
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInserting a lithiation potential gap as a factor for degradation control in aluminum-foil anodes by utilizing roll-bonding processes†
Hongyi
Li
 *a,
Shohei
Nishimura
a,
Yuki
Nakata
b,
Shingo
Matsumoto
b,
Takitaro
Yamaguchi
b,
Hiroaki
Hoshikawa
c,
Toshiaki
Kumagai
c and
Tetsu
Ichitsubo
*a
*a,
Shohei
Nishimura
a,
Yuki
Nakata
b,
Shingo
Matsumoto
b,
Takitaro
Yamaguchi
b,
Hiroaki
Hoshikawa
c,
Toshiaki
Kumagai
c and
Tetsu
Ichitsubo
*a
aInstitute for Materials Research, Tohoku University, Sendai 980-8577, Japan. E-mail: li.hongyi@tohoku.ac.jp; tichi@tohoku.ac.jp
bAdvanced Materials Development Laboratory, Sumitomo Chemical Co., Ltd., Tsukuba 300-3294, Japan
cEnergy and Functional Materials Research Laboratory, Sumitomo Chemical Co., Ltd., Ehime 792-8521, Japan
First published on 29th September 2023
Abstract
During cycles, Al-foil anodes for lithium batteries are degraded by the progressive structure fracture of the Al matrix and the surface passivation by the electrolyte decomposition products. In this work, focusing on the structural design to regulate the degradation of the Al-foil anodes, we compare the lithiation potential of four types of Al foils, including high purity 99.99%Al, Al-1%Si, Mn-based A3003 and Mg-based A5052 foils, and prepare clad Al foils by roll-bonding processes to utilize the potential difference between the foils as a control factor. The alloy additions (Si, Mn, and Mg) in the Al foils produce distinct strain effects during lithiation, resulting in variation in the reaction potential. Incorporating different Al foils into a single clad Al foil creates a lithiation potential gap within the anodes, providing an intelligent method for limiting the reaction depth of the Al-foil anodes.
1. Introduction
Al-foil anodes have been attracting growing attention as promising anode materials for advanced lithium batteries.1–5 Through appropriate adjustment of the composition and hardness,2,6 or prelithiation process,1,4 the Al foils can work as self-contained alloy anodes, combining the functions of both an active material and a current collector. In electrode reactions, the surface layer of the Al-foil anodes undergoes repetitive (de)lithiation as the active material. The main electrode reaction is Li+ + e− + Al = LiAl (∼0.38 V vs. Li)7 with a theoretical capacity of 993 mA h g−1, which is about three times higher than the theoretical capacity of graphite (LiC6). Further lithiation to Li1.5Al and Li2.25Al is usually difficult under the electrochemical conditions due to the low reaction potentials close to Li deposition.8 The remaining unreacted base layer of the Al-foil anodes plays the role of a current collector saving the Cu foil that is usually used in the graphite anodes and the Li-metal anodes. The base layer maintains the structural stability and the electronic conductivity of the Al-foil anodes during cycling. Based on such a two-layer structure, the practical capacity of the Al-foil anodes is determined by the ratio of the surface layer and the base layer. Compared to the commercial graphite anodes, the Al-foil anodes do not need non-active additions, such as polymer binder, conductive carbon black and the as-mentioned Cu current collector. Therefore, the capacity of the Al-foil anodes can be higher than the graphite anodes, and even comparable to the underdevelopment Li-metal anodes in terms of the overall electrode, if the base layer of the Al-foil anodes is thin enough.2 It should be noted that the base layer cannot be absent in the Al-foil anodes. If the Al foil is entirely alloyed with Li, the foil would easily crack into pieces due to the fragility of the LiAl phase (see Fig. S1†).In battery cycling, the degradation of the Al-foil anodes is mainly attributed to the gradual progressions of the structure fracture and the surface passivation by the electrolyte decomposition products. During delithiation, the LiAl phase is transformed to the Al phase accompanied with a −50% volume contraction. Because the Li diffusion is usually inhomogeneous among the grain boundary and the inside of the grains, the Al grains are easily cracked into fine particles during delithiation.2,9 As a result, the surface layer is self-organized to porous structure instead of returning to the bulk structure as illustrated in Fig. 1a. Due to the high reducing activity of the Al-foil anodes, the electrolyte species are spontaneously decomposed on the fresh Al surface and form the so-called solid-electrolyte interphase (SEI). Although