
Structural evolution during solution-based synthesis of Li7P3S11 solid electrolyte by synchrotron X-ray total scattering†
Bowen
Shao
a,
Ratnottam
Das
a,
Yonglin
Huang
a,
Ruihao
Deng
a,
Sara
Seelman
b and
Fudong
Han
 *a
*a
aDepartment of Mechanical, Aerospace and Nuclear Engineering, Rensselaer Polytechnic Institute, Troy, NY 12180, USA. E-mail: hanf2@rpi.edu
bDepartment of Materials Science and Engineering, Rensselaer Polytechnic Institute, Troy, NY 12180, USA
First published on 3rd July 2023
Abstract
Solution-based synthesis has emerged as a promising approach for scalable and efficient synthesis of solid electrolytes (SEs). However, the ionic conductivity of solution-synthesized SEs is much lower than that of SEs synthesized by solid state approaches. The structural evolution during the synthesis of SEs is crucial to explaining the decreased ionic conductivities but has not been well understood. Here, we use synchrotron X-ray total scattering combined with pair distribution function (PDF) analysis to understand the structural evolution during annealing of the solution-processed precursor for the synthesis of Li7P3S11 SEs. Combining Bragg diffraction and diffuse scattering, X-ray total scattering enables detection of structural information including phase transition and crystallinity. The results show that the formation of Li7P3S11 glass-ceramic starts at 230 °C. Increasing the annealing temperature helps to improve the crystallinity, but the degree of crystallization is still lower than 70.3% even after annealing at 290 °C when thermal decomposition of Li7P3S11 to Li4P2S6 has occurred. PDF analysis also revealed the existence of acetonitrile used in the solution-based synthesis in the SEs after annealing at high temperatures. The results rationalize the synthesis–property relationship, explain why annealing at 290 °C provides the highest ionic conductivity at the studied temperatures due to an optimal balance for achieving a high crystallinity while suppressing severe decomposition of Li7P3S11, and provide novel insights to develop approaches for synthesizing superior Li7P3S11 SEs on a large scale.
 Fudong Han | Fudong Han is the Priti and Mukesh Chatter '82 Career Development Chair Assistant Professor in the Department of Mechanical, Aerospace, and Nuclear Engineering at Rensselaer Polytechnic Institute in Troy, New York, USA. He received his PhD degree in chemical engineering from the University of Maryland, College Park in 2018. He was a guest scientist in the Materials for Energy and Sustainable Development Group at the National Institute of Standards and Technology (NIST). He is a recipient of the NSF CAREER Award, the Electrochemical Society Electrodeposition Division Early Career Investigator Award, Electrochemical Society Battery Division Student Research Award, and Materials Research Society Graduate Student Gold Award. His research focuses on developing advanced materials for electrochemical energy storage, particularly solid-state batteries. |
1. Introduction
The development of novel battery systems has mainly transitioned to a solid-state battery (SSB) based concept which is believed to potentially enable safe, high-energy-density, and high-power-density energy storage.1,2 The past three decades have observed significant progress in developing superionic solid electrolytes (SEs).3 Lithium thiophosphate-based ionic conductors, including binary Li2S–P2S5 (LPS),4 Li10GeP2S12 (LGPS),5 and Li6PS5Cl (LPSCl),6 and their derivatives, are considered very promising SEs due to their high ionic conductivities and excellent mechanical properties.7 Nevertheless, full commercialization of SSBs is still challenging due to the difficulties in scalable synthesis and processing of SEs.8 The aspect of upscaling is critical because large amounts of SEs will be needed for producing SSBs on a gigafactory scale. One of the most explored routes to synthesizing SEs is solid-state reaction which is achieved by mixing solid precursors and applying heat to drive their reaction. Such an approach has been used to fabricate some of the most conductive SEs.5 To prevent the release of volatile sulfur species and to eliminate the unwanted reactions with the vessels and ambient air, solid-state reaction is typically done in an evacuated quartz ampoule. Another widely used approach for SE synthesis is the mechanochemical method (often called “ball-milling”) which takes advantage of the influence of mechanical energy in chemical transformation.9 While these approaches are practical for laboratory-scale material discovery and design, both synthetic routes face serious challenges in upscaling. Solid-state reaction involves heating to high temperatures and therefore is an energy-intensive process. The use of quartz ampoules significantly limits the yield (to the order of grams per batch). Mechanochemical synthesis is usually done at room temperature, but to ensure the completion of the reaction, the synthesis is typically performed over a long period (8–50 hours) in a lab-scale ball-milling jar (∼50 mL),10,11 and it is not unreasonable to expect that a longer time will be needed if a large ball-milling jar is used for industrial production.Solution-based synthesis of SEs has recently become an essential field of scientific research due to its great potential for upscaling.12–14 Solution-based synthesis of lithium-thiophosphate based SEs is typically achieved by the reaction of Li2S with P2S5 at varied molar ratios in organic solvent(s) such as tetrahydrofuran (THF), acetonitrile (ACN), and ethyl acetate (EA), with the possible addition of extra Li2S and lithium halides for more complex compositions.15–17 Full dissolution of the precursors may occur in some solvents, but in most cases, the reaction proceeds when the precursors are only partially dissolved. The desired SEs are obtained after solvent removal and possible heat treatment(s). The particle size of SEs synthesized by a solution-based approach is much smaller than that synthesized by solid-state and mechanochemical syntheses,15 and this will help achieve great contact with electrode active materials. The synthesis is typically performed at relatively low temperatures and therefore is energy-efficient. The low vapor pressure of sulfur species also enables solvent-based synthesis to be done in a glass container. Moreover, this approach allows for far more intimate contact between the reactants even when only partial dissolution occurs, thus providing access for efficient synthesis and upscaling. In addition to the upscaling of SE synthesis, the solution-based approach also has distinct advantages for the post-processing of SEs for SSB fabrication, i.e., for scalable processing of SE materials into electrolyte membranes and mixing of SE materials, electrode active materials, and electronically conductive additives to fabricate electrode sheets.18,19
Despite the many benefits of solution-based synthesis, the ionic conductivity of thiophosphate-based SEs synthesized by solution-based approaches is much lower than that of SEs synthesized by solid-state synthesis or mechanochemical approaches, as shown in Fig. S1.† A wide spread of ionic conductivities over a few orders of magnitude can also be observed for the solution-synthesized SEs with identical compositions (Fig. S1†).16,20–24 Multiple reasons have been reported to explain the low ionic conductivity of solution-synthesized SEs. Yamamoto et al. reported that the existence of residual Li2S due to incomplete reaction can decrease ionic conductivity.25,26 Although direct precipitation of the final SE from the solution is desired, the obtained precipitates of the liquid-phase reaction are generally a co-crystallized complex of lithium thiophosphate and the solvent used, i.e., crystalline Li3PS4–solvent complex. Subsequent heat treatment is always needed to remove the solvent in the crystalline phase. While the solvent removal process is usually done at a temperature much higher than the boiling point, the solvent can remain in the solids and the remaining solvent27 or its carbonization product28 after further annealing can also lower the conductivity. In addition, many thiophosphate-based SEs are essentially glass-ceramic materials that contain both crystalline and amorphous phases.29,30 It is well known that crystallinity plays a very important role in the ionic transport properties of thiophosphate-based SEs. For Li7P3S11, higher crystallinity will lead to higher conductivity,31 whereas for Li3PS4, an increase in crystallinity has been reported to decrease its conductivity.32 The variation in the crystallinity of the synthesized SE can also be one reason for the decreased conductivity. In addition, the synthesis of some SEs (e.g., Li7P3S11) will require additional annealing at a high temperature to further drive the solid-state reaction and increase the crystallinity, but prolonged annealing at high temperatures may also lead to decomposition of the SE to poorly conductive phases.31,33 Nevertheless, a concrete reason for the decrease in ionic conductivity for solution-synthesized SEs has not been established, although the dominant reason can be different for different thiophosphate SEs.
