
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMnP4 electrode for Na-ion batteries: a complex and effective electrochemical mechanism†
Julien
Fullenwarth
a,
Bernard
Fraisse
a,
Nicolas
Dupré
 b,
Lorenzo
Stievano
ac and
Laure
Monconduit
*ac
b,
Lorenzo
Stievano
ac and
Laure
Monconduit
*ac
aICGM, Univ. Montpellier, CNRS, ENSCM, 34293 Montpellier Cedex 5, France. E-mail: laure.monconduit@umontpellier.fr
bInstitut des Matériaux Jean Rouxel (IMN), CNRS UMR 6502, Univ. Nantes, 44322 Cedex 3 Nantes, France
cRéseau sur le Stockage Electrochimique de l’Energie (RS2E), FR CNRS 3459, France
First published on 2nd October 2023
Abstract
MnP4 has recently been identified as a possible negative electrode for Li-ion batteries. This study shows that this material can also perform as a negative electrode for Na-ion batteries (SIBs), demonstrating that a suitable electrode formulation allows an exceptional performance, i.e., a stable specific capacity of 1000 mA h g−1 after 50 cycles, much higher than that of hard-carbon-based anodes currently used in SIBs. The combination of operando X-ray diffraction, ex situ Mn K-edge X-ray absorption spectroscopy (XAS) and NMR spectroscopy reveals a complex conversion mechanism with the formation of numerous amorphous sodiated phosphide species. 31P and 13Na NMR were particularly effective in identifying these species, demonstrating that their formation is dependent on the preparation of the electrode, especially the amount of carbon additive. XAS analysis, on the other hand, proved the complete reversibility of the mechanism, including the reformation of MnP4 during the desodiation process.
1. Introduction
Na-ion batteries (SIBs) have been emerging for a decade, and represent a real challenger to Li-ion batteries (LIBs) which have dominated the electrochemical energy storage domain for more than 30 years.1–3 While the natural abundance and low cost of sodium precursors represent a real advantage for SIBs, there are still several issues preventing their large-scale application. For instance, the energy density of a SIB is expected to be lower than that of a LIB due to both the higher standard potential of Na+/Na (2.71 V vs. SHE) compared to Li+/Li and the larger atomic weight of sodium. Moreover, the larger ionic radius of Na+ ions makes their diffusion in solids more difficult.4–6 However, the low cost of SIBs compared to LIBs makes them a viable alternative for some applications, such as large-scale energy storage systems, where energy density is not considered a critical issue.7 Unlike LIBs, graphite does not intercalate Na+ in conventional carbonate-based electrolytes, and therefore cannot be used as the negative electrode in SIBs. In contrast, hard carbon (HC) has been widely used due to its highly disordered structure and large interlayer distance.8 So, the insertion of Na+ into HC carbon layers and micropores can lead to a relatively reversible capacity of only 270 mA h g−1.9,10 In addition to this limited capacity, the disordered structure of HC prevents it attaining high-rate capabilities.Thus, the development of alternative negative electrode materials with both high capacity and low working potential to enhance the energy density of SIBs is currently required. Transition metal phosphides (MPx, where M is a transition metal) were discovered as possible negative electrodes for LIBs more than 10 years ago.11–14 Many pioneering studies by our team have greatly contributed to this research, such as NiP3 or FeP.15–17 Similarly, MnP4 was studied as a promising negative electrode for LIBs, exhibiting a high reversible capacity close to the theoretical one (1800 mA g−1) through the successive formation of Li7MnP4 before its conversion into Mn + 4Li3P.18–20
Very recently, Kim et al. proposed the use of a MnP4/graphene nanocomposite as negative electrode for SIBs, able to deliver a very promising reversible capacity of 446 mA h g−1 after 250 cycles.21 In that work, the conversion of MnP4 to Na3P at the end of discharge could only be tentatively inferred from the large reflections observed in ex situ X-ray diffraction (XRD) patterns. In particular, it was not clear whether the sodiation of MnP4 in the SIB occurs first through an insertion process with the formation of NaxMnP4 (similar to what happens in a LIB) before the complete conversion to Mn + 4Na3P, or directly through a conversion reaction.
In the present work, MnP4 was easily prepared by ball milling and used as a negative electrode for SIBs. Its performance was evaluated vs. Na metal using an appropriate electrode formulation. Excellent capacity retention and rate capability were observed with virtually no capacity fading up to 5 C. A different performance was achieved using two ratios of MnP4/carbon additives in the electrodes. We explored the mechanism in both cases by combining several long-range and local probe techniques, including operando XRD, ex situ X-ray absorption spectroscopy (XAS), as well as 31P and 13Na NMR spectroscopy. We were able to rationalize the different performance observed with different electrode formulations, the latter substantially impacting electronic percolation in the electrode.
2. Experimental section
2.1 Materials synthesis and electrode formulation
Pure MnP4 was synthesized by high-energy ball milling with a Fritsch premium Pulverisette 7. Stoichiometric amounts of manganese metal (Alfa Aesar, 325 mesh, 99.3%) and red phosphorous (Alfa Aesar, 100 mesh, 99%) were loaded in a stainless-steel vial with a volume of 45 mL with 6 balls (4 g, 10 mm diameter) and a ratio powder![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :balls = 1:24. The material was submitted to 150 cycles of 15 min milling followed by a 10 min rest. The obtained MnP4 powder is air stable.
:balls = 1:24. The material was submitted to 150 cycles of 15 min milling followed by a 10 min rest. The obtained MnP4 powder is air stable.
