
New superionic halide solid electrolytes enabled by aliovalent substitution in Li3−xY1−xHfxCl6 for all-solid-state lithium metal based batteries†
Kaiyong
Tuo
 a,
Chunwen
Sun
*a,
C. A.
López
bc,
Maria Teresa
Fernández-Díaz
d and
José Antonio
Alonso
b
a,
Chunwen
Sun
*a,
C. A.
López
bc,
Maria Teresa
Fernández-Díaz
d and
José Antonio
Alonso
b
aSchool of Chemical & Environmental Engineering, China University of Mining and Technology-Beijing, Beijing 100083, P. R. China. E-mail: csun@cumtb.edu.cn
bInstituto de Ciencia de los Materiales de Madrid, CSIC, E-28049, Cantoblanco-Madrid, Spain
cINTEQUI (UNSL, CONICET), Facultad de Química, Bioquímica y Farmacia, UNSL, Chacabuco y Pedernera, San Luis, 5700, Argentina
dInstitut Laue Langevin, BP 156X Grenoble, F-38042, France
First published on 4th July 2023
Abstract
Rechargeable all-solid-state batteries (ASSBs) are considered as promising candidates for next-generation energy storage due to their high energy density and excellent safety performance. However, the low ionic conductivity of the solid-state electrolytes (SSEs) and interfacial issues are still challenging. Herein, we report a series of new mixed-metal halide superionic conductors Li3−xY1−xHfxCl6 (0 ≤ x < 1) with high ionic conductivity up to 1.49 mS cm−1 at room temperature. Using various experimental characterization techniques and bond-valence energy landscape (BVEL) calculations, we gain insights into the aliovalent substitution of Hf for Y in halide Li3YCl6 that influences the local structural environment and the underlying lithium-ion transport. Importantly, it is found that the existence of prevalent cation site disorder and defect structure as well as the synthetically optimized (Y/Hf)Cl6 framework with a more covalent feature in Hf4+-substituted Li3YCl6 strongly benefits the transport properties. In particular, the formation of an infinitely 3D connected Li+ ion diffusion pathway consisting of face-sharing octahedra within the lattice of Hf4+-substituted Li3YCl6 is revealed by structural elucidation and theoretical calculations. Additionally, owing to the exceptional interfacial stability of the as-milled SSEs against high-voltage cathode materials, all-solid-state lithium-ion batteries with a LiCoO2 cathode and Li–In anode exhibit outstanding electrochemical performance.
1. Introduction
The progressively declining fossil fuel reserves and increasing environmental awareness have accelerated the strong upsurge of interest in high-efficiency storage and conversion of green and renewable energy resources.1–3 The rechargeable lithium-ion batteries (LIBs), as one of the most crucially genuine advancements in the field of electrochemical energy storage, undoubtedly have revolutionized the portable electronic industry in the past few decades, which are simultaneously considered as the pivotal technology to satisfy the rigorous demands of large-scale energy storage and upscale consumer electronic devices.4–6 Unfortunately, conventional lithium-ion batteries which are highly reliant on organic liquid electrolytes with inherently limiting properties regarding flammability and incompatibility have faced tremendous challenges due to the burgeoning demand for high energy density and distinguished safety energy-storage systems.7–10 All-solid-state lithium batteries (ASSLBs) exploiting solid-state electrolytes (SSEs) and an ideal lithium metal anode could fundamentally eliminate the potential safety risks caused by liquid electrolytes as well as offering high energy density, which are widely acknowledged to be the prominent innovative direction for next-generation batteries.11–13 Importantly, identifying the appropriate superionic conductive solid electrolytes with wide electrochemical stability windows is highly desirable to realize the impressive properties of ASSBs.Until now, various classes of inorganic SSEs comprising of sulfides and oxides with acceptable ionic conductivities have been developed, but none of them have enabled the incorporated advantages of outstanding electrochemical stability and superior interface wettability. Sulfide electrolytes commonly featuring exceptional ionic conductivity and excellent ductile nature have been a particular focus of research over the past few years.14,15 However, the intrinsically poor oxidative stability renders the sulfides electrochemically or chemically incompatible with conventional 4 V cathode active materials LiMO2 (M = Ni, Co, Mn, Al) in the absence of an insulating protective coating, and the high sensitivity to decomposition of sulfides in the atmosphere causing the emission of toxic H2S gas requires rigorous dry-room conditions for large-scale processability.16,17 Oxide electrolytes such as garnets take advantage of their nominal chemical stability with lithium metal and outstanding electrochemical stability, but these merits could be readily offset by their apparent brittleness, thus making it difficult to process them into electrolyte/active material composite cathodes, along with the high interfacial and grain boundary impedance.18,19 In consequence, this remains a challenge facing the application of ASSBs which can be overcome by identifying the prominent solid electrolytes that could simultaneously satisfy the integrated requirements of superionic conductivity, outstanding electrochemical stability and the mechanically soft properties.
Recently, halide superionic conductors have emerged as an additional class of promising inorganic solid electrolytes due to the concurrent advantages of favorable mechanical deformability and intrinsically high oxidative stability, enabling the direct application of high-voltage layered CAMs to assemble ASSBs through a simple cold-pressing-based technique.15,20 Several lithium metal halide materials, including semi-glassy Li3InCl6, Li3ScCl6, Li3−xM1−xZrxCl6 (M = Y, Er, Yb), Li3MBr1−xClx (M = Y, Er), and poorly crystalline Li3ErCl6 and Li2ZrCl6, have been reported to exhibit such impressive properties,21–27 since Asano et al. reported that the trigonal Li3YCl6 and monoclinic Li3YBr6 feature high room-temperature (RT) ionic conductivities of 0.51 and 1.7 mS cm−1,28 respectively. This is based on the fact that the ionic radius of monovalent halogen anions is relatively larger than those of divalent sulfide or oxygen anions, e.g., r(Cl−) = 167 pm, r(Br−) = 182 pm, r(I−) = 206 pm, r(S2−) = 170 pm, and r(O2−) = 126 pm, and this feature indicates that there is a weaker interaction between halogen anions and lithium ions, as well as the correspondingly long ionic lengths and high polarization, from which a favorable lithium-ion mobility in their sublattice and the credible deformability towards halide electrolytes are expected.29 Furthermore, it is noteworthy that the halogen anions, especially for chlorine and fluorine, could be more electronegative than sulfides, motivating the high electrochemical oxidation stability for chlorides and fluorides, as demonstrated by density functional theory (DFT) calculations.29–31 Nevertheless, very few halide materials have been reported that could experimentally exhibit an intrinsic ionic conductivity over 1 mS cm−1 until now, driving an essential search for novel halide electrolytes with such promising properties.
