffusion and electron transmission. In contrast, the original AgVO layered structure offered fewer Zn2+ intercalation sites due to the interaction between oxide layers and Ag+ ions, blocking the transport of Zn2+ in the cathode. An AgVO/Zn pouch cell with 10 cm2 scaled electrodes was assembled to take advantage of the in situ protection and transition of ZnVO, and it had an initial specific capacity of 400 mA h g−1 with a stable average capacity of 250 mA h g−1 at 0.1 A g−1 after reactivation for 100 cycles, proving the practicality of the in situ reconstruction of the cathode for pouch cells (Fig. S40b†).
2.5. Reaction kinetics after cathodic reconstruction
To explore the reactivation process more comprehensively, the reaction kinetics of state E were studied. CV plots with different scan rates from 0.2 to 1.0 mV s−1 exhibit two newly-emerged redox peaks, representing the electrochemical performance of ZnVO (Fig. 4a). The theoretically calculated b-values for peaks 1 to 4 are 0.59, 0.50, 0.81, and 0.95, respectively (Fig. 4b). The b-values belonging to peaks 1 and 2 are higher than that of the original peaks 1 and 2 of the AgVO cathode. Additionally, the peaks pair at 1.0 V/0.95 V exhibit a b-value approaching 1, implying that the reconstructed ZnVO-based cathode can provide more capacity induced by pseudocapacitance than the original AgVO cathode. Moreover, compared with the capacitive contribution before reactivation, the post-reactive capacitance shows a higher contribution at different scan rates, illustrating an enhanced pseudocapacitive ability after reactivation induced by ZnVO (Fig. 4c). Fig. S41† exhibits the increasing capacitive contribution from 5 to 30 mV s−1. The capacitive contribution at 30 mV s−1 is 89.3% in the reconstructed ZnVO, meaning the predominant contribution is from capacitance at high current densities. To understand the intrinsic reaction kinetics of ZnVO, the pseudocapacitive behavior of hydrothermally-synthesized ZnVO was measured and calculated via CV tests. There are four redox peaks in ZnVO (Fig. 4d), corresponding to the CV result in the reconstructed AgVO, which proves the transition from AgVO to ZnVO (Fig. 4a). The b values of each peak from 1 to 4 are 0.9999, 0.9960, 0.9997, and 0.9993, respectively, which are all close to 1.0, meaning capacitive behavior is dominating in ZnVO (Fig. 4e). The high capacitive contributions and the b values which are almost 1.0 can be ascribed to the uncommon intercalation pseudocapacitive behavior in ZnVO (Fig. 4f).50,60 Unlike the redox reactions happening on the electrode surface, the intercalation pseudocapacitance happens when the charge carriers (Zn2+ or H+) intercalate into the layered structure of the cathode material with a faradaic charge-transfer. The intercalation of Zn2+ is verified by the ex situ XRD and TEM results. As shown in Fig. S42a,† the peak located at 12.29° in the XRD patterns, corresponding to the (001) facet of ZnVO, shifted toward lower degrees during discharging and then shifted back to 12.29° upon charging to 1.4 V, which can be ascribed to the expansion of lattice spacing induced by Zn2+ insertion/extraction. Similarly, the TEM image of ZnVO upon discharging to 0.4 V exhibits an expanded lattice spacing of 7.44 Å for the ZnVO (001) plane, which is higher than that of the pristine (001) plane (7.19 Å) (Fig. S42b†). The above results confirm the intercalation of Zn2+ in the ZnVO layered structure instead of the surface Zn2+ adsorption. Since there is no limitation of solid-state diffusion in the cathode, which shows intercalation pseudocapacitance, the diffusion ability of cations in the cathode material determines the reaction kinetics. Under the assistance of promoted reaction kinetics, the AgVO/Zn battery after full reconstruction delivered an excellent rate performance with a capacity of 100 mA h g−1 at 10 A g−1 and no capacity loss at 0.1 A g−1 after cycling at high current densities (Fig. 4g). In contrast, the battery without reactivation provided a limited capacity at a high current density (around 10 mA h g−1 at 10 A g−1, Fig. S43†), demonstrating enhanced rate performance and fast kinetics provided by the in situ formed ZnVO. | ||