the SEI is necessary to prevent the electrolyte from further decomposition, the Al particles covered by the SEI are easily passivated and lose electric connection. In the subsequent lithiation, partial of the fresh Al matrix in the base layer would be consumed to compensate for the Al particles that are passivated on the surface. As shown in Fig. 1b, following this mechanism, the base layer becomes thinner, and the surface layer thickens during cycling.9 Eventually, the cycle life of the Al-foil anodes would be determined by the timing when electrolytes is almost decomposed to form SEI, or when the Al foil is entirely decomposed to fine particles and lose the electric connection to the external circuit.9
 | ||
| Fig. 1 Structure changes of Al-foil anodes and roll-bonding process to insert a lithiation potential gap in clad Al-foil anodes. (a) Structure changes in the first lithiation and delithiation. In the lithiation, Al matrix with appropriate hardness allows a one-dimensional volume expansion. In the delithiation, porous structure is formed due to the inhomogeneous diffusion of Li in the Al-foil anodes. The electrolyte species are spontaneously decomposed on the Al surface forming the solid-electrolyte interphase (SEI). (b) Structure change after cycles. The surface Al particles are gradually passivated, and the remained fresh Al matrix is involved in the reaction. (c) Preparation of clad Al foils by a roll-bonding process. Alloy additions, such as Mn and Mg, help to form a lithiation potential gap between the surface layer and the base layer, providing a factor to control the Li penetration. | ||
Therefore, to extend the cycle life of the Al-foil anodes, we must minimize the increase of the surface area and the corresponding decomposition of the electrolyte species. Recent studies reported that alloy additions, such as 1%Si,6,10,11 are effective to limit the structure fracture during delithiation and improve the cycle capability. However, at the same time, limiting the increase of the surface area also retards the delithiation kinetics and exacerbates the capacity loss in early cycles caused by a diffusional Li trapping mechanism.12 Therefore, it is important to design the Al alloy to promote Li diffusion in the matrix phase while preventing the structure fracture. For the electrolyte, stable and functional SEI formation would be required to maintain the reaction activity of the surface Al particles, which is a problem shared with other metallic active materials such as Si alloy anodes and Li metal anodes.13,14 In addition, due to the fact that the surface layer and the base layer are composed of the same material, it is difficult to distinguish or discern the surface and base layers in terms of the cutoff potential. In order to enhance the cyclability, we have to manage to keep the base layer unreacted. Thus, incorporating a different physical property between the surface and base layers as a control factor within the rolled foil would contribute to the improvement of the electrode durability.
Parallel to the alloy and electrolyte designs, in this work, we concentrated on a structural design viewpoint to regulate the reaction depth of the Al-foil anodes during cycles. For Al foil with a specific composition, it is challenging to control the location of the Al matrix reacting with the Li from the outside of the cell due to the basically uniform distribution of the lithiation potential in the Al matrix. In general, the strain effects from the alloy additions and manufacturing methods can have a considerable impact on the lithiation potential of the Al matrix.15 Therefore, managing the lithiation potential distribution in the Al-foil anodes would be a potential method for limiting the reaction depth of the anodes during cycles. To demonstrate this idea, here we evaluate the lithiation potential of four types of Al foils, i.e., high purity 99.99%Al, Al-1%Si, Mn-based A3003 and Mg-based A5052, and prepare the clad Al foils, as illustrated in Fig. 1c, by roll bonding processes to create a lithiation potential gap in the Al-foil anodes. The clad Al-foil anodes are succeeded in forming a potential gap >0.1 V during lithiation at a constant current of 1.0 mA cm−2. The cycle tests confirm that the consumption of the Al-foil anodes can be limited by setting appropriate cutoff potential with respect to the lithiation potential gap. This cladding method will be useful for the further development of the Al-foil anodes in combination with the advanced alloy and electrolyte designs.
