
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailored imidazolium tetraphenylborate salts for the design of boron, nitrogen co-doped carbon materials as high-performance anodes for fast-rate monovalent ion batteries†
Soha
Aldroubi
a,
Badre
Larhrib
ab,
Louiza
Larbi
ac,
Ibrahim Bou
Malham
d,
Camelia Matei
Ghimbeu
 ce,
Laure
Monconduit
ae,
Ahmad
Mehdi
*a and
Nicolas
Brun
*a
ce,
Laure
Monconduit
ae,
Ahmad
Mehdi
*a and
Nicolas
Brun
*a
aICGM, Univ. Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: ahmad.mehdi@umontpellier.fr; nicolas.brun@enscm.fr
bUniversité de Pau, Avenue de l'Université, 64012 Pau Cedex, France
cInstitut de Science des Matériaux de Mulhouse, IS2M, CNRS UMR 7361, UHA, France
dLaboratoire Energétique et Réactivité à l’Echelle Nanométrique (EREN), Faculté des Sciences IV, Université Libanaise, Haouch el-Omara, 1801, Zahlé, Lebanon
eRS2E, CNRS, Amiens, France
First published on 11th July 2023
Abstract
We report the synthesis of original imidazolium salts with the tetraphenylborate anion and their use as precursors for boron, nitrogen co-doped carbon materials. In addition to the beneficial effect of the tetraphenylborate anion, we showed that the presence of two benzyl groups on the imidazolium cation increases the carbonization yield (15 wt%), promotes the heteroatom-doping (16 wt% of nitrogen and 9 wt% of boron), improves the microstructure order and protects boron atoms from oxidation. The B, N co-doped carbon prepared from 1,3-bis(benzyl)imidazolium tetraphenylborate demonstrated fast-rate capabilities as an anode material for high-power monovalent ion batteries. After 1400 cycles at a high rate (30C) versus Li+/Li, 80% of the capacity was maintained (112 mA h g−1).
1. Introduction
Carbon-based materials and carbon allotropes are amongst the most attractive materials in the fields of electronics,1 electrocatalysis,2 and electrochemical energy storage and conversion.3 Carbon materials can offer high electronic and thermal conductivities and fine nanostructure control together with high physical and chemical stability, which are the major assets driving their wide use in various fields of nanotechnology.4 In particular, the introduction of heteroatoms into the structure of carbon materials, such as nitrogen, boron, sulphur and phosphorus,5 has been demonstrated as a versatile approach to achieve singular structures with improved properties.6 The insertion of secondary light elements in the sp2-hybridized network of molecular (e.g., graphene and fullerene), graphitic or even turbostratic carbon materials offers a versatile control over the electronic structure (e.g., band gap) and physicochemical properties such as wettability/dispersibility, electrical conductivity,7 catalytic activity8 and electrochemical reactivity.9–12Nitrogen is probably one of the most common dopants used to adjust the properties of carbon-based materials. Nitrogen is a direct neighbour of carbon in the periodic table of elements and thus has a very similar atomic radius. Nitrogen has one more electron than carbon, which leads to n-type semiconductor doping. Thanks to nitrogen's lone pairs of electrons and greater electronegativity (i.e., 3.04 versus 2.55 for carbon), it provides higher (electro)chemical activity to doped carbon materials;8,9 the higher electron density of nitrogen lowers the electron density of the adjacent carbon atoms, which display a high positive charge density.13 In addition, nitrogen-doping has proven to greatly affect the electronic conductivity and charge mobility in carbon-based materials. However, due to multifactorial effects, it is difficult to draw general conclusions. The impact on the electronic conductivity depends on the nature of the carbon material, the level of nitrogen-doping,14 and the nature and the density of chemical and structural defects. For instance, a decrease in electronic conductivity and charge mobility was reported for nitrogen-doped graphene compared with pristine graphene,15 while an increase in electronic conductivity was observed for turbostratic nitrogen-doped carbon materials7 or carbon nanotube (CNT) films16 compared with undoped analogues. Finally, the number of defects generated by nitrogen-doping can provide ion and electrolyte diffusion channels that might be important for electrochemical energy storage applications.17
Although less reported in the literature than nitrogen-doping, boron-doping has been of great interest as it can provide opposite features to doped carbon. Like nitrogen, boron is a direct neighbour of carbon in the periodic table of elements. Unlike nitrogen, boron has one less electron than carbon and displays a smaller electronegativity (i.e., 2.04 versus 2.55 for carbon). Thus, boron provides p-type semiconductor doping to carbon materials due to its electron-deficiency; in contrast to nitrogen-doped carbons, positively charged boron atoms are surrounded by negatively charged carbon atoms. The significant delocalization of electrons affects the electron density distribution that can substantially modify the electronic properties18 as well as the surface reactivity19,20 and chemical sensitivity21,22 of boron-doped carbons. Besides, the introduction of boron may increase the defects in carbon-based nanostructures, which can break inertia and improve reactivity.23–25
More recently, simultaneous doping (i.e., co-doping) with nitrogen and boron has been reported as a promising strategy to combine the benefits of each element, while also obtaining unique electronic structures and/or providing synergies. As mentioned above, boron and nitrogen behave in a divergent manner with respect to carbon; they display a reverse type of doping and reverse electronegativity. Interestingly, these features cause boron and nitrogen to have a very strong affinity for each other. The concomitant presence of these two elements together with carbon therefore makes their respective levels of doping significantly higher.26 Due to singular properties, B, N co-doped carbons have been successfully used as metal-free catalysts27 and electrocatalysts,26,28 electrode materials for metal-ion batteries29,30 and electrochemical double-layer capacitors,31,32 sorbents for hydrogen storage33 and sensors for gas detection.34
Various methods have been used to synthesize B, N co-doped carbon materials such as chemical vapor deposition,33,34 laser ablation35 and conventional pyrolysis of organic precursors such as boric acid, phenylboronic acid and borax as boron precursors, and melamine, urea, imidazole and aniline as nitrogen precursors.26–29,32 The use of such volatile organic precursors presents however major drawbacks and usually leads to doped carbon-based materials with low heteroatom doping levels and/or low mass yields. In this context, the use of molten salts and ionic liquids (ILs) as heteroatom-containing precursors with negligible vapor pressure has proven to be of great interest for the synthesis of doped carbon-based materials.36 In the literature, imidazolium-based ionic liquids (ILs) bearing boron-containing anions have been used as stable precursors to produce B, N co-doped carbon materials. The most common boron-containing anions reported in the literature are tetrafluoroborate, BF4−, and tetracyanoborate, TCB−.37,38 Although commonly used for the preparation of B-doped carbon materials, both anions have their limitations. In the work of Ma et al.,39 1-butyl-3-methylimidazolium tetrafluoroborate, [Bmim][BF4], was treated in an autoclave at 200 °C for 6 h before pyrolysis at 600 °C for 2 h under argon. The resulting material displayed low doping levels in both nitrogen (3.7 at%) and boron (0.6 at%).39 Moreover, handling of the BF4− anion is hampered by the formation of gaseous HF during pyrolysis.40 The formation of undesirable toxic volatile co-products, such as hydrogen cyanide (HCN),38 is also a major problem when using the TCB− anion. However, TCB− has been shown to improve the stability of the doped carbon network under thermal-annealing,38 probably due to the cross-linking of the pending nitrile groups.41 For instance, Fellinger et al.38 reported the pyrolysis of 1-ethyl-3-methylimidazolium tetracyanoborate, [emim][TCB], to prepare B, N co-doped carbons. At 1000 °C (without a plateau), a high carbonization yield of 25.5 wt% was reached.38 However, the authors reported a low carbon content (39.1 wt%) associated with a significant presence of oxygen atoms due to the strong oxophilicity of boron and its partial post-oxidation. The authors had to apply pyrolytic treatment at higher temperature, i.e., at 1400 °C, to avoid partial post-oxidation of boron. Under these conditions, the authors could reach relatively high doping levels in both nitrogen (19 wt%) and boron (17.4 wt%). Beyond the need to work at high temperature to avoid partial post-oxidation of boron, the synthesis of TCB− anions gives relatively low yields and relies on the preparation of K[TCB], which needs an excess of potassium cyanide, KCN, which is highly toxic.42,43
In this context, alternative salts and ILs must be explored. In a recent study, Mehler et al.43 reported the synthesis and the pyrolytic treatment of an original IL, 1-ethyl-3-methylimidazolium tetrakis(1-imidazolyl)borate, [emim][BIm4]. The authors used a salt templated pyrolysis approach and prepared porous B, N co-doped carbonaceous materials with a relatively high doping level of nitrogen (22 wt%) and moderate doping level of boron (3 wt%).43 High carbonization yields of ca. 35 wt% were reached, without specifying the exact pyrolysis temperature at which such a high yield was achieved. These materials were successfully used for the adsorptive removal of methylene blue, a cationic dye.
Herein, we propose another original alternative to common boron-containing anions, i.e., tetraphenylborate anion, BPh4−. We report the synthesis of three original imidazolium tetraphenylborate salts with three different imidazolium cations: 1-butyl-3-methylimidazolium, [Bmim][BPh4]; 1-benzyl-3-methylimidazolium, [(Bn)mim][BPh4]; and 1,3-bis(benzyl)imidazolium, [(Bn)2im][BPh4]. These three salts were pyrolyzed at 900 °C for 7 h under argon yielding B, N co-doped carbon-based materials with relatively high doping levels of both nitrogen (12.8–16.4 wt%) and boron (7.0–9.6 wt%). We showed that the presence of two benzyl groups on the imidazolium cation increases the carbonization yield, promotes the heteroatom-doping (in percentage by weight), improves the microstructure order and protects boron atoms from partial post-oxidation. Besides, the B, N co-doped carbon prepared from [(Bn)2im][BPh4] demonstrated fast-rate capabilities as an anode material for high-power lithium ion batteries.