For electrode formulation, Timcal C65 carbon black and Showa Denko Carbon Fiber VGCF-H were used as conductive additives and carboxymethyl cellulose (CMC) (DS = 0.7, Mw = 250000 Aldrich) was used as the binder. Part of the study (see NMR section) was carried out to determine the impact of the percentage of active material (AM) on the mechanism; thus, the percentages of electrode components were fixed at 63 wt% or 74% of MnP4, 21 or 13 wt% of conductive additive and 16 or 13 wt% of binder. The materials were introduced into a 12 mL agate jar and ground for 1.5 h with a Fritsch Pulverisette 7 planetary ball mill. The MnP4-richer electrode was used for the NMR study. Deionized water (0.6 mL for 140 mg of active material) was added to the composite electrode material. The slurry was tape cast with a 150 μm knife onto a 20 μm thick copper foil, dried first for 12 h at room temperature and then for an additional 2 hours at 100 °C under vacuum.
For operando XRD a self-supported electrode was prepared with an optimized formulation of 70 wt% of MnP4, 12 wt% of conductive additive (6% C65, 6% VGCF) and 18 wt% of binder. The optimization was done to simultaneously promote diffraction by maximizing the amount of MnP4 and good working of the operando electrochemical cell which needs an increased amount of conductive additive.
For XAS analysis a self-supported electrode containing 74 wt% of MnP4, 13 wt% of conductive additive (6.5% C65, 6.5% VGCF) and 13 wt% of binder was used. For the XAS measurements an electrode loading of 3–4 mg cm−2 was used for an optimal signal-to-noise ratio of the collected spectra.
2.2 Materials characterisation
XRD patterns of the synthesized MnP4 were recorded using a zero background sample holder on a Panalytical X'Pert PRO MPD θ–θ powder diffractometer equipped with Co Kα radiation to limit Mn fluorescence in the Bragg–Brentano configuration. Operando measurements were carried out using a specifically developed in situ cell equipped with a 250 μm-thick beryllium window.22 Lattice parameters were obtained by profile matching of the collected patterns using the Fullprof software package.23,2423Na and 31P MAS NMR experiments were carried out at room temperature on a Bruker Avance-500 spectrometer (B0 = 11.75 T, Larmor frequency ν0 (23Na) = 132.29 MHz and ν0 (31P) = 202.45 MHz). MAS spectra were obtained using a Bruker MAS probe with a cylindrical 2.5 mm o.d. zirconia rotor. Spinning frequencies up to 25 and 30 kHz were utilized. 23Na NMR spectra were acquired using an echo sequence to discard the significant contribution from the probe signal, with a π/2 pulse of 1 μs. 31P NMR spectra were acquired by making use of a single π/2 pulse sequence of 4 μs. Recycle delays of 5 s were determined to ensure quantitative measurements for both 23Na and 31P. The isotropic shifts, reported in parts per million, are relative to external 1 M solutions of NaCl and H3PO4 in water set at 0 ppm. NMR integrated intensities were determined by spectral simulation using Demit software.25 The cells were cycled vs. Na metal at a regime of C/10.
Ex situ Mn K-edge XAS experiments were carried out in transmission mode at the ROCK beamline of Synchrotron Soleil26 on selected samples: pristine MnP4, fully discharged MnP4, fully discharged MnP4, and finally MnP4 after a second full discharge. The samples were prepared and stored in an Ar-filled glove box before measurement, protected in an air-tight holder with Kapton windows. Spectra were collected from 6380 to 7400 eV around the Mn K-edge. The energy was calibrated by assigning the first inflection points of the spectrum of manganese metal to 6539 eV. The spectra were analysed using the IFEFFIT software package.27 Fourier transformation of EXAFS oscillations with different k weights was carried out in the k-range from 0.3 to 1.2 nm−1. Fitting was performed in the R-range from 0.1 to 0.37 nm using k1, k2 and k3 weights. EXAFS amplitudes and phase-shifts were calculated with FEFF7,28 starting from the lattice parameters of pure MnP4 and Mn metal. The fitting parameters of the different scattering paths used for fitting the spectra, including interatomic distances and Debye–Waller factors, are detailed in the ESI section.†
Electrochemical sodium insertion/extraction tests were carried out vs. Na metal with an electrolyte of 1 M NaPF6 in EC:PC:3DMC containing 1% VC and 5% FEC and a Whatman glass fibre separator in coin cells assembled in an argon-filled glove box. Sigma-Aldrich 99.9% sodium ribbon was used as counter and reference electrodes. A crimping machine MSK-160 from the MTI company was used to seal the CR2032 coin cells.
Galvanostatic cycling was carried out at a current density of C/2 (where 1C is defined as the reaction of 1 mol of Na per mol of MnP4 per hour, i.e., 150 mA g−1), and with currents from C/20 to C for the rate capability test.
