
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solid oxide electrolysis cells – current material development and industrial application
Stephanie E.
Wolf†
 ab,
Franziska E.
Winterhalder†
cd,
Vaibhav
Vibhu
a,
L. G. J. (Bert)
de Haart
a,
Olivier
Guillon
cd,
Rüdiger-A.
Eichel
ab and
Norbert H.
Menzler
*cd
ab,
Franziska E.
Winterhalder†
cd,
Vaibhav
Vibhu
a,
L. G. J. (Bert)
de Haart
a,
Olivier
Guillon
cd,
Rüdiger-A.
Eichel
ab and
Norbert H.
Menzler
*cd
aForschungszentrum Jülich GmbH, Institute of Energy and Climate Research, IEK-9: Fundamental Electrochemistry, 52425, Jülich, Germany
bRWTH Aachen University, Institute of Physical Chemistry, 52074, Aachen, Germany
cForschungszentrum Jülich GmbH, Institute of Energy and Climate Research, IEK-1: Materials Synthesis and Processing, 52425, Jülich, Germany. E-mail: n.h.menzler@fz-juelich.de
dRWTH Aachen University, Institute of Mineral Engineering (GHI), Department of Ceramics and Refractory Materials, 52074, Aachen, Germany
First published on 10th July 2023
Abstract
Solid Oxide Electrolysis Cells (SOECs) have proven to be a highly efficient key technology for producing valuable chemicals and fuels from renewably generated electricity at temperatures between 600 °C and 900 °C, thus providing a carbon-neutral method for energy storage. The successful implementation of this technology on an industrial level in particular requires the long-term stability of all system components with a concurrent overall degradation rate of a maximum of 0.75% kh−1 or even better 0.5% kh−1, corresponding to a performance loss of 20% over approx. five years under constant operating parameters. However, the materials currently used for SOEC systems have been developed and optimized in recent decades for fuel cell operation. The degradation of these Solid Oxide Fuel Cell (SOFC) materials used in SOECs, however, slows down the technology and market ramp-up. Accordingly, a selection and development of materials specifically for use in SOEC operation, must therefore be based not only on the highest performance but also on the lowest achievable degradation rate. In general, the systematic development of new SOEC materials must be driven towards key performance parameters such as mechanical, thermal, and chemical stability as well as an application-oriented assessment (cost effectiveness, simple manufacturing). This review presents the state-of-the-art materials in current industrial use for planar SOECs as well as future challenges regarding materials design and degradation. Recent advances in material compositions are discussed and evaluated in terms of their performance, stability, and potential for industrial implementation. In addition, a materials selection for interconnects, coatings, and sealants is briefly listed to outline current developments in these areas.
 Stephanie E. Wolf | Stephanie E. Wolf graduated with distinction from RWTH Aachen University, Germany, in 2020 with a degree in chemistry, focusing on catalysis, computational chemistry, and spectroscopy. Since October 2020, she has been working as a PhD candidate at the Institute of Energy and Climate Research (IEK-9: Fundamental Electrochemistry) at Forschungszentrum Jülich. Her fields of research include material synthesis, chemo-physical material characterization, solid oxide cell preparation, electrochemical cell characterization, long-term degradation studies as well as postmortem analyses. |
 Franziska E. Winterhalder | Franziska E. Winterhalder is currently pursuing her PhD in materials engineering at the Institute of Energy and Climate Research (IEK-1: Materials Synthesis and Processing) at Forschungszentrum Jülich under the supervision of Norbert H. Menzler. She received her Master's degree in Materials Sciences from the Friedrich Schiller University Jena in 2021. Her research interests include material development and engineering for solid oxide cells, currently focusing on alternative fuel electrodes. |
 Vaibhav Vibhu | Vaibhav Vibhu studied chemistry and obtained his master's degree from the University of Delhi and the University of Grenoble Alpes. He then completed his PhD in 2016 at the ICMCB-CNRS, University of Bordeaux, France, on “Stability and ageing studies of praseodymium-based nickelates as cathodes for SOFCs”. Afterward, he moved to IEK-9 at Forschungszentrum Jülich, Germany, to continue his work in the field of solid oxide cells (SOFCs/SOECs). His research focuses on the development of electrode materials, materials characterizations and processing, the manufacturing of cells, electrochemical characterizations and the identification of reaction mechanisms of electrodes and degradation phenomena of cells. |
 L. G. J. (Bert) de Haart | L. G. J. (Bert) de Haart (*1958) studied Chemistry at State University of Utrecht (The Netherlands) and obtained his Master degree (Drs.) with specialty Solid State Chemistry in 1981. In 1985 he obtained his PhD degree (Dr) also from the State University of Utrecht. From 1987–1992 he was research assistant (Post-Doc) at the University of Twente (The Netherlands) in the Department of Chemical Technology. In 1992 he joined the Forschungszentrum Jülich (Germany) where he continued his work on the development of Solid Oxide Fuel and Electrolysis Cells (SOFC/SOEC) with emphasis on materials development and cell- and stack-design, -manufacturing and -characterization. |
 Olivier Guillon | Olivier Guillon is a materials scientist and engineer. He became director at the Institute of Energy and Climate Research – Materials Synthesis and Processing (IEK-1, Forschungszentrum Jülich) and professor at the RWTH Aachen University in 2014. He is the spokesperson of the research program “Materials and Technologies for the Energy Transition” within the Helmholtz Association and works on ceramic materials and components for solid oxide fuel and electrolysis cells, gas separation membranes, solid-state batteries and high-temperature applications. He has published more than 300 articles in international referred journals and is member of the World Academy of Ceramics, fellow of the American and European Ceramic Societies. |
 Rüdiger-A. Eichel | Rüdiger-A. Eichel studied physics at the University of Cologne and completed his PhD at the Swiss Federal Institute of Technology (ETH) in Zurich. In 2012, he became a director at the IEK-9 at Forschungszentrum Jülich and simultaneously was appointed full professor for physical chemistry at RWTH Aachen University. His research interests in the field of solid oxide cells focus on the development, characterization, and optimization of high-temperature electrolysis and fuel cell stacks and systems. In his institute, systems and system components are conceived, designed, modeled, built as well as tested, and electrochemically analyzed. |
 Norbert H. Menzler | Norbert H. Menzler studied Materials Science at the University of Erlangen–Nuremberg from where he got his Diploma and his PhD Afterwards, he acted as a Managing Director of a Bavarian research network. In 2000 he joined Forschungszentrum Jülich working now on solid oxide cells. His specialization is the cell materials, their processing, and the post-test analysis of operated stacks. Since 2016 he has been the head of the solid oxide cell department of the Institute of Energy and Climate Research (IEK-1: Materials Synthesis and Processing). In 2019 he received an honorary professorship from the RWTH Aachen University. |
1. Introduction
The detrimental impact of fossil-fuel-based energy carriers on the global climate has led to an increasing interest in carbon-neutral energy technologies such as wind, solar, and biomass to renewably generate electricity and the usage of green hydrogen (H2) as an energy carrier. Hydrogen can also be stored and re-electrified in a fuel cell, as well as be used as a sustainable feedstock for the chemical industry (power-to-X, power-to-fuel, power-to-chemicals, power-to-heat), thus facilitating the large-scale integration of renewables, enabling efficient global energy logistics and locally supporting the balancing of the grid. Due to its versatile application possibilities, hydrogen plays a key role in a future sustainable energy system as a coupling element for different sectors. Therefore, in addition to an intensive expansion of renewably generated power, a flexible hydrogen infrastructure is necessary. Hydrogen is a non-toxic energy carrier with a high mass-based specific energy content that can be produced in either a carbon-neutral or carbon-free manner. The chemical industry and refineries are currently the main hydrogen consumers. Of the total hydrogen production in 2020 (90 Mt), 44% (40 Mt) was used for the production of valuable oil products in refineries through hydrocracking and hydrotreatments for the desulfurization of fossil fuels.1 An additional 45 Mt H2 was consumed in the chemical industry as a basic component for the production of methanol and ammonia.1 The remaining 5 Mt were mostly used for steelmaking with less than 0.02% H2 (<20 kt H2) used for transport. Hydrogen fuel produced from renewably generated power sources could decarbonize these sectors as well as various energy-intensive industries (e.g., steel), the mobility sector, and residential heating demand. Using natural resources also means a reduction in the price of generating electricity. Strategies are being implemented worldwide to build a hydrogen economy and infrastructure, as highlighted in Fig. 1. The political effort to increase hydrogen production includes supporting research and innovation projects, scaling up and decarbonizing existing hydrogen production facilities, expanding logistics infrastructure, and establishing larger-scale storage facilities. Europe is expected to achieve climate neutrality in 2050, which is why a continent-wide hydrogen network is planned. A significant aspect is the repurposing of e.g., existing natural gas pipelines for the global hydrogen trade of gaseous hydrogen to lower the delivery costs. The hydrogen pipeline network may connect industrial end users with large-scale electrolyzers also found outside of the EU. In addition, establishment of new pipeline networks to transport hydrogen as ammonia, methanol, and liquid organic hydrogen carriers (LOHC) is considered.2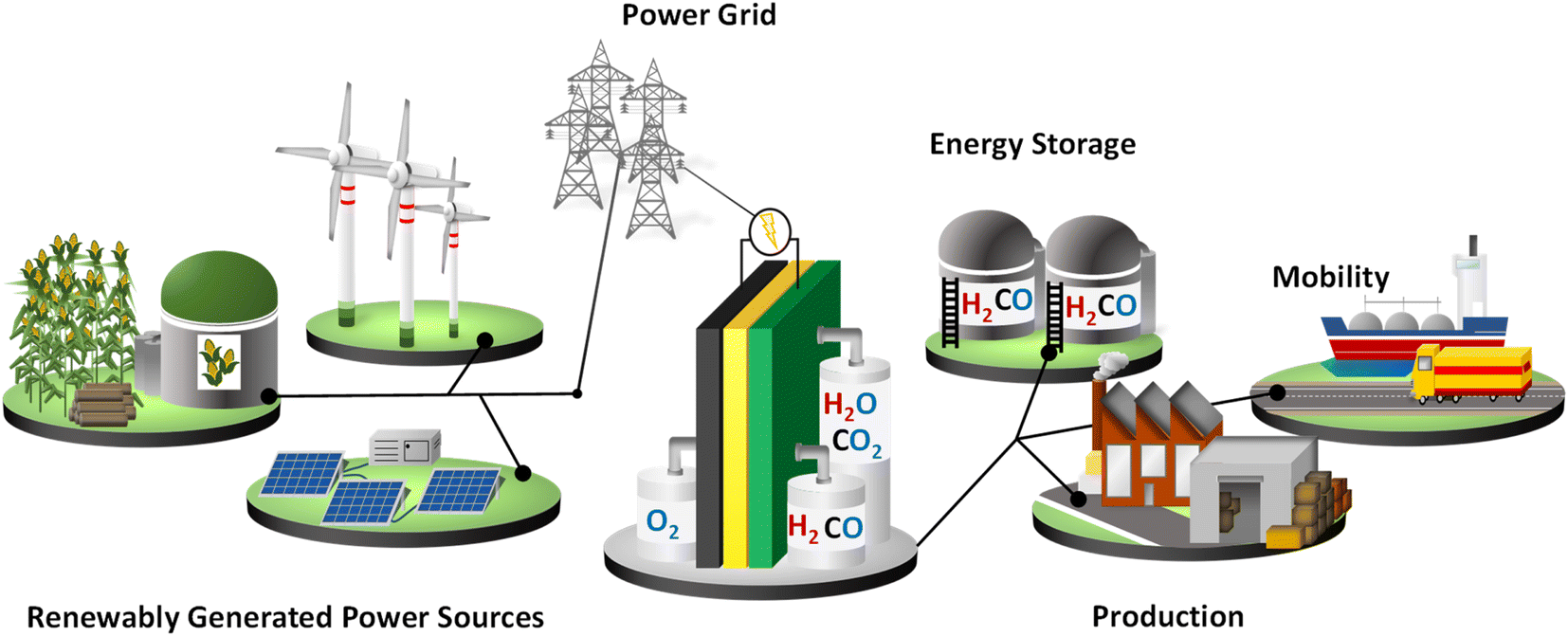 | ||
| Fig. 1 Schematic representation of a hydrogen infrastructure: renewably generated power is used to generate hydrogen through a high-temperature solid oxide electrolysis cell (SOEC). Energy-intensive industries, e.g., production and mobility, are powered by the low-emission energy carrier hydrogen, and surplus chemical energy is stored. | ||
In particular, green hydrogen produced by renewably generated power sources is considered essential for a sustainable transition of the energy system. However, current hydrogen production is mainly based on gray power sources. Hydrogen production routes and sources are shown in Table 1 with the corresponding color encoding according to their present-day classification.1 Hydrogen production via coal gasification results in high carbon emissions and the end product is referred to as black or brown hydrogen depending on the coal type. Hydrogen produced by the steam methane reforming (SMR) of natural gas was the main source of hydrogen in 2020, representing 59% of global production. In naphtha reforming, most refineries produce hydrogen as a by-product, with 21% of global hydrogen production attributed to this route. Obtaining hydrogen from oil oxidation, on the other hand, is negligible at 0.6%. All three synthesis routes produce grey hydrogen and are considered ‘carbon-positive’ technologies. The industrial chloralkali process generates clean, white hydrogen as a by-product, although it does produce CO2 emissions, which stem from electricity produced to operate the electrolyzers. In recent years, low-carbon hydrogen technologies have emerged. Blue hydrogen is classified as production using the SMR process, with the subsequent storage of carbon emissions (CCS). This method still has a low share of global H2 production (0.7%) and is considered a bridging technology. Hydrogen production from fossil fuels has the disadvantage that the gaseous product stream has to be treated in downstream processes to reach an adequate level of purity for further catalytic utilization. Green hydrogen is produced by water electrolysis based on various renewable energy sources. The electrochemical conversion of water to hydrogen results in an almost pure product gas without detrimental impurities. To achieve the goal of net-zero carbon emissions by 2050, water electrolyzer capacities will have to be increased to meet the increasing demand for green hydrogen especially in the additional field of energy logistics.3
| Color | Source | Method | Production in 2020a/% |
|---|---|---|---|
| a Production is calculated as more than 100% in reference. | |||
| Black H2 | Black coal | Gasification | 19% |
| Brown H2 | Brown coal/lignite | Gasification | |
| Grey H2 | Natural gas | Natural gas reforming | 59% |
| Oil | Partial oxidation | 0.6% | |
| By roduct | Naphtha reforming | 21% | |
| White H2 | By product | Chloralkali electrolysis | |
| Blue H2 | Natural gas + carbon capture and storage (CCS) | Natural gas reforming | 0.7% |
| Turquoise H2 | Methane | Pyrolysis | |
| Pink H2 | Nuclear energy | Water electrolysis | |
| Yellow H2 | Mixed grid energy | Water electrolysis | |
| Green H2 | Renewable-generated power sources | Water electrolysis | 0.03% |
Water electrolyzers are classified, depending on their operating conditions and charge carrier type (OH−, H+, O2−), as alkaline electrolyzers, proton exchange-membrane (PEM) electrolyzers, and solid oxide electrolyzers (SOE). Alkaline electrolysis is a commercially mature, low-cost, and stable technology, usually operated at 20–80 °C, up to 30 bar, with process efficiency of about 62–82%.4,5 The charged Raney nickel electrodes are immersed in a liquid alkaline electrolyte, which is commonly potassium hydroxide (KOH). A diaphragm permeable only for OH− anions separates the product gases. Polymer electrolyte membrane (PEM) electrolysis is also commercially available and can operate at high current densities of around −2 A cm−2 to −3 A cm−2 between 50–80 °C. Compared to alkaline electrolyzers, the polymer electrolyte membrane Nafion® is much thinner, provides good proton conductivity (0.12 ± 0.04 S cm−1),6 and has a quicker input response. This results in efficiencies of 67–82% for PEM electrolyzers. The use of expensive noble metal electrodes, for example, Ir, Pt, Ru, and Pr, is essential due to the acidic environment.4 In anion exchange membrane (AEM) electrolyzers, the conventional diaphragm of alkaline electrolyzers is replaced with a quaternary ammonia (polysulfone) AEM permeable for negatively charged OH− ions. These membranes are less expensive than Nafion®-based membranes in PEM electrolysis and enable the use of transition metal catalysts instead of noble metals. AEM electrolysis is still a developing technology operated at 50–70 °C with up to −1.0 A cm−2 at ∼2 V and a current efficiency of ∼40%.7,8 The specific technological comparison of the limitations, advantages, and disadvantages of different electrolyzer systems depends on the application as well as on the economic and ecological point of view. However, this is not the focus point of this paper and is discussed in more detail elsewhere.9–11
Electrochemical energy conversion to generate chemical fuels with high efficiency is possible in traditional oxide ion-conducting electrolysis cells (SOECs) as well as protonic ceramic electrolysis cells (PCECs). Proton-conducting materials have attracted increasing interest due to their lower operating temperatures (450–700 °C).12 As proton-conducting cells are currently still in the development phase and not commercially available, this review focuses on commercially available oxygen ion-conducting electrolysis cells. Solid oxide electrolyzer cells (SOECs) are typically operated between 600 °C and 900 °C, consist of two porous electrodes separated by a dense pure oxide-ion conducting electrolyte, and can achieve technical efficiencies up to 100% due to advantageous thermodynamics and reaction kinetics.13–15 In steam electrolysis, steam (H2O) is electrochemically reduced to hydrogen (H2) at the negatively charged fuel electrode. The resulting oxide ions migrate through the electrolyte to the positively charged oxygen electrode, where they are oxidized to molecular oxygen (eqn (1) and (2)). Green CO production through direct CO2 electrolysis is also feasible in SOECs.16 This process has attracted increasing interest in a possible “closed recycling loop” within the synthetics industry (eqn (3)). A unique advantage of SOECs is also the direct production of syngas (H2 + CO) through co-electrolysis, which is the simultaneous conversion of H2O and CO. H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO ratios from 1:1 to 3:1, which are currently of great importance in the chemical industry, can be achieved using high-temperature SOECs.17 The operating conditions and equilibrium process impact the (reverse) water gas shift reaction ((R)WGS) (eqn (4)) occurring during the co-electrolysis process, with the product ratio on the fuel side being variable. Synthetic natural gas and methane are derived from methanation with an H2:CO ratio of 3:1. In Fischer–Tropsch synthesis, syngas with lower ratios of 1.8:1.2 is used to generate liquid hydrocarbons, for example, synthetic fuels and chemical lubricants.18,19 Additionally, aldehydes synthesized via the “oxo process” hydroformylation are valued products and give way to bulk chemicals such as esters, alcohols, and amines.20
:CO ratios from 1:1 to 3:1, which are currently of great importance in the chemical industry, can be achieved using high-temperature SOECs.17 The operating conditions and equilibrium process impact the (reverse) water gas shift reaction ((R)WGS) (eqn (4)) occurring during the co-electrolysis process, with the product ratio on the fuel side being variable. Synthetic natural gas and methane are derived from methanation with an H2:CO ratio of 3:1. In Fischer–Tropsch synthesis, syngas with lower ratios of 1.8:1.2 is used to generate liquid hydrocarbons, for example, synthetic fuels and chemical lubricants.18,19 Additionally, aldehydes synthesized via the “oxo process” hydroformylation are valued products and give way to bulk chemicals such as esters, alcohols, and amines.20
| 2H2O + 4e− → 2H2 + O2 | (1) |
| O2− → 2e− + O2 | (2) |
| 2CO2 + 4e− → 2CO + O2 | (3) |
| CO2 + H2 ⇄ H2O + CO | (4) |
The high operating temperature is the main advantage of SOECs, which results in lower ohmic losses compared to alkaline and proton exchange membrane electrolyzers as well as advantageous kinetics and thermodynamics. The required process enthalpy ΔH of the endothermic steam reduction process can be supplied in the form of heat and electrical energy (Gibbs energy ΔG). With increasing temperatures, the enthalpy increases slightly for steam reduction while the electrical energy demand ΔG becomes smaller at higher temperatures. The remaining energy demand TΔS can be supplied in the form of heat.21 By integrating the electrolyzer system into pre-existing chemical industry plants, the heat demand can be supplied from the external waste heat of exothermic downstream chemical processes, for example, the production of synthetic fuels, ammonia, or industrial processes such as the production of steel, copper, aluminum, cement, or ceramics. In addition, the internal cell resistances lead to the production of Joule heat, which adds to the enthalpy for educt-to-product conversion. At the thermo-neutral voltage Etn, the exact power required for the endothermic electrolysis process is produced, and therefore no external thermal management is necessary aside from compensating for the heat loss to the surroundings. The energy efficiency of up to 100% and the use of inexpensive catalyst materials make SOEC systems more cost-effective in comparison to alkaline and PEM electrolyzers.22
The current demonstrator systems for SOECs on laboratory and industrial scales have been tested for up to 23000 h at a high current density of −0.9 A cm−2 with a voltage increase of 7.4 mV kh−1 (Table 2). Between 2016 and 2019, the European project GrInHy manufactured an rSOC system (reversible solid oxide cell) with 48 stacks, each with 30 Sunfire cells, arranged in six modules, with a power of 143 kW in electrolysis mode (84%el,LHV) and 30 kW (48%el,LHV) in fuel cell mode.23 The world's most powerful industrial high-temperature electrolyzer (as of 2022) is showcased in the subsequent European collaboration project, GrInHy2.0, in which hydrogen is produced using waste heat from an iron and steel factory.24 The industrial implementation of SOEC technology produces 200 Nm3 h−1 green H2 at 850 °C with a nominal power input of 720 kW and an electrical efficiency of 84%el,LHV.
| Operators | Degrad./% kh−1 | T °C | Time/h | Ref. |
|---|---|---|---|---|
| Electrolyte-supported cell-based stack | ||||
| Topsoe, EIFER | 4.6–5.6 | 820 | 4055 | 32 |
| INL, Ceramtec | 5.6, 4.6 | 800 | 1000 | 33 |
| IKTS | 0.5 | 800 | 2100 | 34 |
| Sunfire, EIFER | 0.8 | 830 | 4224 | 35 |
| Sunfire, DLR | 0.3–1.4 | 820 | 3370 | 36 |
|
||||
| Fuel-electrode-supported cell-based stack | ||||
| Jülich | 0.7 | 800 | 2400 | 37 |
| 1.9 | 700 | 2300 | ||
| 1.9 | 750 | 700 | ||
| 0.4–1 | 800 | 14600 |
||
| Jülich | 1.5 | 810 | 2034 | 31 |
| 810 | 1001 | |||
| Solydera, EIFER | 0.5 | 710 | 3250 | 38 |
| 7450 | ||||
An example of a schematic layout of established stack systems is presented in Fig. 2 for the laboratory scale of the Jülich integrated module.25 All necessary system parts are organized around the integrated module and inlet fuels are kept at room temperature before being fed into the system. On the fuel side, steam and hydrogen can be supplied to the stack by mass flow controllers (MFCs). The steam is generated by the evaporation of demineralized water. The system is connected at the air side to inlet tubes for both compressed air as well as purified ambient air. Fuel and air are heated before being fed into the stack. Post-reaction, the gas mixture on the fuel side is separated into hydrogen and steam in a plate heat exchanger. In a downstream recirculation unit, the off-gas is condensed and compressed with a double-headed prototype diaphragm compressor to overcome the pressure drop of the recirculation loop. The product gas is cooled down and transported out of the system. The reintegration of off-heat and off-gas on the fuel side increases the efficiency of the system.26–29 On the air side, the off-gas is partly recirculated and cooled down with heat exchangers, thus making way for fresh air. The system surrounding the integrated module is permanently ventilated with fresh air to reduce safety risks, for example, the ignition limits of hydrogen.
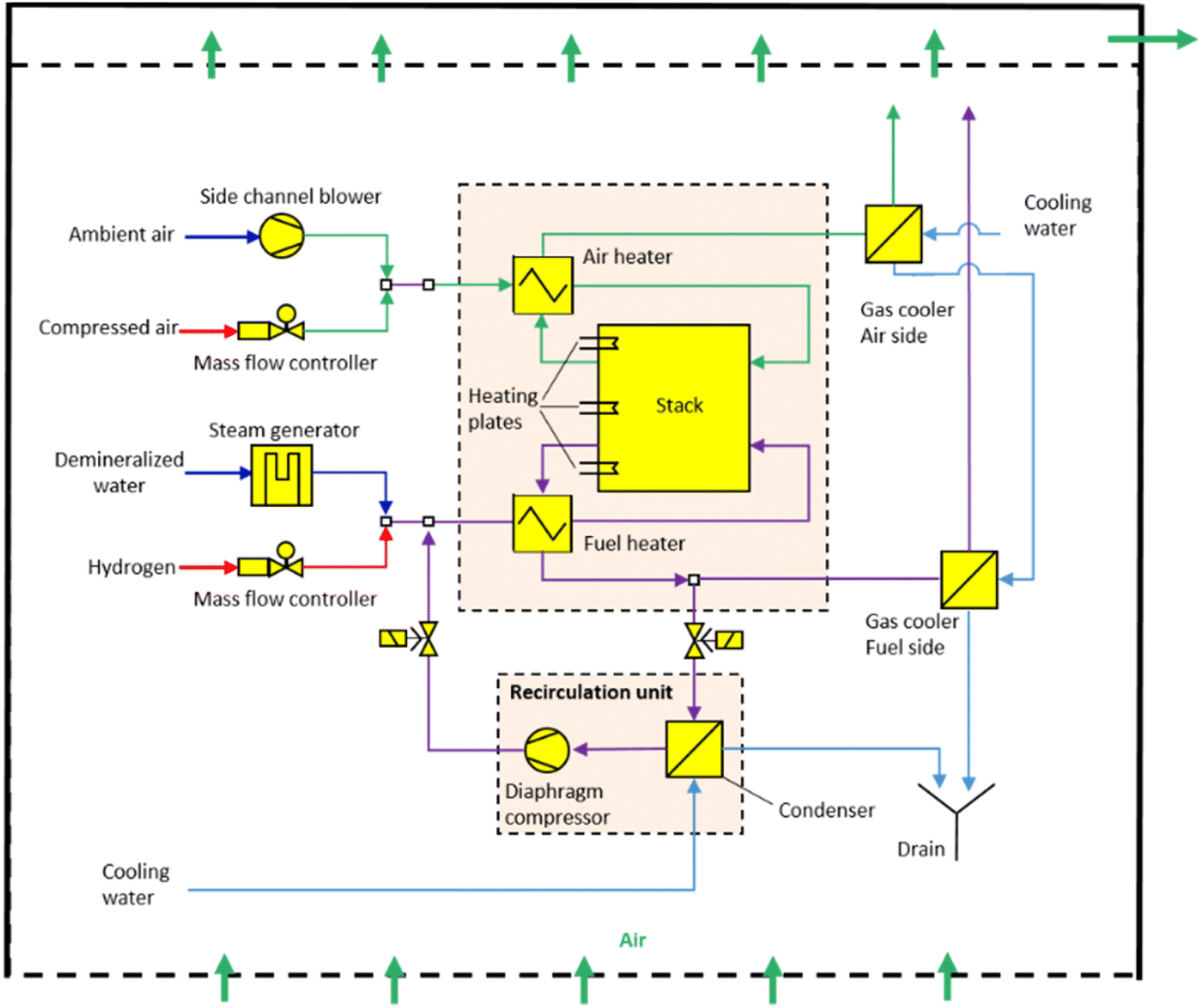 | ||
| Fig. 2 Flow scheme of a laboratory-scale SOC system at Forschungszentrum Jülich. This work is licensed under the terms of the Creative Commons Attribution 4.0 License © 2021 by the authors (CC BY, (https://creativecommons.org/licenses/by/4.0/)).25 | ||
The long-term operation of solid oxide cells in fuel cell mode has been well developed and demonstrated for more than 100000 h with a voltage degradation rate of only 0.5% kh−1.30 In contrast, SOEC stack designs still exhibit higher degradation rates of 0.6–1.5% kh−1, depending on the respective operating conditions applied, for example, current density.31Table 2 provides an overview of long-term SOEC tests with different cell configurations and operating conditions conducted in cooperation between research institutes and industry. Long-term system durability is a key challenge to the increased economic competitiveness and more widespread industrial implementation of SOEC technology in the future.
The reliable long-term performance of an SOEC stack system requires all components to be thermally stable. However, state-of-the-art systems have shown several lifetime issues related to single-stack components. The interconnect plates separate the stacked ceramic cells to give mechanical stability, thus also separating the fuel and the air gas feed and connecting the electrical current. Cr alloys and high Cr-containing ferritic stainless steel (FSS) are currently used due to their good mechanical strength and matching thermal expansion coefficients (TECs) with other SOC stack materials. When operated at high temperatures, the metallic interconnect oxidizes in the presence of steam or oxygen. This promotes the growth of an electronically conductive oxide layer on the interconnect, which may lead to spallation phenomena. Additionally, further chromium oxidation can lead to the formation of Cr6+, which is known to negatively impact the SOC electrodes via Cr evaporation39 from the oxide scale and react either electrochemically or chemically.40 Glass-ceramic sealants are used to separate the gas chambers in the horizontal plane, seal the cell into the interconnect, and ensure gas-tightness to the outer surrounding of the stacks. They require similar properties to the interconnect, for example, thermal expansion match and mechanical robustness. In addition, they must be gas-tight and show good creep resistance. In long-term operation, sealants can show reactivity with other stack components, for example, interdiffusion, and delamination.41 The long-term and large-scale deployment of SOEC technology also currently faces the challenge of electrode and electrolyte durability. The state-of-the-art materials of SOECs are adopted from materials used in solid oxide fuel cells. However, since the operating conditions/reaction mechanisms are different for SOFC and SOEC operation modes, the electrode materials requirements also vary depending on the mode of operation. Compared to SOFC operation, different reactions take place at the electrode/electrolyte interfaces, the materials must be stable in different atmospheres/oxygen partial pressures and under different applied voltages. At the moment, the state-of-the-art materials considered from SOFC research exhibit microstructural degradation as a consequence of the different gas compositions and pollutants present in the gas stream.42,43 For a successful commercial implementation of SOEC technology as part of a future hydrogen infrastructure, the stack costs and the longevity of the system have to be addressed. To meet the industrial goal of a maximum degradation rate of a maximum of 0.75% kh−1 or even better 0.5% kh−1, corresponding to a performance loss of 20% in approx. five years, the materials used in current SOEC systems need to be optimized or new innovative materials engineered that exhibit excellent chemical stability and high electrochemical performance in long-term, high-temperature operation.2
For the oxygen electrode, new perovskite-based materials such as double perovskites e.g., Sr2Fe1.5Mo0.5O6−δ (SFM), PrBaCo2O5+δ (PBC) and GdBaCo2O5+δ (GBC) as well as Ruddlesden–Popper phases such as La2NiO4+δ (LNO), Nd2NiO4+δ (NNO), and Pr2NiO4+δ (PNO) are currently under investigation and have already shown sufficient performance in short-term measurements up to ∼200 h. Doped perovskite oxides based on La0.2Sr0.8TiO3+δ (LST), La0.75Sr0.25Cr0.5Mn0.5O3−δ (LSCM), La0.6Sr0.4Fe0.8Mn0.2O3−δ, (LSFM), or double perovskites system Sr2Fe1.5Mo0.5O6−δ (SFM) or Sr2FeNbO6−δ (SFN) are investigated as alternative fuel electrode materials.44
In this paper, we elaborate on the key challenges in state-of-the-art materials for all essential components, for example, electrodes, interconnects, coatings, sealants, and electrolytes used in commercial SOC stack systems. Novel material classes, their production methods, and the assessment of their potential in terms of real-life use are essential to prolonging the longevity of SOEC stack systems and establishing them within a future hydrogen infrastructure. Special emphasis is placed on the cell materials and the development of the electrode microstructure from the laboratory scale to current commercial applications.