2. Experimental section
2.1. Materials
1-Methylimidazole (C4H6N2, 99%), 1-bromobutane (CH3(CH2)3Br, 99%), benzyl chloride (C6H5CH2Cl, 99%), imidazole (C3H4N2, 99%), sodium hydride (NaH, 60% dispersion in mineral oil), sodium tetraphenylborate (C24H20BNa, 99%), 1-methyl-2-pyrrolidone solvent (NMP, 99%), ethylene carbonate (EC, 99%), diethyl carbonate (DEC, 99%) and potassium hexafluorophosphate (KPF6, 99%) were purchased from Sigma Aldrich. All reagents were used without further purification.Carbon black (Super C65) was purchased from Imerys, Polyvinylidene fluoride (denoted as PVDF, Grade: 5130) from Solef and K ingots (99.95%) from Alfa Aesar. A glass microfiber filter (Whatman, Grade GF/D) was used as the separator in the batteries.
2.2. Synthesis of the imidazolium tetraphenylborate salts
1-Butyl-3-methylimidazolium bromide, 1-benzyl-3-methylimidazolium chloride and 1,3-bis(benzyl)imidazolium chloride were synthesized as described in the ESI.†1-Butyl-3-methylimidazolium tetraphenylborate, [Bmim][BPh4] (Scheme 1), was synthesized by adding an equimolar amount of sodium tetraphenylborate (16.1 g, 0.047 mol) to 1-butyl-3-methylimidazolium bromide (10.3 g, 0.047 mol) previously dissolved in acetonitrile (100 mL), while maintaining the mixture under stirring for 24 h at 25 °C. 1-Benzyl-3-methylimidazolium tetraphenylborate [(Bn)mim][BPh4] and bis(benzyl)imidazolium [(Bn)2im][BPh4] (Scheme 1) were prepared in the same way but starting from 1-benzyl-3-methylimidazolium chloride (9.73 g, 0.047 mol) and 1,3-bis(benzyl)imidazolium chloride (9.61 g, 0.034 mol) and using respectively a mass of 16.0 g (0.047 mol) and of 11.6 g (0.034 mol) of sodium tetraphenylborate. Sodium chloride and sodium bromide were removed by filtration under vacuum on a sintered glass topped with Celite. The acetonitrile was evaporated using a rotary evaporator and the obtained solid salts were washed several times with 50 mL of ultrapure water. After each washing, the aqueous phase was tested with silver nitrate until total elimination of the salt formed, i.e., AgBr or AgCl, which is revealed by the formation of a white precipitate. Absolute ethanol was used to remove remaining traces of sodium tetraphenylborate in products. Lyophilization of the as-synthesized imidazolium salts was performed to fully remove traces of residual water.
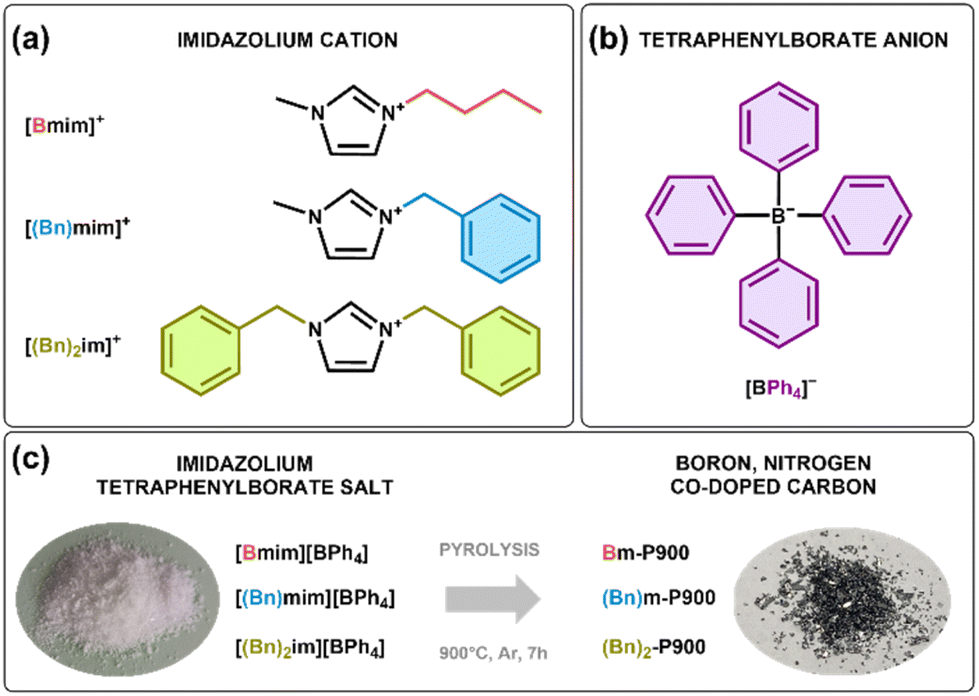 | ||
| Scheme 1 Molecular structure and nomenclature of (a) the imidazolium cations and (b) the tetraphenylborate anion constituting the (c) salts used as precursors (left) for the synthesis of boron, nitrogen co-doped carbon materials (right). | ||
For [Bmim][BPh4] (18.0 g, 0.039 mol; yield of 84.2%). [Bmim][BPh4] is a white powder that melts at 131 °C. Elemental analyses: %Cexp 83.768 (%Ctheo 83.836); %Hexp 7.816 (%Htheo 7.694); %Nexp 6.088 (%Ntheo 6.110). 1H NMR [Bmim][BPh4] (400 MHz, CD3CN) δ 8.17 (s, 1H); 7.69–6.43 (m, 22H); 4.07 (t, 2H); 3.76 (s, 3H); 1.91–1.68 (m, 2H); 1.48–1.23 (m, 2H); 0.97 (t, 3H). 13C NMR [Bmim][BPh4] (101 MHz, CD3CN) δ 165.12–163.65; 136.31; 126.18; 122.36; 49.84; 36.40; 32.13; 19.53; 13.23. 11B NMR [Bmim][BPh4] (128 MHz, CD3CN) δ −6.57.
For [(Bn)mim][BPh4] (16.7 g, 0.028 mol; yield of 60.1%). [(Bn)mim][BPh4] is in the form of very small bright white solid crystals that melt at 150 °C. Elemental analyses: %Cexp 85.492 (%Ctheo 85.362); %Hexp 6.681 (%Htheo 6.754); %Nexp 5.712 (%Ntheo 5.688). 1H NMR [(Bn)mim][BPh4] (400 MHz, CD3CN) δ 7.94 (s, 1H); 7.61–6.77 (m, 27H); 5.16 (s, 2H); 3.65 (s, 3H). 13C NMR [(Bn)mim][BPh4] (126 MHz, CD3CN) δ 164.79–163.79; 136.29; 129.80; 129.09; 126.19; 124.54; 122.36; 53.30; 36.50. NMR 11B [(Bn)mim][BPh4] (128 MHz, CD3CN) δ −6.55.
For [(Bn)2im][BPh4] (11.9 g, 0.021 mol; yield of 62.05%). [(Bn)2im][BPh4] is a white powder that melts at 145 °C. Elemental analyses: %Cexp 86.503 (%Ctheo 86.612); %Hexp 6.583 (%Htheo 6.559); %Nexp 5.016 (%Ntheo 4.926). 1H NMR [(Bn)2im][BPh4] (400 MHz, CD3CN) δ 8.40 (s, 1H); 7.89–6.57 (m, 32H); 5.25 (s, 4H). 13C NMR [(Bn)2im][BPh4] (126 MHz, CD3CN) δ 168.37–160.78; 136.30; 129.83; 129.17; 126.19; 122.36; 117.89; 53.51. 11B NMR [(Bn)2im][BPh4] (160 MHz, CD3CN) δ −6.58.
2.3. Pyrolysis
The three salts (ca. 600 mg) were pyrolyzed at 900 °C for 7 hours at a heating rate of 10 °C min−1 under an argon flow (50 mL min−1) in a tube furnace. The carbonization mass yield, C yield, corresponds to the ratio of the mass of the carbon-based material recovered after pyrolysis to the initial mass of salt. The pyrolytic treatments were carried out three times for each salt in order to determine an average carbonization mass yield. Blackish and shiny powders were obtained (Fig. 1c). | ||
| Fig. 1 TGA and DSC curves of (a) [Bmim][BPh4], (b) [(Bn)mim][BPh4], and (c) [(Bn)2im][BPh4] obtained under argon between 20 and 1100 °C with a rate of 10 °C min−1. | ||
2.4. Characterization
The 1H liquid-state NMR spectra of the three salts were obtained using SiMe4 as a reference on a Bruker 200. 1H solid-state NMR was performed on a VNMR operator at 20 °C. 13C NMR and 11B NMR were performed on a Bruker 200. FTIR spectroscopy was performed in attenuated total reflectance (ATR) mode on a PerkinElmer Spectrum Two spectrometer.Combustion elemental analyses (C, H, N, and O) were performed on a vario MICRO cube analyzer from Elementar. Boron loading for all carbon materials was determined by using a Thermo Fisher ICP-OES (iCap 7400 Duo). Before this analysis, 20 mg of carbonaceous materials were digested in a mixture containing 2 mL of nitric acid and 2 mL of water. This mixture was placed in an autoclave, and then heated at 180 °C for 3 days in an oven. The clear solution was subsequently diluted with water prior to analysis.