Aliovalent substitution of central metals with different ionic radius cations is acknowledged to be a feasible strategy to enhance the ionic conductivity of halide electrolytes through changing the local structural environment and tuning the site occupancy of the metal/vacancy. Nazar and coworkers23 reported that the heterovalent substitution of central metals in Li3MCl6 (M = Y, Er) by Zr4+via solid-state reaction could considerably improve the ionic conductivity up to 1.4 mS cm−1, and confirmed the structural evolution from trigonal to orthorhombic with the gradually increased amount of Zr4+, which is favorable to create more lithium vacancies for effectively promoting the construction of 3D lithium ion transport networks. Jung and coworkers26 indicated that the central metal substitution of Li2ZrCl6 with trivalent Fe3+, V3+ and Cr3+ causes a dramatic increment of the ionic conductivity, and a new halide superionic conductor Li2.25Zr0.75Fe0.25Cl6 with high ionic conductivity approaching 1 mS cm−1 was synthesized, which primarily benefits from the adequately enlarged lithium-ion diffusion channel and the regulated energy landscape for favorable lithium-ion mobility. In addition, mechanochemical milling has been prevailingly employed to synthesize the halide electrolytes with significantly improved ionic conductivity by inducing the generation of an abundant defect structure and partially disordered cation arrangement.32–34 Zeier and coworkers35 pointed out that the employed synthesis method of mechanochemical milling could yield amorphous Li3MCl6 (M = Y, Er) with higher ionic conductivity compared to subsequent crystallization methods as well as typical solid-state syntheses, and thus the prevalent M2–M3 cation site disorder along with the local change of LiCl65− octahedral distortion could be generated in halide electrolytes derived from mechanochemical synthesis which strongly benefits the lithium-ion transport through enlarging bottlenecks for lithium diffusion. In consequence, incorporating multiple effective strategies to optimize the controllable factors, including local structural environment, cation site disorder, and lithium-ion concentration and distribution, is highly feasible for developing novel halide electrolytes with exceptional ionic conductivity.
Inspired by these previous studies, a series of Li3−xY1−xHfxCl6 (0 ≤ x < 1) solid electrolytes are successfully constructed by the aliovalent substitution of Y3+ with tetravalent Hf4+ through the facile mechanochemical milling syntheses, which could exhibit remarkably improved ionic conductivity up to 1.49 mS cm−1 at room temperature. Complementary analyses using X-ray diffraction (XRD), transmission electron microscopy (TEM), neutron powder diffraction (NPD), impedance spectroscopy and bond valence energy landscape (BVEL) calculations are employed to investigate the influence of the aliovalent Hf4+ substitution on the local structural environment and resulting ionic transport properties of halide electrolytes. The appropriate heterovalent metal substitution and mechanochemical milling technique could induce the prevalent site disorder and defect structure as well as an enhanced lithium vacancy concentration which strongly benefits the generation of fast 3D lithium-ion conduction pathways in the structural framework. Furthermore, the exceptional compatibility of Hf4+-substituted Li3YCl6 with LiCoO2 is demonstrated through the ex situ X-ray photoelectron spectroscopy (XPS) results and the corresponding outstanding electrochemical performance.
2. Experimental section
2.1. Materials synthesis
The halide solid electrolytes Li3−xY1−xHfxCl6 (0 ≤ x < 1) were synthesized through a facile mechanochemical milling route. The stoichiometric precursor materials of LiCl (99.9%, Aladdin), YCl3 (99.9%, Aladdin) and HfCl4 (99.9%, Aladdin) were placed into an argon-filled zirconia pot and the powder-to-milling media ratio was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 with the media diameter of 5 and 9 mm. The mixed powder samples were mechanochemically milled at 550 rpm for 50 h with the run procedure of milling lasting 15 min with a subsequent 5 min break for cooling. Such mechanochemical milling directly yields the as-milled halide electrolytes in the main text, while annealing the pellets from these powder samples in a vacuum quartz tube at different temperatures for 5 h results in the highly crystalline halide electrolytes.
:40 with the media diameter of 5 and 9 mm. The mixed powder samples were mechanochemically milled at 550 rpm for 50 h with the run procedure of milling lasting 15 min with a subsequent 5 min break for cooling. Such mechanochemical milling directly yields the as-milled halide electrolytes in the main text, while annealing the pellets from these powder samples in a vacuum quartz tube at different temperatures for 5 h results in the highly crystalline halide electrolytes.
2.2. Electrochemical measurements and materials characterization
The ionic conductivity of the as-synthesized halide electrolytes was measured by cold-pressing the electrolyte powders under ∼3 tons in a home-made poly(ether ether ketone) mold with diameter of 10 mm, and two stainless steel rods as the ionically blocking electrodes. Nyquist plots were recorded at a frequency from 1 MHz to 1 Hz using a Zennium electrochemical workstation to determine the temperature dependence of ionic conductivities from room temperature to 70 °C. The electronic conductivity of the as-prepared halide electrolytes was tested by direct current (DC) polarization employing the same mold cell of SS/halide electrolytes/SS through setting the polarization voltage of 1 V. The electrochemical stability window (ESW) was investigated by cyclic voltammetry (CV) measurement using a CHI604E electrochemical workstation on an asymmetric Li/Li6PS5Cl/halide SE/halide SE + vapor grown carbon fiber (VGCF) cell at the range from 0 to 5 V with a scan rate of 0.5 mV s−1. The weight ratio of VFCF and halide electrolytes to furnish the adequate electronic pathways in the electrode for obtaining the accurate ESW was 30:70.
The X-ray diffraction measurements were performed using a PANalytical Empyrean diffractometer with Cu Kα radiation (λ = 1.5418 Å), and the powder samples were sealed in a sample holder under an Ar atmosphere with Kapton film to avoid air exposure. The morphology and elemental distribution of the as-milled samples were investigated using a field-emission scanning electron microscope (SEM, Hitachi SU8020) and an energy-dispersive spectrometer (EDS). High-resolution TEM (HRTEM) was carried out to investigate the microstructure characteristics of the as-milled halide electrolytes on a JEM2100F at an accelerating voltage of 200 kV. X-ray photoelectron spectroscopy (XPS) data was collected by operating an ESCALAB 250Xi spectrometer (Thermo Fisher Scientific) with Al Kα achromatic X-rays.
2.3. Structural characterization
High-resolution neutron powder diffraction (NPD) patterns were collected for two samples of nominal composition Li3YCl6 (LYC) and Li2.4Y0.4Hf0.6Cl6 (LYHC) using the high-resolution D2B neutron diffractometer of the Institut Laue Langevin (Grenoble-France), with the high-flux mode and a counting time of 4 h each. The samples were contained in vanadium cans and were packed under an Ar atmosphere; the containers were sealed with an indium strip. A wavelength of 1.594 Å was selected from a Ge monochromator; the measurement temperature was 295 K. The patterns were refined by the Rietveld method using the Fullprof refinement program.36,37 A pseudo-Voigt function was chosen to generate the line shape of the diffraction peaks. No regions were excluded in the refinement. In the final run, the following parameters were refined: scale factor, background coefficients, zero-point error, unit-cell parameters, pseudo-Voigt corrected for asymmetry parameters, positional coordinates, and isotropic displacement factors. The coherent scattering lengths for Y, Li, Hf and Cl atoms were 0.7750, −0.1900, 0.7770 and 0.9577 fm, respectively.2.4. Bond-valence energy landscape (BVEL) calculations
The BVEL calculations were performed with the BondStr software included in the Fullprof package.38 The initial structural models of the as-milled halide electrolytes were obtained from the NPD Rietveld refinement results.2.5. Assembly of the batteries
The ASSBs were assembled by employing the as-prepared LYC or LYHC, Li6PS5Cl (LPSC) solid electrolytes in combination with a LiCoO2 (LCO) composite cathode and a Li–In alloy anode in a custom-made cell mold, which comprises two stainless steel rods as the current collectors. Firstly, 40 mg of the LPSC powder were added into a polyether ether ketone (PEEK) cylinder with a diameter of 10 mm and cold pressed under ∼1 ton for 1 min. Then, 70 mg of as-prepared LYHC powder were sequentially filled into the PEEK die and pressed at ∼1 ton to form the solid electrolyte layer. Afterwards, ∼5 mg of the LCO composite cathode powder were dispersed evenly on the halide electrolyte layer and pressed at ∼3 tons for 5 min. The composite cathode for the ASSBs was prepared by mixing the LCO powder and as-synthesized halide electrolytes in the weight ratio of 70:30. Afterwards, thin In foil and Li foil were subsequently attached to the side near the LPSC layer and the cell was subject to a constant uniaxial pressure of ∼1 ton during cycling by employing the screw of a stainless steel framework. For the assembly of the Li/LYHC/Li cell, ∼120 mg of as-milled LYHC powder was cold-pressed into a pellet under ∼3 tons for 5 min. Then, two lithium discs were attached on both sides of the pellet and pressed under ∼1 ton for 10 s. For the assembly of the Li/LPSC/LYHC/LPSC/Li cell, ∼120 mg of as-milled LYHC powder was cold-pressed into a pellet under ∼1 ton for 1 min. Then, ∼30 mg of LPSC powder was evenly dispersed on both sides of the LYHC pellet and pressed under ∼3 tons for 5 min. Afterwards, two lithium discs were attached on both sides of the pellet and pressed under ∼1 ton for 10 s. The fabricated symmetric cells were cycled under a pressure of ∼5 MPa.