3. Results
3.1 Electrode preparation
Unlike previous high-pressure or tin-flux syntheses, MnP4 was synthesised here by ball milling, which produces poorly crystalline MnP4 with particle sizes varying from 1 to 20 μm, formed from aggregated smaller primary 0.1–0.5 μm crystallites (Fig. 1c and d).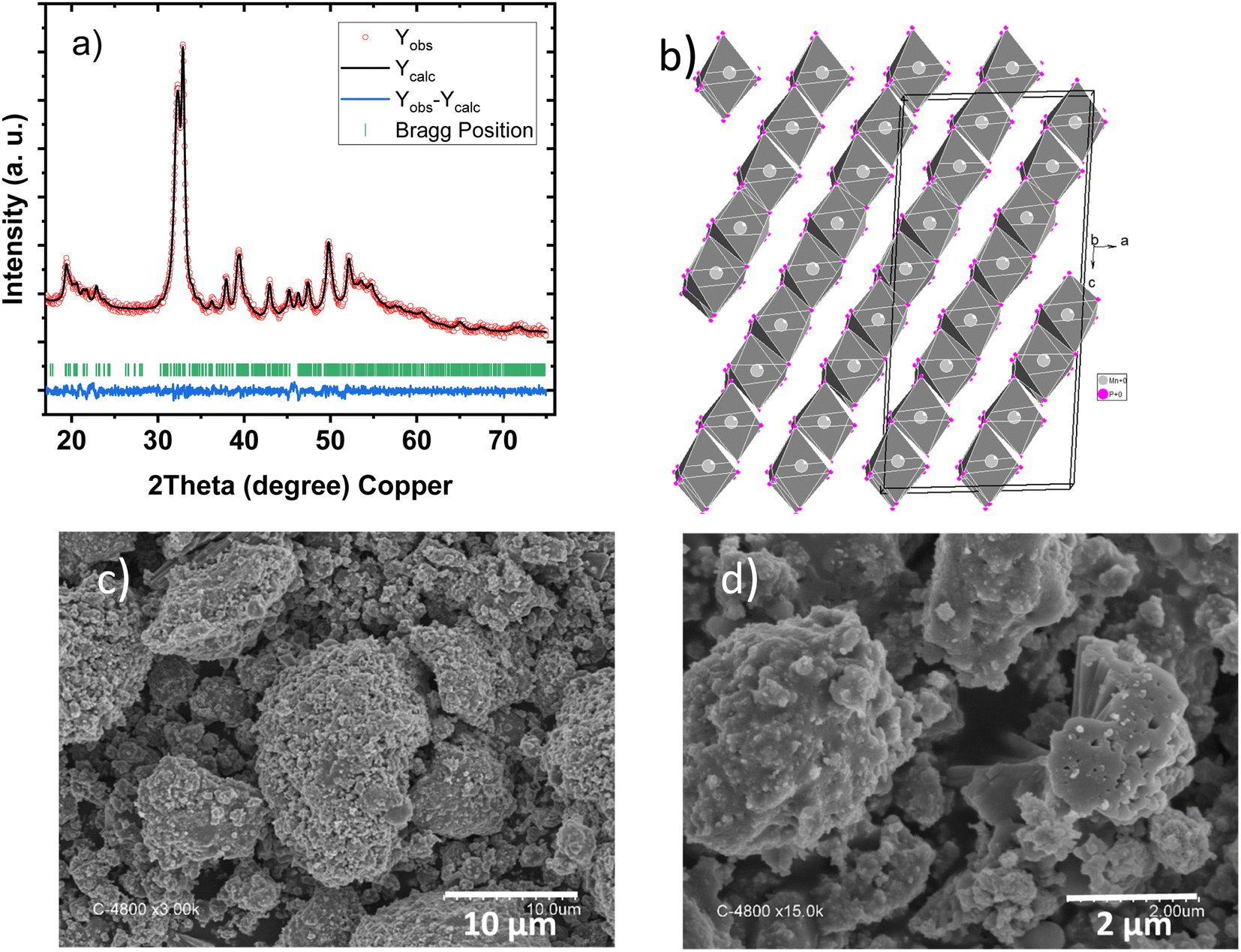 | ||
| Fig. 1 (a) XRD pattern, (b) representation of its crystal structure, (c) and (d) SEM images of pristine MnP4 synthesised by ball milling. | ||
In fact, the XRD pattern collected for the ball milled powder (Fig. 1a) shows large reflections, with peaks at 19.5, 23, 32, 33, 38.5 and 39.5° in agreement with the triclinic lattice of MnP4 (space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , PDF00-044-0944). The cell parameters were refined to a = 16.405(4) Å; b = 5.8714(3) Å, c = 5.1205(3) Å, α = 116.072(3)°, β = 95.261(3)° and γ = 88.869 (3)°. The structure of MnP4 is described by tetramers of edge-sharing octahedra linked to each other by P–P bridges to form a two-dimensional network of interconnected zigzag chains, in the (b,c) plane. As shown in Fig. 1b, this tetramer packing forms a layered-type structure with short interlayer P–P distances (2.27 Å).
, PDF00-044-0944). The cell parameters were refined to a = 16.405(4) Å; b = 5.8714(3) Å, c = 5.1205(3) Å, α = 116.072(3)°, β = 95.261(3)° and γ = 88.869 (3)°. The structure of MnP4 is described by tetramers of edge-sharing octahedra linked to each other by P–P bridges to form a two-dimensional network of interconnected zigzag chains, in the (b,c) plane. As shown in Fig. 1b, this tetramer packing forms a layered-type structure with short interlayer P–P distances (2.27 Å).
3.2 Performance in batteries
MnP4 electrodes formulated with CMC as described in the experimental section were cycled in galvanostatic mode between 1.5 and 0 V (Fig. 2). Such an electrode formulation is rich in conductive carbon, as it contains 21% of a C65/VGCF mixture. To identify the electrochemical contribution of the carbon content, a C65/CMC electrode was prepared, characterized by XRD (Fig. ESI 1†) and cycled under the same conditions as the MnP4 electrode (Fig. ESI 2†). This electrode shows that the electrochemical reaction of the carbon additive with Na is extremely limited and occurs in two voltage regions, around 0.3 and 0.1 V vs. Na+/Na. The charge capacity is even lower, and can thus be considered as negligible compared to that of MnP4. | ||
| Fig. 2 (a) First galvanostatic cycle and (b) corresponding derivative curve (first discharge in red, first charge/second discharge (in black) and 50th cycle in blue) of MnP4/Na (with 63% of MnP4) cycled at C/2. | ||
The galvanostatic discharge of MnP4 is shown for the two active material loadings (63 and 74% in Fig. 2a and ESI 5†, respectively). A sloped curve occurs from 1 to 0.2 V, corresponding to the insertion of 1.5 mol of Na per mol of MnP4, followed by a long plateau corresponding to the reaction of more than 5 mol of Na. The derivative curve allows better definition of the potential of the electrochemical processes (Fig. 2b) with, in the first discharge, two main peaks at 0.15 and 0.05 V. Upon first charge an oxidative peak appears at 0.47 V, a second one at 0.62 V and a third broad one at 0.75 V.
The second discharge curve is strongly modified, with two derivative peaks now centred at 0.4 and 0.2 V, and no peak is visible any more close to 0 V. In spite of these very modest changes, the shape of the derivative curve of further cycles then remains largely unchanged over many cycles (see the 50th cycle in Fig. 2b). No increase in polarisation with the number of cycles is observed.
