
Strategic design of covalent organic frameworks (COFs) for photocatalytic hydrogen generation

Abstract
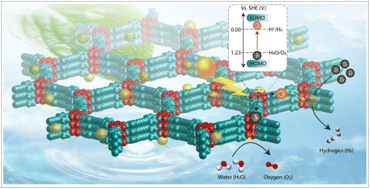
Covalent organic frameworks (COFs) are an emerging class of crystalline materials that are attracting increasing attention due to their high porosity, crystallinity, and tunable properties. Consequently, the strategic design of COF-based photocatalysts for various applications, including energy and environmental remediation, has attracted considerable interest. In particular, the sustainable production of clean fuel – hydrogen (H2) – by water splitting is a promising means to meet the global energy demand and to address the atmospheric CO2 concentration caused by the excessive use of fossil fuels. In this regard, COFs offer potential advantages due to their modular nature, which facilitates their rational design from suitable organic building blocks to achieve optimal properties of visible light harvesting properties and easy charge transport. As a result, extensive research has been devoted to the design of photoresponsive COFs for efficient water splitting to generate hydrogen. Here, we provide a comprehensive review of recent developments in the strategic design of COF-based photocatalysts for solar fuel production via water splitting. The various organic linkers used in the construction of photocatalytic COFs and their structure–property correlations are discussed in detail. The role of bandgap engineering in tuning the hydrogen evolution efficiency of COFs is also discussed. Furthermore, the current challenges and future perspectives of COF-based solid catalysts for green and sustainable clean fuel production are presented. Indeed, this review demonstrates the importance of COF-based photocatalysts for the visible-light-driven hydrogen evolution reaction (HER) and can be beneficial for the future design of photocatalytic systems.
- This article is part of the themed collections: Journal of Materials Chemistry A Recent Review Articles and #MyFirstJMCA
 Please wait while we load your content...
Please wait while we load your content...