3. Results and discussion
To identify the microcrystalline structure of the halide electrolytes Li3−xY1−xHfxCl6 (0 ≤ x < 1) synthesized through a mechanochemical method, XRD measurements were conducted and the corresponding patterns are shown in Fig. 1a. The XRD signals of the as-prepared halide electrolytes well match those of the standard LYC phase with the space group of P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1, and no evident reflections towards the impurity phase were observed in the XRD patterns of the Hf4+-substituted halide electrolyte samples. It is noteworthy that the diffraction peaks of the (031) and (330) crystal planes gradually shift to the high angle as the Hf content increases, without a considerable evolution of new peaks, which implies the substitution of Hf for Y element in the crystal lattice of LYC. Considering the noticeably smaller ionic radius of Hf4+ (71 pm) compared with Y3+ (90 pm),35,39 the substitution of the center metal element in LYC with Hf4+ is capable of shrinking the lattice parameters of the original halide electrolyte, therefore the continuous positive shift in the peaks upon Hf4+-substituted halide samples could be understood.
m1, and no evident reflections towards the impurity phase were observed in the XRD patterns of the Hf4+-substituted halide electrolyte samples. It is noteworthy that the diffraction peaks of the (031) and (330) crystal planes gradually shift to the high angle as the Hf content increases, without a considerable evolution of new peaks, which implies the substitution of Hf for Y element in the crystal lattice of LYC. Considering the noticeably smaller ionic radius of Hf4+ (71 pm) compared with Y3+ (90 pm),35,39 the substitution of the center metal element in LYC with Hf4+ is capable of shrinking the lattice parameters of the original halide electrolyte, therefore the continuous positive shift in the peaks upon Hf4+-substituted halide samples could be understood.
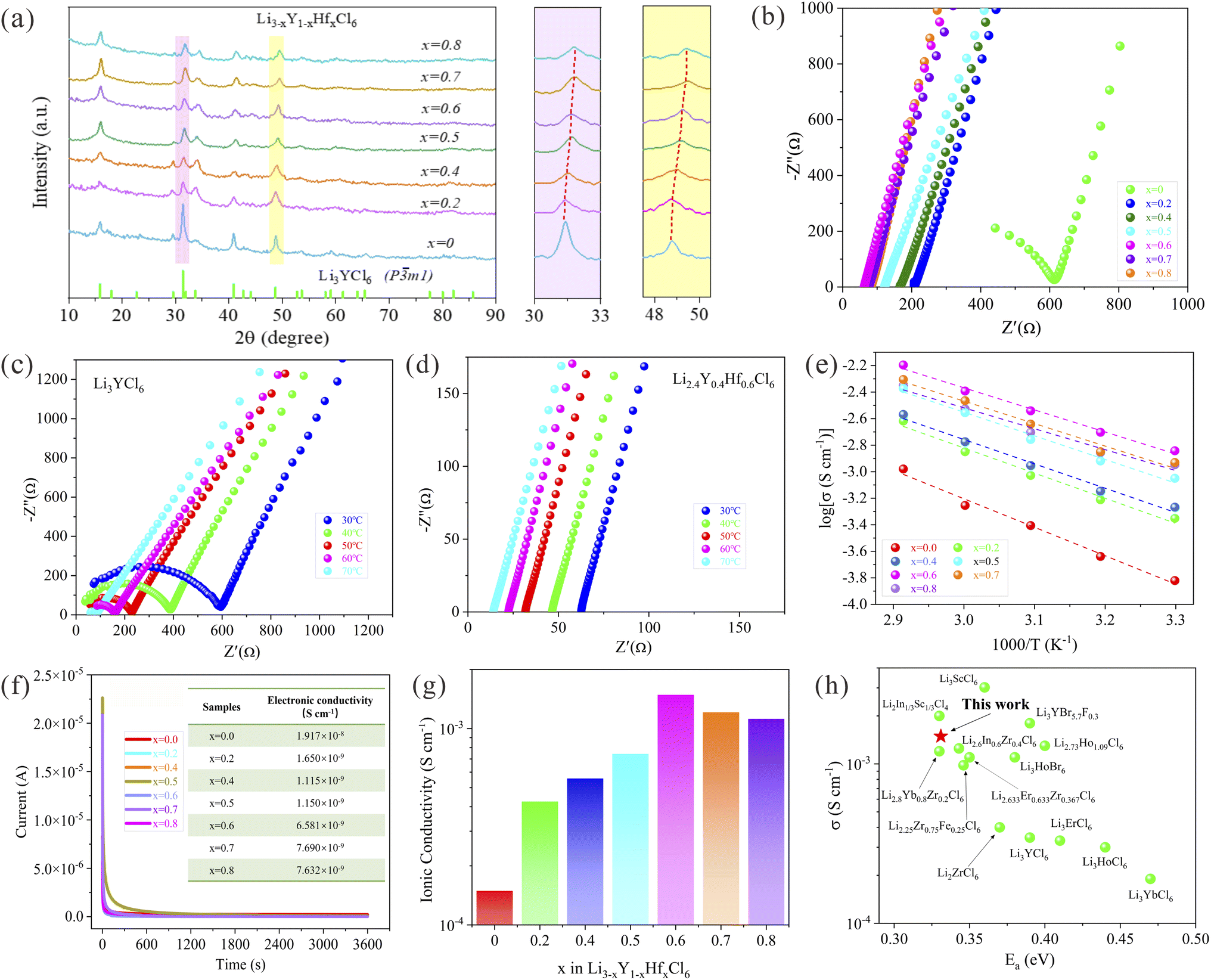 | ||
| Fig. 1 XRD patterns and ionic conductivities of the mechanochemically synthesized Li3−xY1−xHfxCl6 (0 ≤ x < 1) with different processing conditions. (a) XRD patterns of the as-milled Li3−xY1−xHfxCl6 (0 ≤ x < 1) and the corresponding enlarged XRD patterns at 30–33° and 47–49°. (b) Nyquist plots of the as-milled Li3−xY1−xHfxCl6 (0 ≤ x < 1) at room temperature. (c and d) Nyquist plots of LYC (c) and LYHC (d) in the temperature range from 30 to 70 °C. (e and f) Arrhenius plots (e) and DC polarization curves (f) of Li3−xY1−xHfxCl6 (0 ≤ x < 1) using the symmetric cell configuration with an applied voltage of 1 V. (g) Ionic conductivities of Li3−xY1−xHfxCl6 (0 ≤ x < 1) and (h) a comparison of the ionic conductivity and corresponding activation energy of LYHC at room temperature with halide solid electrolytes reported in previous work.22–24,26,28,42–48 | ||
In addition, the surface morphology of halide LYC and the representative Hf-substituted LYHC was characterized by scanning electron microscopy (SEM) as shown in Fig. S1.† It can be found that both of the two samples synthesized through high-energy ball milling present a uniform small particle size less than 10 μm, and a similar micro-morphology is observed for the samples with or without Hf substitution. The energy dispersive spectroscopy (EDS) mapping of the representative LYHC shows that the Y, Hf and Cl elements are uniformly distributed in the halide electrolytes (Fig. S2†), further proving the successful substitution of the Hf elements. The microstructure characteristics of the as-prepared halide electrolytes were investigated through high-resolution TEM (HRTEM).40,41 As displayed in Fig. S3a,† the HRTEM images of LYHC derived from mechanochemical syntheses show plentiful defects upon lattice fringes including disconnections, dislocations and distortions. Certain diffraction rings could be observed in the corresponding FFT diffraction pattern (Fig. S3b†), which demonstrates the small-sized crystal grains as well as the intrinsically low crystallinity for mechanochemically synthesized LYHC.