X-ray diffraction (XRD) was performed on salts and B, N co-doped carbon-based materials on a Bruker D8 Advance diffractometer with a Bragg–Brentano geometry and equipped with a Bruker Lynx Eye detector, with the Kα radiation of Cu (λ = 1.5418 Å) and an angular step size of 0.02° in the 5°–60° interval. For the three carbonaceous materials, X-ray profiles were fitted with a simple Gaussian function (for the peak at 2θ ≈ 24° labelled 002) and a Lorentz function (for the peak at 2θ ≈ 43° labelled 10) using OriginPro® after removing the background using an exponential decay function. We used the as-obtained fitting to determine both the full widths at half maximum (FWHM) and the peak positions. We applied the Debye–Scherrer formula to the peak at 2θ ≈ 43° (10) to estimate the average size of the graphite-like crystalline domains in the basal plane, La. We applied the Debye–Scherrer formula to the peak at 2θ ≈ 24° (002) to estimate the average stack thickness (perpendicular to the basal plane) of the graphite-like crystalline domains, Lc.
Differential scanning calorimetry (DSC) experiments were performed on a STA simultaneous thermal analyser (TGA/DSC) instrument, model 449 F1 Jupiter® (NETZSCH). Samples of ca. 10–15 mg were placed in an enclosure the temperature program of which was finely controlled through a conventional or inversely modulated thermal ramp and exposed to an argon flow atmosphere (50 mL min−1) by heating from 20 to 1100 °C at a rate of 10 °C min−1. TGA-MS experiments were performed by coupling the thermal analyser with a QMS 403 D (NETZSCH) mass spectrometer and using the same experimental conditions as for TGA/DSC experiments: argon flow atmosphere (50 mL min−1) and heating rate of 10 °C min−1.
X-ray photoelectron spectroscopy (XPS) experiments were performed using an Escalab 250 Xi spectrometer with monochromatized Al Kα radiation (hν = 1486.6 eV). Powders were placed on a sample holder using uPVC insulation tape (3 M part number 655) and transferred to an argon-filled glove box connected to the spectrometer. Analysis was performed using the standard charge compensation mode and an elliptic 325 × 650 μm X-ray beam spot. Core spectra were recorded using a 20 eV constant pass energy with a 0.15 eV step size and iterative scans at a dwell time of 500 ms. Using CasaXPS software, the binding energy scale was calibrated from the C–C, CH peak at 285 eV. A non-linear Shirley-type background was used for core peak analysis, while 70% Gaussian–30% Lorentzian Voigt peak shapes (called GL(30)), full width at half maximum, and position constraint ranges were selected to optimize peak positions and areas.
Transmission electron microscopy (TEM) analyses were performed on a JEOL 1200 microscope from the MEA platform, Univ. Montpellier. High-resolution transmission electron microscopy (HRTEM) was performed on a JEOL ARM-200F instrument and a JEOL 2200 FS instrument (from MEA platform, Univ. Montpellier) operating at 200 kV. The samples were ground in a mortar, dispersed in absolute ethanol in an ultrasonic bath for a few minutes and drop-coated on Lacey carbon grids before observation.
CO2 physisorption experiments were carried out at 273 K on a Micromeritics TriStar II Plus apparatus. The carbonaceous materials were degassed at 300 °C for 12 h under high vacuum (ca. 0.1 Pa) on a Micromeritics Smart VacPrep apparatus before physisorption measurements.
Raman spectroscopy was carried out on a Renishaw inVia Raman microscope with a 532 nm laser excitation (IEMM, Univ. Montpellier). The first-order Raman spectra (1000–2000 cm−1) were fitted using the procedure proposed in the literature by A. Sadezky et al. for soot and related carbonaceous materials.44 The curve fitting was performed using OriginPro® after removing the background by using a Gaussian function for the band at 1500 cm−1 and Lorentz functions for the other four bands. We used the as-obtained fitting to determine La using the Tuinstra–Koenig correlation.45,46
TPD–MS analysis which provides a quantitative description of the number of surface groups was performed using a laboratory (IS2M) constructed equipment. A mass of ∼5 mg of (Bn)2-P900 was heated in a quartz tube in a furnace under a secondary vacuum of 10−6–10−7 mmHg from 25 to 950 °C with a heating rate of 5 °C min−1. The released gases were quantitatively detected and analysed using a quadrupole mass spectrometer (MS) that is already calibrated with the following reference gases, i.e., H2, NH3, H2O, CO, N2, NO and CO2. Before each analysis, N2 sensibility measurement was performed to allow further quantification of other gases.47,48 The active surface area (ASA) was determined using the same device. After the first TPD-MS experiment which led to the removal of all functional groups, the material was exposed to dioxygen chemisorption at 300 °C for 10 hours to form new oxygenated surface complexes. Subsequently, another TPD–MS experiment was conducted at 950 °C with a heating rate of 10 °C min−1 to assess the number of oxygenated groups and the ASA.48,49
2.5. Electrode preparation and electrochemical testing
Slurries were formulated to make the electrode materials by combining 90 wt% active material, 5 wt% carbon black to ensure electronic percolation, and 5 wt% binder poly(vinylidene fluoride). As a solvent, N-methyl-2-pyrrolidone (NMP) was added to the powder and mixed by ball milling (PULVERISETTE 7) at 500 rpm for 1 h before spreading the suspension on copper foil and drying it at 60 °C for 8 h under air. The electrodes were cut with a precision puncher with a 11 mm diameter and vacuumed at 80 °C for 12 h before being transferred to an Ar-filled glove box for assembly in coin cells. The active material loading mass was approximately 2 mg cm−2. The CR2032 304L coin cells consisted of the electrode against metallic lithium and potassium as counter and reference electrodes, while LiPF6 and KPF6 dissolved in (1 M and 0.8 M for Li and K, respectively) EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DEC (50:50 by volume) served as the electrolytes. A microporous trilayer (PP/PE/PP, from Celgard) and glass fiber membranes (GF/D, from Whatman) were used as separators. The cycling tests of the assembled coin cells were carried out on a BioLogic VMP3 battery cycler at controlled room temperature, within the potential range 0.01–3 V, and galvanostatic cycling was performed with potential limitation at constant current density (1C corresponding to 230 mA g−1) and various C-rates.
:DEC (50:50 by volume) served as the electrolytes. A microporous trilayer (PP/PE/PP, from Celgard) and glass fiber membranes (GF/D, from Whatman) were used as separators. The cycling tests of the assembled coin cells were carried out on a BioLogic VMP3 battery cycler at controlled room temperature, within the potential range 0.01–3 V, and galvanostatic cycling was performed with potential limitation at constant current density (1C corresponding to 230 mA g−1) and various C-rates.