The capacity retention measured at C/2 rate (Fig. 3a) shows first a loss of capacity between the first discharge and charge, corresponding to a relatively low initial coulombic efficiency (ICE) of 80.5%, likely due to a conspicuous irreversible reaction of the electrolyte to produce a massive solid electrolyte interphase (SEI). A surprising increase in the reversible capacity is observed from the second to the 10th cycle, whereafter a stable capacity of about 1025 mA h g−1 is then well maintained. After 50 cycles a capacity close to 1000 is still measured. The rate capability of the MnP4/Na cell (Fig. 3b) shows the same activation phenomena for the first cycles. An excellent capacity retention of ∼1000 mA h g−1 is observed even when the rate is increased successively from C/20 to C/10, C/5 and C/2. The capacity decreases to ∼900 mA h g−1 only when the rate is increased to C. The CE is especially low at low rates, and is even improved when the rate is increased above C/2, likely due to a decrease in the reduction of the electrolyte at high rate.
 | ||
| Fig. 3 (a) Capacity retention and coulombic efficiency measured at C/2 and (b) the rate capability of the MnP4/Na cell (63%). The empty and filled symbols represent the charge and discharge capacity values. | ||
3.3 Operando XRD analysis
The electrochemical mechanism of MnP4 was followed by in situ XRD during the first discharge at C/20 of an MnP4/Na half-cell (Fig. ESI 3†), and also during the following cycles at C/5 (Fig. 4). | ||
| Fig. 4 Operando XRD of an MnP4/Na cell during the second cycle at C/5 and associated galvanostatic curve. | ||
During the first discharge (Fig. ESI 3†) from 2 to 0.25 V, the strongest Bragg peak of the VGCF conductive additive at 26.5° immediately decreases and totally disappears in the XRD pattern at 0 V. Conversely, during the plateau around 0.1 V, the Bragg peaks of MnP4 decrease (after the insertion of 2 Na) but do not totally disappear at the end of the reduction process. At 0 V, two broad peaks centred at 20–21° and 36–37° grow (see arrows), which could be attributed to the main peaks of Na3P (ICDD #01-073-3917). It is, however, noteworthy that such a very broad XRD pattern cannot exclude the formation of other phosphides such as NaP, Na3P11 or other NaxP species (cf. Fig. ESI 4†). During the following discharges (Fig. 4), the decrease in the contribution of the pattern of MnP4 is accompanied by the appearance of two intense and broad peaks around 36° and 37°, attributed to the formation of Na3P (P63/mmc), but not excluding the formation of other NaxP species (vide infra). At the end of the discharge, although Na3P is the main crystalline phase in the electrode, MnP4 has not totally disappeared, demonstrating that full conversion is not totally achieved with this electrode formulation (70% of AM). During the following charge, Na3P decreases and disappears totally at the end of the process, but it is unclear whether MnP4 reappears simultaneously. The same behaviour is observed during the following cycles.
To obtain a clear view of the chronology of the phase evolution during cycling, and especially the possible formation of reaction intermediates not detected by XRD, the surface of the strongest Bragg peak of each phase was plotted as a function of the sodiation (Fig. 5) during the first cycle. Interestingly, the strongest peak of MnP4 gradually decreases in intensity during discharge, and is at about half of its pristine intensity when those of Na3P start showing up, in line with the possible formation of amorphous intermediates during discharge. During the following charge, the peaks of MnP4 show a slight but significant increase in intensity, indicating the reformation of MnP4 upon desodiation, while those of Na3P disappear. Interestingly, their intensity also seems to increase when those of Na3P have totally disappeared, in line with the possible formation of intermediates in charge, already observed during discharge.
 | ||
| Fig. 5 Evolution of the surface of the strongest Bragg peaks of MnP4 (−111), Na3P (110) and VGCF (002) in the operando XRD patterns collected during the first cycle of a MnP4/C cell. | ||
The poor crystallinity of XRD, however, does not allow a clear description of the formed sodiated phosphides. To go deeper into the identification of these sodiated phases and the chronology of their formation, full 23Na and 31P MAS NMR were carried out.
3.4 23Na and 31P MAS NMR
23Na and 31P MAS NMR spectroscopy measurements were carried out at different stages of cycling to obtain deeper information about the electrochemical mechanism, and especially to verify the formation of intermediate amorphous species impossible to identify by XRD. Fig. ESI 6† displays the 31P MAS NMR spectra of the pristine MnP4 powder, as obtained by ball milling synthesis, and of the formulated MnP4 electrode. Despite a strong decrease in the intensity of the peaks of MnP4, no other peak appears after electrode formulation.Fig. 6 displays the 23Na MAS NMR spectra of the MnP4 electrodes containing 63% active material. The first cycle (first discharge and first charge) and the second discharge for electrodes containing 63% and 74% active material are given in Fig. 2a and ESI 5,† respectively. At the end of the first discharge (0 V), two sets of resonances can clearly be observed. The most shifted one at 65 ppm corresponds unambiguously to Na3P, as previously observed by Morita et al.,29 confirming the XRD results. The higher chemical shift compared to other sodium phosphides is consistent with the absence of P–P bonds in the structure. Although the linewidth seems asymmetrical, no clear quadrupolar line shape can be evidenced in contrast to the Na3P studied by Morita et al., which could suggest a higher degree of disorder in the electrochemically obtained Na3P. The second set of resonances appears between 9 and −20 ppm, and cannot be resolved into separate and well-defined resonances. Overall, the chemical shift range is consistent with the 23Na MAS NMR signal of sodium local environments found in phosphides containing a lower amount of sodium, such as NaP7, Na3P11 or Na3P7.29 In addition, considering the non-zero intensity between the two sets of resonances, the presence of minor signals at approx. 45 ppm, which would correspond to amorphous Na3−γP, cannot be excluded. The 23Na MAS NMR spectrum obtained for an electrode stopped during the first discharge at 120 mV, after the first redox process (Fig. 6, in orange) displays a signal with a low intensity at 46 ppm, suggesting that this sodium local environment may already exist before the end of the reduction.