2. Solid oxide electrolysis cell configurations and designs
There are mainly two different cell configurations used in solid oxide cells: the planar and the tubular type. Both configurations consist of at least three different layers – oxygen electrode, electrolyte, and fuel electrode – as displayed in Fig. 3.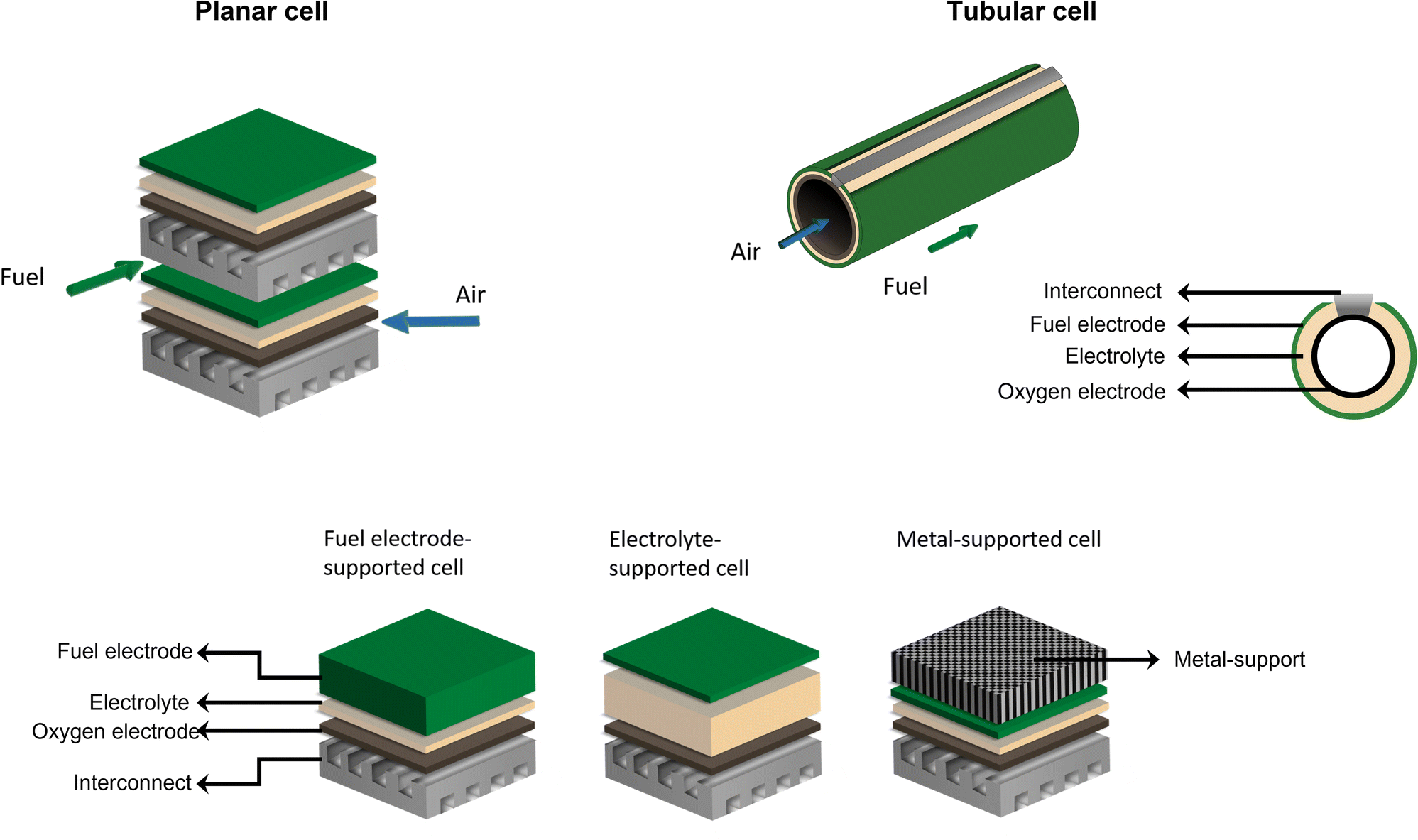 | ||
| Fig. 3 Planar and tubular cell designs and schematic representation of the three different cell support types reported in the literature: (fuel-) electrode-supported cells, electrolyte-supported cells, and metal-supported cells. | ||
Tubular cells are of interest due to their inherent high mechanical stability, quick start-up capability, easier sealing, and improved thermal cycling ability compared to planar cells, however, their application in SOECs and reversible SOCs (rSOCs) is still in the research and development phase.45,46 Therefore, laboratory and industrial applications are predominantly focused on planar cells, as these cells achieve higher current densities as well as being easier to manufacture, for example, by tape casting and screen-printing techniques, thus increasing cost-effectiveness. Furthermore, planar cells exhibit a lower internal resistance as well as higher flexibility in gas flow configurations compared to tubular cells. However, challenges in using the planar cell in stack configurations include more complicated sealing requirements and contact issues between interconnects and cells.45,47–49 Regardless of cell design, each cell requires a thick mechanical support layer, which ensures good handleability during processing and absorbs the stresses that may result from SOC operation. The cell design classification depends on the supportive layer. Industrially manufactured and investigated cell designs are electrolyte-supported cells (ESCs), fuel electrode-supported cells (FESCs), oxygen electrode-supported cells (OESCs), and metal-supported cells (MSCs).45,50,51
2.1. Electrolyte-supported cell
Electrolyte-supported cells typically consist of an electrolyte layer with a thickness of 50–250 μm in between a fuel electrode (5–20 μm) and an oxygen electrode (15–80 μm).50 The advantages of ESCs are their good mechanical robustness and better redox behavior compared to electrode-supported cells.52–54 ESCs for high-temperature electrolysis are currently produced on an industrial scale and sold by, for example, Kerafol, while also being operated in SOEC stacks by Sunfire55 or Bloom Energy. However, they exhibit high ohmic resistances due to the relatively thick electrolyte layer of partially stabilized zirconia, which shows higher strength but lower oxygen-ion conductivity, leading to lower power densities and thus requiring operating temperatures above 800 °C. These high operating temperatures require good thermal insulation and expensive thermostable materials.47,53,562.2. Electrode-supported cell
In electrode-supported cells, a thick, porous electrode layer offers mechanical support. As a cell consists of two different electrodes, a distinction can be made between two types of electrode-supported cells: fuel electrode-supported cells (FESCs) and oxygen electrode-supported cells (OESCs). Here, the electrolyte layer can be significantly thinner (2–20 μm), whereas the supporting layer is 250–1000 μm for FESCs and ∼1 mm for OESCs.50 However, the thickness of the active electrode layers is 5–20 μm for the fuel electrode and 15–80 μm for the oxygen electrode.50 The reduced thickness of the electrolyte in this cell design leads to a decrease in ohmic resistance and the option of lowering the operating temperatures. When comparing FESCs and OESCs, the fuel electrode-supported cells are more favorable due to their higher power densities, especially at lower temperatures, which is why they are mainly used at present.45,47,50 Nevertheless, electrode-supported SOCs exhibit higher electronic resistance and low redox stability, resulting in higher sensitivity for damage caused by redox cycling.53,54,56 At present, fuel electrode-supported cells are key components in Elcogen SOEC stacks as well as in SolydEra solid oxide electrolysis (SOE) systems.572.3. Metal-supported cell
Another possibility for mechanically stabilizing an SOC is the application of metal support. In this design, the electrolyte thickness and the active fuel and oxygen electrode thicknesses correspond to those of electrode-supported cells. The metallic support has a thickness of ∼1 mm.50 Literature reports indicate that MSCs can overcome many of the above-mentioned restrictions for ESCs, FESCs, and OESCs. They exhibit high mechanical robustness, fast start-up times, and high redox tolerance. A further advantage of metal-supported cells is their low material costs thanks to the minimization of the thickness of the expensive, electrochemically active ceramic layers, for example.51,58–62 MSCs can also operate at medium-to-low temperatures (of around 650 °C) in 50% H2O + 50% H2 with a 20% lower area-specific resistance (ASR) and a significantly lower degradation rate compared to state-of-the-art Ni-YSZ fuel electrode-supported cells.58 One major drawback of MSCs might be their complex operating conditions, for example, the differences in the TECs between the metal and ceramic components.51,56,60,63 As MSCs are currently being used successfully in the SOFC systems of Ceres Power, SOEC systems are now being developed.64 However, there is a lack of publications on the long-term operation of MSC-based stacks in both fuel cell and electrolysis mode.3. Solid oxid electrolysis cell components
3.1. Electrolytes
Doped ceria is a fluorite-type material with a higher ionic conductivity than stabilized zirconia at medium temperatures between 600 °C and 800 °C (Fig. 4). The ionic conductivity reaches its maximum with around 15 mol% of dopant.77 A widely used dopant is Gd, while Sm and Y have also been used to a lesser extent.67,68 A major disadvantage of doped ceria electrolytes is the observed reducibility of Ce4+ to Ce3+ in a reducing environment, resulting in possible mechanical instability and electrical conductivity.66,78
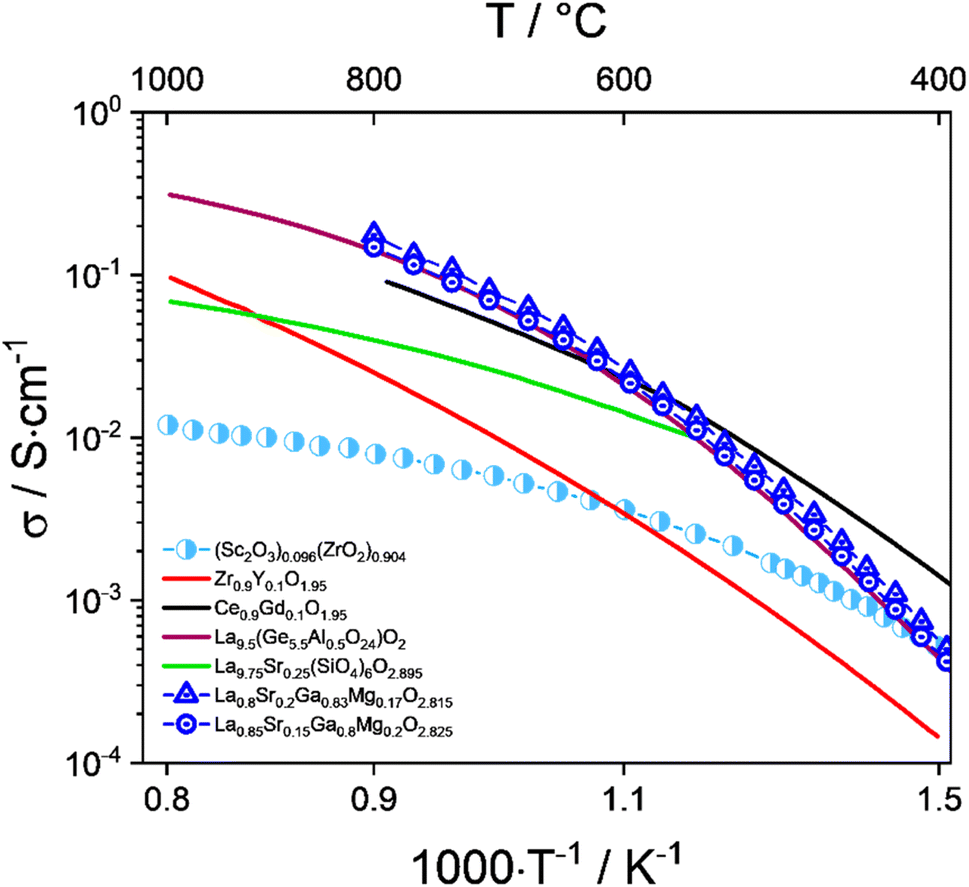 | ||
| Fig. 4 Total conductivity of oxide ion electrolyte materials (Sc2O3)0.096(ZrO2)0.904,104 Zr0.9Y0.1O1.95 (YSZ),105 La9.75Sr0.25(SiO4)6O2.895 (Si-apatite),98 La9.5(Ge5.5Al0.5O24)O2 (Ge-apatite),106 Ce0.9Gd0.1O1.95 (GDC),107 La0.8Sr0.2Ga0.83Mg0.17O2.815, and La0.85Sr0.15Ga0.8Mg0.2O2.825 (LSGM).108 | ||
In addition to its physical characteristics, 8YSZ is still the material of choice due to its cost advantages over scandia-stabilized zirconia (ScSZ) and gadolinium-doped ceria (GDC). Fuel electrode-supported cells with an 8YSZ electrolyte have proven their long-term stability in various tests conducted at Forschungszentrum Jülich,25,37,79,80 Topsoe,81,82 EPFL,38 CEA,83–85 DTU,86 DLR,87,88 and EIFER.89
3.2. Oxygen electrode
The electrode material catalyzes the dissociated oxygen anions to oxygen molecules and surplus electrons (Fig. 5). The mechanism of oxygen evolution at the oxygen electrode includes several steps: ion transfer along the electrode/electrolyte interface, charge transfer, solid-state bulk, surface diffusion, surface desorption, and gas diffusion processes. Among them, bulk transport and surface transport are generally considered to be rate-limiting steps.111 The catalytic activity and usability of an oxygen electrode material are determined by the surface exchange coefficient (k*) and the oxygen self-diffusion coefficient (D*). Adler, Lane and Steele112 proposed the ALS model in 1996 to calculate the chemical reaction contribution Rchem of oxide ion migration in the porous electrode in relation to the oxygen self-diffusion coefficient (D*, cm2 s−1), the surface exchange coefficient (k*, cm s−1), and microstructural parameters such as tortuosity τ, fractional porosity ε, and oxygen surface concentration Co. Taking the charge transfer resistance into account, they expressed the total impedance according to eqn (5) as the sum of chemical impedance derived from the oxygen evolution reaction (OER) and diffusion at the oxygen electrode (zchem), the electrolyte resistance (Relectrolyte), and the electrochemical kinetics at the electrolyte/electrode interface (zinterface). Their models showed that when the surface exchange and solid-state diffusion dominate, the total cell impedance would be reduced to Z = Rchem.
| Z = Relectrolyte + zinterface + zchem | (5) |
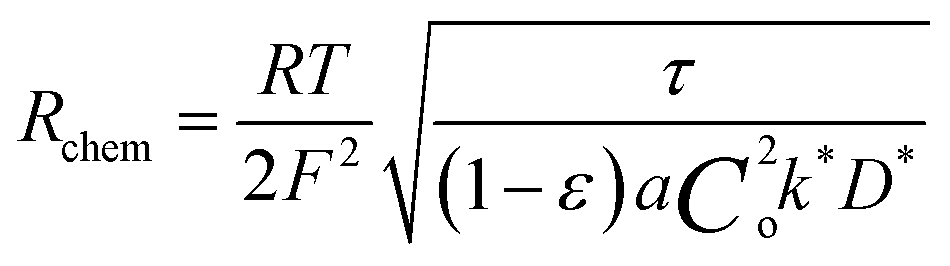 | (6) |
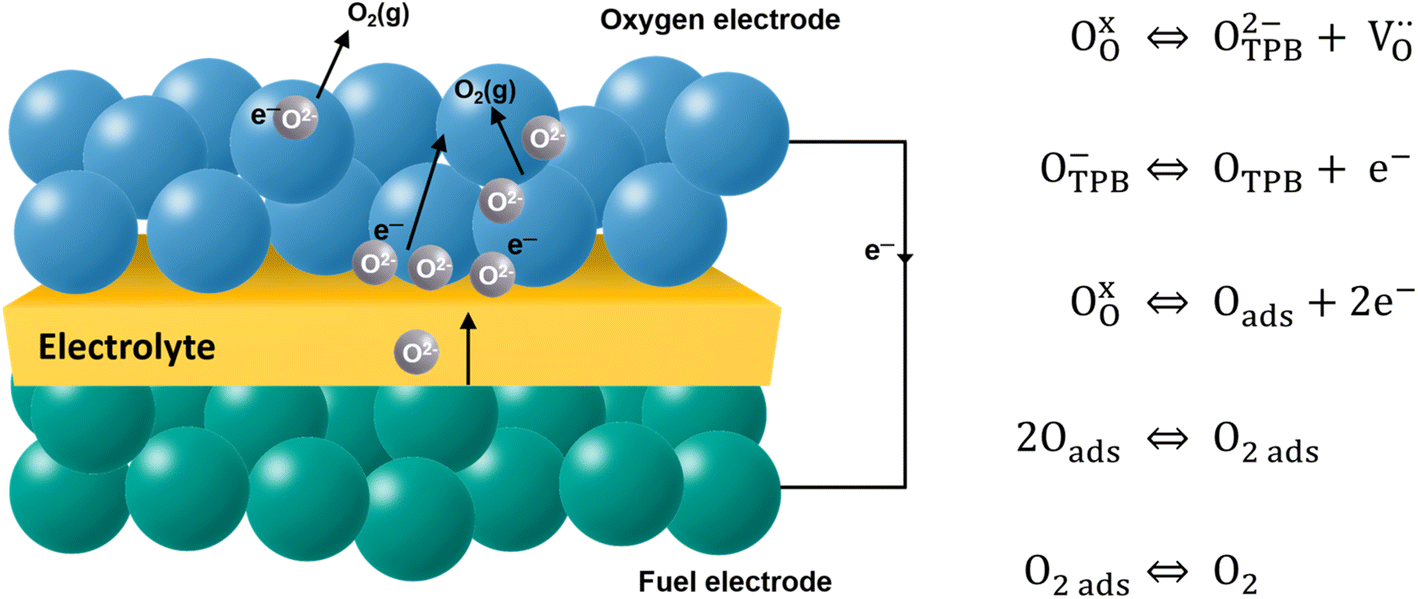 | ||
| Fig. 5 Schematic description of the oxygen evolution reaction (OER) at the oxygen electrode surface. | ||
High surface exchange coefficients (k* ∼10−7 cm s−1 at 700 °C) and a high oxygen diffusion coefficient (D* ∼10−7 to 10−8 cm2 s−1 at 700 °C) are prerequisites for enhanced oxygen transport and oxygen electrode kinetics in addition to sufficient electrode porosity.113 The diffusion and surface exchange properties of the oxygen electrode material are a good measure for the catalytic activity of the oxygen electrode and are therefore investigated by oxygen isotope exchange measurements.
The oxygen electrode material should be compatible with the other cell component materials and possess high chemical stability. The thermal expansion coefficient (TEC) gives the relative expansion with temperature, and a mismatch between the electrode material and electrolyte could pose the risk of delamination. The typical value for common electrolyte materials such as yttrium-stabilized zirconia (YSZ), gadolinium-doped ceria (GDC), strontium, and magnesium-doped lanthanum gallium oxide (LSGM) is around 10–13 × 10−6 K−1.114 A similar or adapted thermal expansion for the electrode thus avoids thermo-mechanical stress and ensures long-term system operation. The thermal expansion coefficient depends on the crystal structure as well as the perovskite cation radii.115 One way of tailoring the TEC is to dope the perovskite material at the A- or B-site. However, this substitution also affects the electrochemical properties of the material.116 It should be noted that the prerequisite of adapted TECs is based on theory, for example, FESCs with an LSC (cf. chapter 3.2.1.1) oxygen electrode, which has a very high TEC (20.5 × 10−6 K−1), are being used by Elcogen®, for instance. They do not show any delamination or other mechanical problems during normal operation and even during thermal cycles.117 High chemical stability under different long-term operating atmospheres is necessary to prevent degradation, which affects efficient oxygen evolution, shortens the lifetime of the cell, and therefore increases costs. High electronic and oxide ion conductivity under an oxidizing atmosphere of over 100 S cm−2 and 10−3 S cm−2, respectively, ensure good oxygen ion transport through the electrode as well as a high conversion rate per area, resulting in low ohmic cell resistance.118
Electrode materials are assigned as either n- or p-type conductors. For the n-type conductor, an oxygen deficiency is required to create electrons, which is why these materials are not stable under oxidizing conditions like ambient air. P-type electronic conductors, on the other hand, are stable in air, as they require oxygen excess to generate holes.119,120 Mixed ionic–electronic conducting (MIEC) single-phase perovskite oxides have increasingly become a field of study due to their advantageous material properties such as faster oxygen diffusion with improved surface exchange kinetics. Due to their improved performance under varied atmospheres and high potential chemical stability, double perovskites with ordering on A- as well as B-sites are a topic of high interest. Ruddlesden–Popper phases have shown conductivities below the targeted 100 S cm−1, but their thermal expansion coefficient matches closely to state-of-the-art electrolytes.
Electrode microstructure plays an important role in the performance of solid oxide fuel and electrolysis cells, as it determines the reactive surface sites. The most important parameters include grain size, grain size distribution, porosity ε, tortuosity τ, and electrode thickness (“active penetration depth”) d. The penetration depth stands for the distance at the electrode/electrolyte interface, in which the electrode is impacted by polarization by the applied potential and takes part in the oxygen evolution reaction. Beyond this distance, the electrode remains in equilibrium with the gas phase. According to Adler, the penetration depth for an MIEC electrode is described by the following equation, where Lc is the characteristic thickness and α the volume-specific surface area of the electrode:112,121
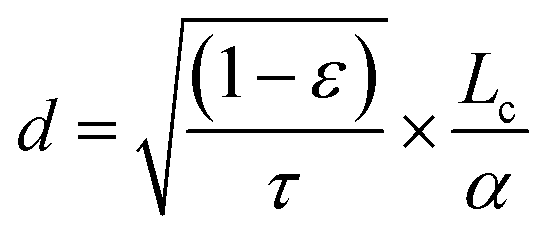 | (7) |
The grain size and the grain size distribution affect grain growth in the sintering process, thus directly influencing electrode performance. The loss of performance can result from microstructural densification (grain size too big) and unpercolated grain particles (grain size too small). The optimization of tortuosity and porosity is aimed at an ideal gas distribution/conversion at the interface between the gas phase, the electron-conducting electrode, and the ion-conducting electrolyte referred to as the triple-phase (TPB) or double-phase boundary (DPB in the case of MIECs) sites of the electrode.
3.2.2.1. Simple ABO3 perovskites. Noble metals such as platinum have previously been selected for the oxygen electrode. However, perovskite materials made of earth-abundant materials are of greater interest due to their better cost-effectiveness and higher compatibility with the electrolyte.123,124 Interest in perovskite materials for high-temperature solid oxide electrolysis cells (SOECs) has continuously increased over the last few decades due to their advantageous properties such as high chemical stability, high conductivity including mixed conductivity, excellent catalytic activity for OER, and good cost-effectiveness. The ideal simple perovskite structure has an ABO3 stoichiometry, as shown in Fig. 6a for the example of SrTiO3. The A-site cations located at the corners of the cubes are coordinated by twelve oxygen ions, while the smaller B-cations are coordinated octahedrally and sit in the cubic center. The A-site cation is commonly characterized by a rare-earth, alkaline, or alkaline-earth ion of low charge and is, therefore, larger than the B-site cation represented by transition metals. Perovskites can exhibit a wide range of physical properties such as ionic conductivity or ferromagnetism, which are influenced by their structure.
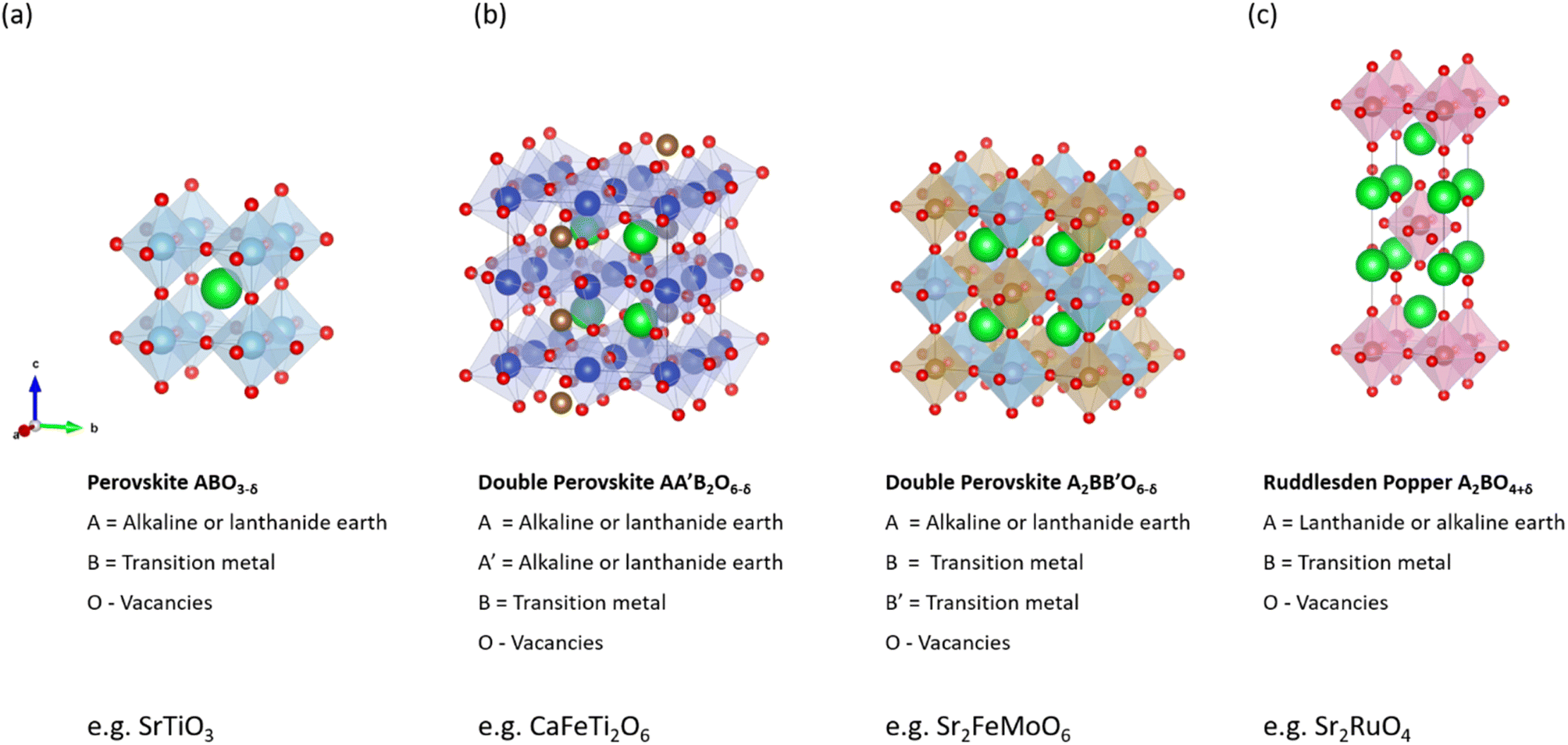 | ||
| Fig. 6 Crystal structure of cathode materials for (a) simple SrTiO3 perovskite, (b) double CaFeTi2O6/Sr2FeMoO6 perovskites, and (c) Ruddlesden–Popper structure Sr2RuO4. Crystal structures produced with VESTA©.122 | ||
Lanthanum manganite (LaMnO3) and lanthanum strontium manganite (La1−xSrxMnO3−δ) crystallize in the simple perovskite structure ABO3 and have attracted widespread research interest due to their high perovskite stability, which improves with increasing A-site cation radii. The partial substitution of the rare-earth cation La3+ with the divalent Sr2+ introduces p-type conductivity due to the principle of electroneutrality: when La3+ is exchanged for Sr2+, an electric hole is formed on the B-site cation.
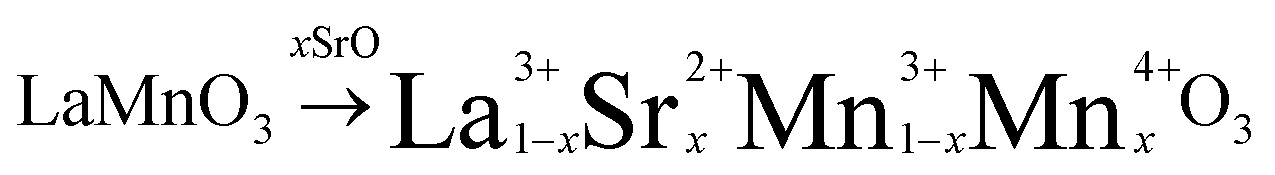 | (8) |
Electronic conductivity increases with an increase in x and a maximum electronic conductivity of 200–490 S cm−1 was obtained for x = 0.5 at 1000 °C.125 The investigation of the oxygen diffusion coefficient and oxygen surface exchange coefficient of Sr-doped LaMnO3 by SIMS technique showed that D* increases slightly with increased A-site doping from 4.8 × 10−12 cm2 s−1 for La0.9Sr0.1MnO3−δ to 1.3 × 10−11 cm2 s−1 measured for the composition La0.8Sr0.2MnO3−δ, as this provided further oxygen vacancies.126,127 In the case of electronic conductors, the reaction sites are located at the interface between the electrode, the electrolyte, and the gaseous phase. To enhance the electrochemically active area, composite LSM-YSZ oxygen electrodes were fabricated.128,129 The combination of ionic conducting YSZ and LSM as an oxygen electrode is applicable through careful tailoring of the stoichiometry of LSM and choosing appropriate sintering conditions of the composite on YSZ. The sintering temperature should ensure good adhesion and no chemical interaction. The combination of the ion-conducting YSZ phase with the p-type electronic-conducting LSM phase results in substantially enhanced electrocatalytic activity for the composite oxygen electrode due to an extended triple-phase boundary (TPB) and enhanced stability. The electrochemical performance can be enhanced by varying the LSM/YSZ ratio130 as well as the microstructure.131 The microstructure is an essential factor for the performance of composite electrodes, as micro-sized particles cause TPB length loss and negatively impact cell performance. Different preparation techniques were therefore employed to synthesize ultrafine nano-sized particles. In addition to the fast solid-state reaction, smaller nanoparticles were targeted by preparation with the combustion method,128 the hydrothermal method,132 spray pyrolysis,129 and sol–gel synthesis.133 Although nanostructuring of the LSM enhances initial performance, high operating temperatures are necessary due to the relatively poor catalysis of the LSM/YSZ mixture. This, in turn, quickly changes the nanostructuring to a micro-sized structure and again decreases the performance. Similar to LSM electrodes, the catalytic activity of the LSM-YSZ composite is enhanced by the application of cathodic polarization but the enhanced performance is lost under SOEC conditions.134 Despite the high electrical conductivity, which made pure LSM a conventional oxygen electrode material in solid oxide cell applications as early as the 1980s and 1990s, the low ionic conductivity and catalytic activity have shifted research to other perovskite materials with mixed ionic and electronic conducting (MIEC) properties.
Cobalt-based lanthanum strontium oxide (La1−xSrxCoO3−δ (LSC)) and lanthanum strontium ferrite oxide (La1−xSrxCo1−yFeyO3−δ (LSCF)) are the current state-of-the-art oxygen electrode materials with MIEC properties that have found widespread interest due to their higher catalytic activity as well as their high ionic and electronic conductivity. In contrast to purely electronic conducting (EC) materials such as LSM, the reaction zone, i.e., the TPB where gas phase, electronic, and ionic conductor meet, is extended to the two-dimensional double-phase boundary (DPB) at the mixed ionic/electronic conductor interface with the gas phase. Compared to LSM, Co-based perovskite electrodes have shown higher structural and performance stability under long-term SOEC operation conditions.135 However, the materials exhibit higher thermal expansion coefficients (TECs) of up to around 20.5 × 10−6 K−1 (30–1000 °C),136 which is higher than TEC values of common electrolyte materials (Table 5). One strategy for lowering the TEC value is to substitute the rare-earth cation La3+ (e.g., La0.4Sr0.6Co0.8Fe0.2O3−δ, 21.4 × 10−6 K−1, 25–1000 °C)137 with the smaller Pr3+ (Pr0.4Sr0.6Co0.8Fe0.2O3−δ, 20.6 × 10−6 K−1, 25–1000 °C) or Gd3+ (Gd0.4Sr0.6Co0.8Fe0.2O3−δ, 17.6 × 10−6 K−1, 25–1000 °C).137 With the substitution of Fe ions for Co, the TEC also decreases.138 LSCF has a high electronic and ionic conductivity (280 S cm−1 and 8.0 × 10−3 S cm−1 at 800 °C) in addition to high oxygen diffusion properties, for example, oxygen self-diffusion coefficient D* (5.00 × 10−7 cm2 s−1, 800 °C) and oxygen surface exchange k* (6.00 × 10−6 cm2 s−1, 800 °C). This is much higher than that of LSM, as shown in Table 3. The electrical and ionic conductivities of LSC surpass LSCF and LSM with an electrical and ionic conductivity of 1585 S cm−1 and 0.22 S cm−1, respectively at 800 °C. The ionic conductivity σi in LSCF depends on the substitution of Co with Fe, with the conductivity decreasing with a higher Fe content. The ionic conductivity σi for La0.8Sr0.2Co0.8Fe0.2O3−δ (4.0 × 10−2 S cm−1, 800 °C) is, therefore, one order of magnitude higher than for La0.8Sr0.2Co0.2Fe0.8O3−δ (2.3 × 10−3 S cm−1, 800 °C).138 Higher Sr and Co contents have shown higher oxygen permeability and an increase in oxide ion mobility D*, which was linked to the smaller oxygen ion binding energy of Co and the increased oxygen vacancy concentration as a result of the substitution of La with Sr.139 The lowest polarization resistance (RP) value obtained for La0.6Sr0.4Co1.05O3−δ (LSC) is 62 mΩ cm2 at 600 °C. Depending on the sintering temperature, LSCF electrodes show resistances of 2.7–4.0 Ω cm2.140 Despite the advantageous oxygen diffusion properties and conductivities for state-of-the-art LSC and LSCF electrode materials, layered perovskites have attracted increased interest as an alternative oxygen electrode material.
| Composition | TEC/10−6 K−1 | σ e/S cm−1 | σ i /S cm−1 | D*/cm2 s−1 | k*/cm s−1 | Ref. |
|---|---|---|---|---|---|---|
| La0.8Sr0.2MnO3−δ | 11.62 (100–900 °C) | 200–300 (900 °C) | 5.93 × 10−7 (1000 °C) | 4.0 × 10−15 (800 °C) | 5.62 × 10−9 (800 °C) | 127 and 141–144 |
| 13.13 (900 °C) | 152 (800 °C) | 4.0 × 10−8 (900 °C) | 1.0 × 10−16 (700 °C) | 1.39 × 10−10 (700 °C) | 145–149 | |
| La0.6Sr0.4CoO3−δ | 20.5 (30–1000 °C) | 1585 (800 °C) | 0.22 (800 °C) | 2.86 × 10−9 (600 °C) | 9.09 × 10−8 (600 °C) | 136, 138 and 150 |
| 1595 (800 °C) | 0.37 (832 °C) | 8.45 × 10−10 (600 °C) | 7.33 × 10−7 (600 °C) | 136, 151 and 152 | ||
| La0.6Sr0.4Co0.2Fe0.8O3−δ | 17.5 (30–1000 °C) | 210 (900 °C) | 8.0 × 10−3 (800 °C) | 5.00 × 10−7 (800 °C) | 6.00 × 10−6 (800 °C) | 136, 138, 144 and 153 |
| 15.3 (100–600 °C) | 280 (800 °C) | 1.0 × 10−2 (1000 °C) | 3.3 × 10−9 (700 °C) | 1.5 × 10−6 (700 °C) | 136, 138 and 154–156 |
3.2.2.2. Double perovskites. Double perovskite compounds are derived from half-substituted B′-site cation places with B′′ cations, for example, resulting in the structure A2 B′B′′X6 with alternatively arranged B′O6 and B′′O6 octahedra (Fig. 6b). In these perovskites, two different transition metal (TM) cations can be found at the B-site, which govern physicochemical material properties such as electrical conductivity and oxygen binding energy, thus offering higher flexibility compared to ABO3-structured perovskites.