3. Results and discussion
We synthesized three original imidazolium tetraphenylborate salts with three different imidazolium cations: 1-butyl-3-methylimidazolium, [Bmim][BPh4]; 1-benzyl-3-methylimidazolium, [(Bn)mim][BPh4] (Fig. S1†) and 1,3-bis(benzyl)imidazolium, [(Bn)2im][BPh4] (Fig. S2†). These salts were fully characterized by 1H (Fig. S3–S5†), 13C (Fig. S6–S8†) and 11B NMR (Fig. S9–S11†), FTIR spectroscopy (Fig. S12–S14†) and X-ray diffraction (Fig. S15†). The three salts were highly pure and crystalline and obtained with reasonable yields, above 60 wt%. We used these tailored imidazolium tetraphenylborate salts as original precursors for the preparation of boron, nitrogen co-doped carbon materials.3.1. Pyrolytic thermal decomposition of the imidazolium tetraphenylborate salts
To better understand the pyrolytic carbonization of the three imidazolium tetraphenylborate salts, we studied their thermal decomposition behaviour by differential scanning calorimetry (DSC) and thermogravimetric analyses (TGA). DSC and TGA were carried out from 20 to 1100 °C with a heating rate of 10 °C min−1 under an argon flow. The three tetraphenylborate salts displayed a clear endothermic peak between 130 and 150 °C (Fig. 1) corresponding to the melting point. The melting of the salt with the less aromatic imidazolium cation, i.e., [Bmim][BPh4], required less heat energy (36.56 J g−1 at 131.1 °C; Fig. 1a) compared with the two other salts bearing at least one benzyl group on the cation, i.e., [(Bn)mim][BPh4] (48.10 J g−1 at 150.4 °C; Fig. 1b) and [(Bn)2im][BPh4] (44.05 J g−1 at 145.1 °C; Fig. 1c and Table 1). Surprisingly, [(Bn)mim][BPh4] displayed a higher melting point and associated heat energy compared with [(Bn)2im][BPh4]. It is worth noting that all salts were ground before analyses; [(Bn)mim][BPh4] was, however, in the form of small crystals while [(Bn)2im][BPh4] appeared as a fine powder. This feature may explain the small differences observed in terms of the melting point and heat of energy. At higher temperature, above 250 °C, the thermal decomposition of the three salts showed clear differences depending on the nature of the imidazolium cation (Fig. 1). The salt with the less aromatic imidazolium cation, i.e., [Bmim][BPh4], underwent one-step thermal decomposition (Fig. 1a), while the other two salts showed two-step decomposition (Fig. 1b and c). One may notice that the benzyl side groups on the imidazolium cations are most probably subject to Hofmann elimination, which may explain the two-step decomposition with a first exothermic peak below 300 °C. Moreover, the thermal decomposition of [Bmim][BPh4] was less exothermic (121.7 J g−1) compared with the other two salts, i.e., 141.3 J g−1 for [(Bn)mim][BPh4] and 251.4 J g−1 for [(Bn)2im][BPh4]. From these values, the stability of the obtained materials increases from Bm-P900 to (Bn)2-P900. The presence of two benzyl groups on the imidazolium cation in [(Bn)2im][BPh4] clearly favors the carbonization of the salt into a very stable carbonaceous structure and allowed reaching a higher carbonization mass yield. [(Bn)2im][BPh4] reached a total weight loss of 87.7 wt% at 600 °C, which is significantly lower than the ones reached for [Bmim][BPh4] and [(Bn)mim][BPh4] at the same temperature, 96.2 and 91.7 wt%, respectively (Table 1). Interestingly, the total weight losses reached at 900 °C for [Bmim][BPh4] and [(Bn)mim][BPh4] were slightly lower than the total weight losses reached at 600 °C. This behaviour suggests a partial oxidation of boron atoms, as mentioned above, and further supports the hypothesis of a more stable carbonaceous structure when starting from a highly aromatic imidazolium cation, namely [(Bn)2im][BPh4]. At higher temperature, between 600 and 800 °C, the three tetraphenylborate salts displayed a clear exothermic peak (Fig. 1). This peak, which is not associated with a weight loss, may be attributed to aromatization and microstructure rearrangement of the carbon-based framework. For the salt bearing two benzyl groups on the cation, i.e., [(Bn)2im][BPh4], the aromatization step started at higher temperature (662 °C) and released less heat energy (175.5 J g−1; Fig. 2c and Table 1) compared with the other two salts. The asymmetry of both [Bmim][BPh4] and [(Bn)mim][BPh4] might be responsible for higher heat energy released during aromatization, 324.4 and 367.3 J g−1, respectively. As mentioned above, the more aromatic character and the high symmetry of [(Bn)2im][BPh4] clearly favour the carbonization of the salt into a very stable carbonaceous structure, most likely more aromatic than the ones obtained from [Bmim][BPh4] and [(Bn)mim][BPh4], as will be discussed later, and allowed reaching a higher carbonization mass yield.| Imidazolium tetraphenylborate salt | Melting (M) | Decomposition (D) | Aromatization (A) | Weight loss (wt%) | ||||
|---|---|---|---|---|---|---|---|---|
| T M (°C) | Q M (J g−1) | T D (°C) | Q D (J g−1) | T A (°C) | Q A (J g−1) | @600 °C | @900 °C | |
| [(Bn)2im][BPh4] | 145.1 | 44.1 | 294 + 353 | 251.4 (126.9 + 124.5) | 662 | 175.5 | 87.7 | 87.6 |
| [(Bn)mim][BPh4] | 150.4 | 48.1 | 300 + 363 | 141.3 (109.6 + 31.7) | 619 | 367.3 | 91.7 | 89.8 |
| [Bmim][BPh4] | 131.1 | 36.6 | 332.5 | 121.7 | 636 | 324.4 | 96.2 | 94.5 |
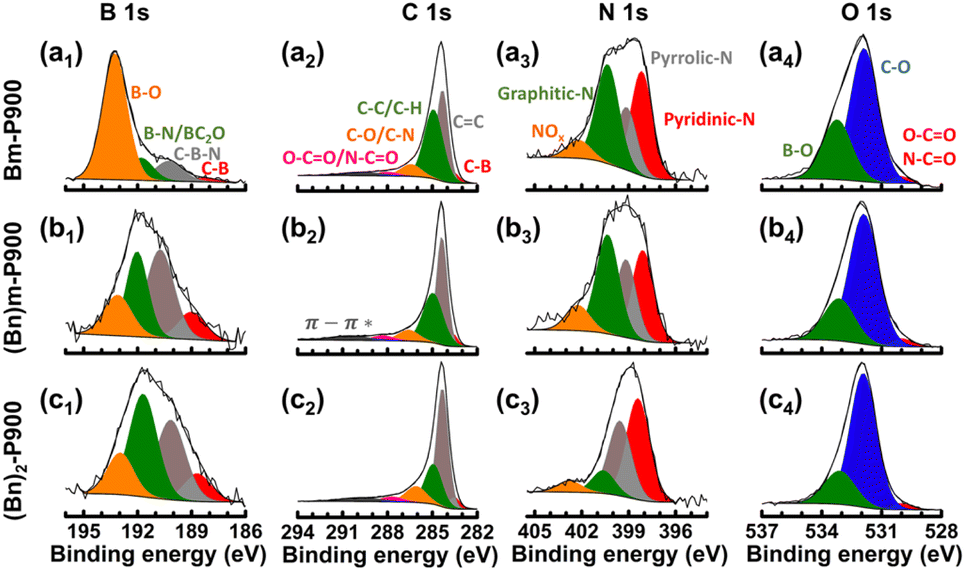 | ||
| Fig. 2 XPS spectra of (a) Bm-P900, (b) (Bn)m-P900 and (c) (Bn)2-P900. | ||
To provide better insights into the thermal decomposition mechanism of the three imidazolium tetraphenylborate salts, we studied their volatile decomposition products by TGA-MS (Fig. S16†). Based on the most prominent masses observed by mass spectrometry, two major volatile decomposition products and their fragments were clearly identified: benzene (m/z = 78, 77, 52, 51, 50, 39) and benzyl derivatives (m/z = 92, 91, 65, 63). The TG-MS curves showing the two most prominent masses, i.e., m/z = 78 (benzene) and 91 (tropylium), are presented in Fig. S16† for the three imidazolium tetraphenylborate salts. While the presence of benzene fragments comes from the decomposition of the tetraphenylborate anions, the presence of benzyl derivatives from the thermal decomposition of [Bmim][BPh4] is questionable. In a previous study on the thermal decomposition of alkylammonium tetraphenylborate salts,50 the authors showed similar results with several exothermic peaks and the presence of benzene and alkylbenzenes in the volatiles. The authors suggested the formation of alkylamine phenylborate intermediates by the dealkylation of the ammonium cations and the subsequent alkylation of the benzene moieties originating from the BPh4− anion. One may note that for [Bmim][BPh4], benzene fragments (m/z = 78; centered at ca. 360 °C) appeared at a lower temperature as compared to tropylium (m/z = 91; centered at ca. 388 °C). This feature is in good agreement with the exothermic signal observed between 330 and 380 °C (Fig. 1), which is most probably related to the formation of the alkylamine phenylborate intermediates. Despite observations made previously by TGA, this feature supports two-step decomposition for [Bmim][BPh4]. For both [(Bn)mim][BPh4] and [(Bn)2im][BPh4], benzene and tropylium fragments appeared concurrently, i.e., at ca. 330 and 315 °C, respectively. This feature is in good agreement with the first exothermic signal observed by DSC for each salt (Fig. 1). For these two salts, the signal related to tropylium lasted up to over 400 °C and might be related to the cyclization of the benzylamine phenylborate intermediates into carbon-rich borazine-like scaffolds, as suggested for alkylammonium tetraphenylborate salts.50 This might explain the second exothermic peak observed by DSC between 350 and 450 °C. Besides, the formation of N-(alkyl/benzyl)imidazolium carbene-borate adducts cannot be ruled out herein. Further characterization will have to be carried out to elucidate the full thermal decomposition mechanism of these three imidazolium tetraphenylborate salts.