 | ||
| Fig. 6 Normalized 23Na MAS NMR spectra of MnP4 electrodes containing 63 wt% of active material, stopped at 120 mV (orange) and 0 V (red) during the first discharge (D1) and at 0 V at the end of the second discharge (blue) (D2). | ||
The corresponding 31P MAS NMR spectrum is shown in Fig. 7. In addition to resonances attributed to the initial MnP4 at 242, 47 and −34 ppm (Fig. ESI 6†), an intense signal at −208 ppm confirms the formation of Na3P at 0 V. A minor signal at −273 ppm seems to confirm the presence of Na3−γP.29 A resonance observed at −316 ppm cannot be assigned to any previously observed signal, to the best of our knowledge. Previous experimental29 or simulation-based30 studies did not investigate species involving phosphorus local environments in sodium phosphides by NMR at chemical shifts below −300 ppm. This particular resonance will be discussed later. Finally, two resonances appear at 5 and 10 ppm and are assigned to surface phosphate species, possibly created by the reaction of the material with the electrolyte. Additional details about the nature of these surface phosphates are given in the ESI section (Fig. ESI 7†).
 | ||
| Fig. 7 Normalized 31P MAS NMR spectra of MnP4 electrodes containing 63 wt% of active material, stopped at 0 V during the first discharge (red) and at 0 V at the end of the second discharge (blue). Spectra were obtained at 28 kHz except first discharge – 0 V which was obtained at 26 kHz. Asterisks correspond to spinning sidebands. The blue areas mark resonances for the initial MnP4. The yellow area marks resonances assigned to surface phosphates. | ||
Both 23Na and 31P MAS NMR spectra measured at the end of the second discharge (i.e., reduction, in blue in Fig. 7) are different from those at the end of the first discharge, indicating that the redox process may follow a different pathway. The 23Na MAS NMR spectrum does not display any more the characteristic signal of Na3P, suggesting that this particular phase is not reached at the end of the second discharge. Only resonances between 9 and −20 ppm, typical of phosphide species poorer in sodium, are detected. The corresponding 31P MAS NMR spectrum confirms the absence of Na3P as no signal is observed at −208 ppm. In addition to the already mentioned resonances at 242, 47, and 34 ppm and at 5 and 10 ppm assigned to pristine MnP4 and surface phosphates, respectively, a dominant signal is observed at −245 ppm which agrees with the formation of Na-poor amorphous phases Na1−αP and Na2−βP already reported at −240 and −235 ppm.29 Overall, the second discharge favours the formation of sodium-poor species. Considering the increase in specific capacity from the first discharge to the second one, this result suggests that sodium ions reach further/deeper into MnP4 particles to form Na-poorer species or regions. This result is supported by the XAS result (see the Mn K-edge XAS section).
Fig. 8 displays the 23Na MAS NMR spectra of electrodes containing more active material with 74 wt% of MnP4 which were stopped at 120 mV after the end of the first redox process (green), stopped at 0 V at the end of discharge (red) and at the end of the second discharge (purple). Clear differences appear compared to the cycling of the film made with 63 wt% of MnP4. Although the set of resonances between 9 and −20 ppm (vide supra) is still present and contributes the majority of the detected NMR signal, no signal at 65 ppm could be detected at the end of the first discharge, indicating that Na3P is not formed. Considering the lower specific capacity delivered by the 74 wt% electrode, this confirms that Na3P is indeed the end product of the sodiation of MnP4 and is only obtained when “full capacity” of MnP4 is achieved. In contrast, at the end of the first discharge, only an additional signal at 46 ppm, already attributed to amorphous Na3−γP, is observed. This particular sodium environment seems also to appear in the spectra of the electrode stopped at 120 mV, but its amount clearly increases on going from 120 to 0 mV. It seems to be still present at the end of the second discharge, but the resonance appears broader, suggesting an even more disordered environment for the corresponding sodium nuclei.
 | ||
| Fig. 8 Normalized 23Na MAS NMR spectra of MnP4 electrodes containing 74 wt% of active material, stopped at 120 mV (green) and 0 V (red) during the first discharge (D1) and at 0 V at the end of the second discharge (D2, purple). | ||
This result is consistent with those obtained for the electrodes containing 63 wt% of active material, and indicates that Na3−γP is one of the intermediate sodium phosphide species formed during the sodiation process. In the case of the electrode containing 63 wt% of MnP4, the higher carbon content allows a better electrochemical reaction of the active material into Na3P to be reached at the end of the first discharge, and thus a higher capacity per gram of MnP4. The Na3−γP-type environments appear for incomplete sodiation. In the case of the electrode containing 74 wt% of MnP4, the overall lower sodium content stops the reaction before the formation of Na3P, and only intermediate sodiated phosphorus species are formed. While amorphous Na3−γP is a sodium-rich species, its presence can be tracked down even at intermediate potential during the discharge process and not only at the end of it.