The Nyquist plots and the Arrhenius curves of nominal composition Li3−xY1−xHfxCl6 over a wide range of x values (0 ≤ x < 1) based on the Li+-blocking symmetric SS/SE/SS cells are displayed in Fig. 1b–e, and the corresponding ionic conductivity as well as the activation energy (Ea) are shown in Fig. 1g and S4.† It can be found that the ionic conductivity of non-substituted LYC is relatively low only achieving 1.39 × 10−4 S cm−1 at RT. As the substituted amount of Hf4+ increased, the volcano-shape trend of ionic conductivity of Li3−xY1−xHfxCl6 is observed which is opposite to the variation tendency of the corresponding activation energy, and the maximum ionic conductivity of 1.49 × 10−3 S cm−1 is reached for LYHC with the lowest activation energy of 0.331 eV. This obtained ionic conductivity of halide electrolyte with aliovalent substitution outperforms most previously reported halide electrolytes derived from mechanochemical milling or other common synthesis routes,22–24,26,28,42–48 as shown in Fig. 1h. Additionally, the subsequent decrease of Li+ conductivity is determined towards the substituted value over 0.6 which implies that the amount of substitution should be accurately controlled to achieve the favorable effect.
The remarkable argument of ionic conductivity upon the superionic conductors Li3−xY1−xHfxCl6 could be interpreted as the positive impact of the heterovalent substitution of Hf4+ for Y3+ in the LYC microcrystalline structure. First of all, the suitable Hf4+ substitution for Y3+, capable of the effective introduction of lithium vacancies into the original LYC crystal structure for the purpose of charge compensation,20 is conducive to facilitating the rapid Li+ migration for enhancing the ionic conductivity. Furthermore, the weaker peak intensity and more diffused peak shape in the XRD pattern of the Hf4+-substituted halide samples were confirmed, in contrast with the strong and sharp reflections of the pristine LYC, which is indicative of the formation of abundant nonperiodic structures in aliovalent substituted samples during the intense ball milling process. The existence of nonperiodic features regarding the cationic site disorder, defect structure and amorphous phase has been demonstrated to render the lithium-ion diffusion more favorable within the halide ionic conductors.35,48,49 In consequence, the introduction of lithium vacancy and the formation of site disorder in the microcrystalline structure of Hf4+ substituted halide samples via high-energy ball milling are responsible for the considerably enhanced ionic conductivity. However, the excessive substitution of Hf4+ would cause an insufficient concentration of mobile charge carriers and a narrowed ionic migration channel in the aliovalent substituted halide ionic conductors, originating from the charge compensation for the introduced tetravalent hafnium cations as well as the smaller ion radius of Hf4+ (71 pm) in comparison to Y3+ (90 pm).35,39 This leads to the decrease of ionic conductivity to a certain extent under high content of aliovalent substitution for halide Li3−xY1−xHfxCl6 (x > 0.6).
The electronic conductivity of mechanochemically prepared Li3−xY1−xHfxCl6 was determined via chronoamperometry measurements with a constant voltage of 1 V as shown in Fig. 1f, and the results indicate that the corresponding values are much lower than that of the ionic conductivities mentioned above. Specifically, the electronic conductivity upon the as-milled LYHC is nearly six orders of magnitude lower than its ionic conductivity, and thus can be regarded as an effective electronic insulator which is capable of sufficient safety guarantee for this material as an outstanding ionic conductor applied in ASSBs. In addition, the electrochemical stability towards halide LYC and the representative Hf4+ substituted LYHC was analyzed through cyclic voltammetry (CV) measurements using the Li/LPSC/halide SEs/halide SSEs + VGCF cells. It is worth noting that both of the two halide samples exhibit rather high oxidation potentials beyond 4 V vs. Li+/Li (Fig. S5†), which enables exceptional compatibility with high-voltage class cathodes for high-energy-density ASSBs.
The subsequent annealing protocol for the mechanochemically derived halide electrolytes commonly exerts different effects regarding the ionic transport properties through altering the crystallization degree or even entirely changing the original microcrystalline structure. In order to investigate the effect of the post-annealing process on the resulting ionic conductivity upon Hf4+ substituted halide ionic conductors, the relevant experimental treatment of the optimal substituted LYHC was carried out by applying elevated temperatures, and the corresponding XRD patterns and the Nyquist plots as well as the ionic conductivity of annealed halide samples under various heat treatment temperatures are displayed in Fig. 2a–c. It can be observed that the primary peaks of the XRD signal for all the annealed LYHC halide samples could still match the standard peaks of hexagonal-close-packed LYC, and the corresponding reflections are dominated by the relatively sharp diffraction peaks with considerably enhanced intensity even at the lower annealing temperature of 200 °C. Despite being an isostructure of mechanochemically prepared LYHC halide, the decrease of ionic conductivity with varying degrees upon post-annealed halide samples was corroborated as the annealing temperature was progressively increased, and those annealed materials exhibit ionic conductivities lower than 10−3 S cm−1 at room temperature. The seeming phenomenon could be achieved for the unsubstituted LYC with the post-annealing process, as observable in Fig. 2d–f. By employing the subsequent crystallization approach, the metastable nonperiodic features composed of the cationic site disorder and local distortion structure within the mechanochemically synthesized halides are subjected to the adverse elimination to severely suppress the ionic transport,20,35,47 which is responsible for the decreasing ionic conductivity in the highly crystallized LYHC samples. Summarizing, the subsequent annealing process for the mechanochemically prepared LYHC could acquire the isostructure counterparts with long-range coherency, which is concomitant with a decrease of the ionic conductivity due to the disappearance of the non-periodic disordered structure. In consequence, the superionic halide conductors directly derived from mechanochemical synthesis are further employed to assemble the halide-based ASSBs in the following experiments.