| Fig. 4 (a) CV curves of AgVO/Zn batteries after 50 cycles at different scan rates (0.2–1.0 mV s−1), (b) the calculated b-values of four redox peaks, and (c) corresponding capacitive contribution at different scan rates. (d) CV curves ZnVO/Zn batteries at different scan rates (0.2–1.0 mV s−1), (e) the calculated b-values of four redox peaks, and (f) corresponding capacitive contribution at different scan rates. (g) Rate performance of AgVO/Zn battery at different current densities after reactivation. (h) Possible diffusion pathways of Zn2+ in ZnVO, and (i) corresponding diffusion energy barriers of Zn2+ and H+. (j) Diffusion coefficients of Zn2+ calculated from GITT measurement after different cycles. (k) Nyquist plots after different cycles. | ||
The faster ion diffusion results in a higher rate performance and faster kinetics. Initially, DFT calculations provide theoretical evidence to support the above results. Fig. 4h and S44a† present the possible pathways for the diffusion of Zn2+ in both AgVO and ZnVO. In AgVO, three Ag+ ions between the V–O layers were deleted to more clearly show the diffusion pathway of Zn2+ (more details can be found in Fig. S45 and S46†). Due to the aforementioned replacement of Ag+ by Zn2+, the diffusion path of Zn2+ is from one Ag+ site (position 1) to another Ag+ site (position 3), where Ag+ is regarded as the significant barrier for the migration path of Zn2+ (Fig. S44a†). Accordingly, the diffusion energy barriers of Zn2+ and H+ from position 1 to position 2 are 2.28 and 1.54 eV, respectively (Fig. S44b†). In contrast, according to the previous XRD and TEM results (Fig. S42†), Zn2+ and H+ intercalate and migrate in the open layer along (001) facet in ZnVO without hindrance (Fig. 4h). To calculate the diffusion energy barrier of the possible diffusion pathway, positions 1 and 3 were selected as the starting and ending sites of diffusion because they are two equivalent sites with the lowest energy in the system. Zn2+ ions are most likely to appear at these sites with the lowest energy. Even though the interlayer spacing of ZnVO (7.2 Å) is smaller than that of AgVO (7.4 Å), the energy barriers of Zn2+ and H+ diffusing from position 1 to position 2, which is the transition state with the lowest energy barrier, is only 0.56 and 0.30 eV, respectively (Fig. 4i). The significantly lower energy barriers of both Zn2+ and H+ are attributed to the open-layered structure of ZnVO, the short diffusion pathway, and the lubrication of water molecules between the layers.61–63 Consequently, Zn2+ and H+ diffuse easier and faster in the ZnVO with a lower migration energy barrier, resulting in fast reaction kinetics and enhanced diffusion coefficient. In contrast, without transformation into ZnVO, NaVO/Zn and V2O5/Zn batteries display poor cycling performance at a high current density of 10 A g−1 with unsatisfying reaction kinetics (Fig. S47†).
Moreover, to experimentally confirm that the enhanced reaction kinetic is due to the improved ion diffusion during phase transition, the diffusion coefficients at different cycles were calculated from GITT results to evaluate the diffusion ability (Fig. S48† and 4j). The diffusion coefficient at state A shows a similar value to previously reported vanadium oxide species.16,64–66 However, following the formation of ZnVO, the diffusion coefficient at state B is significantly higher than that of the state A. After state C, the diffusion coefficients have higher values and become constant after state D, corresponding to the gradual phase transition. The diffusion coefficient at the state E is an order of magnitude higher than that of state A, which indicates that the accumulation of ZnVO remarkably facilitates the diffusion of Zn2+ to provide fast kinetic and high capacity. In addition, from Fig. 4k, EIS plots at different cycles suggested a gradual decrease of impedance during cycling, resulting in a small impedance of 70 Ω after the entire reconstruction, significantly lower than the initial value at the initial state (200 Ω). The impedance reduction suggests increased ionic conductivity in the reconstructed ZnVO and supports the reduced charge transfer resistance demonstrated by the GITT results. The slopes at low frequency after the state B became higher than that of state A, suggesting that the gradual formation of ZnVO facilitates zinc ion transport. The steady structure, large ion diffusion tunnel, and fast diffusion coefficient provide ZnVO with a high capacity and stability.