A comprehensive understanding of the structural evolution of SEs during solution-based synthesis is important to understand the synthesis–property relation but has not been achieved thus far due to the challenges in characterizing the multi-component materials with both amorphous and crystalline structure during the synthesis process. X-ray diffraction (XRD) has been mainly used as the tool to study the structural evolution during synthetic heat treatment,15,16 but no details on the crystallinity of the sample can be determined. The crystallite size of the SE glass-ceramic synthesized by the solution-based method is around tens of nanometers,15 leading to a rather broad peak which can make detailed analysis difficult. Raman spectra were also used to reveal the local structure of P–S polyhedra in solution-synthesized SEs,16 but this surface-specific technique faces challenges in detecting samples in the deep region such as the Li2S core inside an SE shell.25
Here, we report the structural evolution of Li7P3S11 solid electrolyte during annealing of a solution-processed precursor using synchrotron X-ray total scattering and pair distribution function (PDF) analysis. Li7P3S11 was studied because it exhibits one of the highest ionic conductivities (17 mS cm−1 at room temperature) among all thiophosphate SEs.15 The utilization of PDF, which takes into account both Bragg scattering and diffuse scattering, enables detection of structural information, including crystal structure, atomic distances, coordination number, disorder, and size of a particle or coherent domain.34–36 These unique capabilities make PDF very effective in determining the structure of a multi-component system consisting of both crystalline and amorphous phases. Several previous reports have used PDF to study the local structure of Li3PS4 during solution synthesis.25–27,37 However, to our knowledge, no prior study has been reported on the PDF study of the structural evolution of Li7P3S11, which exhibits a much higher ionic conductivity than Li3PS4 and involves more complicated processes (decomplexation, solid-state reaction, crystallization, and decomposition) during synthesis. The structural information revealed from PDF is correlated with the ionic conductivity of SEs annealed at different temperatures. The analysis sheds new light on the principal synthesis–structure–property relationships of Li7P3S11 through solution synthesis and helps to rationalize precise and scalable synthesis of superionic conductive SEs.
2. Results and discussion
2.1. Lab X-ray diffraction and ionic conductivity measurement
To prepare the precursor for the synthesis of Li7P3S11, Li2S and P2S5 with a molar ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 were mixed in ACN at 50 °C for 3 days and the solvent was then removed by drying under vacuum.16 The precursor was then annealed at different temperatures from 170 to 320 °C for 1 hour under argon to synthesize Li7P3S11 SEs. The synthesized SEs were denoted as LPS-170, LPS-200, LPS-230, LPS-260, LPS-290, and LPS-320 based on the annealing temperature. Fig. 1a shows the lab XRD pattern of the synthesized SEs. The peaks for LPS-170 and LPS-200 can be well attributed to the Li3PS4-ACN crystalline complex.38 The existence of Li3PS4-ACN in LPS-170 and LPS-200 is also supported by the Raman spectra (Fig. S2†) which show a strong peak at 429 cm−1, corresponding to PS43−, and three bands at 2927 cm−1, 2265 cm−1 and 366 cm−1 corresponding to the symmetric C–H stretching vibrations, C
:3 were mixed in ACN at 50 °C for 3 days and the solvent was then removed by drying under vacuum.16 The precursor was then annealed at different temperatures from 170 to 320 °C for 1 hour under argon to synthesize Li7P3S11 SEs. The synthesized SEs were denoted as LPS-170, LPS-200, LPS-230, LPS-260, LPS-290, and LPS-320 based on the annealing temperature. Fig. 1a shows the lab XRD pattern of the synthesized SEs. The peaks for LPS-170 and LPS-200 can be well attributed to the Li3PS4-ACN crystalline complex.38 The existence of Li3PS4-ACN in LPS-170 and LPS-200 is also supported by the Raman spectra (Fig. S2†) which show a strong peak at 429 cm−1, corresponding to PS43−, and three bands at 2927 cm−1, 2265 cm−1 and 366 cm−1 corresponding to the symmetric C–H stretching vibrations, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching and C–CN bending of ACN, respectively. The formation of Li7P3S11 starts after annealing at 230 °C. A large decrease in the peak intensity can be observed for the sample annealed at 230 °C and higher temperatures, implying a decrease of the crystallinity and the formation of glass-ceramic SEs. The formation of Li4P2S6 can be clearly observed for samples annealed at 290 and 320 °C due to the decomposition of Li7P3S11. The formation of Li7P3S11 at 230 °C and its decomposition to Li4P2S6 at higher temperatures are also supported by the Raman spectra (Fig. S2†). Fig. 1b shows the impedance plot of SEs measured using two stainless steel blocking electrodes. The semicircle corresponds to the bulk and grain boundary resistance. The calculated ionic conductivity as a function of annealing temperature is shown in Fig. 1c. A sharp increase in the ionic conductivity can be observed after annealing at 230 °C (0.25 mS cm−1). The ionic conductivity increases as the annealing temperature increases, reaches the maximum for LPS-290 (0.70 mS cm−1), and then decreases to 0.35 mS cm−1 for the SE annealed at 320 °C. The maximum ionic conductivity of LPS-290 is still about one order of magnitude lower than that of the Li7P3S11 prepared by the mechanochemical method. It has been well known that the ionic conductivity of Li7P3S11 increases with crystallinity, and the decomposition product Li4P2S6 is a poor ionic conductor.31,39 It should be noted that no apparent changes in the morphology and particle size can be observed for SEs annealed at different temperatures (Fig. S3†), and therefore the evolution in the ionic conductivity is mainly due to the structural evolution in the bulk electrolyte rather than surface or particle boundaries. Correlating with the XRD results, the sharp increase in the ionic conductivity of LPS-230 is due to the formation of crystallites of superionic Li7P3S11. Increasing annealing temperature is expected to increase crystallinity, thus leading to improved ionic conductivity, but annealing at a very high temperature (320 °C) will lead to the formation of poorly conductive Li4P2S6. It should be noted that the maximum ionic conductivity was achieved for the SE annealed at 290 °C where apparent decomposition occurs. The results lead to an important question for further improving the ionic conductivity of solution-synthesized SEs: how can the crystallinity of Li7P3S11be improved without causing decomposition? The results also suggest that crystallization to the desired Li7P3S11 phase (usually determined by lab XRD) is overly simplistic to describe the success of synthesis. More careful and quantitative analysis of the crystallinity of the obtained SE should be done to fully understand the synthesis–property relation for identifying the optimum synthetic condition.
N stretching and C–CN bending of ACN, respectively. The formation of Li7P3S11 starts after annealing at 230 °C. A large decrease in the peak intensity can be observed for the sample annealed at 230 °C and higher temperatures, implying a decrease of the crystallinity and the formation of glass-ceramic SEs. The formation of Li4P2S6 can be clearly observed for samples annealed at 290 and 320 °C due to the decomposition of Li7P3S11. The formation of Li7P3S11 at 230 °C and its decomposition to Li4P2S6 at higher temperatures are also supported by the Raman spectra (Fig. S2†). Fig. 1b shows the impedance plot of SEs measured using two stainless steel blocking electrodes. The semicircle corresponds to the bulk and grain boundary resistance. The calculated ionic conductivity as a function of annealing temperature is shown in Fig. 1c. A sharp increase in the ionic conductivity can be observed after annealing at 230 °C (0.25 mS cm−1). The ionic conductivity increases as the annealing temperature increases, reaches the maximum for LPS-290 (0.70 mS cm−1), and then decreases to 0.35 mS cm−1 for the SE annealed at 320 °C. The maximum ionic conductivity of LPS-290 is still about one order of magnitude lower than that of the Li7P3S11 prepared by the mechanochemical method. It has been well known that the ionic conductivity of Li7P3S11 increases with crystallinity, and the decomposition product Li4P2S6 is a poor ionic conductor.31,39 It should be noted that no apparent changes in the morphology and particle size can be observed for SEs annealed at different temperatures (Fig. S3†), and therefore the evolution in the ionic conductivity is mainly due to the structural evolution in the bulk electrolyte rather than surface or particle boundaries. Correlating with the XRD results, the sharp increase in the ionic conductivity of LPS-230 is due to the formation of crystallites of superionic Li7P3S11. Increasing annealing temperature is expected to increase crystallinity, thus leading to improved ionic conductivity, but annealing at a very high temperature (320 °C) will lead to the formation of poorly conductive Li4P2S6. It should be noted that the maximum ionic conductivity was achieved for the SE annealed at 290 °C where apparent decomposition occurs. The results lead to an important question for further improving the ionic conductivity of solution-synthesized SEs: how can the crystallinity of Li7P3S11be improved without causing decomposition? The results also suggest that crystallization to the desired Li7P3S11 phase (usually determined by lab XRD) is overly simplistic to describe the success of synthesis. More careful and quantitative analysis of the crystallinity of the obtained SE should be done to fully understand the synthesis–property relation for identifying the optimum synthetic condition.