2. Experimental methods
2.1 Materials preparation
Al foils of various alloy additions were prepared by Sumitomo Chemical Co., Ltd. The Al-Si alloy foil was prepared by adding Si chips (1 wt%) into molten Al (purity 99.99%) and then casting ingots (abbreviated as Al-1%Si). The ingots were subsequently rolled to the specified thickness. The Mn-based A3003 and Mg-based A5052 foils were prepared in compliance with their respective compositional standards. Cold-roll bonding was conducted using a rolling machine made by Yoshida Kinen Iron Works Co., Ltd. The bonding side of the Al foils was pre-polished using 100-grit sandpaper.2.2 Electrochemical measurements
Three-electrode batch cells (SB1A, EC-FRONTIER Co., Ltd.) were employed to investigate the lithiation behavior. The structure of the three-electrode batch cells is shown in Fig. S2.† Specifically, galvanostatic intermittent titration technique (GITT) measurements were conducted to evaluate the equilibrium redox potential and the overpotential of the Al foils with different alloy additions. Besides, constant current lithiation experiments were carried out to measure the lithiation potential changes in the clad foils (working electrodes, WE). For a half-cell configuration, Li foil was used as the counter electrode (CE), and the reference electrode (RE). Without a separator, the WE and CE were separated by a distance of 5 mm in the batch cells. 1 M LiPF6/EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMC (50:50 vol%) solution was used as electrolyte, purchased from Sigma-Aldrich Co., LLC. For the cycle tests, three-electrode coin cells (SB7, EC-FRONTIER Co.,) were used with a 25 mm-thick polyethylene (PE) separator to separate the electrodes. The batch and coin cells were assembled in an argon-filled glovebox. The electrochemical measurements were controlled using a potentiostat (VMP-3 or VSP-300, Bio-Logic SAS).
:DMC (50:50 vol%) solution was used as electrolyte, purchased from Sigma-Aldrich Co., LLC. For the cycle tests, three-electrode coin cells (SB7, EC-FRONTIER Co.,) were used with a 25 mm-thick polyethylene (PE) separator to separate the electrodes. The batch and coin cells were assembled in an argon-filled glovebox. The electrochemical measurements were controlled using a potentiostat (VMP-3 or VSP-300, Bio-Logic SAS).
2.3 Materials characterization
Crystal structure of the various Al foils was evaluated by X-ray powder diffraction (XRD) using a diffractometer (SmartLab, Rigaku Co.) with Cu Kα radiation. Chemical composition of the Al foils was measured by inductively coupled plasma optical emission spectroscopy (ICP-OES). The hardness of each Al foil was measured with a micro HV tester (HMV-G21DT, Shimadzu Corporation). A test force of HV0.05 (490.3 mN) was employed and held for 15 s. More than six points were measured for each sample with appropriate intervals. Morphology of the Al foil anodes were observed with a field emission