The introduction of two different B-site cations can lead to disordered as well as ordered A2 B′B′′X6 double perovskites. The key factor in determining the probability of ordering is the oxidation state difference between the B′ and B′′ cations. In addition to the B-site cations, the order degree depends on the size of the A cation and decreases with an increasing A cation radius.157
The Mo-doped SrFeO3−δ is an MIEC perovskite oxide that has shown high sensitivity of the B-site cation order on physical properties such as electrical conductivity.158–160 Oxygen defect formation, electrical properties, and catalytic activity are influenced by the incorporation of the high-valence Mo ions, which influence the oxidation state of iron in the perovskite. With increasing Mo dopant concentration in the brownmillerite structure SrFeO3−δ under an oxidizing atmosphere, the activation energy for oxygen transport decreases.161,162 At higher Mo doping (0.5 ≤ x ≤ 1.5), the structure crystallizes as a double perovskite Sr2Fe2−xMoxO6−δ.163–165 The ferromagnetic perovskite Sr2Fe1Mo1O6−δ, therefore, finds no widespread use as an oxygen electrode. It can only be synthesized under reducing conditions due to the lack of Mo solubility under oxidizing conditions, resulting in a large detectable SrMoO4 phase.166,167
The Sr2Fe1.5Mo0.5O6−δ double perovskite phase, however, has attracted interest due to its high ionic conductivity of 0.13 S cm−1 in air (800 °C).168 This is significantly higher than for current state-of-the-art materials such as La0.6Sr0.4Co0.2Fe0.8O3 (8.0 × 10−3 S cm−1, 800 °C)138 and is comparable to the high-performance oxygen electrode material La0.6Sr0.4CoO3 (0.22 S cm−1, 800 °C).169 In contrast to simple perovskite materials, the p-type conducting material Sr2Fe1.5Mo0.5O6−δ exhibits weak Fe–O bonds and upon removal of the neutral oxygen atom, an extra charge delivered to the lattice is fully delocalized, thus forming a high concentration of oxygen vacancies.170 Values for the total conductivity in air (14.93 S cm−1, 750 °C;171 14.5 S cm−1, ∼450 °C (ref. 172)) do not compare to state-of-the-art oxygen electrode materials.173 They display a thermal expansion coefficient value of 18.1 × 10−6 K−1 for Sr2Fe1.5Mo0.5O6−δ, which is lower than for cobaltites but slightly higher than for common electrolyte materials such as 8 mol% yttrium-stabilized zirconia (8YSZ), gadolinium-doped ceria (GDC), and La0.9Sr0.1Ga0.8Mg0.2O3 (LSGM) in the temperature range of 200 °C to 1200 °C.174 Nevertheless, Sr2Fe1.5Mo0.5O6−δ exhibits a higher TEC compared to Mn-based cathodes such as La0.8Sr0.2MnO3 with a TEC of 11.6 × 10−6 K−1 in air141 but can surpass Co-based materials such as La0.6Sr0.4CoO3 (20.5 × 10−6 K−1),127 which shows a higher value. Similar to other Sr-containing electrode materials, SFM exhibits good chemical compatibility with known electrolyte materials, except for YSZ, with which an insulting secondary phase formation can be observed.166
An investigation of the rate-determining steps of oxygen surface exchange showed that in an oxidizing atmosphere, Sr2Fe1.5Mo0.5O6−δ exhibits high oxygen conductivity and oxygen diffusion, but is limited by the oxygen incorporation rate.175 Diffusion coefficient D* (5.0 × 10−6 cm2 s−1, 750 °C) and surface exchange coefficient k* (2.8 × 10−5 cm s−1, 750 °C) for SFM were determined to be in line with the diffusion properties of La0.6Sr0.4Co0.2Fe0.8O3 (LSCF),176 corresponding to La0.6Sr0.4MnO3+δ.175 However, these values for the high-performance oxygen electrode material La0.6Sr0.4CoO3 are slightly higher than reported for SFM.177 The exchange current density of Sr2Fe1.5Mo0.5O6−δ in air (0.186 A cm−2)87 is higher than that of LSM (0.003–0.004 A cm−2)177,178 at 800 °C and comparable to the Co-based materials (0.005–0.519 A cm−2).174,177,178 These results suggest that SFM may have good electrochemical activity for the oxygen evolution reaction. Table 6 provides examples of electrochemical performance for state-of-the-art materials compared to double perovskite materials such as oxygen electrode material in solid oxide electrolysis cells. To improve the performance of Sr2Fe1.5Mo0.5O6−δ in air, doped SFM materials and composite electrodes have been investigated using symmetrical SFM cells due to their excellent redox stability.179 An excessive Cu doping (x > 0.1) in Sr2Fe1.5−xCuxMo0.5O6−δ (SFCM) resulted in the expansion of the unit cell, which had a detrimental effect on oxygen ion diffusion.172 The maximum total conductivity of 49.3 S cm−1 at 450 °C was observed for 10% mol Cu due to a high electric charge carrier concentration (electron holes). With decreased Cu doping (x < 0.1), the oxygen vacancies increased but could not compensate for the decrease in the electron–hole concentration. Sr2Fe1.4Cu0.1Mo0.5O6−δ also showed the best electrochemical performance with an area-specific resistance (ASR) of 0.26 Ω cm2 compared to undoped SFM (0.63 Ω cm2) and Sr2Fe1.2Cu0.3Mo0.5O6−δ (0.45 Ω cm2) at 800 °C. The electrochemical characterization of composite electrodes of SFC0.1M with Sm0.2Ce0.8O1.9 (SDC) resulted in an ASR of 0.15 Ω cm2 for the mass ratio of 60:40 SFC0.1M to SDC. Measurements in H2O/CO2 co-electrolysis with similar composite SFM-SDC/LSGM/SFM-SDC cells showed a lower polarization resistance of 0.48 Ω cm2 at OCV with an electrolysis current density of −0.73 A cm−2 at 850 °C and 1.3 V. The durability test with −120 mA cm−2 at 800 °C showed an increased cell voltage of 0.13 mV h−1.180,181 Symmetrical cells (SFM/GDC/6Yb4ScSZ/GDC/SFM) tested in SOEC (90% H2O + 10% Ar) and co-SOEC (75% H2O + 25% CO2) mode presented a high current density of −1.4 A cm−2 and −1.1 A cm−2 at 1.3 V.182 Electrochemical impedance spectroscopy also underlined the high performance with an ASR at −0.5 A cm−2 of around 0.31 Ω cm2 in 90% H2O + 10% Ar and 0.42 Ω cm2 in 75% H2O + 25% CO2. The durability in H2O/CO2 co-electrolysis showed promising short-term performance for 24 h (36 mV) at 900 °C.
Doped double perovskites of the formation A′A′′B2X5+δ, with A′ being a lanthanide cation and A′′ an alkaline-earth element such as Sr or Ba exhibit rapid oxygen ion transport. The structural investigation for oxygen-deficient perovskites LnBaCo2O5+δ183 derived from the “112”-type YbaFeCuO5 (ref. 184) showed that the Ba and Ln ions order in alternating layers (001). The oxygen content decreased along the lanthanide series with the cell volume (LaBaCo2O5+δ: δoxygen = 0.806; GdBaCo2O5+δ: δoxygen = 0.698).185–187 An advantage of layered perovskites is the good oxygen diffusion and surface exchange kinetics at rather low temperatures of around 600 °C (k* = 2.8 × 10−7 cm s−1 and D* = 4.8 × 10−10 cm2 s−1 at 575 °C (ref. 188)), which is in the same order of magnitude as simple ABO3-type perovskites187,189 due to the reduced oxygen bonding strength in the [AO] layer and the formation of crystalline channels for enhanced ion transport. The high ionic conductivity was linked to the mixed valence state of Co in the presence of divalent cations (Ba2+, Sr2+) with Co2+/3+ and Co3+/4+ and, therefore, reduced oxygen ion hopping energy.190
These layered materials can also significantly display high electronic conductivity191 that is sufficient for SOEC applications in the range of ∼960 S cm−1 at 470 °C down to ∼500 S cm−1 at around 800 °C.189 However, as with other cobalt-containing perovskites, the A-site cation-ordered perovskites have a high thermal expansion coefficient (24.0 × 10−6 K−1 for PrBaCo2O5+δ (PBC))192 that is not in the range of common electrolytes, such as yttrium-doped zirconia (8YSZ, 10.5 × 10−6 K−1)193 and gadolinium-doped ceria (GDC, 12.5 × 10−6 K−1),193 as listed in Table 5. The secondary alternative electrolyte material, strontium magnesium-doped lanthanum gallate (LSGM, 10.9 × 10−6 K−1),193 offers a similar comparison. These large differences could lead to thermal stress between the cell components. Enhanced electrode reaction kinetics and improved mechanical compatibility have led to the development of composite electrodes consisting of A-site ordered cobalt-containing perovskites and ceria-based electrolyte materials.
PrBaCo2O5+δ is a new, intensely studied material for SOC applications as the electrochemical performance in the LnBaCo2O5+δ series increases from La to Pr as an A-site cation (Pr > Gd > Nd > Sm > La).187 The incorporation of GDC in PBC has an unfavorable effect on electrical conductivity, which decreases with increasing GDC content.194 However, at a typical SOEC operating temperature of 800 °C, the electrical conductivities are sufficient for application with 360 S cm−1, 456 S cm−1, 569 S cm−1, and 682 S cm−1 for x = 20 wt%, 20 wt%, 10 wt%, and 0 wt%, respectively. Symmetrical cell measurements in air have shown a lower RP of 0.86 Ω cm2 at 600 °C compared to 1.2 Ω cm2 for La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) under similar conditions.140,195 Single-cell measurements with a PBC-10 wt% GDC oxygen electrode in a temperature range of 550–850 °C indicate good catalytic activity and improved mechanical stability with the addition of GDC, for example, 0.698 Ω cm2 and 0.550 Ω cm2 at 550 °C for PBC and PBC-10 wt% GDC.194 The A-site cation-deficient PrBaxCo2O5+δ (PBxCO, x = 0.90–1.0)196 has shown a higher performance than (La0.6Sr0.4)1−xCo0.2Fe0.8O3−δ197 and (Ba0.5Sr0.5)1−xCo0.8Fe0.2O3−δ198 PBxCO (x = 0.90–1.0) shows increased electrical conductivity (>600 S cm−1) at the desired operating temperatures and a decrease in the thermal expansion coefficient with Ba deficiency.196 The composition PrBa0.94Co2O5+δ exhibits the highest electrocatalytic activity with a low polarization resistance of 42 mΩ cm2 at 600 °C in addition to a high conductivity of ∼935 S cm−1 (700 °C). An increase in the content of Ba results in an increased TEC of 17.4 × 10−6 K−1 (50–300 °C) for PrBaCo2O5+δ and a higher RP (160 mΩ cm2) in addition to lower electrical conductivity (∼672 S cm−1, 700 °C). Decreasing the Ba doping to PrBa0.90Co2O5+δ also results in less electrical conductivity (∼876 S cm−1, 700 °C), a higher polarization resistance (86 mΩ cm2), and a slightly increased TEC (15.2 × 10−6 K−1, 50–300 °C) compared to PrBa0.94Co2O5+δ.196 Other oxygen electrode materials, for example the cobalt-free perovskite SrNb0.1Fe0.9O3−δ (SNF)199 and the cobalt-containing perovskite Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF)200 exhibit higher resistances than PrBa0.4Co2O5+δ (390 mΩ cm2 and 72 mΩ cm2, respectively). This highlights the potential of PrBa0.4Co2O5+δ for application in electrochemical devices.
3.2.2.3. Ruddlesden–Popper nickelates. Despite the high performance and good durability of perovskite-structured materials such as La1−xSrxCoO3−δ or La1−xSrxCo1−yFeyO3−δ, the search for new materials with a better matching TEC has sparked interest in alternative Ruddlesden–Popper lanthanide nickelates Ln2NiO4+δ (Ln = La, Pr or Nd) presented in Fig. 7 and 6c. They show high surface exchange coefficients (k*) and high anionic bulk diffusion (D*) with good electrical conductivity (∼100 S cm−1 (ref. 201)) as well as good thermal expansion properties matching state-of-the-art electrolyte materials (Tables 5 and 7) and cell components such as metallic interconnects.202–205
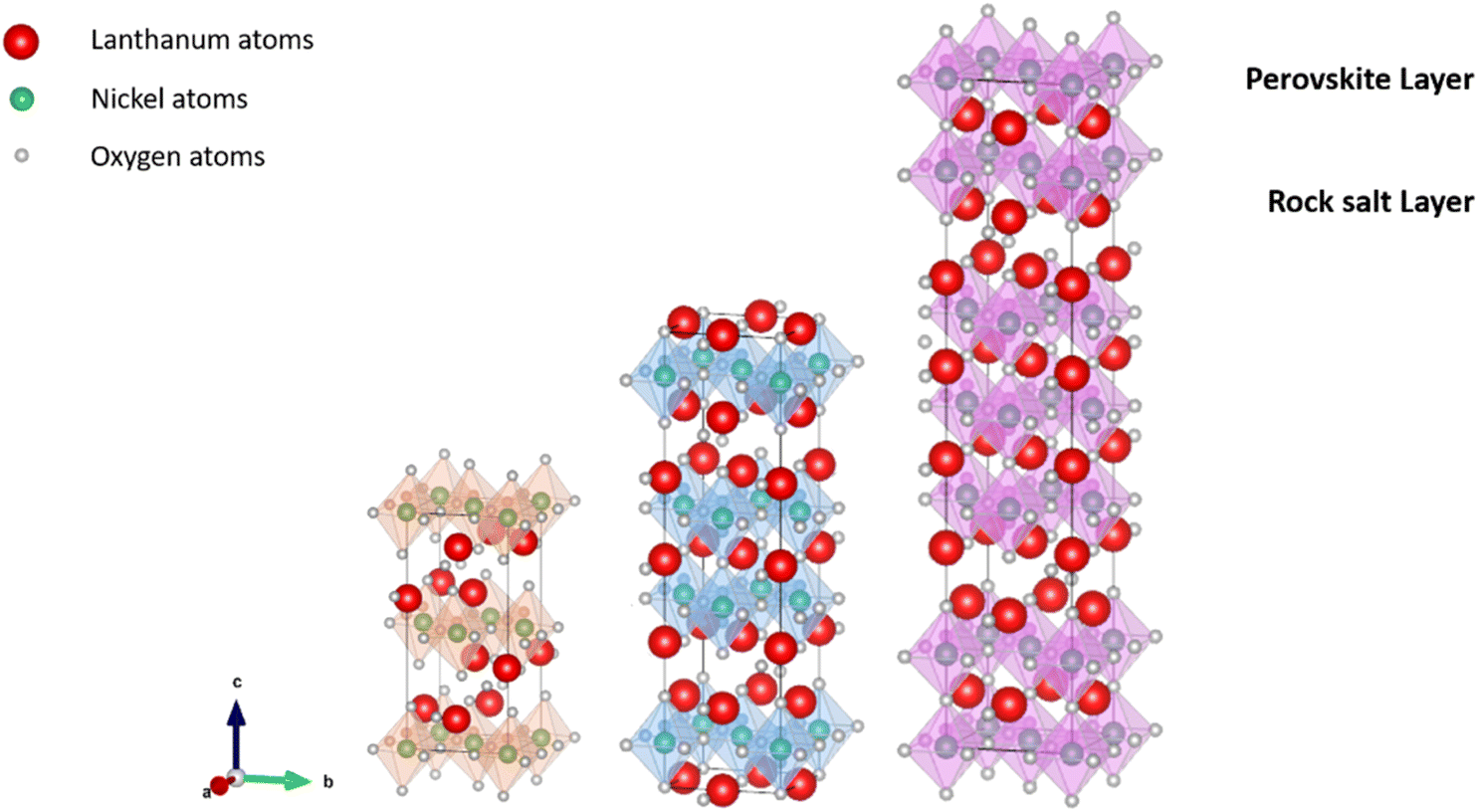 | ||
| Fig. 7 Schematic representation of La2NiO4 (n = 1),206 La3Ni2O7 (n = 2),207 and La4Ni3O10 (n = 3),208 with n-stacked NiO6 octahedral layers sandwiched between rock-salt LaO layers. Crystal structures produced with VESTA©.122 | ||
The nickelate structure Lnn+1NinO3n+1 (n = 1) consists of alternate Ln2O2 rock-salt layers and polyhedral NiO2 square plane layers with interstitial oxygen in the salt layers.209,210 This material accommodates the oxygen excess in the form of interstitial oxygen in the Ln2O2 rock-salt layer, which leads to a mixed-valence of the B-site cation Ni (Ni2+/Ni3+) and, in turn, leads to fast oxygen ion transport.202,211–213 The oxygen ion mobility in the ab-plane is favored and induces anisotropy to the ion conductivity. Any diffusion along the c-axis of the structure involves a less favorable diffusion pathway through the occupied oxygen sites in the perovskite blocks. This is highlighted by Fig. 8, which shows 16O and 18O SIMS images (along the c-axis, Fig. 8a) obtained using the surface scanning method alongside the normalized 18O concentration profile extract along the (a,b)-plane in Fig. 8b for Nd2NiO4+δ. Although the diffusion 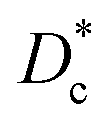 is three orders of magnitude lower than
is three orders of magnitude lower than 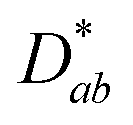 , the overall ionic conductivity of the nickelates is compatible with known SOC oxygen electrode materials and higher than those of LSCF at 700 °C (ref. 202 and 214–216) (Fig. 9).
, the overall ionic conductivity of the nickelates is compatible with known SOC oxygen electrode materials and higher than those of LSCF at 700 °C (ref. 202 and 214–216) (Fig. 9).
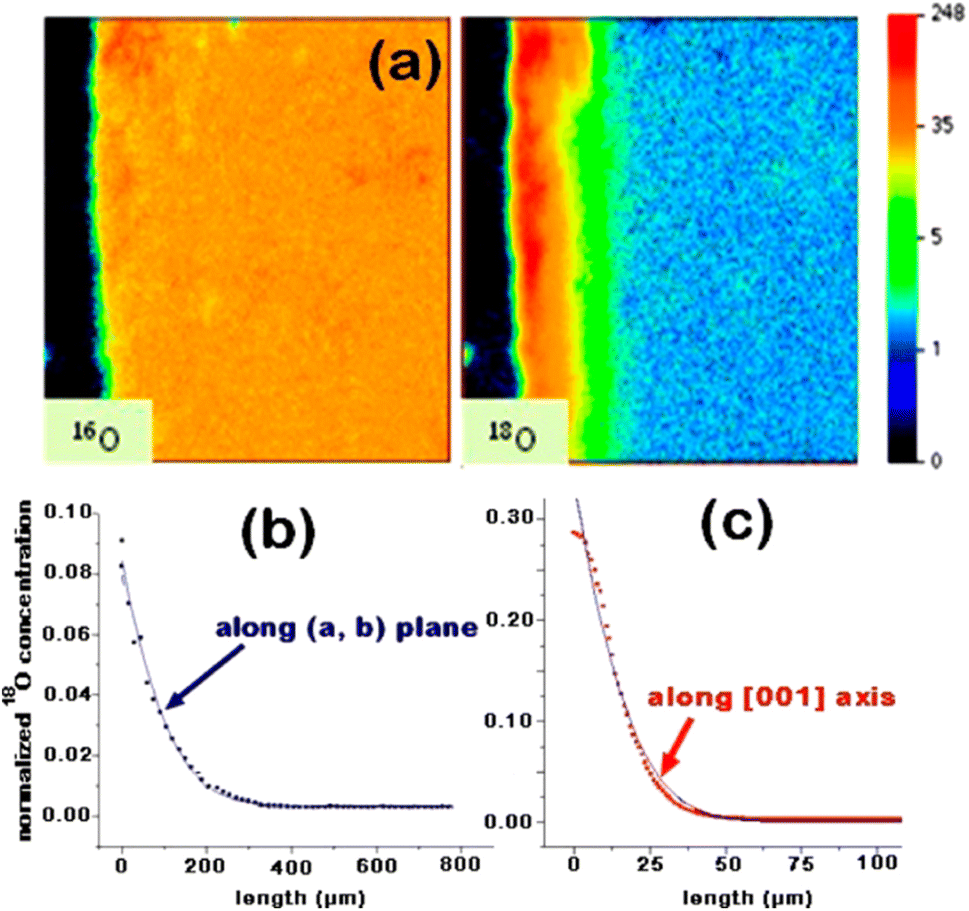 | ||
| Fig. 8 Nd2NiO4+δ crystal exchange experiment at 703 °C for 856 s: (a) surface scanning methods: 16O and 18O SIMS images (along the c-axis). (b) Using the line-scanning method, the normalized 18O concentration profile along the (a,b)-plane was recorded. (c) Normalized 18O concentration profile extract along the c-axis (obtained from image in (a)). Reprinted with permission from ref. 214. Copyright 2013 American Chemical Society. | ||
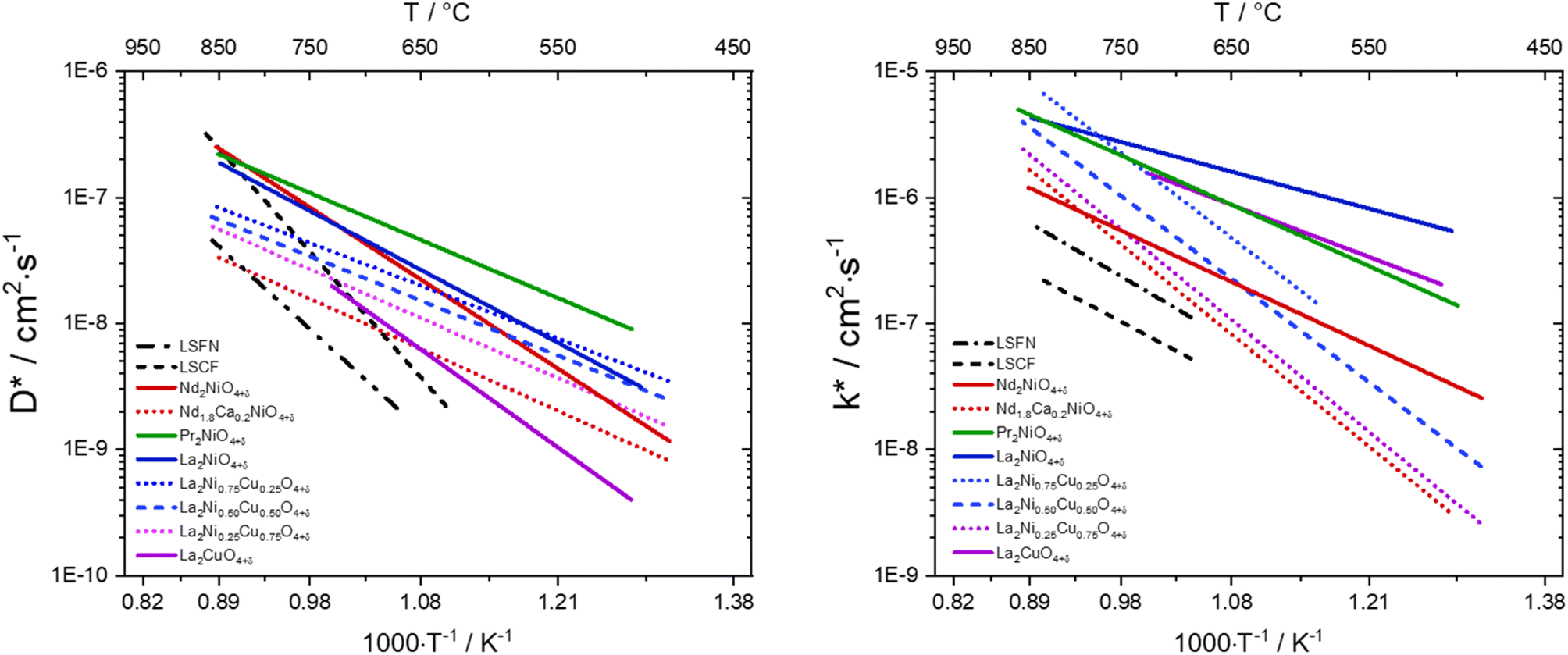 | ||
| Fig. 9 Diffusion properties of different oxygen electrode materials compared to La0.6Sr0.4Fe0.8Ni0.2O3−δ (LSFN), La0.6Sr0.4Fe0.8Co0.2O3−δ (LSFC).201,216 | ||
The investigation of diffusion coefficients for different lanthanide nickelates showed higher diffusion along the ab-plane and lower activation energy for diffusion for Pr2NiO4+δ compared to Nd2NiO4+δ (EPra ∼0.7 eV and ENda ∼1.4 eV). This is in contrast to 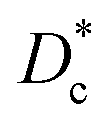 , which is very similar for both materials, with similar activation energies (1.1–1.3 eV) for oxygen diffusion.214 Of the lower order Ruddlesden–Popper phases, La2NiO4+δ (LNO), Pr2NiO4+δ (PNO), and Nd2NiO4+δ (NNO) have been investigated as the most promising oxygen electrode materials for SOC application.217–222 The oxygen over stoichiometry (+δ) indicates the ionic conductivity of the lanthanide nickelates and is highly dependent on the rare-earth cation size. Lanthanum nickelate La2NiO4+δ (LNO) has a lower amount of oxygen interstitials (δ ∼ 0.13)216 and a larger La3+ cation radius (1.16 Å) compared to Pr2NiO4+δ (PNO) (δ ∼ 0.21)216 and the smaller ionic radius of Pr3+ (1.126 Å). Nd2NiO4+δ (NNO) with Nd3+ (0.98 Å) was reported to have the highest oxygen diffusion coefficient above 850 °C and an interstitial oxygen amount with δ ∼ 0.22.216,217,223,224 The cation of lanthanide nickelates also influences material properties. PNO shows the highest electrochemical performance, i.e., the lowest polarization resistance (RP) value, better ionic and electronic conductivity, as well as diffusion properties compared to LNO, especially at intermediate temperatures (600–700 °C).217,225 In comparison, LNO exhibits higher chemical stability. At high temperatures of around 800–950 °C, the decomposition of Pr2NiO4+δ after 6.5 h into PrOx and Pr4Ni3O4+δ was reported.226 In the intermediate temperature range from 600 °C to 800 °C, a decomposition into Pr6O11, PrNiO3−δ, and Pr4Ni3O10+δ was observed.227,228 The comparison of polarization values RP before and after the complete dissociation of PNO as a measure of electrochemical performance showed no change at idc = 0. The second RP nickelate structure (Ln3Ni2O7±δ) is composed of two NiO6 layers connected in the c-axis between rock-salt layers. Ln4Ni3O10±δ (n = 3) has a similar structure to three infinite NiO6 sheets connected between the rock-salt layers. Higher order RP phases have relatively high total conductivity and are oxygen under-stoichiometric in contrast to the n = 1 phases.229 No Ni2+/Ni3+ impurity phase formation for La3Ni2O6.95 and La4Ni3O9.78 was reported in contrast to La2NiO4+δ and La2Ni1.9Co0.1O4+δ at 900 °C after two weeks in air.229–231 Pr4Ni3O10+δ shows metallic behavior232 and higher conductivity than Nd4Ni3O10+δ and La4Ni3O10+δ. Pr4Ni3O10+δ was shown to be an over-stoichiometric MIEC type under air and oxygen.
, which is very similar for both materials, with similar activation energies (1.1–1.3 eV) for oxygen diffusion.214 Of the lower order Ruddlesden–Popper phases, La2NiO4+δ (LNO), Pr2NiO4+δ (PNO), and Nd2NiO4+δ (NNO) have been investigated as the most promising oxygen electrode materials for SOC application.217–222 The oxygen over stoichiometry (+δ) indicates the ionic conductivity of the lanthanide nickelates and is highly dependent on the rare-earth cation size. Lanthanum nickelate La2NiO4+δ (LNO) has a lower amount of oxygen interstitials (δ ∼ 0.13)216 and a larger La3+ cation radius (1.16 Å) compared to Pr2NiO4+δ (PNO) (δ ∼ 0.21)216 and the smaller ionic radius of Pr3+ (1.126 Å). Nd2NiO4+δ (NNO) with Nd3+ (0.98 Å) was reported to have the highest oxygen diffusion coefficient above 850 °C and an interstitial oxygen amount with δ ∼ 0.22.216,217,223,224 The cation of lanthanide nickelates also influences material properties. PNO shows the highest electrochemical performance, i.e., the lowest polarization resistance (RP) value, better ionic and electronic conductivity, as well as diffusion properties compared to LNO, especially at intermediate temperatures (600–700 °C).217,225 In comparison, LNO exhibits higher chemical stability. At high temperatures of around 800–950 °C, the decomposition of Pr2NiO4+δ after 6.5 h into PrOx and Pr4Ni3O4+δ was reported.226 In the intermediate temperature range from 600 °C to 800 °C, a decomposition into Pr6O11, PrNiO3−δ, and Pr4Ni3O10+δ was observed.227,228 The comparison of polarization values RP before and after the complete dissociation of PNO as a measure of electrochemical performance showed no change at idc = 0. The second RP nickelate structure (Ln3Ni2O7±δ) is composed of two NiO6 layers connected in the c-axis between rock-salt layers. Ln4Ni3O10±δ (n = 3) has a similar structure to three infinite NiO6 sheets connected between the rock-salt layers. Higher order RP phases have relatively high total conductivity and are oxygen under-stoichiometric in contrast to the n = 1 phases.229 No Ni2+/Ni3+ impurity phase formation for La3Ni2O6.95 and La4Ni3O9.78 was reported in contrast to La2NiO4+δ and La2Ni1.9Co0.1O4+δ at 900 °C after two weeks in air.229–231 Pr4Ni3O10+δ shows metallic behavior232 and higher conductivity than Nd4Ni3O10+δ and La4Ni3O10+δ. Pr4Ni3O10+δ was shown to be an over-stoichiometric MIEC type under air and oxygen.