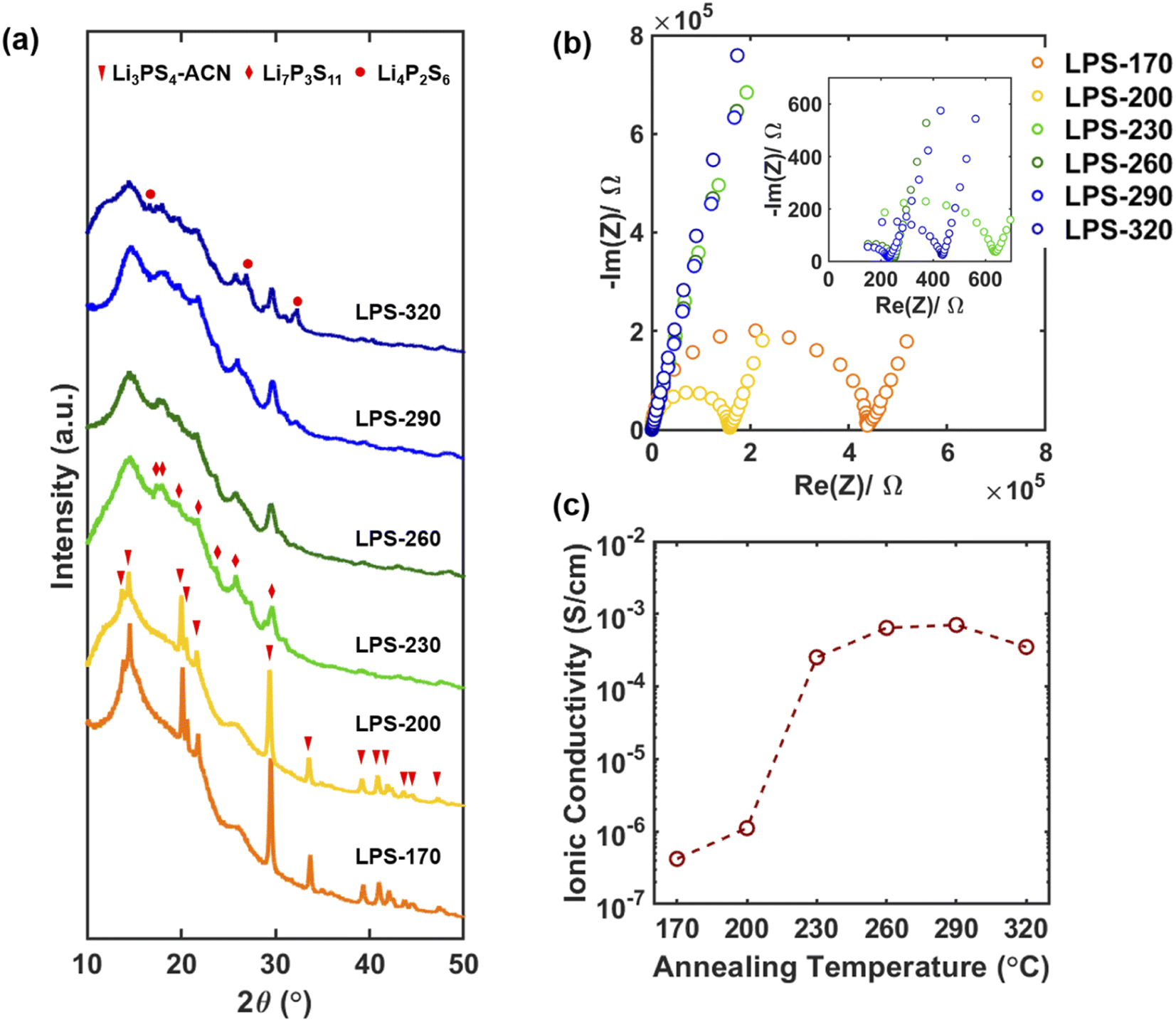 | ||
| Fig. 1 (a) Lab X-ray diffraction patterns of SEs synthesized after annealing at different temperatures. (b) Impedance plots of SEs with stainless steel blocking electrodes measured at room temperature. The impedance measurement was performed from 7 MHz to 10 mHz. (c) Room temperature ionic conductivity of SEs synthesized after annealing at different temperatures. | ||
2.2. Ex situ diffraction and reduced PDFs from synchrotron X-ray total scattering
We use synchrotron X-ray total scattering to better understand the structural evolution of SEs after annealing at different temperatures. Combining both Bragg scattering and diffuse scattering, the total scattering technique can not only provide information on the crystal structure that can be directly correlated with different thiophosphate phases but also reveals the local structure, through PDF analysis, to understand the degree of crystallization. Fig. 2a shows the Bragg data from the total scattering measurement. The diffraction results agree very well with the lab XRD data (Fig. 1a). The conversion between Li3PS4-ACN and Li7P3S11 occurs at 230 °C, and decomposition of Li7P3S11 to Li4P2S6 can be clearly observed after annealing at 290 °C. A decrease in the peak intensity can also be observed after Li7P3S11 glass ceramic has been formed after 230 °C.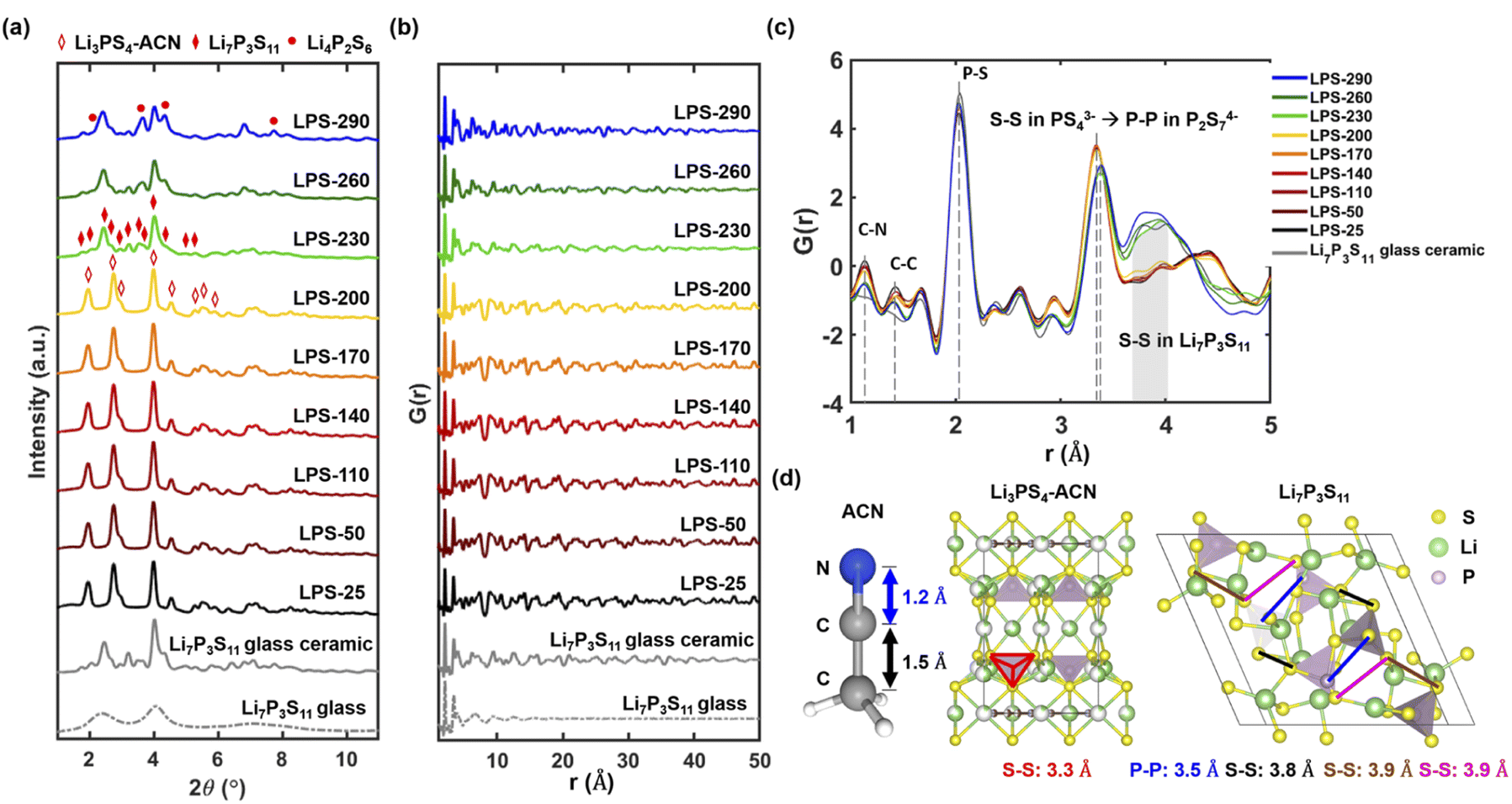 | ||
| Fig. 2 (a) Synchrotron diffraction Bragg data of SEs after annealing at different temperatures. The Bragg data of Li7P3S11 glass and glass-ceramic synthesized by the ball milling method are also provided for comparison. Reduced pair distribution functions, G(r), of SEs after annealing at different temperatures in the range of 1 Å < r < 50 Å (b) and in the range of 1 Å < r < 5 Å (c). The dashed line in (b) shows the evolution of the peak at about 3.8 Å. (d) Structures of ACN, Li3PS4-ACN, and Li7P3S11 with several characteristic bond lengths. | ||
We then analyze the reduced PDF of SEs after annealing at different temperatures. The reduced PDFs in the range of 1 Å < r < 50 Å (Fig. 2b) show very similar profiles for SEs annealed at lower temperatures from 25 to 200 °C. Apparent changes in the reduced PDF profile can be observed after 230 °C and the profile looks similar to that of the Li7P3S11 glass-ceramic, suggesting the formation of Li7P3S11 occurs at 230 °C. The decrease in long-range ordering in the high r range (>40 Å) can also be observed at 230 °C, consistent with a decrease in crystallinity as Li7P3S11 glass-ceramic forms.
The reduced PDFs in a lower range of 1 Å < r < 5 Å are provided in Fig. 2c to reveal more information on the local structure evolution after annealing at different temperatures. All samples show a main peak at 2 Å which can be attributed to the P–S bond in PS44− tetrahedra. While different P–S polyhedra are present in Li3PS4-ACN (PS44−), amorphous Li7P3S11 (PS44−, P2S72−, and P2S64−) and crystalline Li7P3S11 (PS44− and P2S72−), the two main structural units PS44− and P2S72− have very similar short-range structure because P2S72− is essentially two corner-sharing PS44−.14 Another important feature is the peak at 3.4 Å shifts to a slightly higher r value as the annealing temperature increases from 200 °C (yellow) to 230 °C (green), and we believe the shift is caused by changes in the coordination from S–S (3.3 Å) in PS44− of Li3PS4-ACN to P–P (3.5 Å) in the P2S72− in Li7P3S11 (Fig. 2d). A more predominant feature that can be observed is the appearance of a broad peak from 3.8 Å to 4.0 Å for LPS-230, LPS-260, and LPS-290. The broad peak is present in the Li7P3S11 glass-ceramic, and the appearance of this broad peak is attributed to the formation of multiple S–S bonds in Li7P3S11 with a bond length at around 3.8 or 3.9 Å (Fig. 2d). All of these results suggest that the transformation from Li3PS4-ACN to Li7P3S11 occurs at 230 °C which is consistent with the diffraction results (Fig. 1a and 2a).