scanning electron microscope (FE-SEM, JSM-7200F, JEOL Ltd.). For the clad foils, distribution of the alloy additions on the cross section was measured by an energy dispersive X-ray spectrometer attached to the FE-SEM. For the Al-foil anodes after electrochemical tests, the anodes were washed by dropping tetrahydrofuran (99.5%, FUJIFILM Wako Pure Chemical Corporation) and subsequently dried in argon atmosphere to remove the electrolyte.3. Results and discussion
3.1 Characteristics of Al foils with different alloy additions
Chemical composition, thickness, and Vickers hardness (HV) of the four types of Al foils are shown in Table 1. The XRD patterns and the EDX mappings of the main alloy additions are given in Fig. 2. The high purity 99.99%Al foil contains a very slight amount of Si that was contaminated in the manufacture. For the Al-1%Si foil, the Si (111) diffraction peak is clearly detected in the XRD profile (red curve in Fig. 2a) and the Al (111) peak is almost at the same degree compared to the 99.99%Al (enlarged view in Fig. 2b). Besides, the EDX mapping in Fig. 2c shows that finely dispersed Si particles are observed in the Al-1%Si foil, which increases the hardness compared to the 99.99%Al foil (Table 1). For the Mn-based A3003 and Mg-based A5052 foils, the Al (111) peaks distinctively shift to the low 2theta side, indicating that the alloy additions dissolved in the Al phase increase the interlayer distance, which would be connected to the high hardness as indicated in Table 1.| Alloy additions/mass% | Thickness/μm | Vickers hardness/HV | ||||||
|---|---|---|---|---|---|---|---|---|
| Mg | Si | Mn | Fe | Cu | Zn | |||
| 99.99%Al | <0.002 | 0.02 | <0.005 | <0.005 | <0.002 | <0.002 | 300 | 41 |
| Al-1%Si | <0.002 | 0.942 | <0.005 | <0.005 | <0.002 | <0.002 | 300 | 65 |
| A3003 | 0.005 | 0.40 | 1.02 | 0.54 | 0.12 | 0.02 | 150 | 72 |
| A5052 | 2.43 | 0.13 | 0.08 | 0.19 | 0.06 | 0.08 | 150 | 135 |
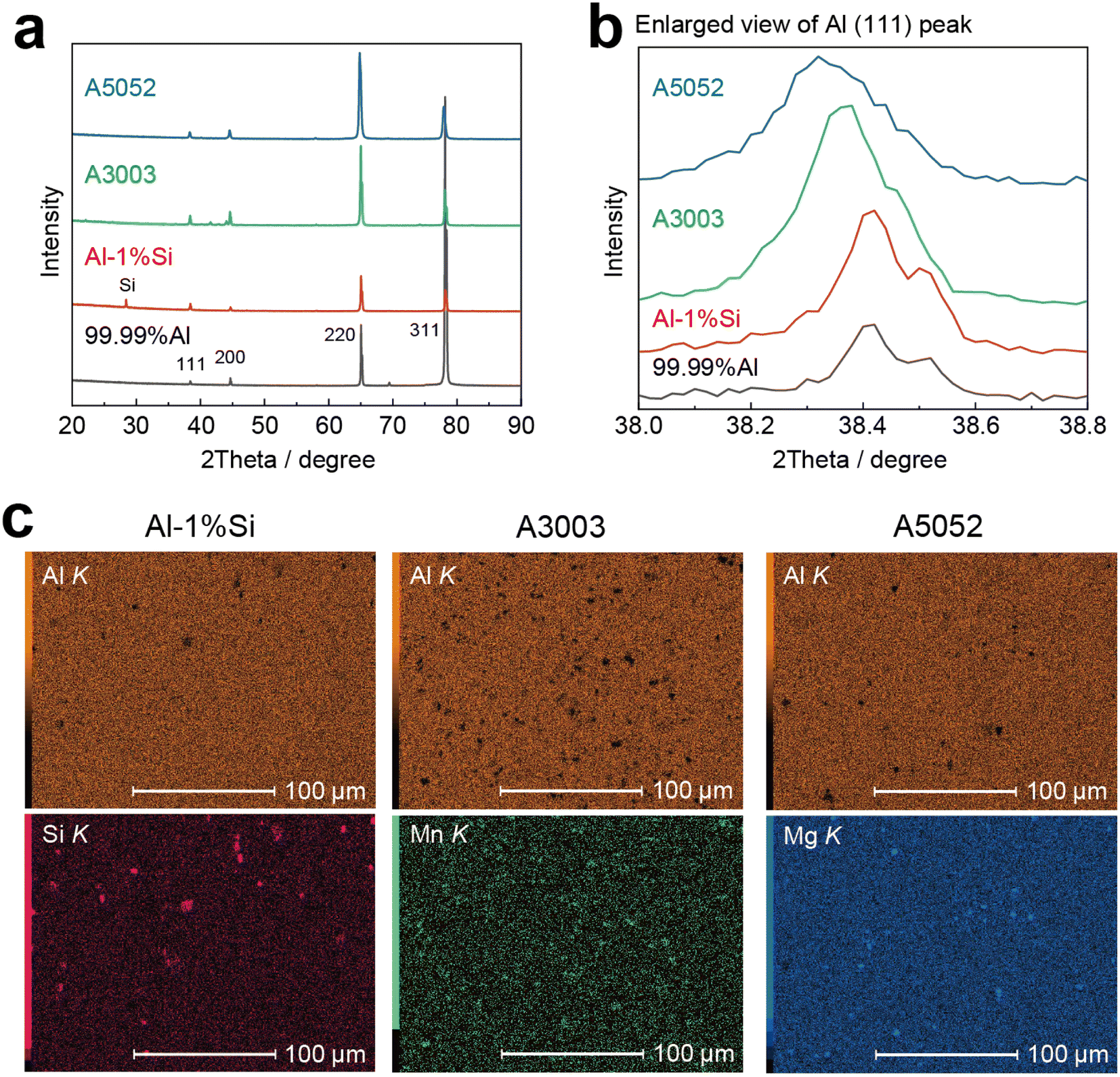 | ||
| Fig. 2 Characteristics of four types of Al foils. (a) X-ray diffraction (XRD) profiles of Al foils with different alloy additions. (b) Enlarged view of the range of Al (111) peak. In A3003 and A5052 foils, the interlayer distance of the Al matrix is expanded by the alloy additions. (c) EDX mapping on the surface of the Al-1%Si, A3003 and A5052 foils. The main alloy additions, Si, Mn and Mg are shown in addition to Al matrix, respectively. | ||
3.2 Lithiation potential of the various Al foils
We conducted GITT measurements with a half-cell configuration to evaluate the lithiation potential of the four types of Al foils. In general, the equilibrium potential of the electrode reaction can be estimated based on the open circuit potential (OCP) in the GITT measurements, and the reaction kinetics can be estimated from the overpotential during reaction progressing. For the lithiation of the Al foils, the electromotive force (emf) can be given as follows.