3.2. Chemical composition of the B, N co-doped carbon materials
In order to produce larger quantities and fully characterize the carbon-based materials, the three salts were pyrolyzed in a tube furnace at 900 °C for 7 h under argon. It is worth noting that the carbonization yields obtained were slightly different from the ones obtained from DSC/TGA. This feature can be explained by the differences in terms of dimensioning of the set-up (sample mass, volume of the enclosure, etc.) and program of the thermal treatment (plateau, heating rate, gas flow rate, etc.). However, the trends were very similar and confirmed the observations made previously from DSC/TGA experiments. Thus, [(Bn)2im][BPh4] led to the highest carbonisation yield of ca. 15 wt% as compared to the other two salts ([(Bn)mim][BPh4] (ca. 3.6 wt%) and [Bmim][BPh4] (ca. 1.1 wt%)). The as-obtained carbon-based materials were named (Bn)2-P900, (Bn)m-P900 and Bm-P900, respectively. In order to evaluate the fate of heteroatoms (B and N) during pyrolysis, elemental analyses were performed on the three carbon-based materials (Table 2). The doping level of nitrogen for (Bn)2-P900 and (Bn)m-P900, i.e., the two materials derived from the salts carrying at least one benzyl group on the imidazolium cation, was ca. 16 wt%. This percentage was slightly lower, ca. 13 wt%, for Bm-P900. Following the same trend, the percentage of boron obtained from ICP-OES analyses was 8.9 and 9.8 wt% for (Bn)2-P900 and (Bn)m-P900, respectively, and only 7 wt% for Bm-P900 (Table 2). Similarly, both N/C and B/C mass ratios determined from elemental analyses (Table 2) were slightly higher for (Bn)2-P900 and (Bn)m-P900 as compared to Bm-P900. We observed another interesting behaviour concerning the oxygen content. While the amount of oxygen in both (Bn)2-P900 and (Bn)m-P900 was negligible, it reached 8.3 wt% for Bm-P900 (Table 2). This behaviour suggests partial post-oxidation of Bm-P900 and further supports the hypothesis of more stable carbonaceous frameworks when starting from highly aromatic imidazolium cations.| Sample | Elemental composition (wt%) | Mass ratio | C yieldg (wt%) | d 002 (Å) | Raman shift (cm−1) | I D/IGj | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ca | Ha | Na | Oa | Bb | N/Cd | B/Ce | O/Cd | G-band | D-band | ||||
| a Data obtained from combustion elemental analyses. b Data extracted from combustion elemental analyses by difference, considering that boron is the only element present in addition to carbon, hydrogen, nitrogen and oxygen. c In brackets: data obtained from ICP-OES analyses. d Mass ratio calculated from combustion elemental analyses' data. e Mass ratio calculated from combustion elemental (for C) and ICP-OES (for B) analyses' data. f Mass ratio calculated from XPS data. g Carbonization yield and standard deviation calculated from the results of three replicates. h Interlayer spacing, d002, determined from XRD patterns applying Bragg's law. i In brackets: XRD (002) peak position determined after fitting (unit: 2θ degree). j Intensity ratio of the D-band and the G-band obtained by Raman spectroscopy. | |||||||||||||
| (Bn)2-P900 | 72.8 | 1.6 | 16.4 | n/a | 9.2 (8.9)c | 0.23 | 0.12 | n/a (0.13)f | 14.7 ± 0.9 | 3.7 (24.3)i | 1586 | 1365 | 1.42 |
| (Bn)m-P900 | 72.1 | 2.5 | 15.7 | n/a | 9.7 (9.6)c | 0.22 | 0.13 | n/a (0.12)f | 3.6 ± 0.5 | 3.8 (23.4)i | 1584 | 1365 | 1.47 |
| Bm-P900 | 68.4 | 2.1 | 12.8 | 8.3 | 8.4 (7.0)c | 0.19 | 0.10 | 0.12 (0.17)f | 1.1 ± 0.1 | 3.8 (23.4)i | 1580 | 1358 | 1.49 |
X-ray photoelectron spectroscopy (XPS) was used to study the surface chemistry of the three B, N co-doped carbon materials. For the three samples, the signals of B 1s (ca. 191 eV), C 1s (ca. 284 eV), N 1s (ca. 399 eV), O 1s (ca. 535 eV) and O KL (ca. 978 eV) were recorded, confirming the co-existence of B, C, N and O atoms (Fig. 2). In particular, the C 1s spectra of the three materials showed a major contribution related to sp2 hybridized carbon atoms (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C)51 at 284.3 eV. This contribution was significantly higher for (Bn)2-P900 (45.9 at%) as compared to (Bn)m-P900 (39.2 at%) and Bm-P900 (31.0 at%). This feature further evidenced the superior aromatic character of the carbon-based framework when starting from highly aromatic imidazolium cations, i.e., [(Bn)2im][BPh4] and, to a lesser extent, [(Bn)mim][BPh4]. Another contribution centred at 286.1 eV was consistently observed, confirming the presence of C–N bonds.52 Besides, N 1s spectra could be deconvoluted into four peaks located at 398.4 eV, 399.6 eV, 400.6 eV and 402.8 eV, which were assigned to pyridinic-N, pyrrolic-N, graphitic-N and NOx respectively.53,54 While the contribution of graphitic-N was the most important for (Bn)m-P900 and Bm-P900, this contribution was minor for (Bn)2-P900. As mentioned above, the relative instability of the benzyl groups on the imidazolium cations of [(Bn)2im][BPh4], which are subject to Hofmann elimination, might be responsible for this clear difference of the environment of the nitrogen atoms in (Bn)2-P900. We also observed clear differences for boron. B 1s spectra could be deconvoluted into four peaks at 188.7 eV, 190.2 eV, 191.7 eV and 193.0 eV corresponding to B–C, C–B–N, B–N and B–O, respectively.55 The contribution of B–O was significantly higher for Bm-P900 (2.80 at%) than for (Bn)2-P900 and (Bn)m-P900 (0.6 at%) (Table S1†), which is in perfect agreement with the observations made earlier. This feature was confirmed by O 1s spectra. It is worth noting that the O/C mass ratios determined by XPS were significantly higher than the ones obtained from elemental analyses. In particular, the oxygen content was clearly overestimated by XPS for (Bn)m-P900 and (Bn)2-P900 (Table 2). This feature might be due to charge effects that can cause partial oxidation of the samples despite charge compensation.
C)51 at 284.3 eV. This contribution was significantly higher for (Bn)2-P900 (45.9 at%) as compared to (Bn)m-P900 (39.2 at%) and Bm-P900 (31.0 at%). This feature further evidenced the superior aromatic character of the carbon-based framework when starting from highly aromatic imidazolium cations, i.e., [(Bn)2im][BPh4] and, to a lesser extent, [(Bn)mim][BPh4]. Another contribution centred at 286.1 eV was consistently observed, confirming the presence of C–N bonds.52 Besides, N 1s spectra could be deconvoluted into four peaks located at 398.4 eV, 399.6 eV, 400.6 eV and 402.8 eV, which were assigned to pyridinic-N, pyrrolic-N, graphitic-N and NOx respectively.53,54 While the contribution of graphitic-N was the most important for (Bn)m-P900 and Bm-P900, this contribution was minor for (Bn)2-P900. As mentioned above, the relative instability of the benzyl groups on the imidazolium cations of [(Bn)2im][BPh4], which are subject to Hofmann elimination, might be responsible for this clear difference of the environment of the nitrogen atoms in (Bn)2-P900. We also observed clear differences for boron. B 1s spectra could be deconvoluted into four peaks at 188.7 eV, 190.2 eV, 191.7 eV and 193.0 eV corresponding to B–C, C–B–N, B–N and B–O, respectively.55 The contribution of B–O was significantly higher for Bm-P900 (2.80 at%) than for (Bn)2-P900 and (Bn)m-P900 (0.6 at%) (Table S1†), which is in perfect agreement with the observations made earlier. This feature was confirmed by O 1s spectra. It is worth noting that the O/C mass ratios determined by XPS were significantly higher than the ones obtained from elemental analyses. In particular, the oxygen content was clearly overestimated by XPS for (Bn)m-P900 and (Bn)2-P900 (Table 2). This feature might be due to charge effects that can cause partial oxidation of the samples despite charge compensation.
Overall, we showed that the three original imidazolium tetraphenylborate salts reported herein allowed synthesizing carbon-rich materials with high nitrogen and boron doping levels as compared to other IL and salt precursors used in the literature.37–39,43 Besides, we showed that the presence of two benzyl groups on the imidazolium cation increased the carbonization yield and could protect boron atoms from partial post-oxidation.
3.3. Microstructure of the B, N co-doped carbon materials
Beyond chemical composition, we studied the impact of the nature of the imidazolium tetraphenylborate salt on the microstructure of the B, N co-doped carbon materials. By DSC (Fig. 1), we observed a clear exothermic peak between 600 and 800 °C that was attributed to microstructure rearrangement of the carbon-based framework and suggested slight differences between the three samples. To provide deeper insights into their textural properties and microstructure, we characterized the carbon-based materials by transmission electron microscopy (TEM and high resolution HR-TEM; Fig. S17† and 3), carbon dioxide sorption at 273 K (Fig. S18†), X-ray diffraction (XRD; Fig. 4a) and Raman spectroscopy (Fig. 4b). The observations made by TEM at low magnification on the three materials (Fig. S17†) show rather dense particles with no visible sign of mesopores (pore diameters between 2 and 50 nm). Regarding the possible presence of micropores (pore diameters below 2 nm), this cannot be visualized by TEM. In this purpose, carbon dioxide sorption at 273 K was performed for each sample. The unusual shape of the as-obtained isotherms, with no adsorption at low relative pressures (Fig. S18†), strongly suggests the absence of micropores. One may, however, note that the total pore volumes of Bm-P900 and (Bn)m-P900 (ca. 0.03 cm3 g−1 at p/p0 = 0.029) are three times higher than the one obtained for (Bn)2-P900 (ca. 0.01 cm3 g−1). | ||
| Fig. 3 HR-TEM micrographs of (a) Bm-P900, (b) (Bn)m-P900, and (c and d) (Bn)2-P900. | ||
 | ||
| Fig. 4 (a) XRD patterns of the three B, N co-doped carbon materials and Raman spectra of (b) Bm-P900, (c) (Bn)m-P900 and (d) (Bn)2-P900. | ||
HR-TEM micrographs obtained at high magnifications for the three materials (Fig. 3) reveal disordered-like microstructures that seem to be embedded in an amorphous carbon matrix. Some short graphenic domains made of 2–3 stacked carbon layers were observed (Fig. 3d). This feature was confirmed by XRD and Raman spectroscopy. The diffraction peak corresponding to the interplane distance (002) is centred at 24.3° (d002 = 3.7 Å) for (Bn)2-P900, at 23.3° (d002 = 3.8 Å) for (Bn)m-P900 and at 23.4° (d002 = 3.8 Å) for Bm-P900 (Table 2, Fig. 4a and S19†). For graphite, this peak is found at about 26.5° (d002 = 3.4 Å). The inter-plane distance (002) is therefore clearly larger for these three carbon-based materials than that usually observed for pristine graphite, supporting disordered carbonaceous microstructures. This shift was more pronounced for Bm-P900 (0.4 Å) and (Bn)m-P900 (0.4 Å) as compared to (Bn)2-P900 (0.3 Å). Once again, this feature further confirms the importance of the benzyl group(s) on the imidazolium cation. The second peak centred at ca. 43° and characteristic of the (10) plane of graphite is broad and not very intense, confirming the above findings.38 The correlation lengths of the graphite-like crystalline domains, La (in the basal plane) and Lc (perpendicular to the basal plane), determined by applying the Debye–Scherrer formula are ca. 3 and 1 nm for each material.