It should also be noted that two peaks at 1.2 Å and 1.4 Å appeared in the reduced PDFs for SEs annealed at all temperatures. Those two peaks cannot be observed in the synchrotron X-ray and neutron PDFs of Li7P3S11 glass and glass-ceramic synthesized by the ball-milling method (Fig. S4†) and are attributed to the C–N and C–C bonds respectively based on the bond lengths of ACN shown in Fig. 2d. While the existence of the local structure of ACN is expected at temperatures below 200 °C because the major phase is the Li3PS4-ACN crystalline complex, it is quite surprising to see that ACN remains in the SE annealed at a very high temperature of 290 °C which is much higher than the boiling point of ACN (82 °C), although the intensity of the peaks decreases as the annealing temperature increases. Overall, the local structure analysis confirms that the transition from Li3PS4-ACN to Li7P3S11 starts from 230 °C. More importantly, it also shows the existence of ACN even for SEs that are annealed at high temperatures. The existence of solvent can have a large influence on the ionic transport properties, but this factor has not been considered in previous literature. It should be noted that the existence of ACN is not observable in the Raman spectra of the SE annealed at temperatures higher than 230 °C because Raman, as a surface specific technique, cannot provide structural information in the bulk SEs, highlighting the unique advantage of the synchrotron total scattering for the current research.
2.3. Structural evolution before Li7P3S11 formation
To further understand the synthetic pathway, we then separately discuss the structural evolution before and after the transition temperature. For the SEs annealed at temperatures lower than 200 °C, the Bragg data (Fig. 3a) agree very well with that of the pure Li3PS4-ACN crystalline complex which is separately synthesized by a solution method with the molar ratio of Li2S to P2S5 being 3:1. No other crystalline phases can be observed. In contrast, the reduced PDF profiles of SEs (Fig. 3b) are quite different from that of Li3PS4-ACN. The intensities of the peaks corresponding to ACN (at 1.2 Å and 1.4 Å) are much lower in the SEs than in pure Li3PS4-CAN while the intensities of the peaks at ∼2.1 Å and ∼3.4 Å, which exist in typical LPS thiophosphate, are much higher in the SEs than in pure Li3PS4-ACN. The results suggest that an additional component that is amorphous in nature and without ACN also exists in the solution processed precursor. Wang et al.16 reported that the composition of the amorphous component is Li2S–P2S5 with a molar ratio of 1:1. This amorphous component, which is presumably the product dried from the supernatant,16 is critical for the successful synthesis because it will react with Li3PS4-ACN to form Li7P3S11. Our Raman spectra of LPS-170 and LPS-230 suggest that the local structure of P in the amorphous Li2S–P2S5 mainly contains P2S64− and P2S72− (Fig. S2†). More careful characterization of the product dried from the supernatant should be done in the future to understand the local structure of the amorphous component in the precursor. Although the intensities for the peaks corresponding to ACN gradually decrease as the annealing temperature increases, no apparent evidence can be observed for the formation of crystalline and amorphous Li3PS4 for LPS-200 because no new crystalline phases can be observed from the diffraction data (Fig. 3a) and the decomplexation from Li3PS4-ACN to amorphous Li3PS4 will lead to apparent changes in the reduced PDFs (Fig. S5†). Therefore, no apparent decomplexation reaction occurs within this temperature range.