μAlLiAl, μAlAl are the chemical potentials of the LiAl phase and Al phase in the Al-foil working electrode, and μAlLi, μLiLi are the chemical potentials of Li in the Al foil electrode and the Li metal (counter electrode), respectively. F is the Faraday constant. Considering the effect of strain energy, the equation can be written as the following form.15

emfs is the electromotive force accounting the strain energy, ΔUs = UsLiAl − UsAl, which is determined by the difference of the strain energy in the LiAl phase (UsLiAl) and in the
Al phase (UsAl). If ΔUs > 0, the strain energy is mainly stored in the LiAl phase and decrease the emfs. Detailed strain effects on the lithiation behavior of foil anodes have been described in our previous studies.2,15
As shown in Fig. 3a and b, 99.99%Al foil and the Al-1%Si foil show very close OCPs at 0.35 V vs. Li in the GITT measurements. This indicates that the Si addition does not bring obvious strain energy during lithiation. On the other hand, the overpotential is slightly increased (see the enlarged view in Fig. S3†), suggesting that the Si additions mainly affect the reaction kinetics, retarding the growth of LiAl phase from the Al matrix and consequently increasing the overpotential. In contrast, the A3003 and A5052 foils show remarkable low OCPs compared to the 99.99%Al foil as shown in Fig. 3c and d, respectively. As mentioned above, the decrease of the OCPs suggests that a larger strain energy is stored in the LiAl phase during lithiation which would originate from the strong constrain from the Al matrix. Besides, the overpotentials of the A3003 and A5052 foils are gradually increased as the lithiation progressing. In particular, the electrode potential of the A5052 foil even fall below 0 V vs. Li, where Li metal deposition is detected on the Al surface (inset in Fig. 3d). The strong elastic stress would increase as the LiAl phase grows into the foil, resulting in the increased overpotential observed in the GITT measurements. To incorporate the lithiation potential difference into a single Al foil, we prepared clad Al foils by cold-roll bonding.