Fig. 9 displays the 31P MAS NMR spectra for the MnP4 electrodes containing 74 wt% of active material stopped at 0 V at the end of the first and second discharges. The two spectra are very similar, in agreement with their 23Na MAS NMR counterparts. The resonances assigned to pristine MnP4, at 242, 47 and −34 ppm are more intense with respect to the 31P MAS NMR spectra of the electrodes containing 63 wt% of active material, consistent with the less extensive sodiation of the active material producing a lower specific capacity for the 74 wt% electrodes. The major resonances assigned to sodium phosphides appear at −316, −245 and −208 ppm, with an additional minor signal at −273 ppm. The previously unreported signal at −316 ppm appears here as one the most intense resonances and therefore refers to one of the major phosphorus environments at the end of discharge of the 74 wt% electrodes (even though it slightly decreases in intensity at the end of the second discharge), while it was barely visible at the end of the first discharge of the 63 wt% electrode. Considering that the corresponding 23Na NMR signals appear in the 9 to −20 ppm chemical shift range, typically corresponding to Na-poor species or environments, we assign the −316 ppm resonance in the 31P MAS NMR spectra to an Na-poor phosphide. Its significant contribution to the spectra of 74 wt% electrodes is also in agreement with the lower capacity delivered and the lower extent of sodiation compared to the 63 wt% electrodes. The weak resonance at −273 ppm matches quite well with reported values for amorphous Na3−γP29 and agrees with the corresponding 23Na MAS NMR signal clearly detected at 46 ppm, albeit with low intensity. The intense and broad resonance at −245 ppm again agrees with Na-poor amorphous species Na1−αP and Na2−βP. More surprisingly, a sharp resonance at −208 ppm (usually assigned to Na3P) is visible even though no 23Na NMR signal could be seen at 65 ppm, suggesting that phosphorus local environments resembling those found in the Na3P phase are present in the active material, maybe corresponding to slightly defective Na3P-like species. Resonances at −316, −244 and −208 ppm disappear completely from the spectrum acquired at the end of the first desodiation (blue spectrum), confirming their attribution to Na–P species formed reversibly upon cycling.
 | ||
| Fig. 9 Normalized 31P MAS NMR spectra of MnP4 electrodes containing 74 wt% of active material, stopped at 0 V at the end of the first discharge (D1, red) and at the end of the second discharge (D2, purple). Asterisks correspond to spinning sidebands. The blue areas mark resonances for the initial MnP4. The yellow area marks resonances assigned to surface phosphates. | ||
Fig. S8† displays the 31P MAS NMR spectra of the 74 wt% and 63 wt% electrodes stopped during the first discharge at 120 mV. The two spectra are quite similar and show several overlapping resonances and spinning sidebands. Resonances assigned to pristine MnP4 are still visible at −36, 47 and 242 ppm as well as the set of resonances at 5 and 10 ppm attributed to surface phosphate impurities. The resonance at −316 ppm, tentatively assigned to an Na-poor phosphide, is clearly visible in both spectra and is more intense in the spectrum of the 63 wt% electrode, in agreement with the higher specific capacity of this electrode for a fixed potential (120 mV). Other resonances at −273 and −245 ppm, already discussed and assigned to amorphous Na3−γP, Na1−αP and Na2−βP, also seem to be present, with the −273 ppm (Na3−γP) resonance being more resolved and the −245 ppm (Na1−αP and Na2−βP) resonance being less resolved in the case of the 63 wt% electrode compared to the 74 wt% electrode, in agreement with the less advanced sodiation of the latter.
A more accurate determination of the phosphorus species in the electrodes is quite difficult because of the presence of the conductive carbon additive, as discussed in ESI (Fig. ESI 6†).
3.5 Mn K-edge XAS
To complete our understanding of the electrochemical sodiation mechanism of MnP4, the chemical state of manganese was followed by ex situ Mn K-edge XAS. The near-edge structure (XANES) of the XAS spectra, shown in Fig. 10, shows only small modifications upon reduction and oxidation of MnP4. The well-defined features of the edge of pristine MnP4 are lost at the end of the first sodiation, and only partially recovered at the end of the first charge. The very broad features of the XANES signal after the first discharge process reflect the low crystallinity or amorphous state of all Mn-containing species in the electrode. While the edge position does not vary substantially, subtle changes in its shape agree well with the formation of small particles of Mn metal (see the ESI section for a detailed description†). These small changes are in line with the slight difference in edge position between phosphidic and metallic manganese (which is set at 6539 eV). | ||
| Fig. 10 Mn K-edge XANES spectra of MnP4 electrodes containing 74 wt% of active material in the pristine state (blue), stopped at 0 V at the end of the first discharge (red), at the end of the first charge (green) and at the end of the second discharge (violet). | ||
More interesting features can be derived from an analysis of the extended fine structure (EXAFS) of the XAS spectra, shown in Fig. 11. In fact, while the main feature in the Fourier transform (FT) of the EXAFS spectra is represented by the peak of the first neighbours centred at ∼1.8 Å, a shoulder grows in the spectrum at the end of the first and second discharges at about 2.4 Å, which corresponds to the appearance of Mn–Mn bonds in Mn metal (vide infra). To verify this hypothesis, the EXAFS spectra were fitted using a combination of the first three shells of MnP4, corresponding to Mn–P, Mn–Mn and Mn–P distances of ∼2.2, ∼3.2 and ∼3.8 Å, respectively, and the first shell of Mn metal, corresponding to Mn–Mn distances of ∼2.7 Å. The fitting was performed by leaving the number of nearest neighbours fixed to the theoretical number for each phase, and fitting their respective amplitude (together with the corresponding Debye–Waller factors and edge shifts) so that their sum is equal to the amplitude reduction factor obtained for pristine MnP4 and Mn metal, here used as reference materials. In this way, the ratio between the fitted amplitude and the amplitude reduction factor provides the concentration percent of each phase in the material. The fitting results are summarised in Table 1, while the fitted EXAFS spectra are gathered in Fig. ESI 11.†