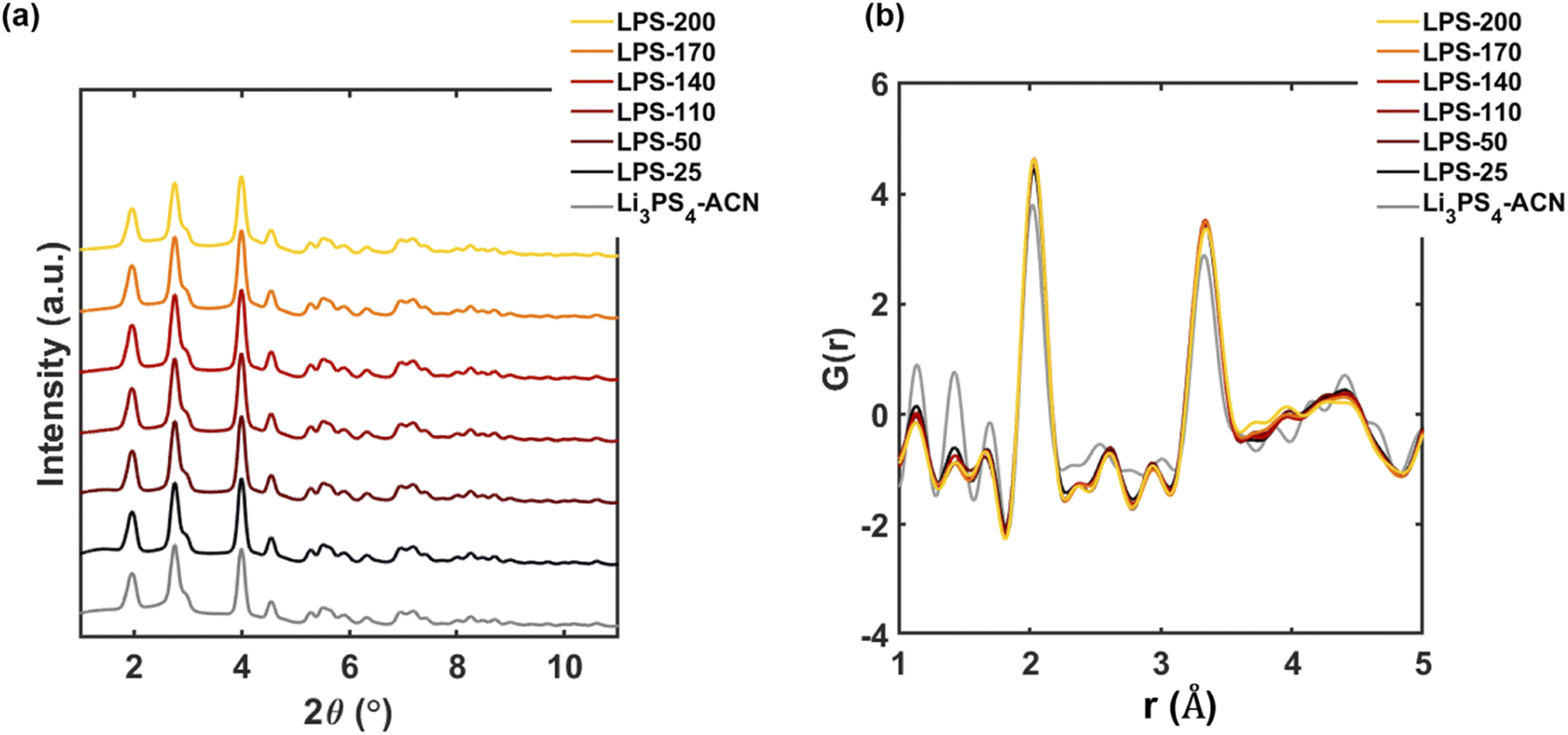 | ||
| Fig. 3 Synchrotron diffraction Bragg data (a) and reduced PDFs (b) of SEs after annealing at temperatures below 200 °C. The data of the pure Li3PS4-ACN crystalline complex is also provided for comparison. | ||
2.4. Structural evolution after Li7P3S11 formation
Fig. 4a–c show the diffraction Bragg data from the total scattering measurement. The reflection peaks for LPS-230 (Fig. 4a) could be attributed to single-phase Li7P3S11. The reflection peaks for SEs annealed at 260 °C (Fig. 4b) and 290 °C (Fig. 4c) can be refined with a mixture of Li7P3S11 and Li4P2S6 phases, indicating the decomposition of Li7P3S11 at higher temperatures. The phase fraction of Li4P2S6 is also determined to increase from 13.35% to 32.15% as temperature increases from 260 °C (Fig. 4b) to 290 °C (Fig. 4c). To quantitatively understand the evolution of crystallinity as the temperature increases, we used a difference PDF method40,41 where the reduced PDFs of SEs annealed at different temperatures, G(r)experimental, were fitted by a linear combination of the reduced PDF of ball-milled Li7P3S11 glass (G(r)glass) and the reduced PDF of Li7P3S11 glass-ceramic (G(r)glass-ceramic) using the following equation:| G(r)experimental = (1 − x)G(r)glass + xG(r)glass-ceramic |
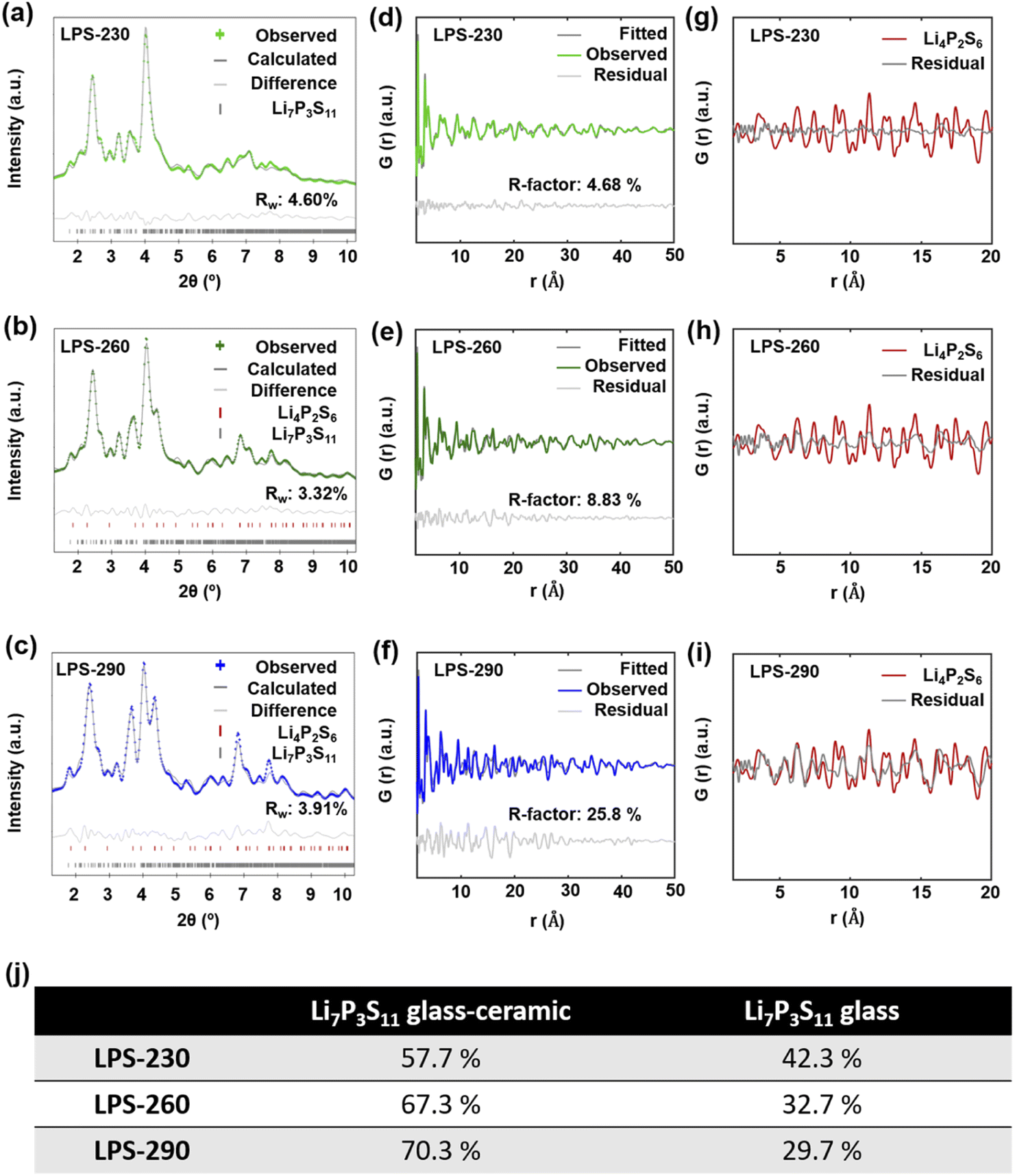 | ||
| Fig. 4 Synchrotron diffraction Bragg data of SEs annealed at 230 °C (a), 260 °C (b), and 290 °C (c). Reflections for LPS-230 could be assigned to crystalline Li7P3S11, and reflections for LPS-260 and LPS-290 were refined with both Li7P3S11 and Li4P2S6 phases. The phase fraction of Li4P2S6 increases as the annealing temperature increases. Reduced PDFs of LPS-230 (d), LPS-260 (e), and LPS-290 (f) in the range of 1.7 Å to 50 Å were fitted by a linear combination of reduced PDFs of ball-milled Li7P3S11 glass and Li7P3S11 glass-ceramic (ball-milled glass annealed at 260 °C for 12 hours) to estimate the crystallinity. The R-factor is labeled on each figure. (g–i) The resulting profile differences of the linear combination fitting of SEs annealed at different temperatures were compared with the calculated reduced PDF of crystalline Li4P2S6. The difference profile in (f) shows reasonably good agreement with the reduced PDF of Li4P2S6. The phase fractions of Li7P3S11 glass and Li7P3S11 glass-ceramic in the SEs annealed at different temperatures for the linear combination fitting are included in the table (j). | ||
Correlating the structural information with the ionic conductivity of SEs annealed at different temperatures, we are able to get a better understanding on the synthesis–structure–property relationship. As shown in Fig. 5, at temperatures lower than 200 °C, the removal of ACN from the Li3PS4-ACN complex cannot occur, leading to the co-existence of the poorly conductive crystalline Li3PS4-ACN complex and amorphous Li2S–P2S5 precursor dried from the supernatant. Increasing the annealing temperature to 230 °C is sufficient for the removal of ACN and the solid-state reaction of the precursors to form Li7P3S11, although based on the current ex situ study we are unable to tell whether the solvent removal process occurs prior to or simultaneously with the solid-state reaction. The formation of Li7P3S11 at 230 °C explains the sharp increase in the ionic conductivity from 200 °C (1.1 × 10−6 S cm−1) to 230 °C (2.5 × 10−4 S cm−1), but the ionic conductivity of Li7P3S11 is still limited due to the low crystallinity (<57.7%), as it has been well known that for Li7P3S11 the ionic conductivity increases with the increase in crystallinity.31,33 Further increase in the annealing temperature can improve crystallinity, which can improve the ionic conductivity, but also leads to the formation of poorly conductive Li4P2S6 due to decomposition of Li7P3S11. Therefore, two competing mechanisms exist during high-temperature annealing. At 260 and 290 °C, the increase in the ionic conductivity due to improved crystallinity (to ∼70%) surpasses the decrease in the ionic conductivity due to Li4P2S6 formation, and therefore the ionic conductivity increases to 7.0 × 10−4 S cm−1 for the SE annealed at 290 °C, but continuous increase to 320 °C leading to severe decomposition of Li7P3S11 that decreases the ionic conductivity.
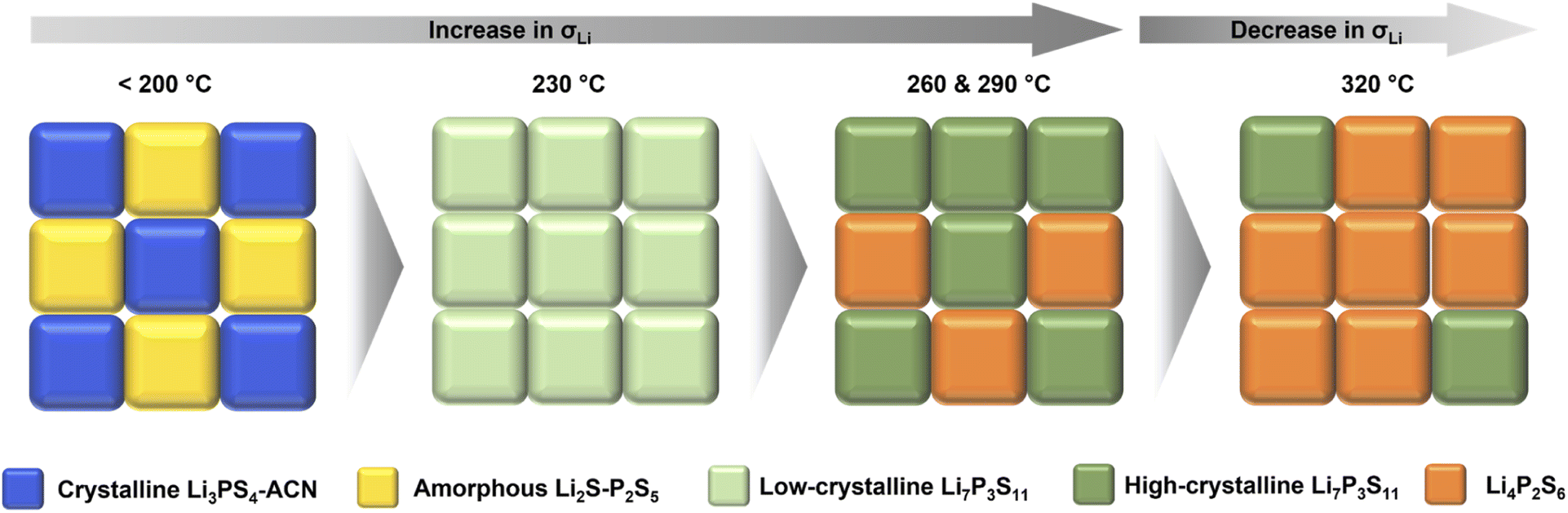 | ||
| Fig. 5 Schematic illustration of the synthesis–structure–property relationship during annealing of the solution-processed precursor. | ||
2.5. Strategies to increase the ionic conductivity of solution-synthesized Li7P3S11