Doped lanthanide nickelates were synthesized to improve diffusivity, surface exchange coefficients, conductivity, and electrochemical performance. The substitution of the A-and/or B-site cation can lead to improved electronic and ionic conductivity. Ni is frequently substituted with Co, and La with Sr and Ca. Atomistic simulations predicted that the substitution of Ni2+ with Co3+ or Fe3+ would increase the oxygen interstitial concentration, but also have a detrimental effect on oxygen ion diffusivity.233 The investigation concerning factors affecting the ionic transport in oxygen-hyperstoichiometric phases with K2NiF4-type structures such as the lanthanum nickelates found that the decreasing radii of the A-site rare-earth cation led to a decrease in ionic transport.212
Electrochemical studies as to the effect of Co doping on Pr2NiO4+δ showed an increase in electrochemical performance and long-term stability of the lanthanide nickelates.217 Increasing the cobalt content (x = 0.0, 0.1, and 0.2) led to an increase in δ, which is one indication for better electrochemical properties and higher diffusivity/ionic conductivity. Symmetrical cell measurements under pO2 attributed the process resistances to the gas diffusion process and the oxygen surface exchange reaction, i.e., the charge transfer process (surface diffusion of the adsorbed/desorbed oxygen atoms and their consequent reduction/evolution).217 The charge transfer process was identified as the rate-limiting step among the electrode processes for these PNO and Co-substituted PNO electrodes. An investigation of single cells (NiO-YSZ/YSZ/GDC/electrode, CeramTec®, ASC-10C type) exhibited very high performance for Pr2Ni0.8Co0.2O4+δ (PNCO20) with an RP value of 118 mΩ cm2 compared to 128 mΩ cm2 (PNO). Stability tests at 800 °C with −1 A cm−2 up to 250 h showed less degradation for cells containing Co-doped nickelates as oxygen electrodes. The degradation of LSCF and PNO cells were similar (∼88 mV kh−1), although Pr2Ni0.9Co0.1O4+δ (PNCO10) and Pr2Ni0.8Co0.2O4+δ (PNCO20) exhibited less than half with 36 mV kh−1 and 22 mV kh−1, respectively. La2Ni0.8Co0.2O4+δ (LNCO20) showed similar degradation (∼30 mV kh−1).234 Post-test analysis showed no severe damage or delamination/cracks for Co-doped nickelate electrodes, but SEM-EDS revealed the reactivity of GDC and PNO in the form of Ce and Pr interdiffusion at the electrode/electrolyte interface. The investigation of thin PNO and PNCO10 films showed an improved performance of PNO with cobalt substitution due to an enhancement in the diffusion coefficient (D*) and surface exchange coefficient (k*).235 La2NiO4+δ shows a relatively lower electrical conductivity of 70–80 S cm−1 between 600 °C and 800 °C. For this reason, attempts to substitute the A-site cation with Ca and Sr were conducted. The investigation of Sr-substituted LNO showed higher oxygen diffusivity of La1.9Sr0.1NiO4+δ than LaxSr1−xCoyFe1−yO3−δ, but lower transport properties than La1−xSrxCoO3−δ223 and La2NiO4+δ. Substituting the A-site cation La3+ with Sr2+ (La2−xNiO4+δ with x = 0.1 and 0.2) leads to a decrease in the diffusion coefficient D* compared to the non-doped La2NiO4+δ due to a decrease in interstitial oxygen atoms.216 This decrease was observed to be less severe for the substitution (Nd, Ca) than for (La, Sr) due to the similar ionic radius of Ca2+ and Nd3+ (1.18 and 1.16 Å) compared to La3+ and Sr2+ (1.22 and 1.31 Å). Similar trends were observed by other authors.212,223,230,236 In contrast, the creation of Nd vacancies on the A-site improved the oxygen diffusivity and had a beneficial effect on k*. The substitution of Ni (B-site) with other transition metal ions (Fe, Cu, Co) gave mixed results regarding the diffusion properties. The substitution of Ni with Co (x ≥ 0.5) leads to slightly higher diffusion and largely enhanced surface exchange coefficients, with remarkably low activation energies of around 20 kJ mol−1 reported.202,237 Although the surface exchange coefficient k* for La2−xSrxNi1−yMyO4+δ (M = Fe, Cu, Co) showed an increase after the substitution of Ni with Co, iron did not have a significant effect and Cu substitution was detrimental to the coefficient k* up to around 800 °C.238 Similar observations were made for Ca-doped NNO (x = 0.2).216 Another strategy to enhance material properties is the development of mixed or composite nickelate electrodes, which have shown improved performance compared to single phases as well as better TEC matching between the electrode material and electrolyte.239 A significant improvement in conductivity σ, δ-values, and electrochemical performance was achieved for mixed composition nickelate electrodes La2−xPrxNiO4+δ (0.0 ≤ x ≤ 2.0) (LPNO).240–243 In this series, the highest RP was observed for LNO with 0.28 Ω cm2 at 700 °C. With increasing Pr content, the RP decreased to 0.03 Ω cm2 for PNO.240 The composite electrodes with a La2NiO4+δ and La4Ni3O10−δ ratio of 40:60 showed the highest conductivity of ∼29 S cm−1 at 700 °C, in the same range as La4Ni3O10−δ (∼30 S cm−1) and with a much lower ASR in symmetrical cell measurements (0.90 Ω cm2) compared to La4Ni3O10−δ (∼1.47 Ω cm2). The best electrochemical performance was observed for a 50:50 wt% mix with an ASR of 0.62 Ω cm2 at 700 °C.244Table 8 summarizes the electrochemical performance of nickelate materials as an oxygen electrode material in solid oxide electrolysis cells.
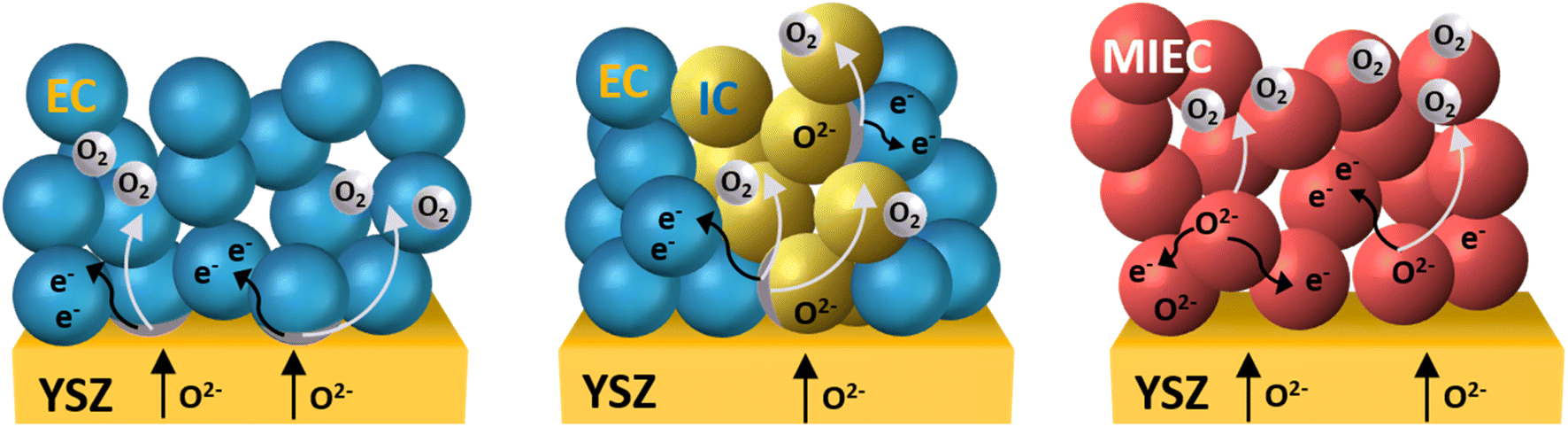 | ||
| Fig. 10 Microstructures for SOC electrodes. (left) Single-phase electron conductor, e.g., LSM; (middle) ion conduction (IC)/electronic conduction (EC) composite structure, e.g., LSM-YSZ. (right) Single-phase MIEC electrode, e.g., LSCF. The oxygen evolution reaction (OER) is restricted to the triple-phase boundaries (TPBs) in the case of a single EC layer as well as for the composite oxygen electrode including both an electronic conductor and an ionic conductor (EC + IC). Materials with mixed ionic and electronic conductivity (MIEC) properties enable the reaction at triple- and double-phase boundaries, thus extending the reaction zone. | ||
The development of the oxygen electrode for solid oxide cells at Forschungszentrum Jülich optimized the electrode material parameters, for example, microstructure, chemical composition, and thickness as well as the processing techniques. Fig. 11 shows cross-sections of the typical fuel electrode-supported O2−-conducting SOC developed over the last few decades.
 | ||
| Fig. 11 Novel SEM micrographs of the fuel electrode-supported single cell cross sections comparable to:248,261 (a) cell manufactured at Forschungszentrum Jülich and characterized by a double-layered La0.65Sr0.3MnO3 (LSM)/LSM-Y2O3-stabilized ZrO2 (YSZ) oxygen electrode applied on a thin YSZ electrolyte. (b) Cell manufactured at Forschungszentrum Jülich with a 5 μm-thick diffusion barrier layer (GDC, gadolinia-doped ceria) and a 40 μm-thick La0.58Sr0.4Co0.2Fe0.8O3−δ (LSCF) oxygen electrode. (c) Pre-test commercial Elcogen® cell made of LSC with a GDC barrier layer reduced at 900 °C.262 | ||
Until 2010, the “state-of-the-art” fuel electrode-supported cells manufactured at Forschungszentrum Jülich were characterized by a double-layered La0.65Sr0.3MnO3 (LSM)/LSM-Y2O3-stabilized ZrO2 (YSZ) oxygen electrode applied on a thin YSZ electrolyte (Fig. 11a).248 These cells were mainly operated in fuel cell mode at temperatures between 800 °C and 900 °C. Subsequent studies focused on the variation of the LSM/YSZ ratio, thickness, and porosity of the oxygen electrode functional layer and current collector layer as well as the grain size distribution of the electrode material.248–250 Early studies on LSM and the composite LSM-YSZ electrode investigated the correlation between microstructure and electrode performance. A correlation between performance and structure was observed for screen-printed LSM on YSZ and identified the length of the TPB and electrode thickness as the main influencing factors.251 The performance of a composite LSM-YSZ layer and two current collecting layers of LSM sintered at different temperatures showed that decreasing sintering temperatures led to smaller grain size and higher electrode porosity.252 Several studies also underlined the decrease in cell resistance with reduced particle size and an optimized current collector layer.121,253,254 Noble-metal-containing oxygen electrodes showed enhanced catalytic activity due to higher surface reactions in comparison to conventional Ni-8YSZ/8YSZ/LSM-8YSZ/LSM cells manufactured at Forschungszentrum Jülich with a 60/40 wt% LSM-8YSZ functional layer.255
The development of mixed ionic–electronic conducting perovskite-type electrode materials, for example, lanthanum strontium cobalt ferrite (LSCF), aimed to achieve higher power densities for fuel electrode-supported cells (Fig. 11b). In addition, the operating temperature could be lowered due to higher electrocatalytic activity and higher oxygen-ion conductivity. The new cells were assembled as the LSM electrode cells in the first approach with equivalent materials and layer thicknesses for the fuel electrode, support, and electrolyte. Later studies optimized the cell geometry in terms of the electrode thickness, material composition, and sintering temperature.256,257 Due to the chemical reactivity of these materials with the YSZ electrolyte, the investigation of thin-film processes for barrier interlayers comprised of GDC gained an increased significance.258 A screen-printed diffusion barrier layer made of GDC is now applied whenever an LSC(F)-based electrode is used for intermediate operating temperatures (∼700 °C). State-of-the-art commercial Elcogen® fuel electrode-supported cells (Fig. 11c) incorporate oxygen electrodes comprised of strontium-substituted lanthanum cobaltite (La1−xSrxCoO3−δ, (LSC)), which exhibits significant ionic conductivity (0.01–0.45 S cm−1) in addition to high electronic conductivity in the operating temperature range of 650–800 °C.259 Lower operating temperatures (<700 °C) in fuel cell mode with LSCF oxygen electrodes require thinner barrier layers applied by, for example, PVD or sol–gel technology.109,260
In recent years, the influence of electrode microstructure was studied by electrostatic spray deposition (ESD).121,129,263–266 Electrospray is a low-cost electrohydrodynamic method used to deposit thin films and coatings with various morphologies. The inorganic or organometallic precursor materials dissolved in organic solvents result in sub-micrometer droplets accelerated towards the substrate. The morphology can be varied depending on the deposition conditions, which enables the fabrication of, for example, nanosponge-like, nanopillar, or dense materials.267
La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) cathode films with different morphologies on a dense Ce0.9Gd0.1O2−δ (GDC) electrolyte were synthesized as shown in Fig. 12.263 The FIB-SEM reconstruction differentiated between cracked, coral, and dense microstructures. The coral microstructures showed distinct features, with solid particles arranged in highly ramified structures and a thickness of up to 25 μm. The cracked films exhibited a thickness varying between 3 μm and 8 μm and contained noncontinuous reticulated surfaces, with gaps of ∼1 μm throughout the surface. All films displayed the three elementary steps identified as the charge transfer process at the LSCF/GDC interface, the diffusion process within the volume of LSCF, and oxygen transfer at the LSCF/gas interface. The lowest polarization resistance RP of 0.82 Ω cm2 at 600 °C was found for the coral sample with a high surface area of around 24.7, normalized to the geometrical area. Nanostructured MIEC electrode materials can therefore be regarded as promising candidates for intermediate temperatures. The influence of different microstructures for two LSCF layers sequentially deposited onto the Ce0.9Gd0.1O2−δ (GDC) substrate was shown as well.121 The first layer of nano-scaled LSCF with a thickness of 7 μm was deposited by electrostatic spray deposition (ESD). A current collector layer of La0.58Sr0.4Co0.2Fe0.8O3−δ with a thickness of ∼45 μm and a larger particle size was deposited by screen printing after annealing. The area-specific resistance (ASR) determined from impedance spectroscopy measurements decreased from 0.82 Ω cm2 (ref. 268) to 0.3 Ω cm2 at 600 °C.
 | ||
| Fig. 12 SEM micrographs of surface and cross-sectional views of dense (a–c), cracked (d–f), and coral (g–i) La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) films. Reprinted from ref. 264, copyright 2012, with permission from Elsevier. | ||
Similar investigations were made into the microstructural design of La2NiO4+δ (LNO) as an oxygen electrode on Ce0.9Gd0.1O2−δ (GDC) electrolyte to enhance electrochemical properties characterized through the polarization resistance (RP). Compact 1–2 μm-thick LNO layers were deposited in addition to a 12 μm porous screen-printed LNO electrode on top (particles in the range of 1–2 μm), thus decreasing the RP value from 7.4 to 1.0 Ω cm2 at 700 °C.269 The thin, compact LNO layer enhanced the contact between the electrode and the electrolyte and therefore also oxygen mobility. Higher sintering temperatures can also increase layer connectivity while simultaneously decreasing the electrode surface area. The insertion of a dense base layer was shown to lower the sintering temperature and enlarge the TPB region. This decreased the ASR at 600 °C by 35% compared to the best porous cell.270
The polarization resistance of La2NiO4+δ electrodes with different microstructures prepared on Ce0.9Gd0.1O2−δ (GDC) by ESD and screen printing (SP) was analyzed (Fig. 13).266 The porous electrode with a 3D coral microstructure and a thickness of 20 μm (sample 1, particle size ∼100 nm) exhibited an intermittent cathode/electrolyte interface coupled with a rough contact between the current collector and the top of the electrode (RP = 40.73 Ω cm2 at 600 °C). The implementation of a thin, dense LNO interlayer with a 3D coral microstructure on top (sample 2, particle size ∼120 nm) improved the cathode/electrolyte interface connectivity, which is crucial for the charge transfer process from cathode to electrolyte (RP = 3.33 Ω cm2 at 600 °C). A screen-printed cathode with a thickness of 20 μm (sample 3, particle size ∼400 nm, RP = 3.53 Ω cm2 at 600 °C) was reported. Vibhu et al. achieved an RP of 0.93 Ω cm2 at 600 °C for a 20 μm thick screen-printed LNO electrode with an average particle size of 0.6 μm on a YSZ electrolyte.240
 | ||
| Fig. 13 Schematic and SEM micrographs of calcined films showing the surface, cross sections, and scheme of the samples. Sample 1 and sample 2 were obtained by electrostatic spray deposition (ESD). Sample 3 was prepared by screen printing (SP). Sample 4 was prepared using a combination of SP and ESD. For the individual figures labelling please refer to the original publication. Reprinted from ref. 266 with permission from Elsevier. | ||
The optimized RP of 0.42 Ω cm2 was achieved at 600 °C by combining the thin, dense LNO interlayer with an LNO 3D coral microstructure and a screen-printed current collecting layer on top (sample 4). The improved electrochemical performance is related to the improved electric contact between the current collector and the electrode, resulting in decreased ohmic resistance. The thin, dense contact enhances the oxygen ion transfer between the electrode and the electrolyte.
Nanostructured materials have been suggested as a means of improving gas diffusivity in the electrode bulk and increasing the specific surface area of the porous electrode structure. Electrospun ceramic one-dimensional (1-D) nanofibers provide a large surface-to-volume ratio with good catalytic activity and high charge mobility. The diameters of the as-spun nanofibers range from 100 nm to 300 nm (ref. 271 and 272) depending on the materials. With a successful synthesis of LSCF nanofibers by electrospinning, a polarization resistance of 0.26 Ω cm2 at 750 °C was achieved.271 The cell performance was further improved by the infiltration of 20 wt% GDC, decreasing the RP to 0.21 Ω cm2 at 750 °C. Sr2Fe1.5Mo0.5O6−δ nanofibers used as electrode material in a symmetrical cell setup with humidified air exhibited a similar performance at 750 °C. Despite the performance of nanostructured materials, the microstructural changes when sintered at elevated temperatures will affect the performance and change the nanofiber porosity.273
3.2.4.1. Chemical stability and delamination. ABO3 perovskite oxides (Fig. 6a), double perovskites like AA′B2O5+δ (Fig. 6b), and Ruddlesden–Popper An+1BnO3n+1 (RP, n = 1 shown in Fig. 6d) exhibit remarkable capabilities as oxygen electrodes for electrolysis, but they are hindered by degradation effects. The fast degradation of the oxygen electrode responsible for the oxygen evolution reaction (OER) has affected the widespread adoption of solid oxide electrolysis cells. Interface evolution is considered one of the most critical factors determining the stability and performance of solid oxide electrolysis cells. The typical changes at the electrode/electrolyte interface are considered detrimental to the cell performance and include microstructural changes, interfacial reactions, micropore formation, element segregation/diffusion, and delamination.
In addition, the segregation of electrode components at the surface leads to the deterioration of surface functionality.274 Surface segregation is influenced by, for example, lattice structure, operating temperature, ion mobility, polarization, and composition/stoichiometry. The difference in ion size is one main factor that leads to enrichment or depletion compared to the bulk phase.275 Perovskites are typically comprised of large A-site (lanthanide and/or alkaline-earth cation) and smaller, catalytically active B-site (transition metal) cations. The segregation of the larger A-site cation to the surface forms insulating AO islands or RP layers,276 which obstruct contact of the B-site cation with reactants and hinder oxygen and charge transfer.277 Recent studies on A-cation segregation suggest that a suitable choice of A-site cations can minimize surface segregation in perovskites.275
The investigation of LSM chemical stability with the typical electrolyte YSZ has shown that cation (Sr and Mn) migration plays a key role in electrode deactivation. Depending on the stoichiometric composition of LSM, a high Sr content leads to Sr depletion and the interfacial formation of SrZrO3. However, it was suggested that a deficiency at the A-site in perovskites leads to Mn diffusion into YSZ and chemical reactivity to La2O3 and of La2Zr2O7 at the electrolyte/electrode interface.278–280 This phase is detrimental to the electrochemical activity and performance of the LSM electrode.281,282 A recent characterization of the YSZ/LSM electrolyte/electrode interface with transmission electron microscopy (TEM) and DFT clarified the interfacial nanostructure, which is essential to improving cell performance and stability.283 After much discussion on the nature of the interface,284–289 an interlayer of self-limited width with partial amorphization and strong compositional gradient was found, which exhibited characteristics of a complexion stabilized between two bulk phases. Images taken with high-magnification EDS are provided in Fig. 14. The dark field STEM (DF-STEM) electron micrograph in Fig. 14a shows the LSM phase on top of the YSZ. The elemental mappings in Fig. 14b for Y and Zr (blueish colors), as well as for La, Sr, and Mn (reddish colors), present the intermixing of elements with YSZ at the grain boundary in a compositionally unique region (white dashed). Monte Carlo simulations and corresponding elemental line profiles are taken across the interface to verify these results and give an average interdiffusion width of 1.25 nm (Fig. 14d). Based on these findings, the origin of Sr surface nucleation after thermal aging was localized by combining force field-based simulations, energy dispersive X-ray spectroscopy (EDS), and multi-variate statistical analysis.290 The graph on the left in Fig. 14 displays the validation through surface-sensitive XPS measurements, showing interfacial Sr enrichment occurring at the LSM/YSZ side of SOEC after aging. The aged sample was sintered at 1200 °C for 1 h and exhibited a slight accumulation of Sr at the surface and an increase in the Sr/La ratio by 11.5%. This corresponds to an increase of 9.7% in the nominal A site occupancy by Sr, which might result from the segregation of strontium oxide.
 | ||
| Fig. 14 Right: interdiffusion region at the YSZ/LSM interface visualized in (a) DF-STEM imaging of the YSZ/LSM interface and (b) EDS elemental mapping with dashed lines highlighting the interdiffusion region with (c) intergranular layer width derived from experiments and simulations. Experimental and theoretical average interdiffusion widths indicated in (d) are qualitatively close. Reproduced from ref. 283, which is licensed under the terms of the Creative Commons Attribution 4.0 License (CC BY, https://creativecommons.org/licenses/by/4.0/), © 2021 by the authors. Left: Sr 3d photoelectron spectra underline the change in Sr/La ratio upon aging at the surface, including the accumulation of Sr. Reproduced from ref. 290, which is licensed under the terms of the Creative Commons Attribution 4.0 License (CC BY, https://creativecommons.org/licenses/by/4.0/), © 2022 by the authors. | ||
Experimental investigations showed that microstructural changes in the oxygen electrode could also lead to oxygen electrode delamination. This electrode delamination was related to the disintegration of the La0.8Sr0.2MnO3 (LSM) particles and the formation of nanoparticles at the electrode/electrolyte interface.291 The nanoparticle formation resulted from the shrinkage of the LSM lattice due to the migration of oxygen ions from the YSZ electrolyte into the LSM grain, thus oxidizing Mn3+/Mn4+ into Mn4+ and creating local tensile stresses. The delamination at the electrode/electrolyte interface is a common failure mechanism attributed to the build-up of high internal oxygen partial pressure close to the interface.292,293 The bubbles evolve under anodic overpotential and penetrate the porous electrode/electrolyte interface, which leads to pressure buildup.294–296 Studies of Ni-YSZ/YSZ/LSM-YSZ electrolysis cells under high current densities exhibited hole/pore formation in the grain boundaries near the oxygen electrode, leading to very high grain boundary resistivity and thus increasing the ohmic resistance of YSZ.297 Densification and delamination of the LSM-YSZ oxygen electrode were attributed to cation migration under a high anodic current (−1.5 A cm−2 at 750 °C).298 Interdiffusion between the two phases was observed as well as an intergranular fracture along YSZ grain boundaries. La, Sr, and Mn cations are reported to segregate along the grain boundary of the electrolyte, thus facilitating the generation of oxygen gas bubbles, which in turn led to electrode delamination. The formation of the second phase La2Zr2O7 is suggested to be the cause of LSM electrode delamination, as shown in eqn (9).299
| LaMnO3+δ + ZrO2 + 1/4O2 ↔ 1/2La2Zr2O7 + MnO2 | (9) |
Due to their high chemical reactivity with zirconia-based electrolytes, cobaltite-based electrodes such as LSC and LSCF are only used with a ceria-based barrier layer. Otherwise, the formation of the electronically insulating phases SrZrO3 and La2Zr2O7 can be observed as a result of Sr segragation.137 This phenomenon was observed at the oxygen electrode/electrolyte interface after the operation of an electrolysis cell for 6100 h at −0.75 A cm−2 between 777 °C and 780 °C.42 The authors theorized from the microscopic results that oxygen electrode demixing, the transport of gaseous Sr species, and Zr mass transport through the grain boundary to the 8YSZ electrolyte are contributing to the degradation of the LSCF electrode. This is driven by the chemical potential gradient of SrZrO3 formation. The relationship between the material destabilization and operating conditions was characterized by comparison of reference samples and cells tested under both fuel cell and electrolysis mode through synchrotron X-ray microdiffraction and micro-fluorescence.43 While pristine samples exhibited strontium zirconate and a Gd-rich interdiffusional layer pre-test/post-manufacturing, the accumulation of SrZrO3 at the interface between the barrier layer and the interdiffusional layer was found for the aged samples. Additionally, the LSCF cell volume evolved due to strontium segregation. Both phenomena were found to be enhanced in electrolysis mode and thermally activated. Ni-YSZ-supported SOEC cells with an LSC-GDC oxygen electrode were tested at −1 A cm−2 in co-electrolysis conditions for 2700 h at 800 °C. The authors observed Sr and Co depletion at the LSC-GDC boundary of the tested cell around 3–5 μm from the electrode interface. As this depletion was not observed in the reference cell, the depletion was attributed to long-term testing.300 The performance and stability were tested of the LSCF electrode for 50 h with and without a GDC interlayer in a Ni-YSZ/YSZ-supported full cell at 800 °C and −0.8 A cm−2.301 The cell without an interlayer showed a large increase in area-specific resistance (ASR) and more severe delamination compared to the measured cell with a GDC interlayer. The degradation of the cells was associated with the phase change of LSCF from rhombohedral to cubic. Half-cells measured in a three-electrode configuration at 800 °C after polarization with an electrolysis current of −1 A cm−2 for 24 h showed relatively stable ohmic resistance RΩ for 18 h, with the resistance increasing abruptly thereafter.302 The YSZ surface characterization showed the formation of an insulating SrZrO3 layer at the LSCF/YSZ interface during the sintering of fresh samples. Post-test analysis suggested that the SrZrO3 interlayer delaminated from the YSZ electrolyte rather than the LSCF electrode from the interlayer. Co diffusion into the SrZrO3 layer and the YSZ electrolyte was confirmed by EDS analysis. The diffusion process enabled the formation of O2 at the interlayer/electrolyte interface, which led to the delamination of the SrZrO3 layer and the LSCF electrode.
Based on the proposed delamination mechanisms as described in Fig. 15, several studies have focused on countermeasures. To minimize the formation of La2Zr2O7,299 manganese-modified yttria-stabilized zirconia (Mn-YSZ) interlayer was utilized on the electrolyte.303 The symmetrical cells were tested at 840 °C at a constant voltage of 0.8 V for 200 h in electrolysis mode. No delamination was observed post-test for Mn-modified cells. Compared to unmodified cells, less Mn migrated from the LSM electrode to the YSZ electrolyte. The authors hypothesized that the porous sol–gel coating averts high oxygen pressure build-up, thus diminishing the electrode delamination. Based on previous investigations about nanoparticle formation in LSM,291 the LSM-infiltrated YSZ (LSM-YSZ) composite electrodes were prepared to prevent reactivity between LSM and the YSZ electrolyte.304 The infiltrated LSM-YSZ oxygen electrodes exhibited high electrocatalytic activity and good stability under SOEC conditions compared to non-composite electrodes. The results underlined that the microstructural stability of the LSM nanoparticles is impacted by two opposing parameters: (a) LSM lattice shrinkage under the anodic polarization and (b) grain growth by thermal sintering. Inserting a Ce0.43Zr0.43Gd0.1Y0.04O2−δ (CZGY) contact layer between the YSZ and GDC led to an increased interface stability and prevented GDC delamination. The CZGY contact layer improved the stability with the neighboring layers and prevented delamination during the short-term (100 h) electrolysis tests (800 °C, −0.8 A cm−2). Cation surface segregation and phase instability also occur on various perovskite oxides for example, layered perovskite oxides and Ruddlesden–Popper oxides except for LSM and LSC/LSCF.
 | ||
| Fig. 15 Proposed delamination mechanisms due to microstructural change: Co migration and formation of SrZrO3. Adapted and reprinted from ref. 302, copyright 2018, with permission from Elsevier. Formation of La2Zr2O7 nanoparticles. Adapted and reprinted from ref. 299, copyright 2012, with permission from Elsevier. The disintegration of LSM due to tensile stress. Adapted and reprinted from ref. 291, copyright 2011, with permission from Elsevier. | ||
Chemical reactivity was also found between double perovskites, for example, Sr2Fe1.5Mo0.5O6−δ and derivatives with commonly used YSZ. This limits the selection of compatible electrolyte materials or necessitates a protective layer of Ce0.8Gd0.2O2−δ or La0.6Ce0.4O2−δ deposited between the electrolyte and the electrode material. Stoichiometric composition is a crucial factor that affects the extent of SrO surface segregation. In addition to the amount of Sr dopant, thermodynamic calculations indicate that the B-site cation type impacts SrO activity in (La,Sr)MO3 (M = Fe, Co, Mn), which decreases in the order of Co > Fe > Mn.305 The surface chemistry of double perovskites PrBaCo2O5+δ (PBC) and GdBaCo2O5+δ (GBC) was investigated using low-energy ion scattering (LEIS) after annealing (400–800 °C).306–308 The results showed segregation and surface domination of the larger A-site cation with B-cation-enriched regions below the surface. Similar observations were made when observing Ba and Co segregation out of the lattice after annealing at high temperatures (>800 °C).309
The chemical reactivity between Ruddlesden–Popper nickelates and commonly used electrolyte materials is an important factor in long-term SOEC application. The formation of an insulating phase can result from excessive chemical reactivity and lead to increased polarization resistance. The stability under operating conditions of lower-order RP is therefore the main issue in the long-term application of nickelates such as LNO, PNO, and NNO. Studies on chemical reactivity with the GDC electrolyte have shown that it has good chemical compatibility with Nd2NiO4+δ.310 With the electrolyte material YSZ, Nd2NiO4 has shown good stability from 700 °C to 900 °C and only a minor formation of Nd2Zr2O7 at 1000 °C.311 Compatibility tests with LSGM electrolytes have shown partial reactivity after 1 h in air at 1150 °C through the formation of Nd4Ga2O9 due to Ga vaporization at high temperatures.310
La2NiO4+δ and Pr2NiO4+δ have been known to react with both YSZ and GDC.311–313 The reactivity test of La2NiO4+δ (LNO) with YSZ shows the formation of an insulating pyrochlore La2Zr2O7 phase at 900 °C after 72 h, which led to increased ASR values over 700 °C.312 For the electrolyte material LSGM, the reactivity test shows good chemical compatibility with LNO, although small traces of decomposed LSGM (≈3%) were observed310 (Fig. 16). Despite the relative stability of LSGM, the authors argued that the very low electrical conductivity of the small impurities could infringe on cell performance. The secondary phases NiO, La2O3, and La3Ni2O7 were observed after performing a reactivity test using GDC as an electrolyte.312
 | ||
| Fig. 16 Left: XRD patterns after heating at 1150 °C for 1 h: (a) GDC, (b) GDC/LSFC, (c) GDC/NNO, (d) GDC/LNO and XRD pattern after heating at 800 °C for 5 days for (e) GDC/PNO. Right: XRD patterns after heating at 1150 °C for 1 h: (a) LSGM, (b) LSGM/LSFC, (c) LSGM/LNO, (d) LSGM/NNO and XRD pattern after heating at 800 °C for 5 days for (e) LSGM/PNO. Reprinted from ref. 310, copyright 2013, with permission from Elsevier. | ||
Pr-based lower-order Ruddlesden–Popper phases were observed decomposing, while the studies of higher order RP such as Pr4Ni3O4+δ showed good structural stability up to 1000 °C.243,314 PNO decomposition in air was observed for example after 24 h at 900 °C and the decomposition product reacted subsequently with YSZ to Pr2Zr2O7 and Pr6O11.311 The formation of the insulating phases was attributed to the thermodynamic instability of PNO above 700 °C and not the chemical reactivity. Pr2NiO4+δ also showed no stability with GDC, decomposing to the higher order RP Pr4Ni3O10 and Pr6O11, although the results did not clarify whether PNO only decomposes above 700 °C or a chemical reaction between PNO and the electrolyte material occurred. Similar results were reported for an 8% PNO phase after sintering a PNO/LSGM sample for five days.310 Higher order RP phases have relatively high total conductivity and are oxygen under-stoichiometric in contrast to the n = 1 phases.229 In addition, the Ni3+ content is increased, which leads to higher chemical stability. Up to 1000 °C, the Pr4Ni3O10+δ phase is fully stable under oxygen and decomposes into Pr2NiO4+δ and NiO at higher temperatures.314
3.2.4.2. Cr poisoning. An additional issue to consider is the interaction of oxygen electrodes and the Cr-forming alloys (i.e., stainless steel), which are used as metallic interconnects in SOEC stacks similar to SOFC. They are inexpensive and easy to produce and exhibit high thermal and electrical conductivity. At high temperatures, volatile chromium species are generated under oxidizing conditions, which can react electrochemically with the LSM electrode and lead to fast material degradation.315–317 The Cr deposition and poisoning of oxygen electrodes by the volatile species CrO3 and CrO2(OH)2 from the protective Cr2O3 scale have been extensively studied under SOFC operation conditions.318–323 It is known that under cathodic polarization, Cr deposition at the LSM/electrolyte interface and on the surface leads to the formation of Cr2O3 and the (Cr,Mn)3O4 spinel, which have a detrimental effect on cell performance.315,324,325 Although Fe–Cr alloys are commonly used as interconnect materials for SOEC, little work has been carried out on electrode poisoning under SOEC conditions. The Cr poisoning of an LSM oxygen electrode under SOEC condition313 is shown in Fig. 17.