 | ||
| Fig. 2 Crystal structures and ionic conductivities of halide electrolytes derived from subsequent crystallization routine. (a–c) Collected X-ray diffraction data and Nyquist plots as well as the ionic conductivities of the as-milled LYHC and corresponding subsequently annealed samples with different elevated temperatures. (d–f) Collected X-ray diffraction data and Nyquist plots as well as the ionic conductivities of the as-milled LYC and corresponding subsequently annealed samples with different elevated temperatures. | ||
A deeper crystallographic analysis of the pristine phase LYC and hafnium-substituted LYHC, obtained purely from mechanochemical procedures at room temperature, was carried out from neutron diffraction and Rietveld refinements. This analysis confirms that both phases crystallize in the trigonal Pm1 space group, according to previous reports.50 The trigonal lattice can be visualized as an arrangement of lithium and yttrium octahedra, in which the LiCl6 octahedra are all connected by sharing faces or edges (Fig. S6†). There are two types of YCl6 octahedra. The first ones are isolated, while the second ones are sharing faces and forming chains along the c axis. Additionally, both Li and Y present a partial disorder in the lattice. Thus, the lithium ions are distributed in 6h and 6g Wyckoff sites, and the arrangement of the lithium ions viewed from various directions is shown in Fig. S7.† There are three crystallographically different Y atoms in two sites, 1a and 2d. Finally, there are three Cl atoms allocated in 6i sites. For the undoped phase, initial refinements were performed based on this crystallographic model50 leading to an adequate fit. However, from a detailed analysis using Difference Fourier Maps (DFM), an additional positive nuclear density was detected along the YCl6 chains, indicating the presence of an additional yttrium cation, which was not previously described. This extra Y3+ was added to the 2d site and their occupation was shared with the rest of the yttrium atoms. This fact supports that the Y3+ can move along the c direction.50 For the phase with Hf it was not possible to discern between Y and Hf due to the similarity of the scattering lengths. Hence the structure obtained for pristine phase was used as starting model by replacing Y by Y0.4Hf0.6 with simultaneously modified lithium redistribution. Thus, an acceptable fitting was achieved in both cases, as illustrated in Fig. 3a and b. The main crystallographic results are listed in Tables S1 (for the parent specimen) and S2† (for the Hf-substituted sample). Fig. 3c–f show schematic views of the crystal structures for Hf-substituted phase.
 | ||
| Fig. 3 Crystal structures of the as-milled halide electrolytes. (a and b) Rietveld refinements against the collected NPD data at room temperature for LYC and LYHC. (c) LYHC unit cell with the building units of (Y/Hf)Cl6 octahedra to form a trigonal unit cell in which Y/Hf occupies Wyckoff 1a and 2d positions. (d) Lithium is found to occupy two different octahedral sites, namely, Wyckoff 6h and 6g positions. (e and f) Six edge-sharing LiCl6 octahedra surround the (Y/Hf)Cl6 octahedron resulting in the formation of a distorted honeycomb-like arrangement along the ab-plane. | ||
Notably, the main interest of the NPD analysis resides in its contribution to the understanding of the high ionic conductivity in these materials upon a crystallographic basis. To deepen into this feature, from the point of view of crystal structure, the possible pathways for Li+ ionic diffusion were analyzed by bond valence methods. Initially, the Bond Valence Energy Landscape (BVEL) maps were calculated for both Li+ and Y3+. Lithium isovalent maps (Fig. 4) show a full 3D connection of Li+ cations into the YCl6 octahedral skeleton. Thus, it is possible to observe that each Li1 at 6h sites can jump into Li2 positions at 6g by moving along +c or −c directions. Besides, the Li1 sites form groups of six connected ions perpendicular to the z axis, located between Y1Cl6 octahedra. Also, each Li2 atom is connected to another Li2 along the a (or b) direction. Altogether, a complex framework of pathways with multiple possibilities for lithium jumps can be unveiled by the BVEL maps. In addition, this 3D mobility of Li+ is equally promoted along the three axes, as evidenced by the calculation of the percolations energy, yielding the same value (2.74 eV) for the three axes. This situation is also observed in the Hf-doped phase with an almost equal percolation energy (2.8 eV), which allows the formation of a favorable 3D lithium-ion diffusion pathway consisting of face-sharing octahedra in microcrystalline structure.
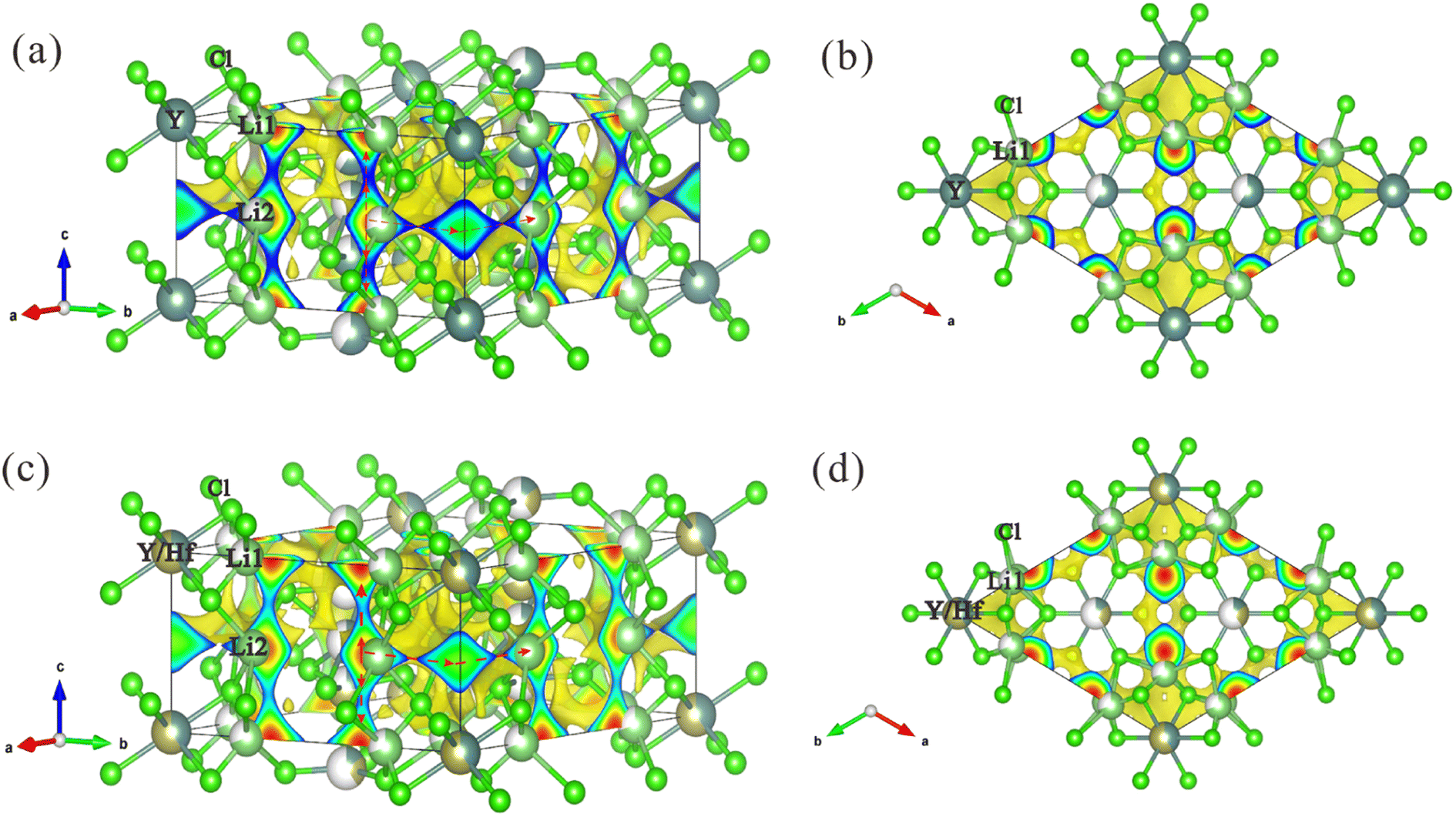 | ||
| Fig. 4 BVEL analysis of Li-ion migration within the crystal structures of the as-milled halide electrolytes. (a and b) Isovalent surfaces of Li-ions obtained from BVEL maps for LYC along the [110] and [001] directions superimposed on the crystal structure. (c and d) Isovalent surfaces of Li-ions obtained from BVEL maps for LYHC along the [110] and [001] directions superimposed on the crystal structure, and the possible Li-ion migration pathways through edge-sharing LiCl6 octahedra in the ab plane as well as the face-sharing LiCl6 along the c-direction. | ||
Regarding the difference of the BVEL maps upon Hf incorporation, a substantial change is observed in LYHC. While in the pristine phase the yttrium mobility is eased along the c axis, in the Hf-doped phase this connectivity seems to be reduced. In addition, there appears to be a possible connectivity between the channels formed by Y2/Hf2, Y3/Hf3 and Y4/Hf4 sites. This fact can be clearly observed in Fig. 5. This is also observed in the percolation energy listed in Table S3,† which suggests an overall reduction in the mobility of Y3+ (or Hf4+) cations that appear after Hf incorporation. A similar analysis can be made from the volume fraction for ionic mobility (see Table S3†). These changes can be explained by the higher polarizing power of Hf4+ with respect to Y3+. This implies that the (Y/Hf)Cl6 framework becomes more covalent, which, besides the intrinsic effect due to the disorder, makes easier the Li+ mobility and diffusion across the structure. Both effects account for the exceptional transport properties here described for the Hf-doped specimen.