2.6. Ag nanoparticles boosted reaction kinetics
It has already been proved that the boosted rate performance is due to the fast ionic diffusion and intercalation pseudocapacitive behavior in the in situ reconstructed ZnVO cathode materials. However, as mentioned above (Fig. S27b†), ZnVO is an electronic insulator with an inferior ability to transfer electrons. In contrast, as one of the most conductive materials, the in situ formed Ag nanoparticles may facilitate the electron exchange between ZnVO and conductive materials to increase the capacity further.67–69 To understand the effect of Ag nanoparticles on the performance of batteries, the hydrothermally-synthesized ZnVO were mixed with Ag nanoparticles (<100 nm) with a mole ratio of ZnVO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ag = 1:0.33 working as the cathode material. Initially, the CV measurement at different scan rates was conducted to understand the influence of Ag nanoparticles on the reaction kinetics (Fig. 5a). As shown in Fig. 5b, all the b values of four peaks are almost 1, indicating the intercalation pseudocapacitance in ZnVO. However, the capacitive contribution has an obvious increase to 98.5% by adding Ag nanoparticles, representing that even a tiny amount of Ag can accelerate the reaction kinetics in the cathode materials (Fig. 5c). The increased kinetics in the cathodes may be due to the facilitated electron transfer via adding the Ag nanoparticles. To prove the effect of Ag nanoparticles on the improvement of electronic conductivity, EIS measurement of ZnVO/Zn and ZnVO + Ag/Zn batteries was conducted. As shown in Fig. 5d, the presence of semicircles at high- and medium-frequency regions in both batteries can be ascribed to the charge-transfer impedance on the electrode/electrolyte interface. It is obvious that ZnVO + Ag/Zn battery exhibits a smaller diameter of the semicircle with an impedance of 99.2 Ω than ZnVO/Zn battery (158.3 Ω), demonstrating a lower charge-transfer impedance of the ZnVO + Ag cathode. This reduced charge-transfer resistance confirms the electronic conductivity of the cathode was enhanced by introducing Ag nanoparticles in the cathode. Additionally, more electrons can be stored in the ZnVO cathodes beneficial from the improved electronic conductivity. Fig. 5e shows the cycling performance of ZnVO + Ag/Zn battery at 10 A g−1 exhibits higher capacity than that of ZnVO/Zn battery, meaning the addition of Ag nanoparticles can efficiently enhance the capacity by increasing the transition of electrons. The rate performance of ZnVO + Ag/Zn battery displays higher capacities at different current densities, reinforcing the existence of Ag nanoparticles can increase the capacity (Fig. 5f). The better rate performance in ZnVO + Ag/Zn battery can be attributed to the pseudocapacitance with higher capacitive contribution than ZnVO/Zn battery, indicating the presence of Ag nanoparticles facilitated the reaction kinetics by accelerating the electron transfer. The above results and observations support the idea that the simultaneous influence of enhanced electronic conductivity from Ag and boosted ionic conductivity from ZnVO promote more efficient charge transfer to further enhance the battery's overall performance.
:Ag = 1:0.33 working as the cathode material. Initially, the CV measurement at different scan rates was conducted to understand the influence of Ag nanoparticles on the reaction kinetics (Fig. 5a). As shown in Fig. 5b, all the b values of four peaks are almost 1, indicating the intercalation pseudocapacitance in ZnVO. However, the capacitive contribution has an obvious increase to 98.5% by adding Ag nanoparticles, representing that even a tiny amount of Ag can accelerate the reaction kinetics in the cathode materials (Fig. 5c). The increased kinetics in the cathodes may be due to the facilitated electron transfer via adding the Ag nanoparticles. To prove the effect of Ag nanoparticles on the improvement of electronic conductivity, EIS measurement of ZnVO/Zn and ZnVO + Ag/Zn batteries was conducted. As shown in Fig. 5d, the presence of semicircles at high- and medium-frequency regions in both batteries can be ascribed to the charge-transfer impedance on the electrode/electrolyte interface. It is obvious that ZnVO + Ag/Zn battery exhibits a smaller diameter of the semicircle with an impedance of 99.2 Ω than ZnVO/Zn battery (158.3 Ω), demonstrating a lower charge-transfer impedance of the ZnVO + Ag cathode. This reduced charge-transfer resistance confirms the electronic conductivity of the cathode was enhanced by introducing Ag nanoparticles in the cathode. Additionally, more electrons can be stored in the ZnVO cathodes beneficial from the improved electronic conductivity. Fig. 5e shows the cycling performance of ZnVO + Ag/Zn battery at 10 A g−1 exhibits higher capacity than that of ZnVO/Zn battery, meaning the addition of Ag nanoparticles can efficiently enhance the capacity by increasing the transition of electrons. The rate performance of ZnVO + Ag/Zn battery displays higher capacities at different current densities, reinforcing the existence of Ag nanoparticles can increase the capacity (Fig. 5f). The better rate performance in ZnVO + Ag/Zn battery can be attributed to the pseudocapacitance with higher capacitive contribution than ZnVO/Zn battery, indicating the presence of Ag nanoparticles facilitated the reaction kinetics by accelerating the electron transfer. The above results and observations support the idea that the simultaneous influence of enhanced electronic conductivity from Ag and boosted ionic conductivity from ZnVO promote more efficient charge transfer to further enhance the battery's overall performance.