Similarly, Raman spectra obtained by irradiation with a 532 nm wavelength laser clearly indicate poorly graphitized microstructures for the three materials (Fig. 4b–d). The first-order Raman spectra (1000–2000 cm−1) were fitted using the procedure proposed in the literature by A. Sadezky et al. for soot and related carbonaceous materials44 (Fig. 4b–d and Table S2†). For each sample, the curve fitting allowed the deconvolution of the signal into five bands: the G-band related to ideal graphitic lattices (E2g-symmetry); the D-band (A1g-symmetry), D′-band (E2g-symmetry) and I-band (A1g-symmetry) related to disordered graphitic lattices; the D′′-band related to amorphous carbon domains. The clear appearance of both the I-band (at 1200 cm−1) and the D′′-band (at 1500 cm−1) for the three samples strongly suggests the presence of a non-negligible fraction of amorphous carbon and/or polyene-like structures.44 As mentioned above, the G-band, at ca. 1580 cm−1, results from the transverse and longitudinal stretching modes of the ideal graphitic lattices; conversely, the D-band, in the range 1300–1350 cm−1, corresponds to second order double resonant modes and is closely related to lattice disturbances at the edges of graphene layers. Thus, the more intense the D-band, the more the sp2-hybridized carbon structure is disturbed by such defects. As a direct consequence, the intensity ratio of the D- and G-bands (ID/IG) is frequently used to evaluate the number of defects and/or the in-plane correlation length (La) in carbon-based microstructures.56 Herein, (Bn)2-P900 showed the lowest ID/IG ratio of 1.42. In contrast, (Bn)m-P900 and Bmim-P-900 showed the highest ID/IG ratios of 1.47 and 1.49, respectively. According to the Tuinstra–Koenig correlation,45,46 these ID/IG ratios correspond to in-plane correlation lengths (La) of ca. 3.6 nm for (Bn)2-P900 and 3.4 nm for (Bn)m-P900 and Bmim-P-900. These values are in good agreement with the ones determined by XRD. Once again, the presence of at least one benzyl group on the imidazolium cation seems to promote aromatization and microstructure rearrangement into an extended π-conjugated network. We also observed significant differences regarding the Raman band position (Table 2). The G-band appeared at 1586 cm−1 for (Bn)2-P900, at 1584 cm−1 for (Bn)m-P900 and at 1580 cm−1 for Bm-P900. Regarding the D-band, it appeared at 1365 cm−1 for (Bn)2-P900 and (Bn)m-P900 and at 1358 cm−1 for Bm-P900. Although the position of these bands is close to the ones usually observed for pristine graphite (i.e., the G-band at ca. 1580 cm−1 and the D-band in the range 1300–1350 cm−1), significant shifts were observed. The G-band underwent a 6 and 4 cm−1 upshift for (Bn)2-P900 and (Bn)m-P900, respectively, i.e., the two materials derived from the imidazolium salts bearing at least one benzyl group; conversely, no significant shift was observed for Bm-P900. Furthermore, the D-band underwent a 15 cm−1 upshift for (Bn)m-P900 and (Bn)2-P900, respectively. A smaller upshift of 8 cm−1 was observed for Bm-P900. Although not yet clarified, the shifts observed in both the G- and D-bands may be attributed to various features. As mentioned by Fellinger et al.,38 the position of these bands is directly related to bond strength and coupling in the carbonaceous framework. According to Hagio et al.57 and Yang et al.,58 these shifts can be attributed to structural defects and new types of disorders inlaid in the carbon sheets. Overall, one may note that these shifts were more pronounced for (Bn)2-P900 and (Bn)m-P900 compared with Bm-P900. Consequently, one may assume that these shifts are related to efficient heteroatom-doping.
3.4. Surface chemistry of (Bn)2-P900
In view of potential applications, in particular as an electrode material for electrochemical energy storage devices, we studied the surface chemistry of (Bn)2-P900 by thermal programmed desorption coupled with mass spectroscopy (TPD-MS). For this study, we selected (Bn)2-P900 which seems to be a more promising candidate as compared to (Bn)m-P900 and Bm-P900. Indeed, (Bn)2-P900 showed better structural organisation and chemical stability, especially with respect to post-oxidation. For the sake of atom economy, (Bn)2-P900 is also more relevant as it gives B, N co-doped carbons with a higher yield.Fig. S20† shows the TPD-MS profile of the desorbed gases during heating under vacuum for (Bn)2-P900. The relative amount of each desorbed gas can be seen in Fig. S20b.† The oxygen-based functional groups were decomposed into CO, CO2, H2O, H2, NO and NH3. The groups were attributed according to their decomposition temperature described in the literature.59 The desorption profiles show a small continuous peak of CO2 desorption between ∼400 and 900 °C indicating decomposition of lactones and anhydrides. The CO desorption exhibits a well-defined and intense peak in the range of 600 to 950 °C coming from the degradation of ethers, phenols and quinones. The total amount of oxygenated groups (CO + CO2) is 3.73 mmol g−1 which is higher than the values reported in the literature for graphite and hard carbon materials47,60 (less than 0.11 and 0.55 mmol g−1, respectively). In view of potential applications as an electrode material for electrochemical energy storage devices, such a high amount of oxygenated groups may cause irreversible capacity due to their interactions with the electrolyte as proved in sodium-ion batteries (NIBs).49 Besides, H2O desorbed in the low temperature region (35–300 °C) resulting from both the desorption of physisorbed H2O and the chemical decomposition of NH4OH into H2O and NH3. It is worth noting that the NH3 desorption presents a maximum at ca. 350 °C and stops at ca. 600 °C, indicating the decomposition of N-based functional groups and confirming the presence of nitrogen species at the surface of the material. The boron signature was not detected using this technique. In addition, H2 was released starting from a relatively low temperature (ca. 500 °C). This feature is most probably due to the cleavage of C–H bonds and confirms the presence of defects, either sp3-hybridized carbon atoms at the edges of disordered graphitic lattices, polyene-like structures or amorphous carbon domains.