 | ||
| Fig. 5 (a and b) Isovalent surfaces of yttrium obtained from BVEL maps for LYC along the [110] and [001] directions superimposed on the crystal structure. (c and d) Isovalent surfaces of yttrium obtained from BVEL maps for LYHC along the [110] and [001] directions superimposed on the crystal structure, in which appears a connectivity between the channels formed by Y2/Hf2, Y3/Hf3 and Y4/Hf4 sites and resulting in the more covalent (Y/Hf)Cl6 framework within the crystal structure. | ||
Cathode composites could be successfully prepared through simply grinding the mixture of as-milled halide electrolytes with a high potential limit and the cathode active material (CAM) particles due to the superior mechanical deformability intrinsic of halide ionic-conductors.20 By controlling the volume ratio of CAM particles and halide electrolytes, the direct electronic and Li-ion percolation pathways within the composite cathode can be established (Fig. 6a), accounting for the inherently high ionic conductivity of the as-milled halide electrolytes and the electronic conduction properties towards CAM particles. The thin layer of the halide electrolytes on the surface of CAM particles is homogeneously coated, which can be observed in the resulting composite cathode based on the as-milled LYHC, and accompanied by the uniform distribution of Co and Cl elements, as revealed by the scanning electron microscopy (SEM) images and energy-dispersive spectroscopy (EDS) maps in Fig. 6b. Thus, the exceptional Li-ion transfer at the interface of halide electrolytes and CAM particles could be realized by the well distributed thin halide coating layer with high ionic conductivity. Simultaneously, the compact contact of the CAM surfaces is capable of providing sufficient electronic connection among the particles in the composite cathodes, and ultimately the optimal interconnected mixed ionic/electronic network is achieved which is vital for realizing the high-performance full cells.
 | ||
| Fig. 6 Ionic and electronic percolation within the composite cathode and electrochemical performance of ASSBs. (a) Schematic diagram of the mixed ionic/electronic network for the LCO composite cathode. (b) SEM image of the LYHC-coated LCO composite cathode after simple grinding, and corresponding EDS mapping of Co and Cl elements. (c) Schematic diagram of halide-based ASSBs in this work. (d) Initial charge/discharge curves of 0.1C with coulombic efficiency denoted. (e) Rate performance at various current densities. (f) Long-term cycling performance at 0.1C. (g) Nyquist plots of the ASSBs before and after 100 cycles at 0.1C. | ||
In addition, the exceptional deformability of the LYC and LYHC prepared through the mechanochemical method was further verified by the flattened and dense surface of the pellets obtained from direct cold-pressing of halide powders at ∼3 tons (Fig. S8†). The surface morphology and the corresponding element distribution of the composite cathode pellet derived from cold-pressing based on the as-milled LYHC are shown in Fig. S9,† and the smooth and compacted surface features could be observed along with the uniformly distributed element on the resulting cathode pellet, which is indicative of the homogeneous distribution of CAM particles and halide ionic conductors to efficiently offer electronic and Li-ion percolation pathways within the composite cathode. Especially, the compact contact between the halide electrolyte layer and composite cathodes could be clearly observed in the cross-sectional SEM image as displayed in Fig. S10.†
The mechanochemically derived LYHC with the optimal aliovalent substitution of Hf4+ in a series of LYHC was integrated as the solid electrolyte in all-solid-state cells with LCO particles as the cathode and the Li–In alloy as the negative electrode. The thin layer of LPSC electrolyte was applied on the top of the Li–In anode to restrain the parasitic reaction between the anode and the halide electrolytes as determined in Fig. S11,† and the schematic diagram of the configuration for the ASSBs is illustrated in Fig. 6c. The applicability of aliovalent substituted LYHC compared to the original LYC prepared by the mechanochemical method for cathodes using LCO was evaluated in all-solid-state cells tested at room temperature (Fig. 6d–g). The first-cycle charge–discharge voltage profiles at 0.1C for LCO electrodes employing mechanochemically synthesized LYHC and LYC are displayed in Fig. 6d. The full cell assembled with halide LYC exhibited distinctly inferior feature in terms of the substantially enormous polarization in the voltage file, and a consequently low discharge capacity of nearly 100 mA h g−1 as well as a poor initial coulombic efficiency of 88.7% were achieved. In contrast, the cell assembled with Hf4+-substituted LYHC shows a high first discharge capacity of 120 mA h g−1 with outstanding initial coulombic efficiency of 93.0%, emphasizing that the considerably enhanced Li-ion conductivity may play a decisive role in achieving the high-performance full cells constructed by halide electrolytes. Furthermore, the long-term cycling at 0.1C of the LCO electrode employing LYC and LYHC derived from ball-milling methods was performed as shown in Fig. 6f. The outstanding cycling stability of the LCO electrodes employing LYHC was confirmed which enabled high capacity retention of 70% after 100 cycles, but significantly lower capacity retention of 25% after 100 cycles was obtained for the LCO electrodes using unsubstituted LYC. The enhanced cycling stability for LYHC-based ASSBs is primarily due to the integrated effects in terms of the exceptional interface stability and the highly efficient ionic migration in the composite cathode. In addition, the rate capability of the LCO electrode employing LYHC was considerably superior to that using LYC (Fig. 6e), and the reversible discharge capacity could achieve 90% of the initial value while the current densities return to 0.1C, which probably accounts for the fact that the LCO does not encounter isolation during the cycling of high current density originating from the intensified ionic and electronic conduction percolation network within the composite cathode.