The structural analysis based on synchrotron X-ray total scattering provides important insights to rationalize the structure–property relation of solution-synthesized Li7P3S11 SEs. While LPS-230 shows single-phase Li7P3S11 from lab XRD, its crystallinity is too low. Increasing the annealing temperature increases the crystallinity which is helpful for improving ionic transport and achieving the highest ionic conductivity after annealing at 290 °C. However, further increasing the annealing temperature to 320 °C will decrease the ionic conductivity because of the formation of the poorly conductive Li4P2S6 phase due to the thermal decomposition of Li7P3S11. Thus, a trade-off exists between improving crystallinity and suppressing decomposition. These fundamental insights also lead to effective strategies for further improving the ionic conductivity of solution-synthesized Li7P3S11 SE by overcoming this trade-off. This can be achieved by a better understanding of the nucleation and growth of Li7P3S11 during annealing. The two-step annealing process reported to facilitate crystallization of a ball-milled glass31,42 could also be applied here. The thermodynamics for the glass to ceramic phase transition is also strongly dependent on the particle size. Decreasing the particle size of the solution-processed precursor, by reducing the particle size of the Li2S reactant or lowering the solvent to solids ratio, might also help to lower the crystallization temperature, although careful characterization should also be done to study the size effect on the thermal decomposition of Li7P3S11. Another important factor that cannot be ignored is the existence of ACN solvent in the resulting SEs. Developing approaches that can effectively remove the solvent in the solids is also expected to increase ionic conductivity. It should also be noted that the improvement in the solvent removal process should not affect other chemical processes during the synthesis. For example, while improving the annealing time might be helpful for solvent removal, it cannot effectively improve ionic conductivity due to the enhanced decomposition of Li7P3S11 to form Li4P2S6 (Fig. S6†). Annealing the solution-processed precursor under vacuum is also effective in removing ACN but leads to the formation of β-Li3PS4 instead of Li7P3S11 at high temperatures (Fig. S7†), possibly due to the loss of the amorphous Li2S–P2S5 precursor under high vacuum. Additional approaches include exploring low boiling point solvents24 and reducing the particle size of the solution-processed precursor to mitigate the diffusion limitation.3. Conclusions
In summary, synchrotron X-ray total scattering and PDF analysis are used to study the structural evolution during annealing the solution-processed precursor for the synthesis of the Li7P3S11 SE at different temperatures. The solution-processed precursor consists of a crystalline Li3PS4-ACN complex and amorphous lithium thiophosphate dried from the supernatant, and no apparent change in the structure of the precursor can be observed below 200 °C. The formation of Li7P3S11 was observed after annealing at 230 °C, but the SE formed after annealing at this temperature is confirmed to be a glass ceramic that contains both amorphous and crystalline Li7P3S11. Increasing the annealing temperature to 260 and 290 °C helps to improve the crystallinity of Li7P3PS11, however, Li4P2S6 was also formed after annealing at these temperatures due to the decomposition of Li7P3S11. The quantitative determination of the crystallinity, based on the difference PDF method, indicates that the crystallinity of the Li7P3S11 SE is lower than 70.3% after annealing at 290 °C. In addition, residual ACN is still present in the SE annealed at 290 °C, although the temperature is much higher than the boiling point of ACN. The obtained structural information helps to rationalize the synthesis–property relationship and explains why the SE annealed at 290 °C demonstrates the highest ionic conductivity based on the balance between improving the crystallinity and suppressing the decomposition. The results suggest that crystallization to a desired phase determined by lab XRD is overly simplistic for describing the success of the synthesis. Several approaches to improving crystallinity and decreasing the residual solvent while suppressing the decomposition were also discussed to develop approaches for increasing the ionic conductivity of solution-synthesized Li7P3S11. Synchrotron X-ray total scattering combined with PDF analysis will be an important tool to identify the optimal synthetic conditions of these approaches.4. Experimental
4.1. Materials synthesis