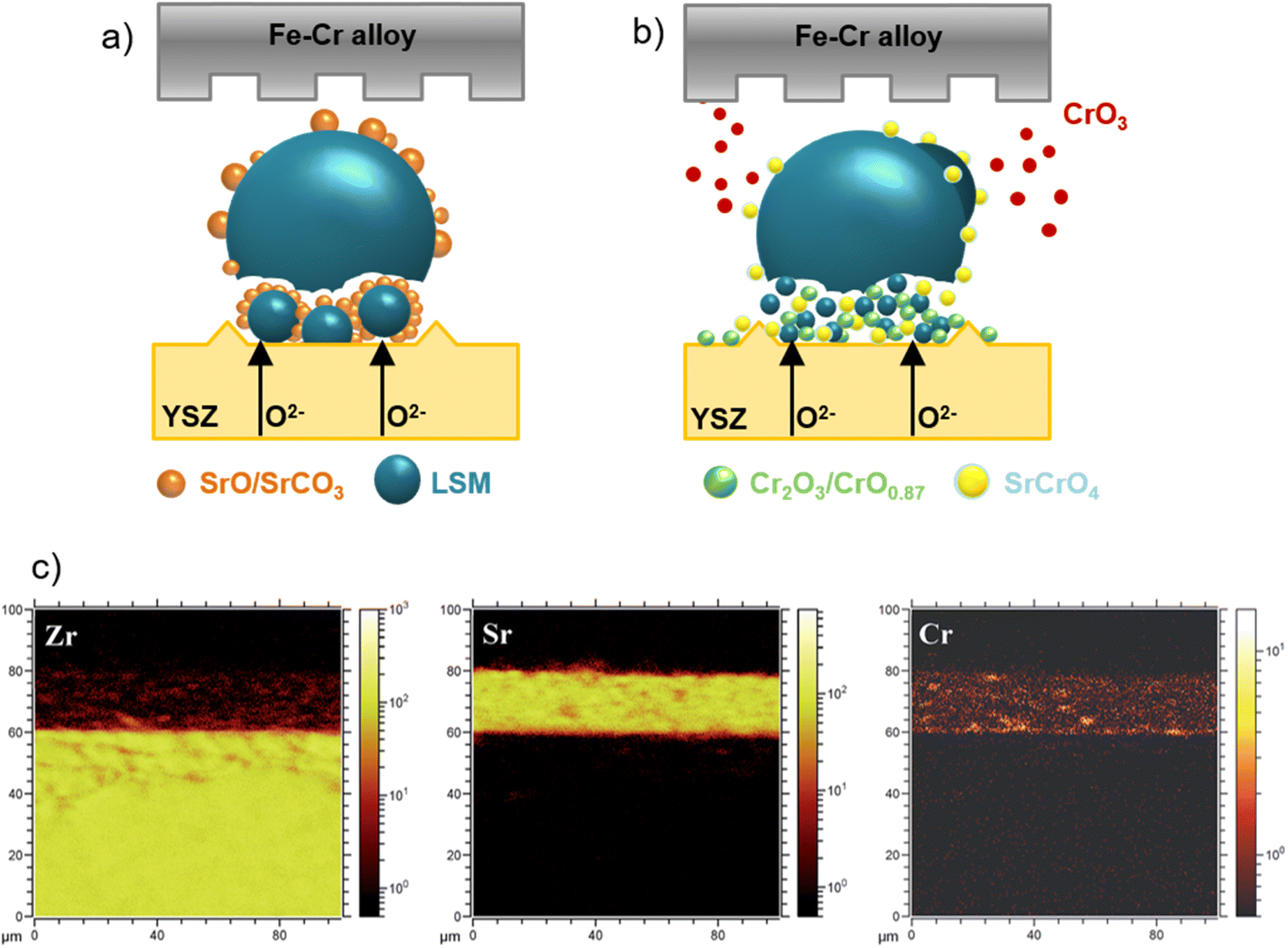 | ||
| Fig. 17 Cr deposition and subsequent poisoning of the LSM oxygen electrodes under SOEC operating conditions. (a) LSM nanoparticle formation at the electrolyte/electrode interface. Under anodic polarization, SrO segregates from the bulk of the electrode to the surface. (b) Formation of SrCrO4 on the LSM surface and Cr deposition on the electrolyte surface and at the electrolyte/electrode interface. (c) SIMS image of the LSM electrode taken under the rib of the Fe–Cr interconnects after 1 h at 800 °C and −0.8 A cm−2. Reproduced and adapted from ref. 326 with permission from the Royal Society of Chemistry. | ||
The LSM electrode partially disintegrates to form LSM nanoparticles at the electrode/electrolyte interface. Under anodic polarization, SrO/SrCO3 species segregate to the electrode surface, which is enhanced by the inhibition of Mn2+ generation. Cr deposition and high activity between Cr and Sr, in turn, lead to the dominant SrCrO4 product on the electrolyte surface and the electrode/electrolyte interface, rather than the (Cr,Mn)3O4 spinels under SOFC conditions. Based on the nucleation theory,327 the reaction of SrO segregation to the surface and the subsequent reaction with the gaseous Cr species is given by eqn (10)–(13).
Sr segregation:
| La0.8Sr0.2MnO3+δ → La0.8Sr0.2−xMnO3+δ + xSrO | (10) |
Cr deposition via nucleation and grain growth route:
| SrO + CrO3 → Cr–Sr–Ox (nuclei) | (11) |
| Cr–Sr–Ox + CrO3 → CrOy + Cr2O3 | (12) |
| Cr–Sr–O + SrO + CrO3 → SrCrO4 | (13) |
Chromium deposition of SrCrO4, CrO2.5, and Cr2O3 phases at the La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) oxygen electrode increased the overpotential of the O2 evolution reaction (OER). The hindered electrocatalytic activity was linked to the significant deficiency of catalytically active Sr in the electrode bulk segregating to the electrode surface. The areas near the rib of the interconnect (Fig. 18) showed significantly higher Cr enrichment compared to the regions of the interconnect channels.328 The dissociation of La0.8Sr0.2CoO3−δ (LSC) was the reason to conclude that Cr-containing phases from the stainless steel interconnects are driven into the contact layer microstructure, thus causing the long-range transport of Sr and Co cations and, consequently, La–Cr–O phases.329
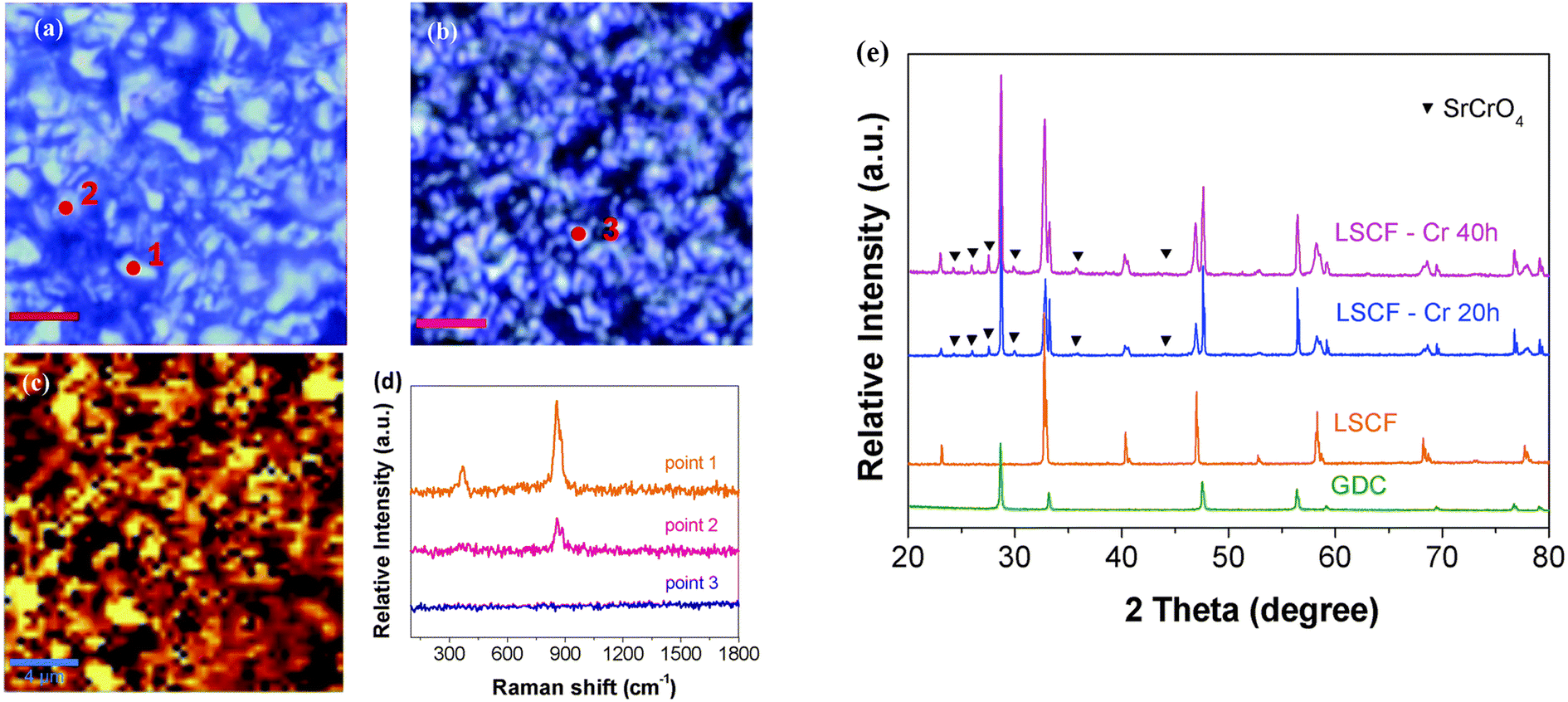 | ||
| Fig. 18 Post-test analysis of an electrode before and after polarization at 900 °C and −0.2 A cm−2 in the presence of Fe–Cr interconnects: (a) optical images of the LSCF electrode surface under the Fe–Cr interconnect after 40 h treatment. (b) As-prepared cell before testing. (c) Raman spectroscopy mapping at ν = 860 cm−1, corresponding to SrCrO4. (d) Raman profiles collected at points 1, 2, and 3. (e) XRD pattern before and after testing of the GDC electrolyte and the LSCF oxygen electrode for 20 h and 40 h at 900 °C and −0.2 A cm−2 in the presence of Fe–Cr interconnects. Reproduced from ref. 328 with permission from the PCCP Owner Societies. | ||
The effects of Cr poisoning upon LSM and LSC(F)-based electrodes have received the highest level of research interest. Although other oxygen electrode materials have also been investigated, the current literature is mostly focused on Cr effects in SOFC, and degradation studies are often not conducted for an extended period (<500 h). Few studies consider double perovskite materials in terms of their chromium poisoning resistance. The durability of strontium iron-molybdenum oxygen (SFM) electrodes was reported to be affected by Cr poisoning (Fig. 19i). Short-term Cr-poisoning tests of around 160 h on symmetrical cells with SFM oxygen electrodes showed Cr species poisoning the electrode surface, resulting in the formation of SrCrO4 after the direct reaction of the Sr with Cr2O3 and gaseous CrO3.330 The reactivity with chromium was studied by looking at SmBaCo2O5+δ (SBC)331 and later PrBaCo2O5+δ (PBC)309 (Fig. 19a–h). At high temperatures, Ba and Co cation diffusion was observed for PBC, forming BaO and Co3O4 precipitates on the surface, mainly located at the grain boundaries. In the presence of Cr, BaCrO4 was formed and had an impact on electrochemical performance.309
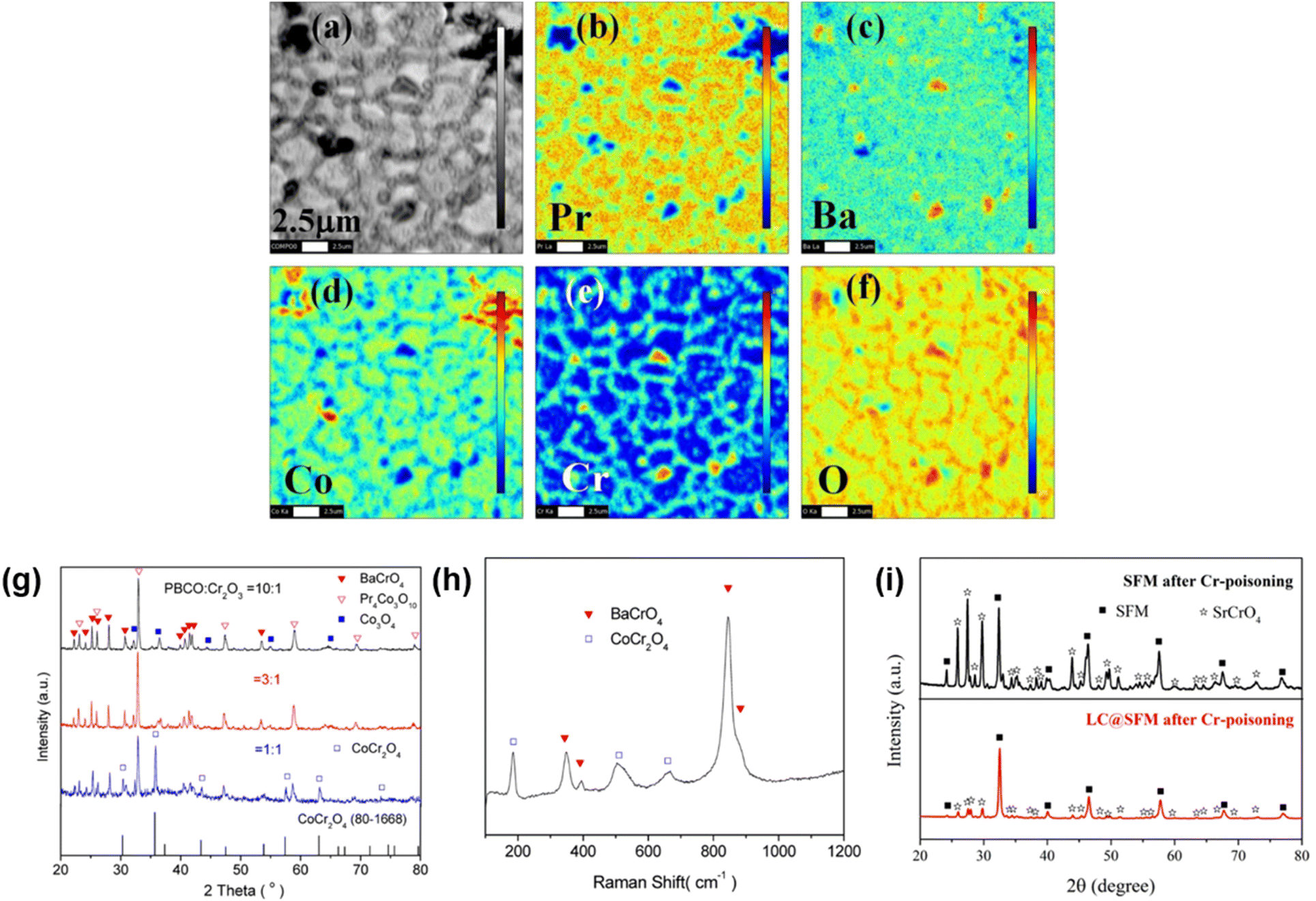 | ||
| Fig. 19 (a) Backscattered electron (BSE) imaging of segregated PBCO surface at 900 °C in 100% O2. Electron microprobe analyzer (EPMA) elemental mappings for (b) Pr, (c) Ba, (d) Co, (e) Cr, and (f) O. Higher concentrations are indicated in red, while lower concentrations with scale bars of 2.5 μm are highlighted in blue. (g) XRD patterns for PBCO–Cr2O3 mixtures after reacting for 5 h in air at 1000 °C, and (h) Raman profile of the PBCO–Cr2O3. Reprinted in part with permission from ref. 309. Copyright 2018 American Chemical Society. (i) XRD patterns after Cr poisoning tests for SFM and LSC@SFM surfaces. Reprinted with permission from ref. 330. Copyright 2019 American Chemical Society. | ||
The electrochemical activity of SmBaCo2O5+δ (SBC) also degraded quickly in the presence of a chromium-forming metallic interconnect. The authors concluded that the Cr deposition was induced by the segregated BaO on the SBCO surface, leading to the formation of BaCrO4.331 Similar results were observed for NdBaCo2O5+δ (NBC) and PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (BSCF), although the authors found that LNO (La2NiO4+δ)-infiltrated PBSCF improved tolerance to Cr poisoning. This result highlights the possibility of Cr-poisoning-resistive oxygen electrodes in the absence of nucleating agents (Sr, Ba, Mn). Long-term measurements over 1000 h revealed that La2NiO4+δ oxygen electrodes exhibited good stability under dry conditions in the presence of Cr.332 However, the oxygen activity declined as soon as humidity was introduced into the system, and the LNO surface layer decomposed. Similar results were found for PNO.333 Although LNO electrodes react readily with Cr2O3 to form LaCrO3 and LaNiO3 reaction products, their chromium tolerance was found to be higher than for LSCF oxygen electrodes.334 Nd2NiO4+δ (NNO) is a potential alternative to standard perovskite materials due to its high catalytic activity and chromium poisoning tolerance.335 No chromium-related compound was observed in either the Nd2NiO4+δ cathode or the NNO/YSZ interface. However, Nd depletion of the nickelate was identified as a result of secondary phase formation with the Cr contaminants.336 The changes in NNO due to Cr poisoning only had a minor influence on the surface exchange properties.183
3.3. Fuel electrode
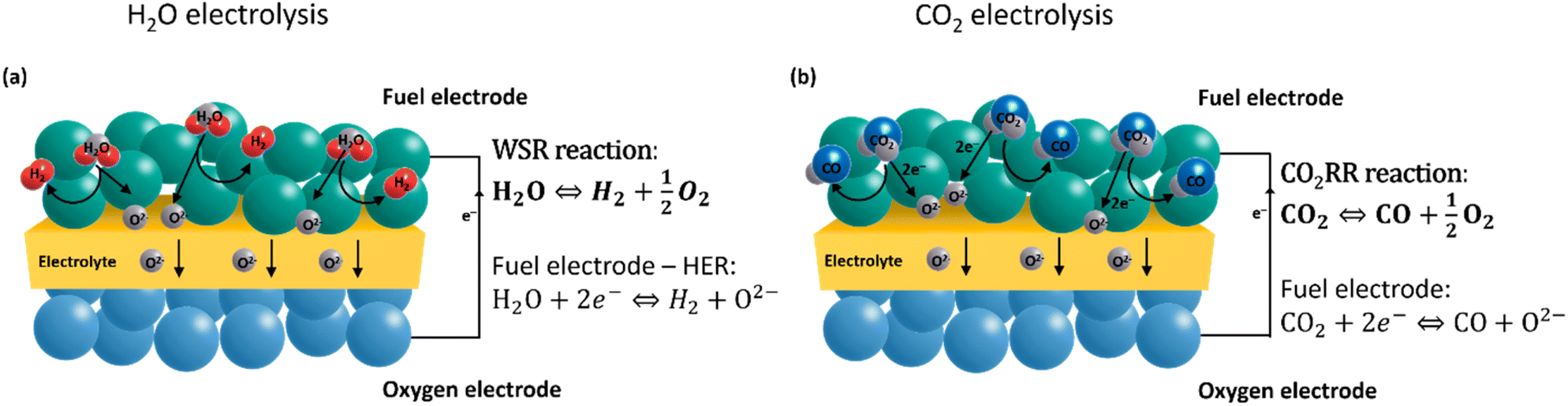 | ||
| Fig. 20 Schematic description of the reactions at the fuel electrode/electrolyte interface during different electrolysis operations: (a) steam electrolysis: water splitting reaction (WSR) including the hydrogen evolution reaction (HER). (b) Carbon dioxide electrolysis: carbon dioxide reduction reaction (CO2RR). | ||
The main advantage of SOECs is the combination of steam and CO2 electrolysis, referred to as co-electrolysis, resulting in the production of industrially crucial syngas (H2 + CO). Here, both the dissociation of water molecules and the reduction of CO2 take place at the active sites of the fuel electrode for the respective reaction. However, depending on the gas composition, the conversion of CO2 into CO takes place mainly due to the reverse water–gas shift reaction (RWGS) between the produced H2 and CO2, and is not electrocatalytically, but catalytically activated.340,341
The fuel electrode also requires long-term chemical stability in humid reducing atmospheres as well as in mixed gases. In particular, the respective gas composition can have a strong influence on the performance and durability of the fuel electrode. The gas mixture, which is partly dependent on the electrolysis mode, can consist of different fractions of reducing agents (e.g., H2), water molecules, carbon-containing molecules (CO2, CO), and possibly carrier gas. The optimization of the electrode material is dependent on the gas mixtures occurring during the electrolysis operation.
An example of how the complexity of the gas composition influences the electrode material has been fairly well investigated for the commonly used Ni-YSZ fuel electrode. The selection of Ni-YSZ as a fuel electrode material for SOECs is based on the fact that the materials used in SOECs were initially adapted from the solid oxide fuel cell. However, the gas mixture at the fuel electrode for SOFCs differs from the composition used for electrolysis cells. For instance, there is a variation in fuel gas properties such as oxygen partial pressure, humidity, and the amount of reducing gases. In the case of Ni-YSZ being used as a fuel electrode in steam electrolysis, an increase in humidity in the fuel gas initially leads to a higher conversion of H2O into H2 and 1/2O2. While at the same time, material degradation and reduced electrode performance are accelerated over time.342,343 Additionally, the observed cell voltage in SOEC operation is higher at 1.1–1.4 V compared to SOFCs (0.6–1.0 V), which leads to a decrease in the local oxygen partial pressure at the fuel electrode side.42
The fuel electrode material also requires preferably high electronic and ionic conduction to extend the electrochemical reaction area (triple-phase boundary (TPB)) as well as matching thermomechanical properties of the electrode material with the other components of the solid oxide cell, especially the electrolyte. A sufficient electrical conductivity for the fuel electrode, which should be achieved throughout the whole operation time, is in the scale of 100 S cm−1.344 The relative expansion depending on temperature given by the thermal expansion coefficient (TEC) is typically considered the first indicator of the thermomechanical compatibility between the electrode and the electrolyte. The TEC values for commonly applied electrolytes such as YSZ or GDC are around 10−13 × 10−6 K−1.114,193 A mismatch between the TECs of the electrode and the electrolyte heightens the possibility of cracks or delamination during the heating and/or cooling of the cell. Adaption or optimization of the thermal expansion coefficient can be achieved by doping the electrode material or by mixing it with other materials, for example, electrolyte material. The microstructure of the respective fuel electrode has a strong impact on its properties and, therefore, on the performance and degradation behavior of the electrode. Microstructure parameters such as grain size, grain size distribution, tortuosity, percolation, and porosity define the resulting electrode properties/performance. For instance, grain size or its distribution affects the percolation paths of the electrode. A specific porosity is required to ensure unimpeded gas diffusion throughout the electrode toward the electrode/electrolyte interface.345,346 On the microstructural level, a well-performing electrode exhibits a triple-connecting network of pores as well as electronic and ionic paths.
3.3.2.1. Ni cermet electrodes. The most commonly used materials for fuel electrodes in SOCs are currently Ni cermets. Adopted from use in SOFCs, the composites of Ni metal and YSZ or GDC are also mainly used as fuel electrodes in high-temperature electrolysis operations. These materials feature a high catalytic activity for electrochemical water reduction and high electronic conductivity, resulting in a good initial performance of Ni-cermet-based fuel electrode SOECs.347 Properties of the Ni cermets such as the thermal expansion coefficient or electrical conductivity depend on the material fabrication process and its resulting microstructure.348,349 For the Ni-GDC cermet, first, a TEC of 14.3 × 10−6 K−1 was approximated.350 The measured TEC for a NiO-GDC (50
:50 wt%) composite in air resulted in 13.5 × 10−6 K−1 for a temperature range of 25–1000 °C.351 The TEC also changes with different amounts of Ni inside the composite material. An increase of Ni from 15 vol% to 30 vol% caused an increase in the TEC from 10.4 × 10−6 K−1 to 13.2 × 10−6 K−1 at 900 °C.352 An investigation regarding the influence of porosity and sintering temperature on the resulting TECs and the electrical conductivity of Ni-YSZ (50:50 wt%) showed that the thermal expansion of Ni-YSZ changes with the addition of pore former.349 The initial value of 11.88 × 10−6 K−1 increased to 12.23 × 10−6 K−1 using 15 wt% of pore former.349 For a 50% Ni-YSZ material fabricated by citric method TEC of 9.1 × 10−6 K−1 was obtained in 5% H2 + 95% Ar atmosphere.353
The electrical conductivity is partly influenced by the particle size of the material, the porosity, the volume fraction of Ni, the distribution of Ni and YSZ or GDC inside the material, and the percolation threshold of Ni (see Section 3.3.2).348
For Ni-YSZ prepared by solid-state reaction, the electrical conductivity in air at 25 °C increased with increasing sintering temperature (3936 S cm−1 at 1250 °C and 9203 S cm−1 at 1450 °C) and decreased with an increasing amount of pore former.349 The conductivities of Ni-YSZ increased to 25 S cm−1 and 4094 S cm−1 using 15 wt% of pore former at the respective sintering temperature. The influence of the Ni content in the electrode material on electrical conductivity showed an increase in conductivity from 0.103 S cm−1 to 989 S cm−1 in hydrogen at 900 °C with a Ni volume change from 15 vol% to 30 vol%.352 Electrical conductivity stability for Ni-YSZ composites (cermets) in dry and wet reducing atmospheres (combinations of H2 + diluting gas) could be confirmed for conductivities of 1100–1200 S cm−1 at 600 °C.344 However, a significant conductivity loss was observed in wet atmospheres for temperatures of 800 °C and above and in dry atmospheres for temperatures ≥850 °C.344 For the electrical conductivity of Ni-GDC composite consisting of 65 wt% NiO and 35 wt% GDC a conductivity of 0.28 × 10−3 S cm−1 was seen at 700 °C. This is slightly higher than the conductivity of a NiO-YSZ (50:50 wt%) composite (0.17 × 10−5 S cm−1).354 The electrical conductivity of NiO-GDC cermets in hydrogen at 610 °C could be increased with Cu doping from 2.4 × 10−3 S cm−1 (pure NiO-GDC) to 4.5 × 10−3 S cm−1 (Cu-doped NiO-GDC).355 The performance of Ni-YSZ and Ni-GDC fuel electrodes in different SOEC operation modes was tested in several studies. The performance of a Ni-YSZ/YSZ/LSM cell has been investigated with a variation of absolute humidity (AH) in steam electrolysis for 800 °C and 900 °C.356 A current density of −1.1 A cm−2 was achieved under thermoneutral voltage with 82 vol% AH at 800 °C. The authors observed decreasing ASR from 0.69 Ω cm2 to 0.42 Ω cm2 with an increase in the absolute humidity from 30 vol% to 82 vol%. The ASR also decreased when increasing temperature up to 900 °C, resulting in values of 0.32–0.20 Ω cm2 for 30–82 vol% AH. At 900 °C and below thermoneutral voltage with 1.1 V, a current density of −1.4 A cm−2 was achieved. The analysis of operating temperature and fuel gas composition on the performance of a Ni-YSZ/YSZ/GDC/LSCF-GDC cell showed an increase in current density at a set voltage of 1.3 V from −0.352 A cm−2 to −0.674 A cm−2 when the temperature was increased from 700 °C to 800 °C. Moreover, a change in the volume ratio of the gas composition at 750 °C (H2 + H2O + CO2) from 20% H2 + 0% H2O + 80% CO2 to 20% H2 + 80% H2O + 0% CO2 led to an increase in the current density of the cell from −0.445 A cm−2 to −0.598 A cm−2.357 The performance of the Ni-YSZ/YSZ/LSCF-GDC cells in dependence of gas composition resulted ASR values of 0.21 Ω cm2, 0.22 Ω cm2, and 0.27 Ω cm2 when operating the cells at 800 °C in 50% H2 + 50% H2O, 25% H2 + 75% CO2, and 25% H2 + 25% CO2 + 50% H2O, respectively.358 For the operation under 25% H2 + 25% CO2 + 50% H2O at 800 °C using a set voltage of around 1.3 V, the authors achieved syngas production rates of ∼7 sccm cm−2. Ni-YSZ/YSZ/LSM-YSZ cells exhibited initial cell voltages of 1.42 V and 1.5 V for current densities of −1.5 A cm−2 and −2.0 A cm−2 at 865–875 °C during co-electrolysis in a 45% H2O + 45% CO2 + 10% H2 atmosphere.359 The syngas conversion was 45% and 60% for −1.5 A cm−2 and −2.0 A cm−2. The effect of Ba infiltration in Ni-YSZ (50:50 wt%) on electrode performance was studied during steam electrolysis, co-electrolysis, and CO2 electrolysis at 800 °C.360 The pure Ni-YSZ electrode at a set voltage of 1.3 V achieved current densities of −0.45 A cm−2, −0.27 A cm−2, and −0.36 A cm−2 for H2O electrolysis, co-electrolysis, and CO2 electrolysis. For the BaCO3-infiltrated Ni-YSZ fuel electrodes, increased current densities of −0.69 A cm−2, −0.55 A cm−2, and −0.62 A cm−2 are observed. Moreover, the interfacial polarization resistance between the electrolyte and the electrode was decreased due to the infiltration of Ba in the Ni-YSZ electrode from 1.01 Ω cm2, 1.59 Ω cm2, and 1.18 Ω cm2 to 0.64 Ω cm2, 0.82 Ω cm2, and 0.70 Ω cm2 for H2O electrolysis, co-electrolysis, and CO2 electrolysis. The difference between using a Ni-YSZ fuel electrode and a Ni-SDC fuel electrode for CO2 electrolysis was tested with cells made of Ni-YSZ/YSZ/LSM and Ni-SDC/YSZ/LSM at a set voltage of 1.6 V at 1000 °C.361 The cells achieved current densities of −1.13 A cm−2 and −0.87 A cm−2 for Ni-SDC and Ni-YSZ electrodes, respectively, in a 45% CO2 + 10% H2 + 45% N2 atmosphere. Analysis of the polarization resistance in gas atmospheres with 25% and 45% CO2 resulted in RP values for Ni-SDC for both gas mixtures around 0.17 Ω cm2, whereas the RP values for Ni-YSZ were 0.46 Ω cm2 and 0.53 Ω cm2 in 25% and 45% CO2 respectively. The performance of a Ni-GDC (65:35 wt%)/YSZ/GDC/LSCF cell configuration under steam and co-electrolysis operation was examined at a set voltage of 1.5 V.362 Current densities in steam electrolysis reached −1.31 A cm−2 at 900 °C and −0.41 A cm−2 at 750 °C, while the corresponding RP increased from 0.061 Ω cm2 to 0.31 Ω cm2 with decreasing temperature. The current density value for co-electrolysis at 900 °C was −1.37 A cm−2, corresponding to an RP of 0.089 Ω cm2. The influence of Mo and Au doping on the electrochemical performance of a Ni-GDC (65:35 wt%) fuel electrode, a YSZ electrolyte, a GDC barrier layer between the electrolyte and the oxygen electrode, and an LSCF oxygen electrode was analyzed in steam electrolysis. The fuel electrode doped with 3 wt% Mo and Au was tested regarding its performance with a pre-reduced fuel electrode in different pH2O/pH2 ratios. The resulting polarization resistances at 900 °C for a pH2O/pH2 of 1, 2.3, and 9 for a pure Ni-GDC fuel electrode were 1.36 Ω cm2, 1.53 Ω cm2, and 1.64 Ω cm2, respectively. Doping with 3 wt% Mo and 3 wt% Au led to a decrease of RP to values of 0.34 Ω cm2, 0.34 Ω cm2, and 0.36 Ω cm2. The authors concluded that for H2O electrolysis, the 3Au–3Mo–Ni-GDC fuel electrode exhibits better performance compared to the undoped material.347 In addition to cell performance, the performance in a long-term test with the use of an applied NiO contact layer was examined. At a set voltage of 1.3 V at 800 °C, the cells without a NiO contact layer achieved a current density of −0.076 A cm−2 in a 50% H2O + 50% H2 atmosphere. Using a NiO contact layer for the same cell configuration and under the same experimental conditions, a current density of −0.361 A cm−2 was reached. A higher current density of −0.780 A cm−2 at 1.3 V and 900 °C was achieved under steam electrolysis with a gas composition of 7% H2 + 30% N2 + 63% H2O.363
3.3.2.2. Perovskite-based electrodes. As materials exhibiting MIEC properties, perovskite oxides have received increased attention in recent years with regard to their use in various components of an SOEC, such as the fuel electrode. The reasons for this increased attention as well as their respective crystal structures are outlined in Section 3.2.1. To optimize and enhance the properties with respect to their respective application, the influence of adding dopants, mixing different phases, and infiltration processes is investigated for all of these possible fuel electrodes.