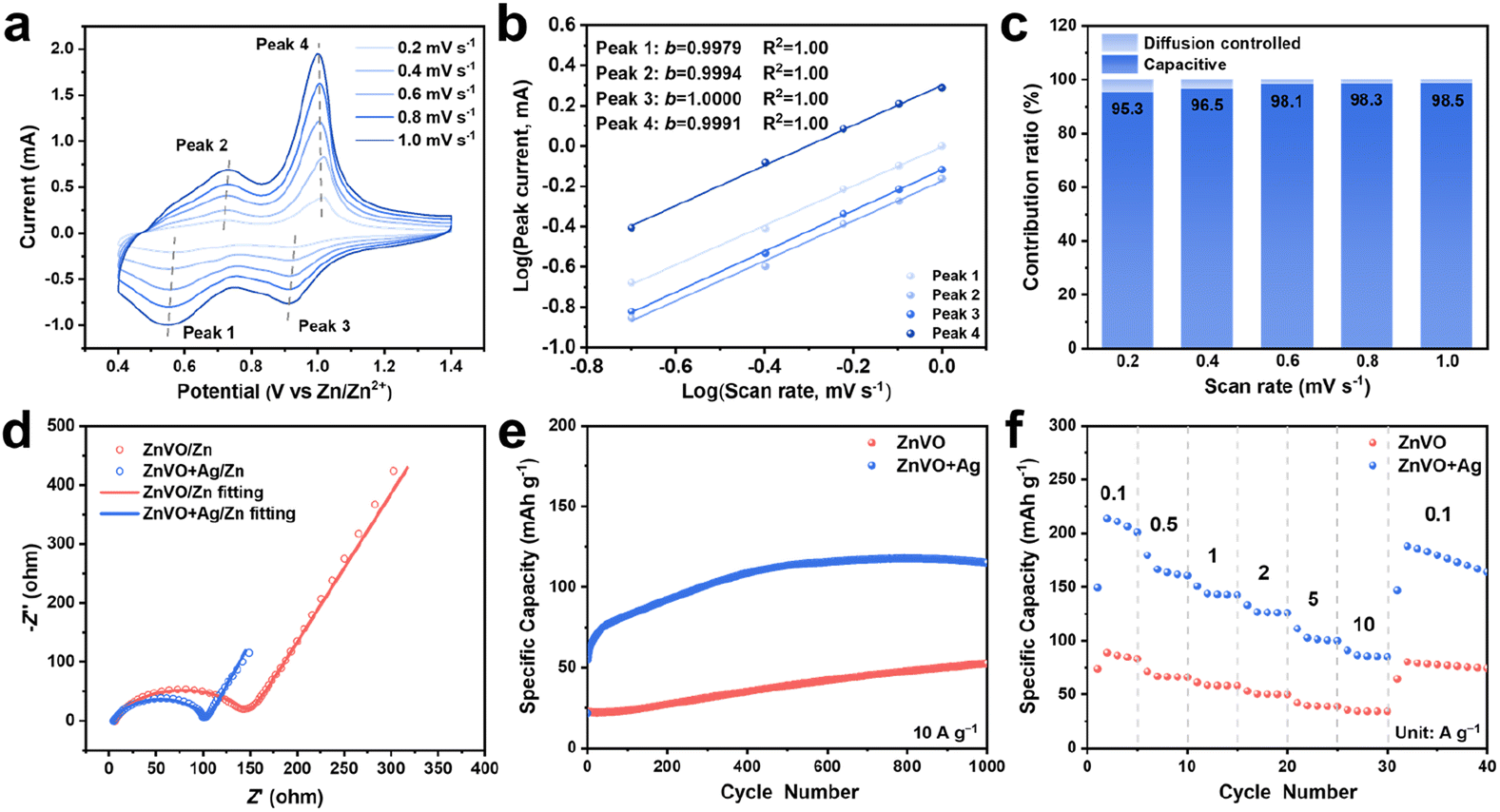 | ||
| Fig. 5 (a) CV curves of ZnVO + Ag/Zn batteries at different scan rates (0.2–1.0 mV s−1), (b) the calculated b-values of four redox peaks, and (c) corresponding capacitive contribution at different scan rates. (d) Nyquist plots and fitting curves of the ZnVO/Zn and ZnVO + Ag/Zn batteries in 3 M Zn(ClO4)2. (e) Cycling performance at 10 A g−1 and (f) rate performance of ZnVO + Ag/Zn batteries and ZnVO/Zn batteries in 3 M Zn(ClO4)2 electrolyte. | ||
3. Conclusions
In summary, we have studied and characterized the Ag0.33V2O5 cathode material during cycling to illustrate the reaction mechanism behind the capacity reactivation. A reconstructed Zn3(OH)2V2O7·2H2O new phase initially forms in situ on the surface of the Ag0.33V2O5 cathode material as a protective layer and grows thicker to fully convert into the active cathode materials, leading to a reactivation process and subsequent high cycling capacity and stability. The in situ formed Zn3(OH)2V2O7·2H2O layer alleviated the dissolution of the vanadium oxide-based cathode in the aqueous electrolyte by inhibiting the direct contact between the cathode and electrolyte due to its electronic insulating property. In addition, the protective Zn3(OH)2V2O7·2H2O nanosheets lowered the activation energy on the electrode surface to aid electrochemical reactions and Zn2+ insertion. Furthermore, due to the intrinsic intercalation pseudocapacitive behavior of the in situ formed Zn3(OH)2V2O7·2H2O, the further reconstruction of the cathode provided enhanced ionic diffusion, low impedance, and fast reaction kinetics, facilitating the capacity and stability of the batteries. DFT calculations showed that the open layered structure of Zn3(OH)2V2O7·2H2O facilitates Zn2+ diffusion. Another in situ formed product, Ag nanoparticles, significantly enhanced the electronic conductivity of the cathode, promoting the electrochemical performance of reconstructed cathode. The reactivation of capacity induced by structural changes could potentially offer a broad explanation for the capacity variations observed in vanadate-based aqueous zinc-ion batteries.Author contributions