 | ||
| Fig. 3 Galvanostatic intermittent titration technique (GITT) profiles of (a) 99.99%Al, (b) Al-1%Si, (c) A3003 and (d) A5052 foils. The duration of constant current lithiation (1 mA cm−2) and open circuit rest were 30 minutes. A photo of the A5052 electrode after GITT is shown in the inset in (d), where the dendritic Li deposition is observed on the surface. Detailed potential profiles in the fourth lithiation and relaxation step are given in Fig. S3† for a clear comparison of the overpotentials. | ||
3.3 Internal lithiation potential gaps in clad Al-foil anodes
Combining the Al foils of different lithiation potentials with cold-roll bonding processes, we prepared four types of clad Al-foil anodes. For convenience, Al/A3003 foil indicates a clad Al foil consisting of 99.99%Al as the surface layer (intended to be alloyed with Li in electrode reaction) and A3003 as the base layer (intended to be unreacted as a current collector). Similarly, Al/A5052, Al-1%Si/A3003, Al-1%Si/A5052 foils were also prepared to investigate the lithiation behavior. The SEM images and EDX mappings of the cross sections of the clad Al foils are shown in Fig. 4a–d. The surface layer and the base layer are smoothly bonded without obvious voids. The thickness of each layer can be approximately determined based on the EDX mapping, and the location of the detected interface is indicated by triangular symbols in each figure. | ||
| Fig. 4 Clad Al foils prepared by cold-roll bonding. (a–d) SEM images and EDX mapping of the main alloy addition on the cross section of (a) Al/A3003, (b) Al/5052, (c) Al-1%Si/A3003, (d) Al-1%Si/A5052 clad foils. The triangle symbols indicate the interfaces between the layers based on the EDX mapping. (e–f) The potential profiles of (e) Al/A3003, (f) Al/5052, (g) Al-1%Si/A3003, (h) Al-1%Si/A5052 clad foils in the constant current 1 mA cm−2 lithiation experiments. The lithiation was preceded from the surface to the base layers. The theoretical capacity of the surface layer in each clad foil was calculated based on the thickness and was indicated with a dashed line and a triangle symbol. Obvious drops in lithiation potential were obtained in all cases of the clad Al foils, where Al/A5052 shows the largest lithiation potential gap. | ||
To exam the lithiation potential difference, constant current experiments (1 mA cm−2) were conducted on the clad Al foils using three-electrode batch cells. The lithiation was proceeded from the surface layer to the base layer to detect the lithiation potential. As shown in Fig. 4e–h lithiation potential gaps are clearly observed in all clad Al foils. Table 2 summarizes the thickness, Vickers hardness and the lithiation potential of the surface and base layers in the clad Al-foil anodes. For the surface layer, 99.99%Al shows a lithiation potential of about 0.29 V vs. Li, and Al-Si1% shows a lithiation potential of about 0.26 V under the constant current condition. As shown in the previous section, the Si additions slightly decrease the reaction kinetics and increase the overpotential during lithiation. On the other hand, for the base layer, A3003 shows lithiation potentials of about 0.25 V vs. Li in Al/A3003 and 0.23–0.20 V in Al-1%Si/A3003. Besides, A5052 shows lithiation potentials of about 0.13 V in Al/A5052 and 0.15–0.11 V in Al-1%Si/A5052, where obvious gaps are obtained in the potential profiles (Fig. 4f and h).
| Surface/base | Surface layer | Base layer | ||||
|---|---|---|---|---|---|---|
| Thickness (μm) | Hardness (HV) | Lithiation potential (V vs. Li) | Thickness (μm) | Hardness (HV) | Lithiation potential (V vs. Li) | |
| a The clad foils correspond to the results shown in Fig. 4. The thickness of each layer is approximately determined based on the EDX mapping. | ||||||
| Al/A3003 | 36 | 40 | 0.29 | 59 | 78 | 0.25 |
| Al/A5052 | 20 | 42 | 0.29 | 29 | 154 | 0.13 |
| Al-1%Si/A3003 | 28 | 75 | 0.26 | 45 | 76 | 0.23–0.20 |
| Al-1%Si/A5052 | 25 | 81 | 0.26 | 45 | 144 | 0.15–0.11 |
3.4 Control factor for limiting the consumption of Al-foil anodes
To examine the effects of the inserted lithiation potential gap, we conducted constant current (1 mA cm−2) cycle tests with Al-1%Si/A5052 foil anodes as an example. Fig. 5a shows the potential profile in the experiment with a maximum lithiation capacity of 2 mA h cm−2 and cutoff potentials of −1 V vs. Li for lithiation and 1 V for delithiation. For comparison, Fig. 5b shows the potential profile with a raised cutoff potential of 0.15 V vs. Li in lithiation, slightly above the lithiation potential of the A5052 base layer as shown in Fig. 4h. The corresponding cyclic capacity is plotted in Fig. 5c. The Al-1%Si/A5052 foil used in the cycle tests consists of a 14 μm-thick Al-1%Si surface layer and a 35 μm-thick A5052 base layer as shown in Fig. 5d. Since the areal capacity of 2 mA h cm−2 corresponds to the theoretical capacity of an 8 