LSCM exhibits a simple perovskite structure with A- and B-site doping of the lattice. It is reported to have excellent redox stability as well as catalytic activity for fuel gases, thus leading to its consideration as a fuel electrode material in SOECs.364–367 Investigations of La0.75Sr0.25Cr0.5Mn0.5O3−δ (LSCM) as a fuel electrode material in H2O electrolysis366,368 and co-electrolysis365 have already been carried out.
The average thermal expansion coefficient of LSCM in a temperature range of 64–956 °C is 9.3 × 10−6 K−1.369 The investigation of the electrical conductivity in air and 5% H2 at different temperatures showed an electrical conductivity of LSCM is 7.7 S cm−1 in air at 320 °C, and 1.4 S cm−1 in 5% H2/Ar.369 With an increased temperature of 900 °C, the values increased to 38.6 S cm−1 in air and 1.49 S cm−1 in 5% H2. In a wet 5% H2 atmosphere at 900 °C, an electrical conductivity of 0.95 S cm−1 was measured for LSCM.370 The authors achieved an increase in conductivity in wet 5% H2 to 1.66 S cm−1 by doping the LSCM electrode with Ce. Work on Sc doping in LSCM (La0.75Sr0.125Ce0.125Cr0.5Mn0.5O3−δ – LSCMS) and its impact on the electrical conductivity in 5% H2 + 95% Ar at 700 °C showed that doping with Sc leads to a decrease in electrical conductivity from 0.639 S cm−1 to 0.185 S cm−1.368 Additionally, doping LSCM with Sc influences the polarization resistance RP of the electrode, which was tested at 700 °C and 1.8 V in a mixture of 5% H2O + 95% Ar. Adding Sc to the material reduces the RP from 9 Ω cm2 (LSCM) to 6.5 Ω cm2 (LSCMS). Performance analysis of LSCM electrodes in co-electrolysis mode at 800 °C and OCV in different ratios of H2O and CO2 revealed that the polarization resistance increases with increasing steam content from 21.49 Ω cm2 (20% H2O + 80% CO2) to 22.17 Ω cm2 (60% H2O + 40% CO2).365 The same observation was made while testing the material at 800 °C with a constant current of −0.12 A cm−2 using different steam concentrations.366 In addition, the study results underline that alongside the polarization resistance, the maximum current density is also dependent on the respective steam content. The steam electrolysis experiments at 850 °C under ∼1.6 V showed that an increase in the steam content from 20 vol% AH to 80 vol% AH leads to a change in the current densities from −0.431 A cm−2 to −0.593 A cm−2.366,367 The maximum current densities of LSCM fuel electrodes for CO2 electrolysis and co-electrolysis (60% H2O + 40% CO2) at 800 °C under a voltage of 2 V achieved were −0.18 A cm−2 and −0.23 A cm−2 for CO2 electrolysis and co-electrolysis, respectively.365 A comparison of the maximum current density for LSCM and Sc-doped LSCM electrodes at 700 °C and 2 V for steam electrolysis (5% H2O + 5% H2 + 90% Ar) showed current densities of −0.4 A cm−2 and −0.75 A cm−2 for LSCM and LSCMS.368 The authors also investigated the hydrogen production rate using the same temperature and voltage, only varying the gas composition. They obtained hydrogen production rates of 0.28 ml min−1 for LSCM and 1.2 ml min−1 for LSCMS in a 5% H2O + 95% Ar atmosphere and similar results for 5% H2O + 5% H2 + 90% Ar.368 For the LSCM electrode, a hydrogen production rate of 561 ml cm−2 h−1 at 850 °C and a voltage of 1.6 V with 80 vol% AH were achieved.367
Fuel electrodes based on LaFeO3−δ perovskite oxide are particularly investigated with regard to their application in SOECs for electrolysis using CO2-containing fuels. LSFM-based fuel gas electrodes are particularly suitable for CO2 electrolysis or co-electrolysis and exhibit a high CO2 selectivity and resistance to C deposition during electrolysis operation.371,372 Pure LSFM electrodes of different stoichiometries were investigated and the authors concluded that the performance of LaFeO3−δ perovskite-based electrodes could be optimized by doping with Sr and Mn.372 LSFM exhibits a TEC of 10.9–12.3 × 10−6 K−1 between 30 °C and 1000 °C in air373 and 18.7 × 10−6 K−1 in H2.374 The maximum conductivity of La0.6Sr0.4Fe0.8Mn0.2O3−δ observed in air at 800 °C was in the range of 35.24 S cm−1 (ref. 375) to 50.3 S cm−1.374 The performance of LSFM fuel electrodes in CO2 electrolysis and co-electrolysis operation was analyzed in several publications.
The cell composed of LSFM/LSGM/BLC (Ba0.6La0.4CoO3–δ) at 1.6 V in 50% CO2 + 1% CO + 49% Ar showed the best performance so far for La0.6Sr0.4Fe0.8Mn0.2O3−δ. With this material, current densities of −0.28 A cm−2 and −0.52 A cm−2 were achieved at 800 °C and 900 °C. The CO2 reduction rate at 900 °C was 153 μmol cm−2 min−1.372 The performance of a composite fuel electrode consisting of La0.6Sr0.4Fe0.8Mn0.2O3−δ and CuFe2O4 (CuF) in a ratio of 1:1 in co-electrolysis was analyzed using the cell configuration of LSFM-CuF/LSGM/BLC.371 At a temperature of 800 °C and a voltage of 1.6 V, the authors achieved a maximum current density of around −1.43 A cm−2 in 30% CO2 + 30% H2O + 40% Ar. The cell performance of an LSFM-GDC (60:40 wt%) composite fuel electrode for CO2 electrolysis was investigated with the LSFM stoichiometry La0.6Sr0.4Fe0.9Mn0.1O3−δ.376 The authors used symmetrical, YSZ electrolyte-supported cells with a GDC barrier layer and achieved current densities of −1.1 A cm−2 at 800 °C and −1.74 A cm−2 at 900 °C using a voltage of 2.0 V. The respective polarization resistances were RP = 0.85 Ω cm2 (800 °C) and RP = 0.48 Ω cm2 (850 °C). At a temperature of 800 °C, they observed a CO production rate of 6.438 ml min−1 cm−2.376,377 Further experiments were conducted in pure CO2 on the La0.6Sr0.4Fe0.9Mn0.1O3−δ fuel electrode side at temperatures of 800 °C and 900 °C and a voltage of 1.6 V. In the operating conditions, current densities of −0.335 A cm−2 at 800 °C and −0.76 A cm−2 at 900 °C could be achieved.378
LST has been categorized as a redox-stable perovskite with great potential as an electrode material in SOECs.379 The material exhibits TEC values of 11–12 × 10−6 K−1 between 30 °C and 1000 °C.380,381
Investigations with regard to the thermal expansion behavior of reduced and non-reduced La0.4Sr0.4TiO3 in different atmospheres between 50 °C and 1000 °C found that the TEC of the material in a 5% H2 + 95% Ar atmosphere with 11.89 × 10−6 K−1 is higher compared to the expansion in air of 10.35 × 10−6 K−1.382 Additionally, the authors observed that reducing the material after sintering could reduce the resulting TEC to 11.60 × 10−6 K−1.382 The conductivity of LST also depends on whether the material is in the pre-reduced or oxidized state. Non-reduced LST (La0.2Sr0.8TiO3+δ) reached a conductivity of 0.001 S cm−1 in air at 800 °C.383 The conductivity of reduced LST and iron-doped LST in air and 5% H2 + 95% Ar at 800 °C achieved conductivities of 0.7 S cm−1 and 0.5–0.6 S cm−1 in air and 1.1 S cm−1 and 1.5 S cm−1 in a reducing atmosphere for LST and Fe-LST.379 In hydrogen, sintered LST has conductivities of 100–500 S cm−1 between 700 °C and 1000 °C under 10−15 < pO2 < 10−20.380 Measured in wet H2, reduced LSTO reached a conductivity of 30 S cm−1 at 700 °C.384
To improve the conductivity and the catalytic activity, LST was doped with Ca and Fe.385 The authors theorized that doping LST on the A-site with Ca should enhance the electrical conductivity of the perovskite oxide. In addition, substituting Ti ions with Fe on the B-site of the crystal lattice of strontium titanate should lead to high mixed ionic–electronic conductivity and high catalytic activity. The investigations revealed a dependency of the conductivity on the Ca amount within the La0.2Sr0.25CaxTi0.95Fe0.05O3−δ material. They determined a maximum conductivity of 5.5 S cm−1 at 850 °C under 97% H2 + 3% H2O with an optimum calcium content of x = 0.45 mol%.385
The combination of titanate and ceria (La0.35Sr0.65TiO3−δ–Ce0.5La0.5O2−δ) was examined as a fuel electrode material in a reversible SOC concerning the material's conductivity.386 The authors discovered that the electrode's conductivity and, in turn, its performance is strongly dependent on the prevailing oxygen partial pressure as well as on the H2O/H2 ratio of the respective gas composition.386 ASRs of 0.2–0.28 Ω cm2 were achieved under a constant polarization loss of 0.1 V in pH2O = 0.5–0.9 using a mixture of LST with Ce0.5La0.5O2−δ as a fuel electrode.386 A composite electrode of LST (La0.2Sr0.8TiO3+δ)-SDC in CO2 electrolysis operation was investigated using a symmetrical cell with a YSZ electrolyte.387 To enhance CO2 selectivity and electrode performance, the LST was doped with Mn (La0.2Sr0.8Ti0.9Mn0.1O3+δ). They observed a decrease in the polarization resistance RP of the fuel electrode with increasing pH2 (10% to 100%). The RP is reduced from 28 Ω cm2 to 9 Ω cm2 for the LST-SDC composite and from 4 Ω cm2 to 2 Ω cm2 for the manganese-doped LSTM-SDC fuel electrode. The authors achieved current densities of −0.12 A cm−2 and −0.25 A cm−2 for LST- and LSTM-based fuel electrodes during operation at 800 °C under 2.0 V. The resulting CO production rates were 0.1–0.2 ml cm−2 min−1 and 1.1 ml cm−2 min−1,387 respectively. Similar experiments using a cell configuration of LST-SDC/YSZ/LSM-SDC achieved a current density of −0.125 A cm−2 during CO2 electrolysis at 700 °C under 2.0 V.384 The CO production rate of 1.23 ml cm−2 min−1 achieved in this work was higher than the LSTO-based fuel electrode results.
SFM, Sr2Fe1.5Mo0.5O6−δ, is a double perovskite, the properties of which are described in detail in chapter 3.2.1.2. It is a mixed ionic–electronic conductor, which also has high catalytic activity.388 The electrical conductivity of SFM (Sr2Fe1.5Mo0.5O6−δ) measured at 800 °C was 32 S cm−1 in H2 and 14.90 S cm−1 at 750 °C in air,180 which is higher than the conductivity of other ceramics such as LSCM or LSTO. Under a 50% CO2 + 50% CO atmosphere at 750 °C, SFM shows an electrical conductivity of 19 S cm−1.182 The conductivity measured at 800 °C decreased from 15.9 S cm−1 to 8.3 S cm−1 by doping with antimony in 5% H2 + 95% Ar.389 Adding Mn to SFM, on the other hand, leads to a decrease in conductivity in air at 800 °C and an increase from 16 S cm−1 to 25 S cm−1 in a 5% H2 + 95% Ar atmosphere for Sr2Fe1.4Mn0.1Mo0.5O6−δ.390 The TEC value of SFMO was found to be around 14.5 × 10−6 K−1 in the temperature range of 200–760 °C in air.174 Regarding the use of SFM fuel electrodes in steam electrolysis, several experiments were conducted using symmetrical SFM/LSGM/SFM cells.179 At 1.3 V and with 40 vol% absolute humidity (AH), current densities of −0.48 A cm−2 and −0.59 A cm−2 were achieved at 800 °C and 900 °C, respectively. Changing the AH from 20 vol% to 60 vol% leads to an improvement in current density from −0.38 A cm−2 to −0.88 A cm−2 at 900 °C. The polarization resistance under OCV conditions with 60 vol% AH RP decreases from 0.83 Ω cm2 to 0.26 Ω cm2 going from 800 °C to 900 °C. The authors reached a hydrogen production rate of 380 ml cm−2 h−1 at 900 °C using a set voltage of 1.3 V and 60 vol% AH.179 The performance of a symmetrical electrolyte (YbScSZ)-supported cell with an SFM electrode tested in steam (90% H2O + 10% Ar) and co-electrolysis (75% H2O + 25% CO2) at 900 °C with a set voltage of 1.3 V resulted in current densities of −1.4 A cm−2 and −1.1 A cm−2, respectively.182
Because the electrical conductivity of pure, single-phase SFM at 800 °C in hydrogen is only 10 S cm−1, the necessity of an enhanced SFM fuel electrode for steam electrolysis was proposed.391 Likewise, the performance in terms of the electrode polarization resistance and current densities achieved at 800 °C and 1.3 V with values of 0.65 Ω cm2 and −0.48 A cm−2 indicate the need for electrode material optimization. The authors suggest that one way of increasing the current density and reducing the polarization resistance of the electrode is to introduce nanocatalysts through infiltration processes or in situ exsolution into the material. Doping the B-site of the perovskite oxide with transition metal elements is one example. After in situ reduction, those elements form exsolved metal catalysts on the surface of the base perovskite grains and also lead to a change in the oxygen vacancy concentration. Using the exsolution process to enhance the performance of SFM-based fuel electrodes is more frequently investigated under fuel cell mode. In the case of SFM-based fuel electrodes in electrolysis operation, only a few publications are available. Here, doping SFM with Ni and/or additional Fe sources such as cobalt was studied concerning the effect of exsolution.181,392–394 In one study, the SFM double perovskite was doped with nickel to obtain the stoichiometry Sr2Fe1.3Ni0.2Mo0.5O6−δ.391 The authors subsequently examined the performance and degradation behavior of cells with two different fuel electrodes: one consisting of pure SFM-SDC and one of the SFMNi-SDC composite in an atmosphere of 42% H2O + 58% H2. In both cases, the ratio between the perovskite oxide and SDC was 60:40 wt%. At 850 °C and a set voltage of 1.3 V, a current density of −0.64 A cm−2 was achieved for the pure SFM-based electrode. For Sr2Fe1.3Ni0.2Mo0.5O6, the current density increased to −1.26 A cm−2 and the resistance at the OCV was reduced from 0.44 Ω cm2 to 0.21 Ω cm2.391 Another study investigated the performance of a Sr1.9Fe1.5Ni0.1Mo0.4O6−δ (SFMN) based fuel electrode for CO2-electrolysis in electrolyte-supported cells composed of SFMN-SDC (Sm0.2Ce0.8O1.9)/SDC/YSZ/LSM-YSZ.392 After annealing at 800 °C in hydrogen, the exsolution of Ni–Fe nanoparticles in the SFMN-SDC composite electrode was observed, which enhanced the chemical adsorption and surface reaction kinetics of the fuel electrode. Moreover, a stability test of the cell for 500 h in CO2-electrolysis mode at 800 °C and 1.3 V showed a relatively stable current density of −1.1 A cm−2. Only a slight current loss at the beginning of the measurement was detected, which the authors ascribe to an initial oxidation of the Ni–Fe particles.392 The use of SFM perovskites with exsolved nanoparticles can therefore improve the catalytic activity of the fuel electrode material. However, the lack of experimental data regarding the long-term stability in electrolysis operation and reversibility of the exsolution process needs to be addressed to evaluate this modification process in more detail. SFM was also doped with Ba to obtain a double perovskite electrode material with improved electrochemical properties.395 As a result of doping the A-site cations with the larger Ba cations, the authors expected an enhanced oxide ion migration due to the expansion of the unit cell. In this publication, electrochemical measurements were performed on half-cells with 0.2 mol% barium-doped SFM as an electrode (Ba0.2Sr1.8Fe1.5Mo0.5O6−δ). The amount of 2 wt% Ba agrees with the results of previous experiments, indicating that this Ba content is advantageous in terms of electrical conductivity and power density.396,397 To prevent a reaction between the YSZ electrolyte and the electrode, a protective SDC layer was applied between the electrolyte and electrode. From their experiments, the authors conclude that the optimal calcination temperature for Ba-doped SFM is 1100 °C, thus avoiding the formation of secondary phases like SrMoO4 during calcination. Furthermore, the authors reported that SOEC operation with a gas composition containing 20% H2O + 80% H2 led to a maximum current density of −0.18 A cm−2 at a set voltage of 0.2 V and −0.36 A cm−2 for 0.4 V.395
SFM-SDC fuel electrodes in a symmetrical LSGM electrolyte-supported cell in co-electrolysis operation were investigated at 850 °C using a set voltage of 1.3 V.180 A current density of −0.734 A cm−2 and a polarization resistance of 0.48 Ω cm2 at OCV and 850 °C were achieved in a 16% H2O + 16% CO2 + 20% H2 + 48% N2 atmosphere. The authors produced syngas with a corresponding CO2 conversion rate of 0.58 and an ideal H2/CO ratio of approx. 2.180 The electrochemical performances of a pure SFM, an Mn-doped SFM, and an SFM-SDC composite fuel electrode were compared in an atmosphere of 50% CO + 50% CO2 at 800 °C.390 The authors obtained polarization resistances of 1.12 Ω cm2, 0.60 Ω cm2, and 0.50 Ω cm2 for SFM, SFMMn, and SFMMn-SDC. Further investigations were conducted using an SFMMn-SDC fuel electrode combined with an LSCF-SDC oxygen electrode and an LSGM electrolyte. Current densities of −1.80 A cm−2 and −1.35 A cm−2 were achieved at 850 °C and 800 °C, respectively, using a set voltage of 1.5 V in pure CO2.390
The double perovskite SFN (Sr2FeNbO6−δ) and the simple perovskite SrFe0.9Nb0.1O3−δ have been investigated several times as an electrode material for SOFCs.398,399 The electrical conductivity of an SFN double perovskite at 850 °C in air is low with 0.05 S cm−1.400,401 The conductivity increases to 2.39 S cm−1 (@900 °C)402 and 2.215 S cm−1 (@850 °C)400 when the atmosphere is changed to the reducing conditions of 5% H2 + 95% Ar and 80% H2O + 20% H2. The effect of Mn doping into the SFN double perovskite on the conductivity in air and in 80% H2O + 20% H2 atmosphere was investigated.401 Substituting 50% Fe with Mn leads to an electrical conductivity value of 0.5 S cm−1 in oxidizing and reducing atmospheres. With an increasing amount of Mn, a maximum conductivity of 1.37 S cm−1 in air was reached for Sr2MnNbO6−δ. The authors concluded that through doping Mn in the B-site of the SFN double perovskite the conductivity increases in air but decreases under reducing atmospheres.401 The observed increase in conductivity in air through doping the B-site of SFN was confirmed by other authors as well.398 They achieved a maximum conductivity of 5.7 S cm−1 at 800 °C for a Co-doped Sr2Fe0.1Co0.9NbO6. The electrical conductivity of the simple SFN perovskite SrFe0.9Nb0.1O3−δ was found to be around 30 S cm−1 (ref. 399) in a reducing atmosphere. Cu doping of SFN in a 5% H2 + 95% Ar atmosphere achieved the highest conductivity of 30–60 S cm−1 between 300 °C and 700 °C for SrFe0.8Cu0.1Nb0.1O3−δ.399 The thermal expansion of pure SFN and a Mo-doped SFN double perovskite in air and in 5% H2 + 95% Ar was investigated in the temperature range of 30–1000 °C.403 The authors obtained average TEC values for SFN and SFNMo of 11.3 × 10−6 K−1 and 12.5 × 10−6 K−1 in air and 12.6 × 10−6 K−1 and 13.5 × 10−6 K−1 in 5% H2 + 95% Ar. The doping of Mo into the SFN double perovskite lattice leads to an increase in thermal expansion in air as well as in a reducing atmosphere.403 The polarization resistances of the electrode at 800 °C in air for pure SFN and Mo-doped SFN using an LSGM electrolyte in air were 1.72 Ω cm2 and 0.469 Ω cm2 for SFN and SFNMo, respectively. The RP values measured in reducing atmospheres (5% H2 + 95% Ar) were 2.97 Ω cm2 and 0.353 Ω cm2 for SFN and SFNMo. Independent of the atmosphere, lower RP values were measured for SFN doped with Mo.403
Only a limited number of studies deal with the use of SFN double perovskite-based fuel electrodes in SOECs.400 The electrochemical performance of an SFN-YSZ (80:20 wt%) composite fuel electrode was compared to a state-of-the-art Ni-YSZ fuel electrode in steam electrolysis operation in a cell composed further of a YSZ electrolyte and an LSM-YSZ oxygen electrode. Applying a set voltage of 1 V at 850 °C in an 80% H2O + 20% H2 atmosphere, a total resistance of 2.84 Ω cm2 was obtained for the SFN-YSZ fuel electrode, which is significantly lower compared to the resistance of the Ni-YSZ fuel electrode (21.42 Ω cm2). The authors concluded that the SFN-YSZ fuel electrode showed a better electrochemical performance compared to the Ni-YSZ electrode in SOEC operation under the same conditions. In addition, they observed the high catalytic activity of SFN for H2O dissociation. Through chemical absorption on the SFN surface, the reaction process was accelerated and the energy barrier for charge transfer was lowered.400
PBFM ((PrBa)0.95(Fe0.9Mo0.1)2O5+δ) is an A-site layered double perovskite with the general stoichiometry of LnBaM2O5+δ (Ln = lanthanides, M = transition metals) and has great potential as a fuel electrode material in SOECs for steam electrolysis.404 The crystal structure can be described as the stacking of transition metal oxide layers in between Ba oxide and lanthanide oxide layers: (BaO)-(MOx)-(LnOδ)-(MOx)-(BaO).405–407 Through this lattice structure, two-dimensional oxygen diffusion via oxygen vacancies formed inside the three inside layers can be observed.405 In most literature studies, praseodymium is used as the lanthanide dopant. Here, the Pr3+ and Ba2+ ions are ordered in alternating layers in the (001) direction.407 This stacking of layers results from the difference in the ion sizes of barium (135 pm) and praseodymium (109 pm). Most of the oxygen vacancies inside the layered structure are located at the lanthanide oxide layer, leading to a high tendency to form ordered structures under changing atmospheres (pO2 decrease).407 Due to this structure, higher conductivities and better electrochemical performance can be obtained compared to simple perovskites.405,407 PBFM reaches high electrical conductivities of 217 S cm−1 and 59.2 S cm−1 in air and 5% H2 + 95% Ar at 800 °C.407 A TEC of 11.96 × 10−6 K−1 was measured for PBFM.407 The performance of a composite PBFM-SDC (70:30 wt%) fuel electrode was also tested in steam electrolysis.404 The authors investigated the dependence of the resulting current density and the electrode polarization resistance on the operating temperature at a set voltage of 1.3 V in a 3% H2O + 97% H2 atmosphere. They observed an increase in current density from −0.28 A cm−2 to −0.82 A cm−2 with a simultaneous decrease in RP from 3.34 Ω cm2 to 0.73 Ω cm2 as the temperature increased from 750 °C to 850 °C.404
The electrode porosity must ensure good gas diffusion in addition to sufficient percolation for conductivity and mechanical stability. Compared to the gas diffusion through the fuel electrode in SOFC operation, the transport of a larger amount of steam through the electrode material during steam electrolysis is more complex.410 The values considered to offer optimal porosity for fuel electrodes, therefore, vary depending on the operation: 35 vol% for SOFCs and 45 vol% for SOECs.345,346,411–413
To increase the porosity, pore formers such as polymethyl methacrylate (PMMA), corn/potato starch, graphite, and carbon black can be introduced into the electrode material. The resulting porosity and performance (electrical conductivity, ohmic resistance) are highly dependent on the pore former used, its particle size, and its shape. Large pores tend to lead to a reduction of the TPB length and a decrease in performance, while very small pores hinder fuel gas diffusion. PMMA used as a pore-forming agent was found to be the most promising in terms of SOEC application.345 This is due to the uniform pore size distribution, the accelerated charge transfer in the active area due to the generally smaller pores, and the simultaneously sufficient porosity compared to other pore-forming agents.345,346 Grain size and microstructure changes impact the degradation behavior and electrode performance in the case of Ni-YSZ fuel electrodes.412,413 The coarser initial microstructure led to Ni loss during SOEC operation at the electrocatalytically active layer. This, in turn, led to a significant decrease in electrode performance. In contrast, electrodes with a finer microstructure exhibit electrode degradation dominated by Ni agglomeration or coarsening. Long-term tests with cells supported by a Ni-YSZ fuel electrode indicate that the Ni loss during SOEC operation is relatively low after 10700 h for the cells with finely structured electrodes.412,413Fig. 21 displays a cross-section of the Ni/YSZ fuel electrode/electrolyte interface using a finer structured electrode after 1000 h operation in electrolysis mode with an applied current density of −1.25 A cm−2 in 90% H2O + 10% H2. The SEM images show the degradation of the electrode/electrolyte interface only in the form of Ni–Ni and Ni-YSZ contact losses and the segregation of impurities inside the nickel particles.
 | ||
| Fig. 21 SEM images of the electrode/electrolyte interface of a Ni-YSZ fuel electrode-supported cell with a fine microstructure tested under electrolysis for 1000 h at −1.25 A cm−2 (ref. 413) (a) SEM image of the tested cell, (b) in-lens low-voltage image, (c) and (d) microstructure changes denoted by blue squares for Ni–Ni contact loss, red arrows for Ni-YSZ loss, and red circles for impurities. Reprinted from ref. 413, copyright 2016, with permission from Elsevier. | ||
In contrast, for the electrode with a coarse microstructure, a clear Ni loss at the electrolyte/electrode interface can already be seen after an operating time of 1000 h.412 These results indicate that the number of TPBs and a higher specific surface area between the two phases for a finer microstructure have a decisive influence. An electrode with a finer microstructure thus mitigates Ni migration by limiting the overpotential at a given current density.412,413 Matching particle size distributions of the individual electrode components improves the mixing and dispersion of nickel, YSZ, and porosity. Well-mixed and homogeneously distributed phases in the fuel gas electrode can restrict Ni migration through the YSZ backbone.413 Another possibility for optimizing certain microstructural properties of the Ni-YSZ fuel electrode in terms of long-term stability is to design the electrode with the highest possible density. The densification of the Ni-YSZ electrode enhances the tortuosity of the Ni particles percolation, increases the number of percolating paths, and benefits the Ni-YSZ interface.413 It was suggested that a suitable choice of the above-mentioned parameters can prevent irreversible degradation during long-term application under a high current and high pH2O. By optimizing the microstructure and the operating parameters, the authors achieved a significant improvement in the degradation rate. Table 4 shows the results of cell tests for SOECs with and without a modified fuel electrode microstructure as well as results for SOFCs together with their respective operating parameters. Due to the obtained low degradation rate of 0.3–0.4% kh−1, the authors expect a prolonged lifetime of an SOEC with such a modified Ni-YSZ fuel electrode microstructure of more than five years under cell operation near the thermoneutral point.413 The design of the electrode microstructure is expected to depend highly on the desired application. For fuel electrodes applied in SOECs at high current densities, a finer particle size and a denser structure are most suitable. If, however, only low current densities are required or the operation takes place at very high temperatures, an electrode with a coarser microstructure is sufficient and more cost-effective.413
| Operation mode | Fuel electrode | Degradation rate | Current density | Temperature | Gas composition | Ref. |
|---|---|---|---|---|---|---|
| SOEC | Microstructure modified Ni-YSZ | 0.3–0.4% kh−1 | −1 A cm−2 | 800 °C | pH2O/pH2 = 0.9/0.1 | 413 |
| SOEC | Ni-YSZ | 2% kh−1 | −0.5 A cm−2 | 850 °C | pH2O/pH2 = 0.5/0.5 | 414 |
| SOFC | Ni-YSZ | <1% kh−1 | 1 A cm−2 | 850 °C | — | 415 |
| SOEC | Ni-YSZ | 6% kh−1 | −1 A cm−2 | 950 °C | pH2O/pH2 = 0.9/0.1 | 414 |
| SOFC | Ni-YSZ | 2% kh−1 | 1.7 A cm−2 | 950 °C | — | 415 |
 | ||
| Fig. 22 SEM images of fuel electrode/electrolyte interfaces of fuel electrode-supported cells: pristine (as-reduced) and after operation at 800 °C for 1000 h in 80% H2O + 20% H2 at OCV and three different current densities (−0.5 A cm−2, −1.0 A cm−2, and −1.5 A cm−2).52 Reprinted from ref. 52, copyright 2018, with permission from Elsevier. | ||
Ni migration toward the fuel electrode substrate is only observed at gas humidities of ≥80% and temperatures of ≥800 °C.52 If the operating temperature of the SOEC is 950 °C, the direction of Ni migration changes and the particles migrate toward the electrolyte. The migration of the particles takes place along the H2O gradient (pO2).411 This also leads to a loss of electrode performance, since the porosity near the electrolyte/electrode interface is clogged and the TPB is therefore reduced.52 The displacement of Ni particles toward the electrolyte at a temperature of 950 °C during electrolysis operation corresponds with observations of Ni migration of an SOFC stack operated at 700 °C for a long period (>10 years).411
The theories presented below are intended to explain and reconcile the above observations. However, the underlying mechanisms of Ni migration are not yet fully resolved.52,411,412 The various literature sources at least agree that the locally prevailing polarization, or the local overvoltage/overpotential, plays a major role in this process.52,412,417,424 Sun et al.424 found that the degradation rate of an SOEC generally increases with increasing polarization of the fuel electrode. There are different hypotheses as to how the particles detach from the composite material and thus migrate. Trini et al.417 suggest that the contact angle, which changes under different polarization, is the driving force for the release of the Ni particles from the composite. According to their hypothesis, the strongly polarized zone close to the electrolyte leads to a high contact angle between Ni/YSZ and a de-wetting of the material occurs. The nickel particles then move from the area at the electrolyte/electrode interface with a low pO2 towards higher pO2 values at the outer and less strongly polarized areas of the electrode. The less strong polarization in these areas results in a lower contact angle between Ni and YSZ, and there is better wetting of the material between them.417 Sun et al. also describe the tendency of Ni to detach from YSZ during strong (cathodic) polarization due to de-wetting processes and to migrate toward the less polarized regions of the electrode substrate.424 High cathodic polarization results in an accumulation of vacancies, which in turn leads to a decomposition of the Ni/YSZ interface and thus to a dissolution of Ni particles from the composite. The migration of nickel is explained here by the formation of gaseous Ni(OH)x species due to the influence of the partial vapor pressure.412 Hoerlein et al. suggest that volatile nickel hydroxides under cathodic polarization follow the gradient of Ni(OH)x activity from non-percolating to percolating nickel.52 Mogensen et al. suggest that Ni migration is driven by electric polarization by the surface diffusion of Ni(OH)x species below 800 °C and by gaseous nickel hydroxides at temperatures ≥900 °C. The main parameters affecting migration are the local overpotential of the Ni particles, the local pH2O, and the activity of the Ni(OH)x species. In the case of an SOEC with an operating temperature of 800–900 °C, a strong cathodic overpotential leads to a loss of contact between polarized Ni particles and YSZ. In the case of simultaneous contact loss among Ni particles, the isolated Ni particles adopt the potential of the local gas composition.411 The local pH2O around the particles becomes higher, as more H2O molecules migrate towards the electrolyte without coming into contact with the polarized Ni particles at the TPB. The pH2O at the active TPB is minimized. The activity of the Ni(OH)x species is proportionate to the potential and is lowest where the potential is also lowest. The dissolved Ni particles thus migrate locally towards the region with a lower pH2O (the new active TPB) and deposit on the polarized nickel at the displaced TPB. The nickel hydroxides thus seem to migrate against the electrochemical potential or the global pH2O/pO2 gradient of the fuel gas electrode.411
Degradation processes also occur due to the segregation of impurities at the TPB. Ni-YSZ composites almost always have thin monolayers of impurities or segregated components at YSZ surfaces, Ni-YSZ interfaces, or grain boundaries.411 Impurities can be introduced into the system by raw materials, the gas flow, or evaporating species from the interconnects and sealings. As an example of an observed process, the precipitation of SiO2 at the TPB was reported. A possible source of Si-containing fuel gas impurities are the vaporizers made of glass containers for water vapor generation. Carbon deposition has also been observed when carbon-containing reactant gases were used.52,411
Several stability studies on Ni cermet fuel electrodes used in SOEC operation have been carried out.79,426–428 The degradation of a Ni-YSZ/YSZ/GDC/LSCF cell in steam electrolysis was carried out at a temperature of around 780 °C.426 The authors used a reducing atmosphere with an AH of 80 vol% and operated the cell for 9000 h at a current density of −1.0 A cm−2. During operation, they observed a voltage degradation rate of 3.8% kh−1. After cell operation, the microstructure was analyzed by SEM images. The authors detected a weakened contact between the electrolyte and the hydrogen electrode and varying surface roughness of the Ni particles. No impurity segregation was observed at the interface. An SOEC stack consisting of Ni-YSZ/YSZ/GDC/LSCF cells was tested under 50% humidified H2 at a current density of −0.5 A cm−2 at 800 °C for 2300 h, followed by 1800 h at 700 °C.79 A voltage and an ASR degradation rate of 0.7% kh−1 and 10.1% kh−1 were observed during the first 2300 h of operation at 800 °C. At 700 °C, a voltage and ASR degradation of 1.9% kh−1 and 9.0% kh−1 were detected. The authors also conducted a long-term electrolysis test of an SOEC stack using the cell configuration mentioned above and the same conditions for around 20000 h. At 800 °C, they achieved an average voltage degradation rate of 0.6% kh−1 after 18000 h of electrolysis operation.419 The same cell configuration was galvanostatically tested for 9300 h at a set current density of −1.0 A cm−2, which resulted in an overall voltage degradation rate of 3.8% kh−1.135 The long-term stability test of a Ni-YSZ/YSZ/GDC/LSCF-GDC cell configuration at 750 °C with a current density of −0.3 A cm−2 in a 20% H2 + 40% H2O + 40% CO2 atmosphere was conducted for 1000 h.357 A voltage increase led to a degradation rate of 10.69% kh−1. The stability of a Ni-YSZ/YSZ/LSM-YSZ cell configuration at current densities of −2.0 A cm−2 and −1.5 A cm−2 in 45% H2O + 45% CO2 + 10% H2 for 700 h exhibited voltage degradation rates of 0.452 V kh−1 and 0.275 V kh−1 respectively for the period between 400 h and 700 h.359 Post-test analysis showed severe changes in the microstructure of the fuel electrodes, such as the loss of Ni percolation, contact loss between YSZ and Ni, and the decomposition of YSZ. Additionally, the formation of nano-zirconia inside the Ni-YSZ electrode was detected.359
The chemical stability of a Ni-YSZ fuel electrode in CO2 electrolysis tested in pure CO2 at 700 °C and a set voltage of 1.5 V for 60 h achieved a relatively stable current density of −0.330 A cm−2 throughout the testing period.429 XRD measurements of the fuel electrode subsequently showed only slight changes in the patterns and no NiO peak. In addition to the XRD, the authors analyzed the post-test microstructure using SEM images. They observed the agglomeration of Ni particles inside the fuel electrode but no carbon deposition on the surface or cross-section of the fuel electrode.429 The stability of a Ni-GDC fuel electrode operated for 80000 current on/off cycles was investigated under steam electrolysis for one year at a temperature of 856–860 °C.430 The authors used the following ESC with a configuration of Ni-GDC/GDC/YSZ/GDC/LSCF. The set current densities were −0.7 A cm−2 (ON) and −0.07 A cm−2 (OFF) in an atmosphere of 75% humidified H2. The resulting voltage degradation rate over the whole period of operation was 0.4% kh−1, with an ASR degradation of 7.3 mΩ cm2 kh−1.