Ziqing Wang: conceptualization, data curation, formal analysis, methodology, writing – original draft, writing – review and editing; Jiefeng Diao: data curation, formal analysis, methodology, software,writing – review and editing; Kenta Kawashima: data curation, formal analysis, methodology, writing – review and editing; Jason A. Weeks: data curation, formal analysis, methodology, writing – review and editing; Rinish Reddy Vaidyula: data curation, formal analysis, methodology, writing – review and editing; Raul A. Marquez: data curation, formal analysis, methodology, writing – review and editing; Nathaniel Miller: data curation, formal analysis, methodology, writing – review and editing; Graeme Henkelman: funding acquisition, software, supervision, writing – review and editing; C. Buddie Mullins: conceptualization, data curation, formal analysis, funding acquisition, methodology, project administration, resources, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Welch Foundation for their generous support through grant no. F-1436 (C. B. M.) and F-1841 (G. H.).References
- N. Zhang, X. Chen, M. Yu, Z. Niu, F. Cheng and J. Chen, Chem. Soc. Rev., 2020, 49, 4203–4219 RSC.
- G. Fang, J. Zhou, A. Pan and S. Liang, ACS Energy Lett., 2018, 3, 2480–2501 CrossRef CAS.
- Z. Xing, Y. Sun, X. Xie, Y. Tang, G. Xu, J. Han, B. Lu, S. Liang, G. Chen and J. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202215324 CrossRef CAS PubMed.
- R. Yi, X. Shi, Y. Tang, Y. Yang, P. Zhou, B. Lu and J. Zhou, Small Struct., 2023, 2300020 CrossRef.
- C. Liu, Z. Luo, W. Deng, W. Wei, L. Chen, A. Pan, J. Ma, C. Wang, L. Zhu, L. Xie, X.-Y. Cao, J. Hu, G. Zou, H. Hou and X. Ji, ACS Energy Lett., 2021, 6, 675–683 CrossRef CAS.
- D. Chao, C. (Rose) Zhu, M. Song, P. Liang, X. Zhang, N. H. Tiep, H. Zhao, J. Wang, R. Wang, H. Zhang and H. J. Fan, Adv. Mater., 2018, 30, 1803181 CrossRef PubMed.