μm-thick Al matrix, the lithiation reaction can occur within the Al-1%Si layer. For the cycle tests between cut off potential of −1 V to 1 V (Fig. 5a), the lithiation potential in the initial cycles is around 0.2–0.3 V vs. Li, which confirms that the Al-1%Si surface layer works as the active material. The capacity loss of about 40% in the first cycle would be mainly attributed to the Li content stuck in the Al matrix.12 Between the cycle 16 and 26, the lithiation potential is gradually decreased to around 0.15 V, indicating that the lithiation reaction shift to the A5052 layer. In contrast, for the cycle tests between cutoff potential of 0.15 V to 1 V (Fig. 5b), the lithiation capacity is decreased around cycle 20, indicating that the lithiation reaction is confined to the Al-1%Si layer and that the A5052 layer is not consumed. Due to the thin original surface layer compared to the fully lithiated case, it appears that increasing the cutoff potential decreases the cycle numbers. Since the cycle numbers for the clad Al-foil anodes are dependent on the thickness of the Al-1%Si layer, thickening the surface layer would increase the cycle numbers proportionally. Fig. 5e and f show the corresponding photos and SEM images of the clad anode after cycle tests, respectively. The Al-1%Si surface layer becomes very thick (about 171 μm) because of the involvement of electrolyte decomposition products, which collaborates to the results in previous studies.9,16 In contrast, the A5052 base layer exhibits no discernible changes after 40 cycles. As shown in Fig. 5g–j, for a uniform 50 μm-thick Al-1%Si foil, the Li alloying progresses along the out-of-plane direction and the unreacted Al matrix (base layer) rapidly decreases to about 23 μm after 15 cycles. The significant changes in the clad foil thus demonstrate that the lithiation potential gap can be used as a control factor to regulate the lithiation depth during cycles. | ||
| Fig. 5 Cycle capability of Al-1%Si/A5052 clad foil electrodes. (a and b) Potential profiles in the cycle tests with cutoff potentials of (a) −1 V vs. Li in lithiation and 1 V in delithiation and (b) 0.15 V in lithiation and 1 V in delithiation. The maximum lithiation capacity was set to 2 mA h cm−2, corresponding to the theoretical capacity of 8 μm-thick surface layer. (c) Lithiation and delithiation capacity in the cycle tests with Al-1%Si/A5052 clad foils. (d) SEM image and EDX mapping on the cross section of the clad Al-1%Si/A5052 foil used in the cycle tests. The interface between the layers is indicated by triangle symbols. (e) Photos of the clad foil electrode after the cycle test in (b). The surface layer and the base layer are detached after cutting. (f) SEM images of the cross section of the Al-1%Si surface layer and the A5052 base layer after cycle tests. Consumption of the A5052 layer is limited by controlling cutoff potential. (g) Potential profiles in a cycle test using a uniform 50 μm-thick Al-1%Si foil for 15 cycles. (h) Lithiation and delithiation capacity in the cycle tests with the uniform Al-1%Si foil. (i and j) SEM image on the cross section of (i) the as-rolled Al-1%Si foil and (j) after 15 cycles. The base layer is significantly consumed compared to the Al-1%Si/A5052 clad foil that contains a lithiation potential gap. | ||
4. Conclusions
In this work, we conducted roll-bonding processes to prepare clad Al-foil anodes with different alloy additions. The alloy additions, such as Si, Mn, and Mg, modified the mechanical properties and the corresponding strain effects significantly altered the lithiation potential of the Al foils. For Al-1%Si foil, Si additions were dispersed among the Al matrix, which did not obviously affect the equilibrium lithiation potential of the Al matrix but retarded the diffusion reaction kinetics. In contrast, for the Mn-based A3003 foil and Mg-based A5052 foil, the LiAl phase was constrained by the hard matrix, resulting to the strong strain effects remarkably decreasing the lithiation potential. Using the high purity 99.99%Al or Al-1%Si as the surface layer and the A3003 or A5052 as the base layer, we created a lithiation potential gap in the clad Al-foil anodes. By controlling the cutoff potential, the lithiation potential gap was demonstrated to prevent Li penetration into the base layer.This work provides a method for intelligently controlling the reaction depth of Al-foil anodes, which will support the advanced alloy and electrolyte designs for radically extending the cycle life of the Al-foil anodes. Future fundamental improvement will be conducted to preserve the activity of the surface layer. Adjusting the microstructure of Al matrix through alloy design can minimize the surface area variations. In addition, the use of suitable electrolyte compositions will effectively suppress the formation of electrolyte decompositions passivating the Al surface.