Ni-GDC fuel electrodes were tested with the same cell composition in co-electrolysis and steam electrolysis operation at 900 °C with a current of −0.5 A cm−2 for 500 h.362 In co-electrolysis conditions, the resulting degradation rate of 308 mV kh−1 was lower than the degradation observed for steam electrolysis (499 mV kh−1). The polarization resistance over time also increased from 0.07 Ω cm2 to 0.28 Ω cm2 in steam electrolysis, and from 0.10 Ω cm2 to 0.16 Ω m2 in co-electrolysis operation. In the microstructural post-test analysis, the authors compared the average Ni particle size of the as-reduced cell with the operated cell and observed an increase in the Ni particle size. The as-reduced cell featured a Ni particle size of 1.37 μm, whereas the Ni particle sizes increased to 2.19 μm (62%) and 2.86 μm (109%) during steam and co-electrolysis. Besides the Ni agglomeration/particle growth, Ni depletion, and a loss of GDC percolation with GDC-covered Ni particles were found.362 A long-term stability test of a 3Mo-3Au-Ni-GDC/YSZ/GDC/LSCF cell configuration was conducted for 1700 h at a temperature of 900 °C and in a 7% H2 + 30% N2 + 63% H2O atmosphere with a set current density of −0.3 A cm−2.363 During the 1700 h of operation, an increase in cell voltage from 0.99 V to 1.05 V was observed with an overall degradation rate of 33 mV kh−1. Post-test analysis showed no delamination or cracks occurring, but Ni particle coarsening and Ni depletion at the fuel electrode/electrolyte interface were detected. In this study, a comparison between an as-reduced cell and an operated cell led to the conclusion that the operation in electrolysis mode increases the tendency to coalesce and build clusters. Furthermore, a loss of GDC percolation and the coverage of Ni particles by GDC was discovered as well as an increase in pore size and pore fraction.345
3.3.5.1. Perovskite-based fuel electrode materials. Perovskites have a high potential for application as fuel electrode materials in high-temperature electrolysis cells. A prerequisite for successful application in SOECs is the long-term stability of the electrode under the applied operating conditions.
The chemical stability of LSCM-based fuel electrodes for SOECs has been tested in different reducing atmospheres.364,366,367 After exposing pure LSCM powder to 40 sccm H2 (ref. 366) and 30 sccm H2 (ref. 367) with an absolute humidity (AH) of 80 vol% at 900 °C for 24 h, the subsequent XRD analysis did not show secondary phases or peak splitting. The authors conclude that LSCM is chemically stable in a reducing atmosphere combined with a high AH.366,367
The chemical stability of an LSCM-GDC composite (ratio 1:1) electrode in a humid CO2 atmosphere (50% H2O + 50% CO2) at 850 °C for 20 h has been confirmed.364 The LSCM-YSZ composite electrode material was tested in terms of its chemical stability during heat treatment at 1400 °C in air for 4 h and under a reducing atmosphere (30 sccm H2, 80 vol% AH, 900 °C, 24 h).367 No secondary phases or peak splitting were detected for both samples, which is in agreement with the results for pure LSCM. Furthermore, changes to the electrode microstructure of a fuel electrode-supported cell, which occurred during electrolysis operation for 103 h at 850 °C with a current density of −0.33 A cm−2 in 45 vol% AH and a 30 sccm H2 atmosphere were analyzed. Coarsening of the interface and a minor crack between the LSCM-YSZ electrode and the YSZ electrolyte were observed. The authors attributed this to the increasing loss of lattice oxygen in the LSCM crystal throughout the operation, leading to an alteration in the thermal expansion behavior of the LSCM electrode and, therefore, mechanical stresses at the electrode/electrolyte interface.367 Initial short-term durability tests for LSCM fuel electrodes in steam electrolysis operations have been already conducted. Cells tested at 850 °C for 35 h under galvanostatic operation using a current density of −0.2 A cm−2, a gas mixture of 60 vol% AH, and 40 sccm H2 as a carrier gas exhibited a relatively stable voltage of 1.22 V throughout the test.366
At a higher current density of −0.6 A cm−2 and a lower temperature of 800 °C in the gas mixture of 13% H2O + 6.5% H2 + 80.5% N2, the cell reached a voltage of 1.7–1.75 V during 25 h with a degradation rate of 0.022% h−1.431 In co-electrolysis mode, short-term stability tests with LSCM fuel electrodes showed a decrease of the current density from −0.1031 A cm−2 to −0.09761 A cm−2 during a test period of 24 h at 800 °C under a set voltage of 1.5 V in 60% H2O + 40% CO2.365 An LSCM-GDC composite electrode with a set current density of −0.3 A cm−2 at 800 °C using 50% H2O + 50% CO2 exhibited an initial voltage increase during the first 8 h, and a stable voltage of 1.5 V was achieved during the following 100 h of operation.364
The chemical compatibility of LSFM (La0.6Sr0.4Fe0.8Mn0.2O3−δ) and YSZ has been investigated for a heat-treated mixture of LSFM and YSZ (50:50 wt%) for one week in a temperature range of 600–950 °C.375 No chemical reaction was detected for temperatures of 600–850 °C, whereas secondary phase formation in the form of SrZrO3 was identified for temperatures of 900–950 °C. The authors concluded that LSFM and YSZ show good compatibility in an intermediate temperature range. The compatibility between LSFM and GDC was also investigated at 800 °C in air and CO2 with respective heat treatment, and no impurity phases were observed in both tested atmospheres.376
The stability of an LSFM (La0.6Sr0.4Fe0.9Mn0.1O3−δ) electrode was investigated in CO2 electrolysis with 50% CO2 + 1% CO + 49% Ar.378 Post-test analysis of the fuel electrode microstructure with SEM and XRD was performed. No significant changes in the electrode microstructure and only small peaks of secondary phases in the XRD pattern were found. The short-term stability of an LSFM-GDC fuel electrode during CO2 electrolysis was tested at 800 °C under a constant current of −0.4 A cm−2. The authors observed a voltage increase from 1.30 V to 1.37 V after 40 h of operation. However, the post-test analysis showed no impurities in the electrode material and no significant structural changes in the microstructure of the electrode.376 The short-term stability of a fuel electrode consisting of LSFM (La0.6Sr0.4Fe0.8Mn0.2O3−δ)-CuF (1:1) was analyzed using an LSGM electrolyte and a BLC (Ba0.6La0.4CoO3–δ) oxygen electrode. After 100 h at a constant current density of −0.765 A cm−2 in an atmosphere with 50% CO2 + 10% H2O + 40% Ar at 800 °C, the cell exhibited a voltage of approx. 1.3 V with a corresponding degradation rate of 0.038% h−1.371
The chemical stability in reducing atmospheres and structural changes of pure and doped LST (La0.2Sr0.8TiO3+d) electrode material has been characterized by several studies.379,384,385,387
Partly chemical reduction of the Ti4+ ions to Ti3+ due to heat treatment at 800 °C for three hours in 5% H2 + 95% Ar followed by an increase in the lattice parameters was observed.379 No phase changes through reduction were detected. Additionally, the authors examined the stability of iron-doped LST in reducing atmospheres. Here, a partial change from Ti4+ to Ti3+ is also observed as well as a reduction from iron oxide to metallic Fe. Redox cycling experiments (800 °C, 5% H2 + 95% Ar/pure air, for 10 h) regarding the conductivity of the respective material confirmed the chemical stability of LST and iron-doped LST in operation in alternating oxidizing and reducing atmospheres.379 The stability of LST (La0.2Sr0.8TiO3−δ) in wet H2 at 1400 °C for 10 h was investigated and XRD results confirmed the preservation of the crystal structure of LSTO during the reduction.384 The authors detected a minimal decrease in the cell parameters after the reduction process. This was attributed to the fact that only a small amount of Ti4+ was reduced to Ti3+ and, therefore, the loss of oxygen likely prevents an increase of the average radii of Ti.384 After heat treatment of LSTO and Mn-doped LSTO (La0.2Sr0.8Ti0.9Mn0.1O3+δ) at 1300 °C for 10 h in 5% H2 + 95% Ar to analyze chemical stability, the unit cell parameters increased due to the reduction of Ti4+ and, in the case of the Mn-doped LSTO, a partial Ti4+ reduction as well as a reduction of Mn4+ to Mn3+.387 Post-test characterization confirmed no phase transition had occurred.387 A Ca- and Fe-doped LSTO (La0.2Sr0.35Ca0.45Ti0.95Fe0.05O3−δ (LSCTF-45)) is chemically stable under a 100% H2 atmosphere for 50 h at 1000 °C.385 The XRD patterns in Fig. 23 show no apparent differences between the as-prepared LSCTF-45 and the reduced LSCTF-45 powder. Furthermore, the authors observed lattice parameter decrease of LSCTF with a higher Ca ratio.385
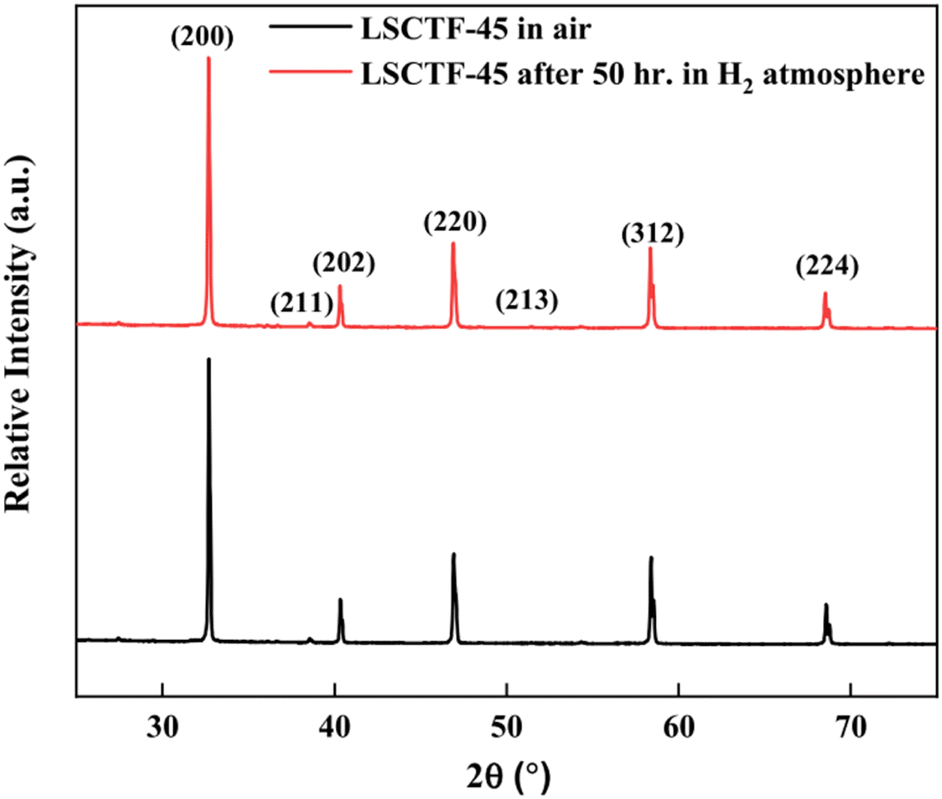 | ||
| Fig. 23 LSCTF-45 diffraction patterns: black: as-prepared LSCTF-45 powder, red: reduced LSCTF-45 powder (1000 °C, 50 h, 100% H2).385 Reprinted from, ©The Electrochemical Society. Reproduced by permission of IOP Publishing Ltd. All rights reserved. | ||
Several studies were conducted to investigate the stability of the LST-based fuel electrode during electrolysis operation. A composite electrode of LST (La0.2Sr0.8TiO3+δ)-Ce0.8Sm0.2O2−δ (SDC) (ratio 65:35 wt%) has been investigated in very short experiments of 21 min each in SOEC CO2 electrolysis with a YSZ electrolyte and an LSM-SDC oxygen electrode.387 The cells were characterized at three different voltages (1.2 V, 1.6 V, and 2.0 V) at 800 °C in pure CO2 atmosphere and exhibited a decrease in current density above 1.2 V. The short-term stability of a composite LST-SDC fuel electrode, using a ratio of 50:50 wt%, was investigated for 0.5 h at 700 °C using a constant voltage of 2 V with cells composed of LST-SDC/YSZ/LSM-SDC.384 The authors tested the stability of the fuel electrode material under CO2 electrolysis operation and achieved a constant current density of −0.125 A cm−2.384 In steam and CO2 electrolysis, a composite LST (La0.2Sr0.8TiO3+δ)-GDC (50:50 wt%) fuel electrode was tested at 700 °C with a voltage of 2 V in different atmospheres (3% H2O + 97% N2 and CO2 = 100%) on a YSZ electrolyte-supported cells with an LSM-GDC oxygen electrode. During steam electrolysis of around 20 h, they achieved a relatively stable current density of −0.090 A cm−2 as well as a current density of −0.080 A cm−2 for 0.7 h CO2 electrolysis.383
Pure SFM (Sr2Fe1.5Mo0.5O6−δ) and Sb-doped SFM (Sr2Fe1.5Mo0.5Sb0.1O6−δ) electrode material were examined at 800 °C in a humidified H2 atmosphere for 5 h.389 The results are shown in Fig. 24 and indicate that both SFM and Sb-SFM preserved the cubic crystal structure under these conditions. Despite a slight shift of some diffraction peaks (Fig. 24b) to lower angles, there are no significant XRD pattern changes. The peak shift is attributed to an increased cell volume correlating with a decrease in Fe and Mo valences. A subsequent microstructure analysis of the Sb-SFM using TEM-EDS (Fig. 24c) confirmed the stability of this electrode material in humidified reducing atmospheres. Similar results were obtained by studying the structural stability of Mn-doped SFM (Sr2Fe1.4Mn0.1Mo0.5O6−δ) at 850 °C for 12 h in a 5% H2 + 95% Ar atmosphere.390 To obtain information about the stability of an SFM fuel electrode in a symmetrical cell with LSGM electrolyte in steam electrolysis, short-term experiments at 850 °C with a set voltage of 1.2 V for 100 h in a 60 vol% AH atmosphere were conducted.179 Throughout the 100 h test, the current density was between −0.5 A cm and −0.7 A cm−2, and only a slight decrease in the current density was detected during the first 10 h. Furthermore, the short-term co-electrolysis stability of a symmetrical cell, consisting of SFM electrodes and a ytterbium scandium stabilized zirconia (YbScSZ) electrolyte, was investigated at 900 °C and a constant current density of −0.5 A cm−2.182 The fuel gas composition consisted of 75% H2O + 25% CO2. After 24 h of, the gas composition of the symmetrical cell was reversed at the two electrodes and the electrolysis was operated for a further 18 h. A slight increase of 1 mV h−1 in voltage over the first 24 h was observed, indicating degradation processes inside the material.182 The stability of an SFM-SDC composite electrode (60:40 wt%) was characterized in co-electrolysis mode and a symmetrical, LSGM electrolyte-supported cell.180 The authors conducted galvanostatic experiments at a current density of −0.12 A cm−2 and a temperature of 800 °C in a 16% H2O + 16% CO2 + 20% H2 + 48% N2 atmosphere. They observed a degradation rate of 0.00013 V h−1 over 103 h of testing. The microstructural analysis of the SFM-SDC electrode before and after electrolysis operation using SEM and EDX analysis subsequently showed no obvious grain growth, new phase formations, or carbon deposition.180 The stability of an Mn-doped SFM-SDC composite electrode in CO2 electrolysis was examined using cells consisting of SFMMn-SDC/LSGM/LSCF-SDC. At 750 °C and a constant voltage of 1.3 V, the authors achieved a stable current density of −0.6 A cm−2 for 50 h.390 Post-test analysis with SEM and Raman spectroscopy confirmed good adherence between the electrode and the electrolyte, the stability of the fuel electrode under the tested conditions, and the fact that there was no carbon deposition on the Mn-doped SFM fuel electrode during pure CO2 electrolysis.390
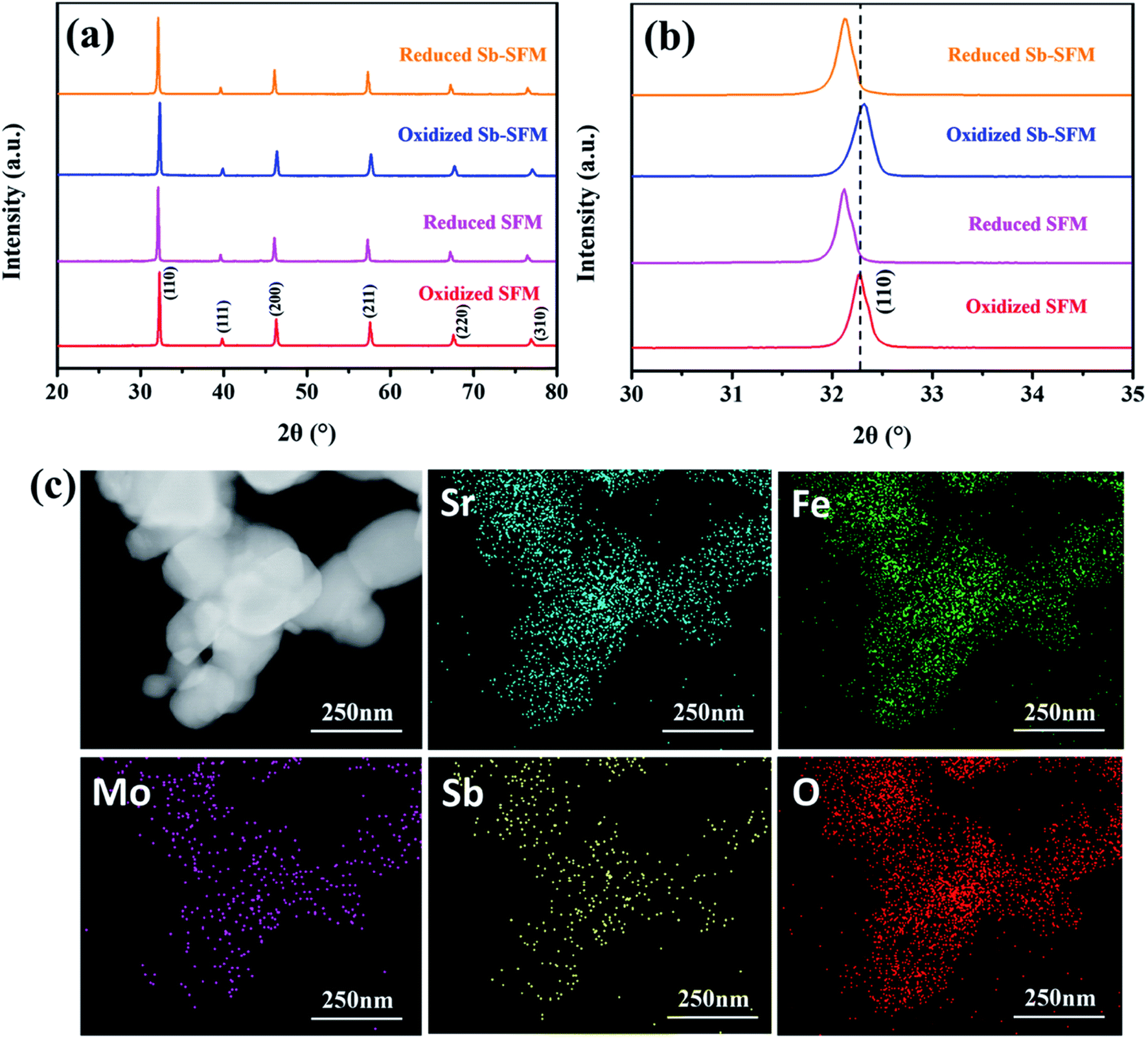 | ||
| Fig. 24 Chemical stability analysis for SFM-based material: (a) and (b): XRD analysis of pure SFM and Sb-SFM in an oxidized and reduced state, (c): TEM-EDS of Sb-SFM after reduction; reduction parameters: humidified H2, 5 h, 800 °C. Reprinted with permission from ref. 389 with permission from the Royal Society of Chemistry. | ||
The chemical stability of Mn-doped SFN double perovskites after heat treatment in a reducing atmosphere (80% H2O + 20% H2/air, 900 °C, 24 h) was analyzed by XRD measurements.401 The oxidized and reduced Mn-SFN powders showed no apparent differences in the XRD patterns for a low doping content of 0 ≤ x ≤ 0.2. Nevertheless, a shift from high-angle peaks to smaller angles was observed with increasing Mn content. According to the authors, this shift correlates to an expansion of the crystal lattice.401 The reactivity of Co-doped SFN (Sr2Fe0.1Co0.9NbO6) with YSZ and GDC using powder mixtures with a weight ratio of 1:1 was tested by sintering at 1200 °C for 24 h.398 The authors were not able to detect any new, additional peaks or peak shifting in the resulting XRD patterns.398 For the simple perovskite SrFe0.9Nb0.1O3−δ, its chemical stability in reducing atmospheres was confirmed after reducing the powder in 5H2 + 95% Ar at 700 °C for 10 h.399 After the reduction of SrFe0.9Nb0.1O3−δ doped with Cu (x = 0.1), the perovskite main phase in addition to a peak of metallic iron was detected in the XRD pattern. With increasing Cu content, Cu-rich and Fe-rich alloys were formed. Cycling tests to establish the reversibility of this exsolution process showed that the exsolved metal ions dissolved back into the crystal lattice during re-oxidation and exsolved again during reduction.399
The stability of (PrBa)0.95(Fe0.9Mo0.1)2O5+δ under reducing conditions after heat treatment for 200 h at 1000 °C was investigated via XRD.407 Besides a slight shift in the diffraction peaks, the authors were unable to detect secondary phase formation in the XRD pattern. The chemical compatibility between the PBFM and LSGM at 1000 °C for 100 h was also studied and no chemical reactions were observed.407 The short-term stability of a PBFM-SDC fuel electrode (70:30 wt%) was tested under steam electrolysis conditions with a cell composed of PBFM-SDC/LSGM/SDC-PBFM. The cells achieved a current density of −0.51 A cm−2 at 1.3 V and 800 °C in a 3% H2O + 97% H2 atmosphere for 6 h.404
3.3.5.2. Summary of fuel electrode materials. The state-of-the-art fuel electrode materials for SOECs are the cermets electrodes composed of Ni-YSZ and Ni-GDC. These materials exhibit sufficient performance and stability in electrolysis operation and are currently used in commercially available SOECs. Nevertheless, especially in the case of fuel electrode-supported SOECs, severe degradation of these fuel electrode materials during electrolysis operation occurs. The main degradation processes inside the Ni cermet fuel electrode are Ni particle agglomeration/coarsening and Ni migration. These mechanisms change the microstructure of electrode and lead to a reduction of active sites and a thicker electrolyte layer during electrolysis operation. Subsequently, this leads to a severe performance loss with increasing duration and operating time.
To overcome the challenges regarding cell longevity and costs, new fuel electrode materials and/or microstructures must be developed. Currently, many materials, including La0.75Sr0.25Cr0.5Mn0.5O3−δ (LSCM), La0.6Sr0.4Fe0.8Mn0.2O3−δ (LSFM), La0.2Sr0.8TiO3+δ (LST) or double perovskites as Sr2Fe1.5Mo0.5O6−δ (SFM) or Sr2FeNbO6−δ (SFN) are investigated as alternative fuel electrode materials. These alternative electrodes still have certain limitations regarding their catalytic activity and/or ionic and electronic conductivity or stability under operating conditions. These limitations lead to insufficient performance and durability of the cells. Therefore, studies are carried out to improve the properties and enhance the material's stability using composite materials, infiltration of active catalysts in perovskite-based electrode scaffolds, and/or exsolution processes on the electrode's surface. However, none of these alternative materials reached the technological maturity for commercialization, and no long-term (>5000 h) durability tests have been carried out yet.
3.4. Further stack components
Other stack components such as interconnects, sealants, contact layers, and Cr retention layers are also crucial to the development of high-performance and durable SOECs. However, in current SOEC research, the components used and their related materials have been adapted from extensively studied and optimized SOFC research. They have already been proven to fulfill all the requirements of SOEC stacks for high and stable performance during medium-term operation.31,35,73,80,432,433 The longest example of operation amounts to ∼20000 h.37 In this section, a brief overview of these additional components and the materials used will be presented.
3.4.1.1. Ceramic interconnects. The most common ceramic interconnect material is perovskite-based lanthanum chromite LaCrO3. It is a p-type semiconductor material, which was previously used due to its sufficient electrical conductivity (∼1 S cm−1 at 1000 °C),434 good material stability, and its average thermal expansion coefficient of 9.5 × 10−6 K−1 near YSZ (10.5 × 10−6 K−1). However, it is difficult to obtain dense LaCrO3 layers during fabrication and processing, which limits the attainable geometries. Additionally, lanthanum is considered an expensive rare-earth material for large-scale production. The most prominent use of doped LaCrO3 as an interconnect is the old tubular-type Westinghouse/Siemens SOFC.436 In recent years, the use of LaCrO3 as a ceramic interconnect material has declined with typical operating temperatures decreasing from ∼1000 °C to 600–900 °C, with the focus of research thus shifting to metallic interconnect materials.