- Y. Zhang, F. Wan, S. Huang, S. Wang, Z. Niu and J. Chen, Nat. Commun., 2020, 11, 2199 CrossRef CAS PubMed.
- Y. Song, P. Ruan, C. Mao, Y. Chang, L. Wang, L. Dai, P. Zhou, B. Lu, J. Zhou and Z. He, Nano-Micro Lett., 2022, 14, 218 CrossRef CAS PubMed.
- X. Chen, P. Ruan, X. Wu, S. Liang and J. Zhou, Acta Phys.-Chim. Sin., 2022, 38, 2111003 Search PubMed.
- X. Chen, X. Shi, P. Ruan, Y. Tang, Y. Sun, W.-Y. Wong, B. Lu and J. Zhou, Small Sci., 2023, 3, 2300007 CrossRef CAS.
- T. Xiong, Z. G. Yu, H. Wu, Y. Du, Q. Xie, J. Chen, Y.-W. Zhang, S. J. Pennycook, W. S. V. Lee and J. Xue, Adv. Energy Mater., 2019, 9, 1803815 CrossRef.
- J. Huang, Z. Wang, M. Hou, X. Dong, Y. Liu, Y. Wang and Y. Xia, Nat. Commun., 2018, 9, 2906 CrossRef PubMed.
- J. Li, N. Luo, L. Kang, F. Zhao, Y. Jiao, T. J. Macdonald, M. Wang, I. P. Parkin, P. R. Shearing, D. J. L. Brett, G. Chai and G. He, Adv. Energy Mater., 2022, 12, 2201840 CrossRef CAS.
- Z. Yang, X. Pan, Y. Shen, R. Chen, T. Li, L. Xu and L. Mai, Small, 2022, 18, 2107743 CrossRef CAS PubMed.
- P. Ruan, X. Chen, L. Qin, Y. Tang, B. Lu, Z. Zeng, S. Liang and J. Zhou, Adv. Mater., 2023, 35, 2300577 CrossRef CAS PubMed.
- L. Wang, K.-W. Huang, J. Chen and J. Zheng, Sci. Adv., 2019, 5, eaax4279 CrossRef CAS PubMed.
- M. Liao, J. Wang, L. Ye, H. Sun, Y. Wen, C. Wang, X. Sun, B. Wang and H. Peng, Angew. Chem., Int. Ed., 2020, 59, 2273–2278 CrossRef CAS PubMed.
- G. Yang, Q. Li, K. Ma, C. Hong and C. Wang, J. Mater. Chem. A, 2020, 8, 8084–8095 RSC.
- B. Deka Boruah, A. Mathieson, S. K. Park, X. Zhang, B. Wen, L. Tan, A. Boies and M. De Volder, Adv. Energy Mater., 2021, 11, 2100115 CrossRef CAS.
- M. H. Alfaruqi, V. Mathew, J. Song, S. Kim, S. Islam, D. T. Pham, J. Jo, S. Kim, J. P. Baboo, Z. Xiu, K.-S. Lee, Y.-K. Sun and J. Kim, Chem. Mater., 2017, 29, 1684–1694 CrossRef CAS.
- Z. Wang, M. Zhou, L. Qin, M. Chen, Z. Chen, S. Guo, L. Wang, G. Fang and S. Liang, eScience, 2022, 2, 209–218 CrossRef.
- T. Yimtrakarn, Y.-C. Liao, A. S. MV, J.-L. Chen, Y.-C. Chuang, N. Lerkkasemsan and W. Kaveevivitchai, Mater. Today Commun., 2023, 34, 105231 CrossRef CAS.
- Q. Liu, Z. Ma, Z. Chen, M. Cui, H. Lei, J. Wang, J. Fei, N. He, Y. Liu, Q. Liu, W. Li and Y. Huang, Chem. Commun., 2022, 58, 8226–8229 RSC.
- Y. Zeng, X. F. Lu, S. L. Zhang, D. Luan, S. Li and X. W. (David) Lou, Angew. Chem., Int. Ed., 2021, 60, 22189–22194 CrossRef CAS PubMed.
- S. Zhang, S. Long, H. Li and Q. Xu, Chem. Eng. J., 2020, 400, 125898 CrossRef CAS.