 | ||
| Fig. 11 Mn K-edge EXAFS spectra (top) and corresponding modulus of the Fourier transform (bottom) of MnP4 electrodes containing 74 wt% of active material in the pristine state (blue), stopped at 0 V at the end of the first discharge (red), at the end of the first charge (green) and at the end of the second discharge (violet). | ||
| Sample | Species | Shell | N | Dist. (Å) | σ 2 | Conc. (%) |
|---|---|---|---|---|---|---|
| MnP4 pristine | MnP4 | Mn–P | 6 | 2.268(5) | 0.006(1) | 100 |
| Mn–Mn | 2 | 3.25(4) | 0.03(2) | |||
| Mn–P | 8 | 3.87(2) | 0.015(3) | |||
| MnP4 EOD | MnP4 | Mn–P | 6 | 2.28(2) | 0.009(5) | 49(5) |
| Mn–Mn | 2 | 3.33(4) | 0.02(2) | |||
| Mn–P | 8 | 3.90(2) | 0.021(6) | |||
| Mn metal | Mn–Mn | 8 | 2.73(4) | 0.021(3) | 51(5) | |
| MnP4 EOC | MnP4 | Mn–P | 6 | 2.290(7) | 0.010(1) | 100 |
| Mn–Mn | 2 | 3.24(3) | 0.016(4) | |||
| Mn–P | 8 | 3.84(3) | 0.024(5) | |||
| MnP4 EOD2 | MnP4 | Mn–P | 6 | 2.26(2) | 0.011(5) | 35(5) |
| Mn–Mn | 2 | 3.26(4) | 0.012(2) | |||
| Mn–P | 8 | 3.87(2) | 0.024(6) | |||
| Mn metal | Mn–Mn | 8 | 2.68(4) | 0.012(3) | 65(5) |
The global decrease in the intensity of the first shell after the first discharge is a combination of the partial transformation of pristine MnP4 into a mixture of Mn metal nanoparticles and disordered MnP4, as confirmed by the significant increase in the Debye–Waller factors. A rather random variation in the Debye–Waller factor for the Mn–Mn shells of MnP4 can be ascribed to the very low intensity of this specific contribution, which gives rise to large errors in its determination. At the end of the first discharge, about half of the starting MnP4 is transformed into relatively disordered or most probably nanosized Mn metal, as expected for classical conversion reactions.31 The Mn metal is fully reconverted to MnP4 at the end of the first charge. However, the strong increase in the Debye–Waller factor compared to pristine MnP4 shows that the initial crystallinity is not recovered, and that also the reformed MnP4 is nanosized and/or partly disordered. Interestingly, the second discharge shows that an increasing amount of MnP4 is transformed into Mn metal, indicating that the first conversion was not complete and that the reaction of MnP4 is gradually activated during the first cycle, in line with the observed increase in capacity in the first cycle.
4. Summary of the sodiation mechanism and conclusion
The (de)sodiation mechanism of an electrode for SIBs based on MnP4 is schematised in Fig. 12. During the first sodiation process, both Na-poor species or environments are formed along the first plateau at approx. 0.2 V. Disordered Na1−αP, Na2−βP and local environments resembling those found in NaP7, Na3P11 and Na3P7 are observed by 23Na and 31P MAS NMR at 120 mV for both 63% and 74% electrodes. In addition, an Na-rich Na3−γP phase appears to be already present at 120 mV and therefore does not seem to be an end product of the sodiation. We tentatively hypothesise the presence of this disordered phase close to the surface. Such Na-rich environments could form close to the surface of MnP4 particles due to a combination of sodium intake and possibly low diffusion of Na into the bulk of active material particles. This hypothesis would be consistent with the fact that even at the end of reduction (close to 0 V) some MnP4 remains unreacted (as observed by XRD, XAS and 31P MAS NMR). In the case of the 63% electrode, further sodiation seems to lead to higher capacity, corresponding to an uptake of 8 Na per formula unit, thanks to a better electronic percolating network. Although full sodiation is not achieved (MnP4 is still detected), 23Na MAS NMR indicates clearly the formation of Na3P along with the remaining contributions of Na3−γP and Na-poor environments. In the case of the 74% electrode, due to a less efficient percolating network, a lower capacity and overall sodiation are reached, leading to the formation of only Na-poor Na1−αP, Na2−βP type environments. | ||
| Fig. 12 Schematics of the sodiation process in MnP4-based electrodes containing 63% and 74% active material. Neither surface impurities nor the extruded Mn metal nanoparticles are represented for the sake of clarity. However, the latter are supposed to be present together with the sodiated phosphide species, as expected for typical conversion reactions.31 | ||
As shown by XAS and XRD, the sodiation process is reversible and a more disordered type of MnP4 is formed at the end of oxidation along with the disappearance of sodiated species in the 23Na NMR spectra.
During the second discharge, the sodiation process seems to be facilitated, as inferred from the specific capacity increase observed during the early stage of cycling. Consequently, a larger ratio of MnP4 reacts with sodium, as shown from the XAS results. In the case of the 63% electrode, however, the reaction of a high amount of Na and MnP4 does not lead to a high amount of Na3P but rather to a large amount of Na-poor species. The facilitated diffusion of Na+ in the “disordered” MnP4 may prevent too high a localised sodium concentration and the nucleation of Na3P. Na-rich Na3−γP is also not observed any more and the second discharge seems to lead to better homogenization of the sodiation reaction in the bulk of the active material. In the case of the 74% electrode, no significant change is observed at the end of the second discharge, as Na3−γP as well as the various Na-poor species are detected, probably as a result of the less efficient percolating network and the lower Na uptake. Finally, the 70% electrode used for operando XRD seems to display behaviour closer to that of the 63% electrode since Na3P or Na3−γP is detected at the end of discharge.