The diagnostic electrochemical analysis was performed to investigate the interfacial stabilities for the full cells assembled by LYC and LYHC through the electrochemical impedance spectroscopy (EIS) measurements, and the corresponding Nyquist plots before and after 100 cycles are displayed in Fig. 6g. Two typical semicircles followed by a low-frequency Warburg tail were observed in the Nyquist plots. The first semicircle in the high-frequency region represents the impedance of the solid electrolyte layer, and the other semicircle in the middle-frequency region is associated with the interface resistance and the charge transfer resistance, while the Warburg impedance in the ultra-low frequency region originates from the Li-ion diffusion in the electrode.51,52 The equivalent circuit diagram is shown in Fig. S12,† which is used to fit the EIS spectra of the full cells after 100 cycles, and the corresponding fitting results are displayed in Table S4.† The interfacial resistance of the cell assembled with LYC suffers from a dramatical increment after cycling which is nearly 7 times higher than that observed when using LYHC, corroborating the superior interfacial stability of the aliovalent substituted LYHC compared with the original LYC.
The underlying interfacial (electro)chemistry of the LCO electrodes with LYHC and LYC was investigated for the initial electrodes and after 100 cycles through the XPS measurements (Fig. S13 and S14†). It can be observed that marginal changes are exhibited in the corresponding spectra of Y 3d and Cl 2p, which adequately confirmed the exceptional compactness and high-voltage tolerance of aliovalent substituted LYHC. Besides, a similar phenomenon could also be found for the LCO electrodes employing LYC, which is in accordance with the satisfactory oxidation stability of LYC determined by CV measurements. Thus, the conditioned ionic transport in the solid electrolyte layer and attenuated Li-ion percolation pathways within the composite cathode caused by intrinsically lower ionic conductivity of the as-milled LYC could be responsible for the inferior performance of ASSBs employing LYC. In consequence, the electrochemical analysis and XPS measurements unambiguously corroborate the strengthened interfacial contact and the outstanding stability of LYHC combined with the LCO electrolyte which is conducive to achieving the high-performance halide-based ASSBs.
4. Conclusion
In summary, we report a new series of mixed-metal halides Li3−xY1−xHfxCl6 (0 ≤ x < 1) with high ionic conductivity up to 1.49 mS cm−1 over a wide compositional range. Highly amorphous halide electrolytes for Hf4+-substituted LYC could be obtained by mechanochemical milling, and the formation of the prevalent cation site disorder and defect structure induced by aliovalent substitution of Y for Hf in LYC strongly benefits the favorable lithium-ion diffusion in its sublattice due to enlarged Li+ migration channels and reduced energy landscape. The subsequent annealing under elevated temperature results in the crystallization of the Hf4+-substituted halides with an effective temperature dependence of the ordering, accompanied with the gradual elimination of the defect structures obtained from mechanochemical synthesis, which in turn causes the observed reduction of ionic conductivity for the corresponding annealed samples. Importantly, the appropriate heterovalent substitution of Hf for Y in LYC provides the infinitely 3D connected Li+ ion diffusion pathway along with balancing the concentration of Li+ carriers, which is favorable for facilitating the adequate improvement of ionic conductivity. Notably, the prominent electrochemical performance is demonstrated for bulk-type ASSBs with Hf4+-substituted LYC in combination with Li–In anodes and 4 V-class LCO cathodes. Namely, high initial coulombic efficiencies of 93.0%, as well as the satisfactory rate capability and cycling performance are obtained. Finally, the EIS and ex situ XPS results indicate the existence of the excellent interfacial stability between the CAM and as-milled halide electrolytes, confirming the good chemical compatibility of Hf4+-substituted halides in contact with a high voltage CAM oxide. We believe these findings offer an intriguing viewpoint for designing novel halide superionic conductors and shed light on the future prospects and directions for constructing practical advanced ASSBs.Author contributions
Kaiyong Tuo: conceptualization, methodology, investigation, data curation, writing – original draft, review & editing. Chunwen Sun: supervision, conceptualization, resources, writing – review & editing, funding acquisition and validation. C. A. López: formal analysis, investigation, software and data curation, visualization and review. Maria Teresa Fernández-Díaz: formal analysis, investigation and data curation. José Antonio Alonso: conceptualization, resources, writing – review & editing and funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the financial support from the Foundation of Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, 21C Innovation Laboratory, Contemporary Amperex Technology Ltd under project No. 21C-OP-202212, Foundation of State Key Laboratory of Highly efficiency Utilization of Coal and Green Chemical Engineering (Grant No. 2022-K15), China University of Mining & Technology (Beijing), Foundation of Top-notch Innovative Talents Cultivation (Grant No. BBJ2023031), China University of Mining & Technology (Beijing), and the National Natural Science Foundation of China (No. 51672029 and 51372271). We also acknowledge the financial support of the Spanish Ministry of Science and Innovation to the project PID2021-122477OB-I00. We are grateful to ILL (France) for making all facilities available.Notes and references
- J. B. Goodenough, Energy Environ. Sci., 2014, 7, 14–18 RSC.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- C. Sun, J. Liu, Y. Gong, D. P. Wilkinson and J. Zhang, Nano Energy, 2017, 33, 363–386 CrossRef CAS.
- G. Harper, R. Sommerville, E. Kendrick, L. Driscoll, P. Slater, R. Stolkin, A. Walton, P. Christensen, O. Heidrich and S. Lambert, Nature, 2019, 575, 75–86 CrossRef CAS PubMed.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS.
- R. Chen, Q. Li, X. Yu, L. Chen and H. Li, Chem. Rev., 2019, 120, 6820–6877 CrossRef.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- J. Xie and Y.-C. Lu, Nat. Commun., 2020, 11, 1–4 CrossRef PubMed.
- Y. Ding, Z. P. Cano, A. Yu, J. Lu and Z. Chen, Electrochem. Energy Rev., 2019, 2, 1–28 CrossRef CAS.
- Y. Zhang and C. Sun, ACS Appl. Mater. Interfaces, 2021, 13, 12099–12105 CrossRef CAS.
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 1–4 Search PubMed.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 1–16 Search PubMed.
- J. C. Bachman, S. Muy, A. Grimaud, H.-H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart and P. Lamp, Chem. Rev., 2016, 116, 140–162 CrossRef CAS.
- A. Mauger, C. M. Julien, A. Paolella, M. Armand and K. Zaghib, Materials, 2019, 12, 3892 CrossRef CAS.
- X. Li, J. Liang, X. Yang, K. R. Adair, C. Wang, F. Zhao and X. Sun, Energy Environ. Sci., 2020, 13, 1429–1461 RSC.