The solution-processed precursor for the synthesis of Li7P3S11 SEs is prepared through a previously reported method.16 Li2S (Sigma-Aldrich, 99.98%) and P2S5 (Sigma-Aldrich, 99%) with a molar ratio of 7:3 were mixed in acetonitrile (ACN) solvent (Fisher Scientific, 99.8%) at 50 °C for 3 days. After removing all the solvents by drying under vacuum at 80 °C for 9 hours, the precursor was recovered and ground before heat treatment at different temperatures from 25 to 320 °C under argon for 1 hour. Pure Li3PS4-ACN crystalline complex was prepared through the same procedure but the molar ratio of Li2S to P2S5 is 3:1. Li7P3S11 glass and glass-ceramic synthesized by a solid-state method are used as a reference sample in the work, while Li7P3S11 glass was prepared by high-energy ball milling of the Li2S and P2S5 with a molar ratio of 7:3 at 510 rpm for 50 hours and the Li7P3S11 glass-ceramic was synthesized by annealing the ball-milled glass at 260 °C for 3–12 hours.
4.2. Materials characterization
Lab XRD measurements were performed on a PanAlytical X'Pert diffractometer using a Cu Kα X-ray source from 15° to 70° 2θ, at a scan rate of 1.25 min−1. Synchrotron X-ray total scattering measurements were performed at beamline 11-ID-B (λ = 0.21150 Å) of the Advanced Photon Source (APS) at Argonne National Laboratory. The samples were loaded in Kapton capillaries and sealed with epoxy. The neutron total scattering measurement was performed at beamline NOMAD at the Spallation Neutron Source (SNS), Oak Ridge National Laboratory. Neutron PDF samples were loaded in the quartz tubes provided by Oak Ridge National Laboratory and sealed with epoxy. Raman spectroscopy was performed on a Renishaw Raman spectrometer with a 514 nm laser beam. Scanning electron microscopy (SEM) images were taken on a Carl Zeiss Supra 55 field emission SEM.4.3. Data analysis
GSAS-II was used to calibrate and extract two-dimensional intensity vs. 2θ histograms from the diffraction images.43 We also used GSAS-II to perform Rietveld refinement of the histograms and obtain the pair distribution functions from the scattering data. A maximum scattering vector (Qmax) of 30 Å−1 was employed in the Fourier transform. PDFgui was used to calculate the reduced PDF of Li4P2S6 based on known structure models.39,44 The linear combination fitting of reduced PDFs was done using Athena.45 The structures were visualized using VESTA.464.4. Electrochemical measurement
The ionic conductivities of SEs were determined from EIS measurements (BioLogic VSP-3 potentiostat). 150 mg SE, annealed at different temperatures, was densified at 400 MPa for 3 min in a polyether ether ketone (PEEK) housing, and two stainless steel rods were applied at each side of the SE to construct a two-blocking electrode cell. The impedance measurement was performed from 7 MHz to 10 mHz with an amplitude of 10 mV at room temperature.Author contributions
F. Han conceived the idea and supervised the project. B. Shao synthesized the materials and performed the electrochemical measurements. R. Das analyzed the synchrotron X-ray total scattering data. Y. Huang and R. Deng assisted with the electrochemical tests. S. Seelman assisted in the experimental work during the revision. B. Shao, R. Das, and F. Han wrote the manuscript with the input from all the co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the US National Science Foundation (award no. 2223217) and US Department of Energy (DOE), Advanced Research Projects Agency for Energy (ARPA-E), IONICS program (Award No. DE-AR0000781). F. H. also acknowledges the support from the Priti and Mukesh Chatter Career Development Chair Professorship at the Rensselaer Polytechnic Institute. This research used resources of the Advanced Photon Source, a U.S. DOE Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The mail-in program at beamline 11-ID-B contributed to the data. This research used resources at the Spallation Neutron Source (SNS), a Department of Energy Office of Science User Facility operated by the Oak Ridge National Laboratory (ORNL). Neutron PDF measurements were done at the NOMAD beamline of SNS. We also thank Dr Jue Liu at ORNL for the assistance in collecting and analyzing neutron PDF.References
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 1–4 CrossRef.
- C. Sun, J. Liu, Y. Gong, D. P. Wilkinson and J. Zhang, Nano Energy, 2017, 33, 363–386 CrossRef CAS.
- K. Tuo, C. Sun and S. Liu, Electrochem. Energy Rev., 2023, 6, 17 CrossRef CAS.
- F. Mizuno, A. Hayashi, K. Tadanaga and M. Tatsumisago, Adv. Mater., 2005, 17, 918–921 CrossRef CAS.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed.
- H.-J. Deiseroth, S.-T. Kong, H. Eckert, J. Vannahme, C. Reiner, T. Zaiß and M. Schlosser, Angew. Chem., Int. Ed., 2008, 47, 755–758 CrossRef CAS PubMed.
- A. Sakuda, A. Hayashi and M. Tatsumisago, Sci. Rep., 2013, 3, 1–5 Search PubMed.
- A. Banik, T. Famprikis, M. Ghidiu, S. Ohno, M. A. Kraft and W. G. Zeier, Chem. Sci., 2021, 6238–6263 RSC.