Conclusion
Working through a complex and electrode-formulation-dependent mechanism, MnP4 proves to be a promising negative electrode material for a SIB with a stable high capacity of about 1025 mA h g−1 maintained over 50 cycles. Moreover, an excellent capacity retention of around 1000 mA h g−1 is observed up to a rate of C/2 and is maintained around 900 mA h g−1 at a rate of C.We demonstrated that the formation of more or less sodium-rich intermediate phases is directly dependent on electronic percolation in the electrode. In fact, it is well known that conversion mechanisms suffer from kinetic limitations,31 and that electrode formulation plays a key role in reducing this limitation. In recent work, Hong and co-workers clearly demonstrated the efficiency of combining an MnP4/graphene nanocomposite and a good porous CMC electrode formulation to improve both electronic and ionic conductivities, respectively, and in consequence the cycling.21 These results demonstrated that MnP4, which is a semiconductor material, with low conductivities and kinetic limitation, can be a very good electrode material provided that good electronic percolation is ensured in the electrode. To continue this study, it would be interesting to examine in more detail the sodiated species formed in the case of graphene/MnP4 composites like those proposed by Hong and co-workers.21
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
ANR is acknowledged for funding through the project Labex STORE-EX (Grant ANR-10-LABX-76-01). Synchrotron Soleil is acknowledged for providing beamtime at beamline ROCK (proposal no. 20190882). Antonella Iadecola (RS2E) is gratefully acknowledged for technical and logistic help in the preparation and the realisation of the measurement session at synchrotron Soleil. Stéphanie Belin (Synchrotron Soleil) is acknowledged for technical help.References
- C. P. Grey and J. M. Tarascon, Nat. Mater., 2017, 16, 45–56 CrossRef PubMed.
- A. Bauer, J. Song, S. Vail, W. Pan, J. Barker and Y. Lu, Adv. Energy Mater., 2018, 8, 1702869 CrossRef.
- J. Deng, W.-B. Luo, S.-L. Chou, H.-K. Liu and S.-X. Dou, Adv. Energy Mater., 2018, 8, 1701428 CrossRef.
- A. Ponrouch and M. R. Palacín, Philos. Trans. R. Soc., A, 2019, 377, 20180297 CrossRef CAS PubMed.
- X. Pu, H. Wang, D. Zhao, H. Yang, X. Ai, S. Cao, Z. Chen and Y. Cao, Small, 2019, 15, 1805427 CrossRef PubMed.
- E. Goikolea, V. Palomares, S. Wang, I. R. Larramendi, X. Guo, G. Wang and T. Rojo, Adv. Energy Mater., 2020, 10, 2002055 CrossRef CAS.
- T. M. Gür, Energy Environ. Sci., 2018, 11, 2696–2767 RSC.
- C. Del Mar Saavedra Rios, A. Beda, L. Simonin and C. Ghimbeu Matei, in Na-ion batteries, ed. L. Monconduit and L. Croguennec, ISTE & John Wiley and Sons, London & New York, 2020, pp. 101–146 Search PubMed.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 1271–1273 CrossRef CAS.
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara, Adv. Funct. Mater., 2011, 21, 3859–3867 CrossRef CAS.
- L. Yue, J. Liang, Z. Wu, B. Zhong, Y. Luo, Q. Liu, T. Li, Q. Kong, Y. Liu, A. M. Asiri, X. Guo and X. Sun, J. Mater. Chem. A, 2021, 9, 11879–11907 RSC.
- L. Zeng, L. Huang, J. Zhu, P. Li, P. K. Chu, J. Wang and X. Yu, Small, 2022, 18, 2201808 CrossRef CAS PubMed.
- Y. Fu, Q. Wei, G. Zhang and S. Sun, Adv. Energy Mater., 2018, 8, 1703058 CrossRef.
- L. Monconduit, J. Phys. Chem. C, 2014, 118, 10531–10544 CrossRef CAS.
- J. Fullenwarth, A. Darwiche, A. Soares, B. Donnadieu and L. Monconduit, J. Mater. Chem. A, 2014, 2, 2050–2059 RSC.
- S. Boyanov, J. Bernardi, F. Gillot, L. Dupont, M. Womes, J.-M. Tarascon, L. Monconduit and M.-L. Doublet, Chem. Mater., 2006, 18, 3531–3538 CrossRef CAS.
- S. Boyanov, M. Womes, L. Monconduit and D. Zitoun, Chem. Mater., 2009, 21, 3684–3692 CrossRef CAS.
- D. C. S. Souza, V. Pralong, A. J. Jacobson and L. F. Nazar, Science, 2002, 296, 2012–2015 CrossRef CAS PubMed.
- F. Gillot, L. Monconduit and M.-L. Doublet, Chem. Mater., 2005, 17, 5817–5823 CrossRef CAS.
- F. Gillot, L. Monconduit, M. Morcrette, M.-L. Doublet, L. Dupont and J.-M. Tarascon, Chem. Mater., 2005, 17, 3627–3635 CrossRef CAS.
- K. Kim and S. Hong, Adv. Energy Mater., 2021, 11, 2003609 CrossRef CAS.
- M. Morcrette, Y. Chabre, G. B. M. Vaughan, G. G. Amatucci, J.-B. Leriche, S. Patoux, C. Masquelier and J.-M. Tarascon, Electrochim. Acta, 2002, 47, 3137–3149 CrossRef CAS.
- J. Rodríguez-Carvajal, Phys. B Condens. Matter, 1993, 193, 55–69 CrossRef.
- T. Roisnel and J. Rodríguez-Carvajal, Mater. Sci. Forum, 2001, 378–381, 118–123 CAS.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- V. Briois, C. La Fontaine, S. Belin, L. Barthe, T. Moreno, V. Pinty, A. Carcy, R. Girardot and E. Fonda, J. Phys.: Conf. Ser., 2016, 712, 012149 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- A. L. Ankudinov and J. J. Rehr, Phys. Rev. B, 1997, 56, R1712–R1716 CrossRef CAS.
- R. Morita, K. Gotoh, M. Dahbi, K. Kubota, S. Komaba, K. Tokiwa, S. Arabnejad, K. Yamashita, K. Deguchi, S. Ohki, T. Shimizu, R. Laskowski and H. Ishida, J. Power Sources, 2019, 413, 418–424 CrossRef CAS.
- M. Mayo, K. J. Griffith, C. J. Pickard and A. J. Morris, Chem. Mater., 2016, 28, 2011–2021 CrossRef CAS.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacín, Adv. Mater., 2010, 22, E170–E192 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta02329j |
| This journal is © The Royal Society of Chemistry 2023 |