- D. Kundu, P. Oberholzer, C. Glaros, A. Bouzid, E. Tervoort, A. Pasquarello and M. Niederberger, Chem. Mater., 2018, 30, 3874–3881 CrossRef CAS.
- H. Cui, T. Wang, Z. Huang, G. Liang, Z. Chen, A. Chen, D. Wang, Q. Yang, H. Hong, J. Fan and C. Zhi, Angew. Chem., Int. Ed., 2022, 61, e202203453 CAS.
- E. Karapidakis and D. Vernardou, Materials, 2021, 14, 2310 CrossRef CAS PubMed.
- H. Geng, M. Cheng, B. Wang, Y. Yang, Y. Zhang and C. C. Li, Adv. Funct. Mater., 2020, 30, 1907684 CrossRef CAS.
- H. Chen, J. Huang, S. Tian, L. Liu, T. Qin, L. Song, Y. Liu, Y. Zhang, X. Wu, S. Lei and S. Peng, Advanced Science, 2021, 8, 2004924 CrossRef CAS PubMed.
- C. Liu, Z. Neale, J. Zheng, X. Jia, J. Huang, M. Yan, M. Tian, M. Wang, J. Yang and G. Cao, Energy Environ. Sci., 2019, 12, 2273–2285 RSC.
- D. Zhao, X. Wang, W. Zhang, Y. Zhang, Y. Lei, X. Huang, Q. Zhu and J. Liu, Adv. Funct. Mater., 2023, 33, 2211412 CrossRef CAS.
- Y. Xu, G. Fan, P. X. Sun, Y. Guo, Y. Wang, X. Gu, L. Wu and L. Yu, Angew. Chem., Int. Ed., 2023, 62, e202303529 CrossRef CAS PubMed.
- G. Zhang, T. Wu, H. Zhou, H. Jin, K. Liu, Y. Luo, H. Jiang, K. Huang, L. Huang and J. Zhou, ACS Energy Lett., 2021, 6, 2111–2120 CrossRef CAS.
- L. Shan, Y. Wang, S. Liang, B. Tang, Y. Yang, Z. Wang, B. Lu and J. Zhou, InfoMat, 2021, 3, 1028–1036 CrossRef CAS.
- P. He, G. Zhang, X. Liao, M. Yan, X. Xu, Q. An, J. Liu and L. Mai, Adv. Energy Mater., 2018, 8, 1702463 CrossRef.
- L. Shan, Y. Yang, W. Zhang, H. Chen, G. Fang, J. Zhou and S. Liang, Energy Storage Mater., 2019, 18, 10–14 CrossRef.
- H. Qin, L. Chen, L. Wang, X. Chen and Z. Yang, Electrochim. Acta, 2019, 306, 307–316 CrossRef CAS.
- R. Li, H. Zhang, Q. Zheng and X. Li, J. Mater. Chem. A, 2020, 8, 5186–5193 RSC.
- N. Zhang, Y. Dong, M. Jia, X. Bian, Y. Wang, M. Qiu, J. Xu, Y. Liu, L. Jiao and F. Cheng, ACS Energy Lett., 2018, 3, 1366–1372 CrossRef CAS.
- J. Zhou, L. Shan, Z. Wu, X. Guo, G. Fang and S. Liang, Chem. Commun., 2018, 54, 4457–4460 RSC.
- C. Wu, S. Gu, Q. Zhang, Y. Bai, M. Li, Y. Yuan, H. Wang, X. Liu, Y. Yuan, N. Zhu, F. Wu, H. Li, L. Gu and J. Lu, Nat. Commun., 2019, 10, 73 CrossRef CAS PubMed.
- J. Ding, Z. Du, B. Li, L. Wang, S. Wang, Y. Gong and S. Yang, Adv. Mater., 2019, 31, 1904369 CrossRef CAS PubMed.
- J. Huang, X. Xie, K. Liu, S. Liang and G. Fang, Energy Environ. Mater., 2023, 6, e12309 CrossRef CAS.
- C. Xia, J. Guo, P. Li, X. Zhang and H. N. Alshareef, Angew. Chem., Int. Ed., 2018, 57, 3943–3948 CrossRef CAS PubMed.
- H. Luo, B. Wang, F. Wang, J. Yang, F. Wu, Y. Ning, Y. Zhou, D. Wang, H. Liu and S. Dou, ACS Nano, 2020, 14, 7328–7337 CrossRef CAS PubMed.