- Q. Zhang, D. Cao, Y. Ma, A. Natan, P. Aurora and H. Zhu, Adv. Mater., 2019, 31, 1901131 CrossRef CAS.
- J. Wu, L. Shen, Z. Zhang, G. Liu, Z. Wang, D. Zhou, H. Wan, X. Xu and X. Yao, Electrochem. Energy Rev., 2021, 4, 101–135 CrossRef CAS.
- C. Sun, J. A. Alonso and J. Bian, Adv. Energy Mater., 2021, 11, 2000459 CrossRef CAS.
- J. Van Den Broek, S. Afyon and J. L. Rupp, Adv. Energy Mater., 2016, 6, 1600736 CrossRef.
- K. Tuo, C. Sun and S. Liu, Electrochem. Energy Rev., 2023, 6, 17 CrossRef CAS.
- X. Li, J. Liang, J. Luo, M. N. Banis, C. Wang, W. Li, S. Deng, C. Yu, F. Zhao and Y. Hu, Energy Environ. Sci., 2019, 12, 2665–2671 RSC.
- J. Liang, X. Li, S. Wang, K. R. Adair, W. Li, Y. Zhao, C. Wang, Y. Hu, L. Zhang and S. Zhao, J. Am. Chem. Soc., 2020, 142, 7012–7022 CrossRef CAS PubMed.
- K.-H. Park, K. Kaup, A. Assoud, Q. Zhang, X. Wu and L. F. Nazar, ACS Energy Lett., 2020, 5, 533–539 CrossRef CAS.
- B. Helm, R. Schlem, B. Wankmiller, A. Banik, A. Gautam, J. Ruhl, C. Li, M. R. Hansen and W. G. Zeier, Chem. Mater., 2021, 33, 4773–4782 CrossRef CAS.
- Z. Liu, S. Ma, J. Liu, S. Xiong, Y. Ma and H. Chen, ACS Energy Lett., 2020, 6, 298–304 CrossRef.
- H. Kwak, D. Han, J. Lyoo, J. Park, S. H. Jung, Y. Han, G. Kwon, H. Kim, S. T. Hong and K. W. Nam, Adv. Energy Mater., 2021, 11, 2003190 CrossRef CAS.
- W. Ji, D. Zheng, X. Zhang, T. Ding and D. Qu, J. Mater. Chem. A, 2021, 9, 15012–15018 RSC.
- T. Asano, A. Sakai, S. Ouchi, M. Sakaida, A. Miyazaki and S. Hasegawa, Adv. Mater., 2018, 30, 1803075 CrossRef PubMed.
- M. Jiang, S. Mukherjee, Z. W. Chen, L. X. Chen, M. L. Li, H. Y. Xiao, C. Gao and C. V. Singh, Phys. Chem. Chem. Phys., 2020, 22, 22758–22767 RSC.
- K. Kim, D. Park, H.-G. Jung, K. Y. Chung, J. H. Shim, B. C. Wood and S. Yu, Chem. Mater., 2021, 33, 3669–3677 CrossRef CAS.
- B. Liu, D. Wang, M. Avdeev, S. Shi, J. Yang and W. Zhang, ACS Sustainable Chem. Eng., 2019, 8, 948–957 CrossRef.
- R. Schlem, C. F. Burmeister, P. Michalowski, S. Ohno, G. F. Dewald, A. Kwade and W. G. Zeier, Adv. Energy Mater., 2021, 11, 2101022 CrossRef CAS.
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS.
- E. Sebti, H. A. Evans, H. Chen, P. M. Richardson, K. M. White, R. Giovine, K. P. Koirala, Y. Xu, E. Gonzalez-Correa and C. Wang, J. Am. Chem. Soc., 2022, 144, 5795–5811 CrossRef CAS.
- R. Schlem, S. Muy, N. Prinz, A. Banik, Y. Shao-Horn, M. Zobel and W. G. Zeier, Adv. Energy Mater., 2020, 10, 1903719 CrossRef CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- J. Rodríguez-Carvajal, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 192, 55–69 CrossRef.
- S. Adams, Solid State Ionics, 2006, 177, 1625–1630 CrossRef CAS.
- A. Omote, S. Yotsuhashi, Y. Zenitani and Y. Yamada, J. Am. Ceram. Soc., 2011, 94, 2285–2288 CrossRef CAS.
- O. Sheng, J. Zheng, Z. Ju, C. Jin and X. Tao, Adv. Mater., 2020, 2000223 CrossRef CAS.
- O. W. Sheng, C. B. Jin, Z. J. Ju, J. H. Zheng, T. F. Liu, Y. J. Liu, Y. Wang, J. M. Luo, X. Y. Tao and J. W. Nai, Nano Lett., 2022, 22, 8346–8354 CrossRef CAS PubMed.
- L. Zhou, T.-T. Zuo, C. Y. Kwok, S. Y. Kim, A. Assoud, Q. Zhang, J. Janek and L. F. Nazar, Nat. Energy, 2022, 7, 83–93 CrossRef CAS.
- T. Yu, J. Liang, L. Luo, L. Wang, F. Zhao, G. Xu, X. Bai, R. Yang, S. Zhao and J. Wang, Adv. Energy Mater., 2021, 11, 2101915 CrossRef CAS.
- J. Liang, E. van der Maas, J. Luo, X. Li, N. Chen, K. R. Adair, W. Li, J. Li, Y. Hu and J. Liu, Adv. Energy Mater., 2022, 12, 2103921 CrossRef CAS.
- J. Park, D. Han, H. Kwak, Y. Han, Y. J. Choi, K.-W. Nam and Y. S. Jung, Chem. Eng. J., 2021, 425, 130630 CrossRef CAS.
- X. Shi, Z. Zeng, M. Sun, B. Huang, H. Zhang, W. Luo, Y. Huang, Y. Du and C. Yan, Nano Lett., 2021, 21, 9325–9331 CrossRef CAS.
- K. Wang, Q. Ren, Z. Gu, C. Duan, J. Wang, F. Zhu, Y. Fu, J. Hao, J. Zhu and L. He, Nat. Commun., 2021, 12, 1–11 CrossRef.
- S. Muy, J. Voss, R. Schlem, R. Koerver, S. J. Sedlmaier, F. Maglia, P. Lamp, W. G. Zeier and Y. Shao-Horn, iScience, 2019, 16, 270–282 CrossRef CAS PubMed.
- N. Flores-González, N. Minafra, G. Dewald, H. Reardon, R. I. Smith, S. Adams, W. G. Zeier and D. H. Gregory, ACS Mater. Lett., 2021, 3, 652–657 CrossRef.
- R. Schlem, A. Banik, S. Ohno, E. Suard and W. G. Zeier, Chem. Mater., 2021, 33, 327–337 CrossRef CAS.
- X. Li, M. Liang, J. Sheng, D. Song, H. Zhang, X. Shi and L. Zhang, Energy Storage Mater., 2019, 18, 100–106 CrossRef.
- Q. Shao, C. Yan, M. Gao, W. Du, J. Chen, Y. Yang, J. Gan, Z. Wu, W. Sun and Y. Jiang, ACS Appl. Mater. Interfaces, 2022, 14, 8095–8105 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta02781c |
| This journal is © The Royal Society of Chemistry 2023 |