- R. Schlem, C. F. Burmeister, P. Michalowski, S. Ohno, G. F. Dewald, A. Kwade and W. G. Zeier, Adv. Energy Mater., 2021, 11, 2101022 CrossRef CAS.
- A. Hayashi, S. Hama, H. Morimoto, M. Tatsumisago and T. Minami, J. Am. Ceram. Soc., 2001, 84, 477–479 CrossRef CAS.
- S. Boulineau, M. Courty, J.-M. Tarascon and V. Viallet, Solid State Ionics, 2012, 221, 1–5 CrossRef CAS.
- A. Miura, N. C. Rosero-Navarro, A. Sakuda, K. Tadanaga, N. H. H. Phuc, A. Matsuda, N. Machida, A. Hayashi and M. Tatsumisago, Nat. Rev. Chem., 2019, 3, 189–198 CrossRef CAS.
- M. Ghidiu, J. Ruhl, S. P. Culver and W. G. Zeier, J. Mater. Chem. A, 2019, 7, 17735–17753 RSC.
- Ö. U. Kudu, T. Famprikis, B. Fleutot, M.-D. Braida, T. Le Mercier, M. S. Islam and C. Masquelier, J. Power Sources, 2018, 407, 31–43 CrossRef.
- Z. Liu, W. Fu, E. A. Payzant, X. Yu, Z. Wu, N. J. Dudney, J. Kiggans, K. Hong, A. J. Rondinone and C. Liang, J. Am. Chem. Soc., 2013, 135, 975–978 CrossRef CAS PubMed.
- Y. Wang, D. Lu, M. Bowden, P. Z. El Khoury, K. S. Han, Z. D. Deng, J. Xiao, J.-G. Zhang and J. Liu, Chem. Mater., 2018, 30, 990–997 CrossRef CAS.
- N. H. H. Phuc, M. Totani, K. Morikawa, H. Muto and A. Matsuda, Solid State Ionics, 2016, 288, 240–243 CrossRef CAS.
- K. Lee, S. Kim, J. Park, S. H. Park, A. Coskun, D. S. Jung, W. Cho and J. W. Choi, J. Electrochem. Soc., 2017, 164, A2075 CrossRef CAS.
- T. Ates, M. Keller, J. Kulisch, T. Adermann and S. Passerini, Energy Storage Mater., 2019, 17, 204–210 CrossRef.
- M. Calpa, N. C. Rosero-Navarro, A. Miura and K. Tadanaga, RSC Adv., 2017, 7, 46499–46504 RSC.
- S. Ito, M. Nakakita, Y. Aihara, T. Uehara and N. Machida, J. Power Sources, 2014, 271, 342–345 CrossRef CAS.
- R. C. Xu, X. H. Xia, Z. J. Yao, X. L. Wang, C. D. Gu and J. P. Tu, Electrochim. Acta, 2016, 219, 235–240 CrossRef CAS.
- B. Fan, Q. Zhang, Z. Luo, X. Zhang, H. Ma, P. Fan and B. Xue, Solid State Ionics, 2019, 343, 115073 CrossRef CAS.
- H. Gamo, A. Nagai and A. Matsuda, Sci. Rep., 2021, 11, 21097 CrossRef PubMed.
- K. Yamamoto, M. Takahashi, K. Ohara, N. H. H. Phuc, S. Yang, T. Watanabe, T. Uchiyama, A. Sakuda, A. Hayashi, M. Tatsumisago, H. Muto, A. Matsuda and Y. Uchimoto, ACS Omega, 2020, 5, 26287–26294 CrossRef CAS PubMed.
- K. Yamamoto, S. Yang, M. Takahashi, K. Ohara, T. Uchiyama, T. Watanabe, A. Sakuda, A. Hayashi, M. Tatsumisago, H. Muto, A. Matsuda and Y. Uchimoto, ACS Appl. Energy Mater., 2021, 4, 2275–2281 CrossRef CAS.
- M. Takahashi, S. Yang, K. Yamamoto, K. Ohara, N. H. H. Phuc, T. Watanabe, T. Uchiyama, A. Sakuda, A. Hayashi, M. Tatsumisago, H. Muto, A. Matsuda and Y. Uchimoto, Solid State Ionics, 2021, 361, 115568 CrossRef CAS.
- J. E. Lee, K.-H. Park, J. C. Kim, T.-U. Wi, A. R. Ha, Y. B. Song, D. Y. Oh, J. Woo, S. H. Kweon, S. J. Yeom, W. Cho, K. Kim, H.-W. Lee, S. K. Kwak and Y. S. Jung, Adv. Mater., 2022, 34, 2200083 CrossRef CAS PubMed.
- M. Tatsumisago, S. Hama, A. Hayashi, H. Morimoto and T. Minami, Solid State Ionics, 2002, 154–155, 635–640 CrossRef CAS.
- A. Hayashi, K. Minami and M. Tatsumisago, J. Solid State Electrochem., 2010, 14, 1761–1767 CrossRef CAS.
- M. R. Busche, D. A. Weber, Y. Schneider, C. Dietrich, S. Wenzel, T. Leichtweiss, D. Schröder, W. Zhang, H. Weigand, D. Walter, S. J. Sedlmaier, D. Houtarde, L. F. Nazar and J. Janek, Chem. Mater., 2016, 28, 6152–6165 CrossRef CAS.
- F. Marchini, B. Porcheron, G. Rousse, L. Albero Blanquer, L. Droguet, D. Foix, T. Koç, M. Deschamps and J. M. Tarascon, Adv. Energy Mater., 2021, 11, 2101111 CrossRef CAS.
- Y. Seino, M. Nakagawa, M. Senga, H. Higuchi, K. Takada and T. Sasaki, J. Mater. Chem. A, 2015, 3, 2756–2761 RSC.
- T. Egami and S. J. L. Billinge, Underneath the Bragg Peaks: Structural Analysis of Complex Materials, Newnes, 2012 Search PubMed.
- K. W. Chapman, MRS Bull., 2016, 41, 231–240 CrossRef.
- X. Wang, S. Tan, X.-Q. Yang and E. Hu, Chin. Phys. B, 2020, 29, 028802 CrossRef CAS.
- K. Ohara, N. Masuda, H. Yamaguchi, A. Yao, S. Tominaka, H. Yamada, S. Hiroi, M. Takahashi, K. Yamamoto, T. Wakihara, Y. Uchimoto, F. Utsuno and S. Kimura, Phys. Status Solidi B, 2020, 257, 2000106 CrossRef CAS.
- M. Calpa, H. Nakajima, S. Mori, Y. Goto, Y. Mizuguchi, C. Moriyoshi, Y. Kuroiwa, N. C. Rosero-Navarro, A. Miura and K. Tadanaga, Inorg. Chem., 2021, 60, 6964–6970 CrossRef CAS PubMed.
- C. Dietrich, M. Sadowski, S. Sicolo, D. A. Weber, S. J. Sedlmaier, K. S. Weldert, S. Indris, K. Albe, J. Janek and W. G. Zeier, Chem. Mater., 2016, 28, 8764–8773 CrossRef CAS.
- J. Peterson, J. TenCate, T. Proffen, T. Darling, H. Nakotte and K. Page, J. Appl. Crystallogr., 2013, 46, 332–336 CrossRef CAS.
- S. Shiotani, K. Ohara, H. Tsukasaki, S. Mori and R. Kanno, Sci. Rep., 2017, 7, 6972 CrossRef PubMed.
- K. Minami, A. Hayashi and M. Tatsumisago, J. Am. Ceram. Soc., 2011, 94, 1779–1783 CrossRef CAS.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- C. L. Farrow, P. Juhas, J. W. Liu, D. Bryndin, E. S. Božin, J. Bloch, T. Proffen and S. J. L. Billinge, J. Phys.: Condens. Matter, 2007, 19, 335219 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta02326e |
| This journal is © The Royal Society of Chemistry 2023 |