- Y. Lu, T. Zhu, W. van den Bergh, M. Stefik and K. Huang, Angew. Chem., Int. Ed., 2020, 59, 17004–17011 CrossRef CAS PubMed.
- T. He, J. Li, Z. Luo, Y. Zhang, Y. Zhao, X. Zhang and Y. Chen, ChemElectroChem, 2022, 9, e202200178 CAS.
- X. Li, X. Zhu, Z. Cao, Z. Xu, J. Shen and M. Ye, Small, 2022, 18, 2105325 CrossRef CAS PubMed.
- Y. Tao, D. Huang, H. Chen and Y. Luo, ACS Appl. Mater. Interfaces, 2021, 13, 16576–16584 CrossRef CAS PubMed.
- H. Huang, X. Xia, J. Yun, C. Huang, D. Li, B. Chen, Z. Yang and W. Zhang, Energy Storage Mater., 2022, 52, 473–484 CrossRef.
- X. Wang, Z. Zhang, B. Xi, W. Chen, Y. Jia, J. Feng and S. Xiong, ACS Nano, 2021, 15, 9244–9272 CrossRef CAS PubMed.
- F. Wan, S. Huang, H. Cao and Z. Niu, ACS Nano, 2020, 14, 6752–6760 CrossRef CAS PubMed.
- J. Hyoung, J. W. Heo, B. Jeon and S.-T. Hong, J. Mater. Chem. A, 2021, 9, 20776–20782 RSC.
- S. Guo, G. Fang, S. Liang, M. Chen, X. Wu and J. Zhou, Acta Mater., 2019, 180, 51–59 CrossRef CAS.
- C. Xia, J. Guo, Y. Lei, H. Liang, C. Zhao and H. N. Alshareef, Adv. Mater., 2018, 30, 1705580 CrossRef PubMed.
- J. Winiarski, W. Tylus, K. Winiarska, I. Szczygieł and B. Szczygieł, J. Spectrosc., 2018, 2018, 2079278 Search PubMed.
- Z. Pan, J. Yang, J. Yang, Q. Zhang, H. Zhang, X. Li, Z. Kou, Y. Zhang, H. Chen, C. Yan and J. Wang, ACS Nano, 2020, 14, 842–853 CrossRef CAS PubMed.
- S. Li, D. Yu, L. Liu, S. Yao, X. Wang, X. Jin, D. Zhang and F. Du, Chem. Eng. J., 2022, 430, 132673 CrossRef CAS.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 RSC.
- M. Yan, P. He, Y. Chen, S. Wang, Q. Wei, K. Zhao, X. Xu, Q. An, Y. Shuang, Y. Shao, K. T. Mueller, L. Mai, J. Liu and J. Yang, Adv. Mater., 2018, 30, 1703725 CrossRef PubMed.
- J. Zeng, Z. Zhang, X. Guo and G. Li, J. Mater. Chem. A, 2019, 7, 21079–21084 RSC.
- X. Li, L. Ma, Y. Zhao, Q. Yang, D. Wang, Z. Huang, G. Liang, F. Mo, Z. Liu and C. Zhi, Mater. Today Energy, 2019, 14, 100361 CrossRef.
- Y. Yang, Y. Tang, G. Fang, L. Shan, J. Guo, W. Zhang, C. Wang, L. Wang, J. Zhou and S. Liang, Energy Environ. Sci., 2018, 11, 3157–3162 RSC.
- H. Liu, J.-G. Wang, H. Sun, Y. Li, J. Yang, C. Wei and F. Kang, J. Colloid Interface Sci., 2020, 560, 659–666 CrossRef CAS PubMed.
- B. Tang, J. Zhou, G. Fang, F. Liu, C. Zhu, C. Wang, A. Pan and S. Liang, J. Mater. Chem. A, 2019, 7, 940–945 RSC.
- F. Strauss, D. Stepien, J. Maibach, L. Pfaffmann, S. Indris, P. Hartmann and T. Brezesinski, RSC Adv., 2020, 10, 1114–1119 RSC.
- X. Qi, B. Blizanac, A. DuPasquier, M. Oljaca, J. Li and M. Winter, Carbon, 2013, 64, 334–340 CrossRef CAS.
- V. Aravindan, K. Karthikeyan, K. S. Kang, W. S. Yoon, W. S. Kim and Y. S. Lee, J. Mater. Chem., 2011, 21, 2470–2475 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta04030e |
| This journal is © The Royal Society of Chemistry 2023 |