Surface defects can be involved in the storage mechanism of lithium-ion batteries (LIBs).60 Therefore, quantification of the edge defects was done by the measurement of the active surface area (ASA) of (Bn)2-P900. An ASA of ca. 10 m2 g−1 was measured, which is higher than the ones reported in the literature for graphite and some hard carbon materials.47,60 However, such values might be explained: on one hand by the nature of the precursor used, usually precursors rich in heteroatoms leading to higher ASA values;44 and on the other hand by the low pyrolysis temperature (900 °C), which is known to result in materials with higher active surface areas.61 Note that a high ASA usually causes high irreversibility in the first cycle in battery cycling.49,60
3.5. Electrochemical performance of (Bn)2-P900 as a negative electrode versus Li+/Li
Due to singular properties, B, N co-doped carbons have been anticipated as serious candidates for applications as electrode materials, especially in metal-ion batteries.29,30 To evaluate the electrochemical properties of the B, N co-doped carbons reported herein, electrochemical tests were carried out versus Li+/Li within a potential range of 0.01–3 V. For the reasons mentioned above, we selected (Bn)2-P900 to carry out the electrochemical study. Composite electrodes were prepared as described in the Experimental part. Fig. 5a shows the electrochemical behavior of (Bn)2-P900 at a 1C rate in different discharge–charge cycles. A high initial discharge capacity was delivered, reaching 476 mA h g−1. However, the discharge capacity decreased to 221 mA h g−1 in the second discharge–charge cycle, corresponding to a low first cycle coulombic efficiency (CE) (54%). Then the capacity gradually stabilized for the subsequent cycles. The low first cycle CE is linked to the solid electrolyte interphase (SEI) formation, which corresponds to the electrolyte decomposition on the electrode surface. In Fig. 5a, the shape of the discharge–charge profiles changed after the first charge as confirmed by the differential capacity dQ/dV profiles of the 1st and 2nd cycles (Fig. S21†), with three irreversible cathodic peaks that disappeared during the 2nd cycle. In addition, the shape of the charge–discharge curves presented a main distinct slope (3 to 0.3 V), which could indicate a pseudo-capacitive behavior of (Bn)2-P900. Furthermore, the half-cell was cycled at a fast rate of 1C for 300 cycles to evaluate the negative electrode material's long-term cycling durability. The results are given in Fig. 5b. (Bn)2-P900 demonstrated a high reversible capacity of 240 mA h g−1 after 300 cycles, and the coulombic efficiency (CE) was always greater than 99.6%, after the first cycle (CE = 54%). The expected high conductivity, low specific surface area and chemical stability of (Bn)2-P900 most probably contributed to the excellent cycling performance.62 As mentioned previously, the carbon dioxide sorption isotherm recorded for (Bn)2-P900 at 273 K (Fig. S17†) strongly suggests the absence of micropores, which could avoid SEI growth. Additionally, the high heteroatom co-doping level observed in (Bn)2-P900 can enhance electronic percolation in the carbon-based scaffold, enabling fast charging and making it suitable for high-power electrochemical energy storage devices. | ||
| Fig. 5 (a) Galvanostatic discharge/charge profiles at 1C of the 1st, 3rd, 10th, 100th, 200th, and 300th cycles, (b) capacity retention and coulombic efficiency upon 300 cycles at 1C, (c) rate performance and (d) capacity retention and coulombic efficiency upon 1400 cycles at 30C obtained between 3 and 0.01 V for (Bn)2-P900 versus Li. | ||
The first 200 cycles presented a slight increase in capacity, which could be assigned to two main reasons behind the activation process. First, the limited active surface area of (Bn)2-P900 is likely progressively impregnated by the electrolyte, and second, with continuous insertion–extraction of Li ions, the interlayer spacing of the crystallized part probably expanded, which is beneficial to further Li ion transport between the electrode and electrolyte; this feature has been observed in previous reports.63–65
To further highlight the promising electrochemical properties of (Bn)2-P900, rate performance was evaluated as shown in Fig. 5c. The rate capability tests were performed at incremental C-rates from C/2 to 30C, with ten discharge–charge cycles for each C-rate. (Bn)2-P900 shows fast-rate capabilities, with 234 mA h g−1 at C/20 and 128 mA h g−1 at very high 30C (6.9 A g−1), which is consistent with high electronic conductivity and Li ion transport. Fig. 5c also shows that at a low rate (C/2) the CE takes more cycles to stabilize, corresponding to the SEI growth (10 cycles to reach a CE of 99%). These results confirm that (Bn)2-P900 could be adapted for high-power applications. Based on the comparative electrochemical performance analysis presented in Table S3,† it is evident that the (Bn)2-P900 material exhibits very high rate capability with a capacity of 128 mA h g−1 at an ultra-high current density of 6.9 A g−1 (which is the highest current density reported in Table S3†). (Bn)2-P900 also exhibits remarkable capacity retention of 80% after 1400 cycles, demonstrating its robustness. Although its capacity is slightly lower than that of B-doped graphene2 and N-doped carbon xerogels,4 it outperforms B-doped carbon fibers and N-doped graphene in terms of capacity retention. It should be noted, however, that direct comparison of electrochemical performance across different materials could be challenging due to the several parameters affecting the performance, especially in the electrode preparation and electrochemical testing conditions. For a more accurate comparison, we prepared a boron-free, nitrogen-doped carbon starting from 1,3-bis(benzyl)imidazolium chloride, namely [(Bn)2im]Cl. After pyrolysis at 900 °C, [(Bn)2im]Cl yielded a carbon material, namely (Bn)2-Cl-P900, with 11.7 wt% of nitrogen, with almost no oxygen and without boron (Table S4†). (Bn)2-Cl-P900 was tested as a negative electrode versus Li+/Li (Fig. S22†). It showed a good rate capability of 115 mA h g−1 at 10C, compared to 130 mA h g−1 for (Bn)2-P900. However, (Bn)2-Cl-P900 showed a poor rate capability below 150 mA h g−1 at C/2, compared to ca. 225 mA h g−1 for (Bn)2-P900. Moreover, the capacity recovery at C/2 was only 90% after 70 cycles for (Bn)2-Cl-P900, while it exceeded 109% for (Bn)2-P900 (Fig. S22†). The higher capacity obtained for (Bn)2-P900 might be due to the aromatic structure of the precursors and the effective co-doping with nitrogen and boron atoms that might enhance electronic percolation in the carbon-based scaffold. This offers easy lithium insertion/adsorption without significant structural damage. Besides, the increasing capacity retention during cycling and the more pronounced capacity loss between rate changes observed for (Bn)2-P900 could be explained by a slower electrochemical activation process as compared to (Bn)2-Cl-P900. This feature was previously observed by L. Qie et al.66 and can be explained by high heteroatom-doping levels.
As depicted by Fig. 5d, for the first 200 cycles at 30C, (Bn)2-P900 showed an excellent coulombic efficiency, with a value oscillating between 98.9 and 100%. After 1400 cycles, a capacity of 112 mA h g−1 is kept, corresponding to a retention of 80% of the starting capacity. Besides, a capacity of 78 mA h g−1 was measured at 1C versus K to compare to that measured versus Li, i.e., 248 mA h g−1 after 300 cycles (Fig. S23†). The high stripping/plating polarization of the K metal counter electrode can also limit the high-rate electrochemical performance of (Bn)2-P900, and better performance could be obtained in KIB without the presence of K metal.67 Moreover, electrolyte formulation should be studied to improve performance in K-ion batteries.
4. Conclusion
We reported the synthesis of original imidazolium salts with the tetraphenylborate anion and their use as precursors for boron, nitrogen co-doped carbon materials. We showed that the presence of two benzyl groups on the imidazolium cation increased the carbonization yield (up to 15 wt% after pyrolysis at 900 °C for 7 h under argon), promoted the heteroatom-doping (16 wt% of nitrogen and 9 wt% of boron), improved the microstructure order and protected boron atoms from oxidation. In particular, the B, N co-doped carbon prepared from 1,3-bis(benzyl)imidazolium tetraphenylborate, i.e., (Bn)2-P900, demonstrated fast-rate capabilities as an anode material for high-power monovalent ion batteries. After 1400 cycles at a high rate (30C) versus Li+/Li, a capacity of 112 mA h g−1 corresponding to a retention of 80% of the initial capacity was achieved. This result demonstrates that the materials reported herein could be adapted for fast-rate monovalent ion batteries and high-power applications, paving the way for the use of tailored aromatic imidazolium salts to engineer heteroatom-doped carbon materials with singular properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SA is grateful to Lebanese University for financial support. Several characterization experiments were performed with the support of the “Balard Plateforme d’Analyses et de Caractérisation” (PAC Balard). The authors would like to thank Amine Geneste for the DSC experiments, Emmanuel Fernandez for the solid-state 1H NMR experiments, Erwan Oliviero (MEA platform, Université de Montpellier) for HR-TEM analyses, Bernard Fraisse for the XRD analyses and Lucie Koeller for the ICP-OES analyses.Notes and references
- R. Rathanasamy, S. Sahoo, J. H. Lee, A. K. Das, M. Somasundaram, S. K. Palaniappan and S. Sivaraj, J. Electron. Mater., 2021, 50, 1845–1892 CrossRef CAS.
- H. Huang and X. Wang, J. Mater. Chem. A, 2014, 2, 6266–6291 RSC.
- Q. Wu, L. Yang, X. Wang and Z. Hu, Adv. Mater., 2020, 32, 1904177 CAS.
- C. Thamaraiselvan, J. Wang, D. K. James, P. Narkhede, S. P. Singh, D. Jassby, J. M. Tour and C. J. Arnusch, Mater. Today, 2020, 34, 115–131 CrossRef CAS.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839–2855 RSC.
- Y. Li, Y. Zhao, H. Cheng, Y. Hu, G. Shi, L. Dai and L. Qu, J. Am. Chem. Soc., 2012, 134, 15–18 CrossRef CAS PubMed.
- J. P. Paraknowitsch, J. Zhang, D. S. Su, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 87–92 CrossRef CAS PubMed.
- Y. H. Cao, H. Yu, J. Tan, F. Peng, H. J. Wang, J. Li, W. X. Zheng and N. B. Wong, Carbon, 2013, 57, 433–442 CrossRef CAS.
- Y. Zheng, Y. Jiao, M. Jaroniec, Y. G. Jin and S. Z. Qiao, Small, 2012, 8, 3550–3566 CrossRef CAS PubMed.
- Q. Yang, W. Xu, A. Tomita and T. Kyotani, J. Am. Chem. Soc., 2005, 127, 8956–8957 CrossRef CAS.
- Y. Shao, S. Zhang, M. H. Engelhard, G. Li, G. Shao, Y. Wang, J. Liu, I. A. Aksay and Y. Lin, J. Mater. Chem., 2010, 20, 7491–7496 RSC.
- H. J. Burch, J. A. Davies, E. Brown, L. Hao, S. A. Contera, N. Grobert and J. Ryan, Appl. Phys. Lett., 2006, 89, 143110 CrossRef.
- J. Wu, Z. Pan, Y. Zhang, B. Wang and H. Peng, J. Mater. Chem. A, 2018, 6, 12932–12944 RSC.
- H. C. Chen, F. G. Sun, J. T. Wang, W. C. Li, W. M. Qiao, L. C. Ling and D. H. Long, J. Phys. Chem. C, 2013, 117, 8318–8328 CrossRef CAS.
- D. C. Wei, Y. Q. Liu, Y. Wang, H. L. Zhang, L. P. Huang and G. Yu, Nano Lett., 2009, 9, 1752–1758 CrossRef CAS PubMed.