Author contributions
T. I. and H. L. conceived the study. H. L. and S. N. designed the research plan. Y. N., S. M., T. Y., H. H., and T. K. prepared the Al foils with various alloy additions and provided important knowledge on the experiments. S. N. conducted the roll bonding, electrochemical experiments, structural and composition analyses. H. L. wrote the manuscript. All authors discussed the results and contributed to the study.Conflicts of interest
S. M., T. Y., T. I., H. L. and S. N. are inventors on patent application PCT/JP2022/044565. Y. N., S. M., T. Y., H. H., and T. K. are employees of Sumitomo Chemical Co., Ltd.Note added after first publication
This article replaces the version published on 29 September 2023, which contained errors in Fig. 3 and 4.Acknowledgements
The authors thank the Analytical Research Core for Advanced Materials, Institute for Materials Research, Tohoku University, for the ICP analyses. This work is a joint research project contracted between Tohoku University and Sumitomo Chemical Co., Ltd. This work is partly supported by Grant-in-Aid for Scientific Research (S) number 23H05452 commissioned by Japan Society for the Promotion of Science (JSPS).References
- H. Xu, S. Li, C. Zhang, X. L. Chen, W. J. Liu, Y. H. Zheng, Y. Xie, Y. H. Huang and J. Li, Energy Environ. Sci., 2019, 12, 2991–3000 RSC.
- H. Li, T. Yamaguchi, S. Matsumoto, H. Hoshikawa, T. Kumagai, N. L. Okamoto and T. Ichitsubo, Nat. Commun., 2020, 11, 1584 CrossRef PubMed.
- B. T. Heligman and A. Manthiram, ACS Energy Lett., 2021, 6, 2666–2672 CrossRef CAS.
- D. Li, F. Chu, Z. He, Y. Cheng and F. Wu, Mater. Today, 2022, 58, 80–90 CrossRef CAS.
- M. Peng, K. Shin, L. Jiang, Y. Jin, K. Zeng, X. Zhou and Y. Tang, Angew. Chem., Int., 2022, 61, e202206770 CrossRef CAS PubMed.
- S. S. Sharma, P. J. Crowley and A. Manthiram, ACS Sustainable. Chem. Eng., 2021, 9, 14515–14524 CrossRef CAS.
- T. R. Jow and C. C. Liang, J. Electrochem. Soc., 1982, 129, 1429 CrossRef CAS.
- M. N. Obrovac and V. L. Chevrier, Chem. Rev., 2014, 114, 11444–11502 CrossRef CAS PubMed.
- C. Wang, T. Chen, Y. Liu, D. H. Kang, D. Majumdar, R. Gopalaswamy and M. T. McDowell, ACS Energy Lett., 2023, 8, 2252–2258 CrossRef CAS.
- Sumitomo Chemical Co., Ltd., PCT/JP2019/040910, 2019.
- Sumitomo Chemical Co., Ltd. and Tohoku University, PCT/JP2020/047004, 2020.
- P. J. Crowley, K. P. Scanlan and A. Manthiram, J. Power Sources, 2022, 546 CrossRef CAS.
- H. Liu, X. B. Cheng, J. Q. Huang, S. Kaskel, S. L. Chou, H. S. Park and Q. Zhang, ACS Mater. Lett., 2019, 1, 217–229 CrossRef CAS.
- Z. Z. Tong, B. Bazri, S. F. Hu and R. S. Liu, J. Mater. Chem. A, 2021, 9, 7396–7406 RSC.
- T. Ichitsubo, S. Yagi, T. Doi, S. Yukitani, K. Hirai and E. Matsubara, J. Electrochem. Soc., 2011, 159, A14–A17 CrossRef.
- T. Chen, A. C. Thenuwara, W. Yao, S. E. Sandoval, C. Wang, D. H. Kang, D. Majumdar, R. Gopalaswamy and M. T. McDowell, Batteries Supercaps, 2023, 6, e202200363 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta03687a |
| This journal is © The Royal Society of Chemistry 2023 |