- Z. Pan, J. Ren, G. Guan, X. Fang, B. Wang, S. G. Doo, I. H. Son, X. Huang and H. Peng, Adv. Energy Mater., 2016, 6, 1600271 CrossRef.
- W. H. Shin, H. M. Jeong, B. G. Kim, J. K. Kang and J. W. Choi, Nano Lett., 2012, 12, 2283–2288 CrossRef CAS PubMed.
- B. Q. Wei, R. Spolenak, P. Kohler-Redlich, M. Ruhle and E. Arzt, Appl. Phys. Lett., 1999, 74, 3149–3151 CrossRef CAS.
- Y. Xia, X. H. Zhao, C. Xia, Z. Y. Wu, P. Zhu, J. Y. T. Kim, X. W. Bai, G. H. Gao, Y. F. Hu, J. Zhong, Y. Y. Liu and H. T. Wang, Nat. Commun., 2021, 12, 4225 CrossRef CAS PubMed.
- X. M. Yu, P. Han, Z. X. Wei, L. S. Huang, Z. X. Gu, S. J. Peng, J. M. Ma and G. F. Zheng, Joule, 2018, 2, 1610–1622 CrossRef CAS.
- R. X. Wang, D. J. Zhang, Y. M. Zhang and C. B. Liu, J. Phys. Chem. B, 2006, 110, 18267–18271 CrossRef CAS.
- J. A. Talla, Phys. B, 2012, 407, 966–970 CrossRef CAS.
- G. Keru, P. G. Ndungu and V. O. Nyamori, Mater. Chem. Phys., 2015, 153, 323–332 CrossRef CAS.
- J. Saloni, W. Kolodziejczyk, S. Roszak, D. Majumdar, G. Hill Jr and J. Leszczynski, J. Phys. Chem. C, 2010, 114, 1528–1533 CrossRef CAS.
- F. Ye, Y. Shi, W. Sun, K. Pang, M. Pu, L. Yang and H. Huang, Chem. Eng. J., 2023, 454, 140148 CrossRef CAS.
- A. Camisasca, A. Sacco, R. Brescia and S. Giordani, ACS Appl. Nano Mater., 2018, 1, 5763–5773 CrossRef CAS.
- H. Wang, Y. Cui, J. Shi, X. Tao and G. Zhu, Appl. Catal., B, 2023, 330, 122457 CrossRef CAS.
- X. Y. Li, X. P. Wang, G. Z. Xiao and Y. Zhu, J. Colloid Interface Sci., 2021, 602, 799–809 CrossRef CAS.
- Y. P. Hu, L. L. Shen, X. H. Wei, Z. Long, X. Q. Guo and X. Q. Qiu, Chemistryselect, 2019, 4, 6445–6450 CrossRef CAS.
- J. L. Zhu, P. C. Wei, J. Key, S. B. Yin, S. J. Ma and P. K. Shen, Sustainable Energy Fuels, 2019, 3, 478–487 RSC.
- Z. Chen, L. Q. Hou, Y. Cao, Y. S. Tang and Y. Li, Appl. Surf. Sci., 2018, 435, 937–944 CrossRef CAS.
- D. Guo, B. Ding, X. Hu, Y. H. Wang, F. Q. Han and X. L. Wu, ACS Sustainable Chem. Eng., 2018, 6, 11441–11449 CrossRef CAS.
- S. V. Sawant, S. Banerjee, A. W. Patwardhan, J. B. Joshi and K. Dasgupta, Int. J. Hydrogen Energy, 2020, 45, 13406–13413 CrossRef CAS.
- J. J. Adjizian, R. Leghrib, A. A. Koos, I. Suarez-Martinez, A. Crossley, P. Wagner, N. Grobert, E. Llobet and C. P. Ewels, Carbon, 2014, 66, 662–673 CrossRef CAS.
- R. Yuge, S. Bandow, M. Yudasaka, K. Toyama, S. Iijima and T. Manako, Carbon, 2017, 111, 675–680 CrossRef CAS.
- J. P. Paraknowitsch, A. Thomas and M. Antonietti, J. Mater. Chem., 2010, 20, 6746–6758 RSC.
- A. Aijaz, T. Akita, H. Yang and Q. Xu, Chem. Commun., 2014, 50, 6498–6501 RSC.
- T.-P. Fellinger, D. S. Su, M. Engenhorst, D. Gautam, R. Schlögl and M. Antonietti, J. Mater. Chem., 2012, 22, 23996–24005 RSC.
- R. Ma, B. Y. Xia, Y. Zhou, P. Li, Y. Chen, Q. Liu and J. Wang, Carbon, 2016, 102, 58–65 CrossRef CAS.
- Y. Chen, L. Fu, Z. Liu and Y. Wang, ChemCatChem, 2016, 8, 1782–1787 CrossRef CAS.
- J. S. Lee, H. M. Luo, G. A. Baker and S. Dai, Chem. Mater., 2009, 21, 4756–4758 CrossRef CAS.
- E. Bernhardt, M. Berkei, H. Willner and M. Schürmann, Z. Anorg. Allg. Chem., 2003, 629, 677–685 CrossRef CAS.
- J. Mehler, M. Ermer, U. Paap, B. S. Heller, F. Maier, H.-P. Steinrück, M. Hartmann, C. Korte, P. S. Schulz and P. Wasserscheid, Journal of Ionic Liquids, 2021, 1, 100004 CrossRef.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Carbon, 2005, 43, 1731–1742 CrossRef CAS.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- P. Kasaira Mubari, T. Beguerie, M. Monthioux, E. Weiss-Hortala, A. Nzihou and P. Puech, J. Carbon Res., 2022, 8, 4 CrossRef.
- A. Beda, F. Rabuel, M. Morcrette, S. Knopf, P.-L. Taberna, P. Simon and C. M. Ghimbeu, J. Mater. Chem. A, 2021, 9, 1743–1758 RSC.
- P. Brender, R. Gadiou, J.-C. Rietsch, P. Fioux, J. Dentzer, A. Ponche and C. Vix-Guterl, Anal. Chem., 2012, 84, 2147–2153 CrossRef CAS.
- A. Beda, C. Vaulot, F. Rabuel, M. Morcrette and C. M. Ghimbeu, Energy Adv., 2022, 1, 185–190 RSC.
- M. N. Akhtar and S. U. Sheikh, J. Therm. Anal., 1994, 41, 105–114 CrossRef CAS.
- M. Endo, C. Kim, K. Nishimura, T. Fujino and K. Miyashita, Carbon, 2000, 38, 183–197 CrossRef CAS.
- Y. Tang, X. Wang, J. Chen, X. Wang, D. Wang and Z. Mao, Energy Technol., 2020, 8, 2000361 CrossRef CAS.
- Y. Shang, Y. Ma, X. Chen, X. Xiong and J. Pan, Mol. Catal., 2017, 433, 128–135 CrossRef CAS.
- Z. S. Wu, A. Winter, L. Chen, Y. Sun, A. Turchanin, X. Feng and K. Müllen, Adv. Mater., 2012, 24, 5130–5135 CrossRef CAS.
- A. B. Bourlinos, G. Trivizas, M. A. Karakassides, M. Baikousi, A. Kouloumpis, D. Gournis, A. Bakandritsos, K. Hola, O. Kozak and R. Zboril, Carbon, 2015, 83, 173–179 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- T. Hagio, M. Nakamizo and K. Kobayashi, Carbon, 1989, 27, 259–263 CrossRef CAS.
- Q. H. Yang, P. X. Hou, M. Unno, S. Yamauchi, R. Saito and T. Kyotani, Nano Lett., 2005, 5, 2465–2469 CrossRef CAS.
- J. L. Figueiredo, M. Pereira, M. Freitas and J. Orfao, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
- C. M. Ghimbeu, C. Decaux, P. Brender, M. Dahbi, D. Lemordant, E. Raymundo-Pinero, M. Anouti, F. Béguin and C. Vix-Guterl, J. Electrochem. Soc., 2013, 160, A1907 CrossRef CAS.
- C. M. Ghimbeu, J. Górka, V. Simone, L. Simonin, S. Martinet and C. Vix-Guterl, Nano Energy, 2018, 44, 327–335 CrossRef.
- W. Lei, L. Han, C. Xuan, R. Lin, H. Liu, H. L. Xin and D. Wang, Electrochim. Acta, 2016, 210, 130–137 CrossRef CAS.
- L. Wang, C. Yang, S. Dou, S. Wang, J. Zhang, X. Gao, J. Ma and Y. Yu, Electrochim. Acta, 2016, 219, 592–603 CrossRef CAS.
- H. Lu, R. Chen, Y. Hu, X. Wang, Y. Wang, L. Ma, G. Zhu, T. Chen, Z. Tie and Z. Jin, Nanoscale, 2017, 9, 1972–1977 RSC.
- L. Wang, W. Guo, P. Lu, T. Zhang, F. Hou and J. Liang, Front. Chem., 2019, 7, 832 CrossRef CAS.
- L. Qie, W.-M. Chen, Z.-H. Wang, Q.-G. Shao, X. Li, L.-X. Yuan, X.-L. Hu, W.-X. Zhang and Y.-H. Huang, Adv. Mater., 2012, 24, 2047–2050 CrossRef PubMed.
- B. Larhrib, L. Madec, L. Monconduit and H. Martinez, Electrochim. Acta, 2022, 425, 140747 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta02611f |
| This journal is © The Royal Society of Chemistry 2023 |