3.4.1.2. Metallic interconnects. Due to their superior characteristics, metallic alloys have replaced LaCrO3 as the preferred interconnect material. Their mechanical strength supports all ceramic components in the stack and the easy, low-cost processing enables the construction of gas channels with innovative flow configurations (e.g., counter-flow, cross-flow, and/or co-flow). The interconnects are commonly comprised of high electronic and thermal conductive metal alloys containing Cr and/or Cr and Fe, which grow electrically conducting oxide layers (Cr2O3) during operation. However, excessive pure Cr-oxide layer growth is certain for higher chromium contents (compared to e.g., Cr-containing spinel layers) and results in electrode poisoning and an undesirable high ASR. A Cr content of 20–25% is necessary for a continuous oxide layer in operation.437 Fe–Cr alloys such as Crofer22APU, Crofer22H, SUS 430, and similar metals are more ductile than nearly pure Cr-based alloys like Cr5FeY2O3, but both types show great promise as interconnect materials in stacks due to their optimized thermal expansion coefficient (Fe–Cr alloys for FESCs and Cr alloys for ESCs) and easy processing methods.438 While the Cr alloys form pure Cr2O3 scales under both oxidizing and reducing conditions, Fe–Cr alloys – developed especially for SOFC use – form an oxide double layer (e.g., Crofer types). This double layer consists of an inner pure chromia scale and an outer Cr–Mn spinel layer. Mn thus has to be added as an alloying element for the build-up of this double layer. The formation of an outer spinel layer reduces the evaporation of volatile Cr species from the oxide scale, thus reducing the number of available molecules which can interact with the oxygen electrode materials and, in turn, leading to reduced Cr-induced degradation. However, even these double layers evaporate too much Cr for long-term SOC operation and additional Cr retention layers therefore have to be added (see Section 3.4.2).439–442
000 h.320,454
Contact layers reduce the interfacial contact resistance between the electrode and the interconnect, act as a gas distribution element (on the fuel side), ensure proper contacting by minimizing/leveling layer surface roughnesses and manufacturing-related differences (e.g., height, layer thickness), and might also act as Cr getter layers.455 On the airside, they are typically applied on the cells or the interconnect and sintered in situ during stack assembly and sealing at appropriate temperatures. On the fuel electrode side, the combination of either a Ni contact paste or current collection layer (e.g., for ESCs) or the Ni-containing support (FESCs) and a Ni mesh/felt/foam forms a kind of metallurgical bond with the interconnect, which lowers the electrical resistance and is therefore currently used exclusively by industry and research for state-of-the-art Ni cermet electrodes.456–459 The contact layer for the oxygen electrode has to be chemically compatible with the Sr-doped electrode material and Cr-forming interconnects, and must therefore be oxidation-resistant. Additional challenges include the high resistance of the metal/ceramic interface and the thermal gradients across the cell, which can lead to contact layer delamination. The materials are selected according to their thermal expansion behavior, their reactivity, and conductivity at operating temperatures. Commonly used contact layer materials listed in Table 5 include La0.7Sr0.3CoO3/La0.6Sr0.4Co0.8Fe0.2O3 LSC(F), La0.98Ni0.6Fe0.4O3 (LNF), La0.65Sr0.3MnO3 (LSM), and La0.7Sr0.3FeO3 (LSF).460 In many cases, the difference between the real air-side electrode and the contact layer, which might be made of the same material, is their microstructure and overall thickness. While the oxygen electrode is developed for high electrochemical performance and thus has a fine structure for the most part (high amount of triple phase boundaries and/or high available surface area) and has a thickness adapted to the resistances within the overall cell, the contact layer has only one electrical task, which is an appropriately less-resistant conduction of electrodes. The grains can thus be coarser, the microstructure can be more open-pored, and the thickness is typically higher than that of the electrode.
| Material | Component | TEC/10−6 K−1 | Ref. | |
|---|---|---|---|---|
| 30–800 °C | 30–1000 °C | |||
| BaO–Al2O3–Nd2O3–SiO2 | Glass sealing | 13.3 | 13.6 | 193 |
| Al2O3–MgO–CaO–BaO–SiO2–B2O3 | Glass sealing | 12.3 | 13.3 | 193 |
| Al2O3–MgO–SiO2–B2O3 | Glass sealing | 11.4 | 12.0 | 193 |
| X 10 CrAl 18 | Interconnect | 12.9 | 13.9 | 193 |
| La0.9Sr0.1CrO3 | Interconnect | — | 10.7 | 69 |
| Cr Fe5 Y2031 | Interconnect | 11.3 | 12.0 | 193 |
| La0.7Ca0.3Cr0.5Ti0.503 | Interconnect | 9.6 | 10.1 | 193 |
| Ni-8YSZ (40% Ni-60%YSZ) | Cermet electrode | 12.5 | 12.6 | 193 |
| Zr0.85Y0.15O1.93 (8YSZ) | Electrolyte | 10.5 | 10.8 | 193 |
| Zr0.82Y0.18O1.91 (10YSZ) | Electrolyte | 10.6 | 11.0 | 193 |
| Zr0.85Sc0.15O1.93 (8ScSZ) | Electrolyte | 10.3 | 10.4 | 462 |
| Zr0.80Sc0.19Al0.02O1.90 (10ScSZ) | Electrolyte | 10.5 | 10.9 | 193 |
| Ce0.8Gd0.2O1.90 (GDC) | Electrolyte | 12.5 | 12.7 | 193 |
| La0.9Sr0.1Ga0.8Mg0.2O3 (LSGM) | Electrolyte | 10.9 | 11.4 | 193 |
| La0.79Sr0.2MnO3 (LSM) | Perovskite | 10.8 | 11.1 | 463 |
| La10Si6O27 | Electrolyte | 9.0 (800 °C) | — | 464 |
| La9.5 (Ge5.5Al0.5O24)O2 | Electrolyte | 8.9 (500–900 °C) | — | 106 |
| LaMn0.45Co0.35Cu0.2O3 (LCC10) | Interconnect | ∼14 | — | 465 and 466 |
| La0.8Sr0.2CoO3 (LSC) | Electrode | — | 19 | 373 |
| La0.58Sr0.4Co0.2Fe0.8O3 (LSCF) | Contact layer, electrode | — | 13–16 | 146 and 373 |
| La0.8Sr0.2Co0.5Mn0.5O3 (LSMC) | Contact layer | — | 12–16 | 373 and 467 |
| Mn1.0Co1.9Fe0.1O4 (MCF) | Protective coating | ∼13 | — | 468 and 469 |
| Crofer22 APU | Interconnect | 11.9 | — | 455 |
| La0.98Ni0.6Fe0.4O3 (LNF) | Contact layer | 13.5 | — | 460 |
| La0.65Sr0.3MnO3 (LSM) | Contact layer | 12.8 | — | 460 |
| Structure | Test conditions | i@ 1.3 V/A cm−2 | R P@OCV/Ω cm2 | Ref. |
|---|---|---|---|---|
| SFM/LSGM/SFM | 40% H2O + 60% H2, 800 °C | −0.48 | 0.68 | 179 |
| 40% H2O + 60% H2, 900 °C | −0.59 | 0.23 | 179 | |
| Ni-YSZ/YSZ/YSZ-LSM | 50% H2O + 50% H2, 800 °C | −0.3 | 1.0 | 470 |
| 50% H2O + 50% H2, 900 °C | −0.7 | 0.44 | 470 | |
| Ni-YSZ/YSZ/YSZ-LSM | 60% H2O + 40% H2, 800 °C | −0.24 | — | 471 |
| 60% H2O + 40% H2, 900 °C | −0.38 | 0.71 | 471 | |
| SFM-SDC/LSGM/SFM-SDC180 | 16% H2O + 16% CO2 + 20% H2 + 48% N2, 850 °C | −0.734 | 0.48 | 180 |
| Ni-YSZ/YSZ/LSM-YSZ | 45% H2O + 10% H2 + 45% CO2, 850 °C | −1.000 | 0.42 | 472 |
| SFM/YbScSZ/SFM | 90% H2O + 10% Ar, 900 °C | −1.4 | 182 | |
| 75% H2O + 25% CO2, 900 °C | −1.1 | 182 | ||
| SFM-LSGM/LSGM/SFM-LSGM | 100% CO2, 800 °C | −0.92 | 0.226 | 473 |
| SFM/LSGM/SFM | 60% H2O + 40% H2, 900 °C | −0.88 | 0.26 | 179 |
| Ni-SDC/SDC/PrBaCo2O5+δ | 100% H2, 650 °C | 0.05 | 195 | |
| Ni-GDC/LCO/LSGM/PBC-10 wt% GDC | H2 + 3% H2O, 850 °C | 0.168 | 194 | |
| H2 + 3% H2O, 550 °C | 0.550 | 194 | ||
| Ni-GDC/LCO/LSGM/PBC | H2 + 3% H2O, 850 °C | 0.136 | 194 | |
| H2 + 3% H2O, 550 °C | 0.698 | 194 | ||
| La0.4Ce0.6O2 (LCO) | ||||
| La0.8Sr0.2Ga0.87Mg0.13O3 (LSGM) | ||||
| (ZrO2)0.9(Yb2O3)0.06(Sc2O3)0.04 (6Yb4ScSZ) | ||||
| Sm0.2Ce0.8O1.9 (SDC) | ||||
| PrBaCo2O5+δ-Gd0.1Ce0.9O2−δ (PBC-GDC) | ||||
| Nickelates | δ TGA | δ iodo | δ mean value | Thermal expansion coefficient/K−1 from | Ref. | |
|---|---|---|---|---|---|---|
| Dilatometry 10−6 | XRD 10−6 | |||||
| La2NiO4+δ | 0.16 | 0.16 | 0.16 | 13.0 | 12.8 | 201, 216 and 240 |
| 0.14, 0.11 | 201 | |||||
| La2Ni0.75Cu0.25O4+δ | 0.16, 0.13 | 0.13 | 0.14 | — | — | 201 |
| La2Ni0.50Cu0.50O4+δ | 0.11, 0.09 | 0.09 | 0.10 | 12.8 | 201 | |
| La2Ni0.25Cu0.75O4+δ | 0.05 | 0.05 | 0.05 | 201 | ||
| La2CuO4+δ | — | 0.01 | 0.01 | 0–250 °C: 8.6 | 12.8 | 201 |
| 250–900 °C: 13.8 | ||||||
| La2Ni0.9Co0.1O4+δ | 0.18 | — | 0.18 | 474 | ||
| La2Ni0.8Co0.2O4+δ | 0.20 | — | 0.20 | 474 | ||
| Pr2Ni0.9Co0.1O4+δ | 0.27 | — | 0.27 | 474 | ||
| La1.5Pr0.5Ni0.9Co0.1O4+δ | 0.23 | — | 0.23 | 474 | ||
| La1.5Pr0.5Ni0.8Co0.2O4+δ | 0.25 | — | 0.25 | 401 and 474 | ||
| La1.5Pr0.5NiO4+δ | 0.16 | 0.17 | 0.17 | 240 | ||
| LaPrNiO4+δ | 0.19 | 0.19 | 0.19 | 240 | ||
| La0.5Pr1.5NiO4+δ | 0.22 | 0.20 | 0.21 | 240 | ||
| 0.21 | 0.20 | 0.205 | 475 | |||
| Pr2NiO4+δ | 0.25 | 0.25 | 0.25 | 240 | ||
| 0.23, 0.19 | 0.21 | 0.21 | 216 | |||
| 0.25 | 0.23 | 0.24 | 217 | |||
| Pr4Ni3O10+δ | 0.10 | — | — | 12.0 | 10.0 | 314 |
| Pr2Ni0.9Co0.1O4+δ | 0.27 | 0.27 | 0.27 | 217 | ||
| Pr2Ni0.8Co0.2O4+δ | 0.30 | 0.28 | 0.29 | 217 | ||
| Nd2NiO4+δ | 0.23, 0.21 | 0.21 | 0.22 | 12.7 | 11.9 | 216 |
| Nd1.9Ca0.1NiO4+δ | — | 0.20 | 0.20 | 216 | ||
| Nd1.8Ca0.2NiO4+δ | — | 0.10 | 0.10 | 216 | ||
| Nd2Ni0.9Co0.1O4+δ | 0.26 | — | 0.26 | 474 | ||
| Nd2Ni0.8Co0.2O4+δ | 0.29 | — | 0.29 | 474 | ||
| Nickelates | σ e in air/S cm−1 | Conditions | R P OCV/Ω cm2 | i@ 1.5 V/A cm−2 | Ref. |
|---|---|---|---|---|---|
| Ni-YSZ/YSZ/GDC/Pr2Ni0.8Co0.2O4+δ | 60 (700 °C) | 50% H2 + 50% H2O, 800 °C | 0.118 | −1.90 | 474 and 476 |
| 50% H2 + 50% H2O, 900 °C | −3.00 | 234 and 476 | |||
| Ni-YSZ/YSZ/GDC/Pr2Ni0.9Co0.1O4+δ | 50% H2 + 50% H2O, 800 °C | −1.75 | 474 | ||
| 50% H2 + 50% H2O, 900 °C | −2.37 | 234 and 474 | |||
| Ni-YSZ/YSZ/GDC/Pr2NiO4+δ | 93 (850 °C) | 50% H2 + 50% H2O, 800 °C | 0.128 | −1.62 | 474 and 476 |
| 50% H2 + 50% H2O, 900 °C | −2.09 | 234 and 476 | |||
| Ni-YSZ/YSZ/GDC/La2Ni0.8Co0.2O4+δ | 51 (850 °C) | 50% H2 + 50% H2O, 800 °C | −1.60 | 474 and 476 | |
| Ni-YSZ/YSZ/GDC/La2NiO4+δ | 43 (850 °C) | 50% H2 + 50% H2O, 800 °C | −1.51 | 474 and 476 | |
| Ni-YSZ/YSZ/GDC/LSCF | 50% H2 + 50% H2O, 800 °C | −1.50 | 476 | ||
| 50% H2 + 50% H2O, 900 °C | −2.37 | 234 and 476 | |||
| Ni-YSZ/YSZ/GDC/Nd2NiO4+δ | 50% H2 + 50% H2O, 800 °C | −1.62 | 474 | ||
| Ni-YSZ/YSZ/GDC/Nd2Ni0.8Co0.2O4+δ | 50% H2 + 50% H2O, 800 °C | −1.80 | 474 | ||
| Ni-YSZ/YSZ/GDC/La1.5Pr0.5NiO4+δ | 50% H2 + 50% H2O, 800 °C | −1.60 | 474 | ||
| Ni-YSZ/YSZ/GDC/La2Ni0.8Co0.2O4+δ | 50% H2 + 50% H2O, 800 °C | −1.79 | 474 | ||
| La2NiO4+/GDC/8YSZ/GDC/La2NiO4+δ | 36 (800 °C) | 100% air | 0.142 | 240 | |
| La2Pr0.5NiO4+δ/GDC/8YSZ/GDC/La2Pr0.5NiO4+δ | 48 (800 °C) | 100% air | 0.044 | 240 | |
| La2Pr1.0NiO4+ δ/GDC/8YSZ/GDC/La2Pr1.0NiO4+δ | 73 (800 °C) | 100% air | 0.017 | 240 | |
| La2Pr1.5NiO4+δ/GDC/8YSZ/GDC/La2Pr1.5NiO4+δ | 81 (800 °C) | 100% air | 0.015 | 240 | |
| La2Pr2NiO4+δ/GDC/8YSZ/GDC/La2Pr2NiO4+δ | 88 (800 °C) | 100% air | 0.010 | 240 | |
| Pr2NiO4+δ/GDC/8YSZ/GDC/Pr2NiO4+δ | 100% air | 0.010 | 227 | ||
| Pr4Ni3O4+ δ/GDC/8YSZ/GDC/Pr4Ni3O4+δ | 86 (800 °C) | 100% air | 0.16 (600 °C) | 314 | |
| La0.6Sr0.4Fe0.8Co0.2O3−δ/8YSZ/La0.6Sr0.4Fe0.8Co0.2O3−δ | 100% air | 0.05 | 477 | ||
| La2NiO4+δ/8YSZ/La2NiO4+δ | 100% air | 0.37 | 477 | ||
| Pr2NiO4+δ/8YSZ/Pr2NiO4+δ | 100 (800 °C) | 100% air | 0.14 | 477 |
3.4.3.1. Compressive sealants. Gastight seals are essential for SOC stack operation. They are fixed at the edges of each cell, at the edges of all stack layers, and at the gas manifold, thus separating the two gas atmospheres and electrically insulating the individual stacking layers. Compressive sealants require an external compressive load and a frame for even load distribution during SOC operation. Applying load on an SOC stack composed of brittle ceramic components can lead to crack formation. The sealant frames are exposed to an oxidative atmosphere and are therefore liable to material oxidation. Commonly used compressive sealants are mica-based and composed of SiO4 sheets and metal gaskets (e.g., Au m.p. 1063 °C), which can deform easily at high temperatures under load.25 Additionally, compressive seals can be used between the stack itself and an adapter plate that connects the stack to either the SOC housing in a system or the furnace. Those sealants must be demountable in case of stack maintenance or replacement.
3.4.3.2. Glass (-ceramic) sealants. Sealants made of glass and glass-ceramic are commonly used in high-temperature SOC applications due to their good electrical insulation, chemical stability in reducing and oxidizing atmospheres, and appropriate thermal expansion behavior. Alkaline-earth metal oxides containing silicate or borosilicate glasses, for example, BaO–SiO2–CaO, have been considered. However, they can exhibit interdiffusion with the Cr-containing interconnector when containing more than 40 mol% alkaline-earth metal oxides. This leads to the formation of an electrical conductive ACrO4 (A = Ca, Sr, Ba) phase that is detrimental to the sealing properties and can be avoided through the additional thin coating on the interconnector.41,461 Typically, these sealants are applied as a paste on the components and de-bindered, melted, and crystallized during stack heat-up. Glass-ceramics are preferred to pure amorphous glasses, as they have a higher strength and are not prone to flowing under load (either for securing gas tightness by external load or when overpressure is applied internally).
4. Conclusion
In this paper, the current progress in SOEC technology and the development of component materials are reviewed. The results show that high-temperature SOEC operation with a suitable current density and cell resistance can now be achieved with state-of-the-art materials as well as with novel materials. Up to now, most of the cell or stack component materials, such as electrodes, electrolytes, interconnects, and sealing and coating materials, have been considered in SOFC research. Therefore, the cells containing state-of-the-art oxygen and fuel electrode materials exhibit larger degradation in electrolysis mode compared to fuel cell mode. Nevertheless, alongside huge R&D efforts, start-up companies are currently developing HT-SOEC stacks (both FESCs and ESCs) and systems containing the state-of-the-art YSZ or GDC electrolytes, LSC/LSCF oxygen electrodes with a GDC barrier layer, and Ni-YSZ or Ni-GDC fuel electrodes capable for operating at 700–900 °C with >0.5–1.0 A cm−2. Systems and stacks based on MSCs operated at medium-to-low temperatures (of around 650 °C) are currently under development due to significantly lower degradation rates.One of the major challenges during HT-SOC operation is the long-term stability of electrodes during SOEC operation. Therefore, novel electrode materials and manufacturing methods for performance optimization (e.g., exsolution) are investigated concerning performance and long-term stability.
On the oxygen electrode side, perovskite-type LSCF is currently used but exhibits Sr segregation due to the migration of the volatile SrO from the LSCF electrode to the GDC/electrolyte interface. There it forms an insulating SrZrO3 phase, which in turn leads to cell degradation and performance loss. Alternative oxygen electrodes with layered perovskites (Ruddlesden–Popper nickelates, e.g., Ln2NiO4+δ, Ln = La, Pr or Nd) and a double perovskite structure (Sr2Fe2−xMoxO6−δ and LnBaCo2O5+δ where Ln = La, Pr, Nd, Gd, Sm) have attracted a lot of scientific attention mainly due to their higher oxide ion diffusivity and surface exchange activity compared to conventional perovskite-based materials. For example, the performance of single cells containing Pr2NiO4+δ and co-doped Pr2NiO4+δ oxygen electrodes exhibit a higher current density compared to the LSCF electrode. In particular, the Pr2Ni0.8Co0.2O4+δ electrode shows a lower degradation rate of 22 mV kh−1 compared to the LSCF cell (88 mV kh−1). Moreover, the microstructure of the oxygen electrode strongly affects the performance of the cell, for example, the La2NiO4+δ electrode with a coral microstructure prepared by the electrostatic spray deposition technique shows a lower polarization resistance (0.42 Ω cm2) than the conventional screen-printed electrodes (0.93 Ω cm2) at 600 °C. In addition, further improvement of cell performance can be achieved by using nanofibers and nanostructured electrodes. However, reproducibility tests, scale-up to stack-relevant sizes, and stack tests are still lacking.
With respect to the fuel electrode, Ni migration and agglomeration are the major concerns for the state-of-the-art Ni-YSZ electrode as well as the Ni-GDC electrode, during SOEC operation. These mechanisms detrimentally impact the electrode's microstructure and subsequently, lead to severe performance loss with increasing duration and operating time.
To avoid such concerns, several alternative perovskite electrode materials based on mixed ionic and electronic conductivity are proposed, such as lanthanum strontium chromium manganite (LSCM), lanthanum strontium iron manganite (LSFM), lanthanum strontium titanate (LST), strontium iron molybdate (SFM), strontium iron niobate (SFN), and praseodymium-doped barium iron molybdate (PBFM). These materials show good thermal expansion behavior, chemical and redox stability, and conductivity at high operating temperatures and under reducing conditions. However, these new materials still exhibit limitations in catalytic activity, therefore material development and initial cell performance testing have to be conducted further. Regardless of promising initial tests of the cell performance for selected materials, their long-term stability is still unknown, which is the focus point for their large-scale and system-level implementation.
Despite the sufficient performance for state-of-the-art materials in industrial high-temperature SOEC stacks, the aspect of the materials' lifetime and stability remains a challenge, especially with regard to future widespread applications and increased market interest. Therefore, to achieve improved SOEC system lifetime, existing materials need to be modified and highly active materials need to be developed. This review of alternative electrode materials underlined the lack of long-term (>5000 h) durability tests, which will be necessary to assess their technology readiness level for future commercialization.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors gratefully acknowledge funding provided by the German Federal Ministry of Education and Research (BMBF) within the SOC-Degradation 2.0 project: transfer of knowledge into products for a “green hydrogen” vector—impedance analysis of intrinsic and extrinsic degradation mechanisms in SOC cells and repeat units (FKZ 03SF0621A). The authors would also like to thank S. B. C. Lehmann for the graphical support.References
- Global Hydrogen Review 2021, Paris, 2020 Search PubMed.
- Clean Hydrogen Partnership, Annual Work Programme for 2023, 2023 Search PubMed.
- M. Wappler, D. Unguder, X. Lu, H. Ohlmeyer, H. Teschke and W. Lueke, Int. J. Hydrogen Energy, 2022, 47, 33551–33570 CrossRef CAS.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- F. M. Sapountzi, J. M. Gracia, C. J. Weststrate, H. O. Fredriksson and J. W. Niemantsverdriet, Prog. Energy Combust. Sci., 2017, 58, 1–35 CrossRef.
- S. Slade, S. A. Campbell, T. R. Ralph and F. C. Walsh, J. Electrochem. Soc., 2002, 149, A1556 CrossRef CAS.
- I. Vincent and D. Bessarabov, Renewable Sustainable Energy Rev., 2018, 81, 1690–1704 CrossRef CAS.
- H. A. Miller, K. Bouzek, J. Hnat, S. Loos, C. I. Bernäcker, T. Weißgärber, L. Röntzsch and J. Meier-Haack, Sustainable Energy Fuels, 2020, 4, 2114–2133 RSC.
- M. Nasser, T. F. Megahed, S. Ookawara and H. Hassan, Environ. Sci. Pollut. Res. Int., 2022, 29, 86994–87018 CrossRef CAS PubMed.
- H. Lange, A. Klose, W. Lippmann and L. Urbas, Int. J. Hydrogen Energy, 2023, 48, 15771–15783 CrossRef CAS.
- M. E. Ivanova, R. Peters, M. Müller, S. Haas, M. F. Seidler, G. Mutschke, K. Eckert, P. Röse, S. Calnan, R. Bagacki, R. Schlatmann, C. Grosselindemann, L.-A. Schäfer, N. H. Menzler, A. Weber, R. van de Krol, F. Liang, F. F. Abdi, S. Brendelberger, N. Neumann, J. Grobbel, M. Roeb, C. Sattler, I. Duran, B. Dietrich, M. E. C. Hofberger, L. Stoppel, N. Uhlenbruck, T. Wetzel, D. Rauner, A. Hecimovic, U. Fantz, N. Kulyk, J. Harting and O. Guillon, Angew. Chem., Int. Ed., 2023, e202218850 CAS.
- J. Cao, Y. Ji and Z. Shao, Energy Environ. Sci., 2022, 15, 2200–2232 RSC.
- S. R. Foit, I. C. Vinke, L. G. J. de Haart and R.-A. Eichel, Angew. Chem., Int. Ed., 2017, 56, 5402–5411 CrossRef CAS PubMed.
- Y. Zheng, J. Wang, B. Yu, W. Zhang, J. Chen, J. Qiao and J. Zhang, Chem. Soc. Rev., 2017, 46, 1427–1463 RSC.
- S. E. Wolf, L. Dittrich, M. Nohl, T. Duyster, I. C. Vinke, R.-A. Eichel and L. de Haart, J. Electrochem. Soc., 2022, 169, 034531 CrossRef.
- S. Foit, L. Dittrich, T. Duyster, I. Vinke, R.-A. Eichel and L. G. J. de Haart, Process, 2020, 8, 1390 CrossRef CAS.
- L. Dittrich, M. Nohl, E. E. Jaekel, S. Foit, L. de Haart and R.-A. Eichel, J. Electrochem. Soc., 2019, 166, F971–F975 CrossRef CAS.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- R. Schlögl, ChemSusChem, 2010, 3, 209–222 CrossRef PubMed.
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed.
- S. H. Jensen, P. H. Larsen and M. Mogensen, Int. J. Hydrogen Energy, 2007, 32, 3253–3257 CrossRef CAS.
- C. Graves, S. D. Ebbesen, M. Mogensen and K. S. Lackner, Renewable Sustainable Energy Rev., 2011, 15, 1–23 CrossRef CAS.
- GrInHy – Green Industrial Hydrogen via Reversible High-Temperature Electrolysis, 2016–2019.
- GrInHy2.0 – Green Industrial Hydrogen via steam electrolysis. Erzeugung von erneuerbarem Wasserstoff in einem Stahlwerk, 2019–2022.
- R. Peters, M. Frank, W. Tiedemann, I. Hoven, R. Deja, N. Kruse, Q. Fang, L. Blum and R. Peters, J. Electrochem. Soc., 2021, 168, 14508 CrossRef CAS.
- M. Frank, R. Deja, R. Peters, L. Blum and D. Stolten, Appl. Energy, 2018, 217, 101–112 CrossRef CAS.
- R. Peters, R. Deja, M. Engelbracht, M. Frank, N. van Nguyen, L. Blum and D. Stolten, J. Power Sources, 2016, 328, 105–113 CrossRef CAS.
- R. Peters, W. Tiedemann, I. Hoven, R. Deja, N. Kruse, Q. Fang, L. Blum and R. Peters, ECS Trans., 2021, 103, 289–297 CrossRef CAS.
- N. Kruse, W. Tiedemann, I. Hoven, R. Deja, R. Peters, L. Blum and R. Peters, ECS Trans., 2021, 103, 555–560 CrossRef CAS.
- Q. Fang, L. Blum and D. Stolten, J. Electrochem. Soc., 2019, 166, F1320–F1325 CrossRef CAS.
- V. N. Nguyen, Q. Fang, U. Packbier and L. Blum, Int. J. Hydrogen Energy, 2013, 38, 4281–4290 CrossRef CAS.
- J. Schefold, A. Brisse, M. Zahid, J. P. Ouweltjes and J. U. Nielsen, ECS Trans., 2011, 35, 2915–2927 CrossRef CAS.
- X. Zhang, J. E. O'Brien, R. C. O'Brien, J. J. Hartvigsen, G. Tao and G. K. Housley, Int. J. Hydrogen Energy, 2013, 38, 20–28 CrossRef CAS.
- S. Megel, C. Dosch, S. Rothe, C. Folgner, N. Trofimenko, A. Rost, M. Kusnezoff, E. Reichelt, M. Jahn, A. Michaelis, C. Bienert and M. Brandner, ECS Trans., 2017, 78, 3089–3102 CrossRef CAS.
- A. Léon, A. Micero, B. Ludwig and A. Brisse, J. Power Sources, 2021, 510, 230346 CrossRef.
- M. Lang, S. Raab, M. S. Lemcke, C. Bohn and M. Pysik, ECS Trans., 2019, 91, 2713–2725 CrossRef CAS.
- Q. Fang, C. E. Frey, N. H. Menzler and L. Blum, J. Electrochem. Soc., 2018, 165, F38–F45 CrossRef CAS.
- G. Rinaldi, S. Diethelm, E. Oveisi, P. Burdet, J. van Herle, D. Montinaro, Q. Fu and A. Brisse, Fuel Cells, 2017, 17, 541–549 CrossRef CAS.
- M. Bianco, J. P. Ouweltjes and J. van Herle, Int. J. Hydrogen Energy, 2019, 44, 31406–31422 CrossRef CAS.
- A. Beez, K. Schiemann, N. H. Menzler and M. Bram, Front. Energy Res., 2018, 6, 70 CrossRef.
- K. Singh and T. Walia, Int. J. Energy Res., 2021, 45, 20559–20582 CrossRef CAS.
- D. The, S. Grieshammer, M. Schroeder, M. Martin, M. Al Daroukh, F. Tietz, J. Schefold and A. Brisse, J. Power Sources, 2015, 275, 901–911 CrossRef CAS.
- F. Monaco, D. Ferreira-Sanchez, M. Hubert, B. Morel, D. Montinaro, D. Grolimund and J. Laurencin, Int. J. Hydrogen Energy, 2021, 46, 31533–31549 CrossRef CAS.
- Z. Li, M. Peng, Y. Zhu, Z. Hu, C.-W. Pao, Y.-C. Chang, Y. Zhang, Y. Zhao, J. Li and Y. Sun, J. Mater. Chem. A, 2022, 10, 20350–20364 RSC.
- S. Zarabi Golkhatmi, M. I. Asghar and P. D. Lund, Renewable Sustainable Energy Rev., 2022, 161, 112339 CrossRef CAS.
- S. K. Sahu, D. Panthi, I. Soliman, H. Feng and Y. Du, Energies, 2022, 15, 3536 CrossRef CAS.
- N. Q. Minh, in High-Temperature Solid Oxide Fuel Cells for the 21st Century, Elsevier, 2016, pp. 255–282 Search PubMed.
- N. M. Sammes, Y. Du and R. Bove, J. Power Sources, 2005, 145, 428–434 CrossRef CAS.
- M. Ni, M. Leung and D. Leung, Int. J. Hydrogen Energy, 2008, 33, 2337–2354 CrossRef CAS.
- C. Lenser, D. Udomsilp, N. H. Menzler, P. Holtappels, T. Fujisaki, L. Kwati, H. Matsumoto, A. G. Sabato, F. Smeacetto, A. Chrysanthou and S. Molin, in Advanced Ceramics for Energy Conversion and Storage, Elsevier, 2020, pp. 387–547 Search PubMed.
- F. Grimm, PhD thesis, Schriften des Forschungszentrums Jülich, Reihe Energie & Umwelt/Energy & Environment, RWTH Aachen University, 2020,vol. 498.
- M. P. Hoerlein, M. Riegraf, R. Costa, G. Schiller and K. A. Friedrich, Electrochim. Acta, 2018, 276, 162–175 CrossRef CAS.
- L. Wehrle, D. Schmider, J. Dailly, A. Banerjee and O. Deutschmann, Appl. Energy, 2022, 317, 119143 CrossRef CAS.
- D. Sarantaridis and A. Atkinson, Fuel Cells, 2007, 7, 246–258 CrossRef CAS.
- C. Walter, O. Posdziech and M. Boltze, in EFCF 2022 Proceedings of the Conference. A Sessions, ed. O. Bucheli, G. Geisser, F. Moore and M. Spirig, 2022, p. A0501 Search PubMed.
- M. C. Tucker, J. Power Sources, 2010, 195, 4570–4582 CrossRef CAS.
- M. Noponen, H. Granö-Fabritius, S. Pylypko and E. Õunpuu, in EFCF 2022 Proceedings of the Conference. A Sessions, ed. O. Bucheli, G. Geisser, F. Moore and M. Spirig, 2022, p. A0503 Search PubMed.
- A. Hagen, R. Caldogno, F. Capotondo and X. Sun, Energies, 2022, 15, 2045 CrossRef CAS.
- M. Zhang, E. Wang, J. Mao, H. Wang, M. Ouyang and H. Hu, Front. Energy Res., 2022, 10, 888787 CrossRef.
- G. Schiller, R. Costa and K. A. Friedrich, in Ceramics for Energy Conversion, Storage, and Distribution Systems, ed. T. Pfeifer, J. Matyáš, P. Balaya, D. Singh and J. Wei, John Wiley & Sons, Inc, Hoboken, NJ, USA, 2016, pp. 19–27 Search PubMed.
- D. Udomsilp, J. Rechberger, R. Neubauer, C. Bischof, F. Thaler, W. Schafbauer, N. H. Menzler, L. G. de Haart, A. Nenning, A. K. Opitz, O. Guillon and M. Bram, Cell Rep. Phys. Sci., 2020, 1, 100072 CrossRef.
- F. Grimm, N. H. Menzler and O. Guillon, J. Power Sources, 2020, 451, 227607 CrossRef CAS.
- M. C. Tucker, Int. J. Hydrogen Energy, 2020, 45, 24203–24218 CrossRef CAS.
- J. Harman, P. Hjalmarsson, J. Mermelstein, J. Ryley, H. Sadler and M. Selby, ECS Trans., 2021, 103, 383–392 CrossRef CAS.
- B. Zhu, I. Albinsson, C. Andersson, K. Borsand, M. Nilsson and B.-E. Mellander, Electrochem. Commun., 2006, 8, 495–498 CrossRef CAS.
- N. Mahato, A. Banerjee, A. Gupta, S. Omar and K. Balani, Prog. Mater. Sci., 2015, 72, 141–337 CrossRef CAS.
- A. J. Jacobson, Chem. Mater., 2010, 22, 660–674 CrossRef CAS.
- J. Zhang, C. Lenser, N. H. Menzler and O. Guillon, Solid State Ionics, 2020, 344, 115138 CrossRef CAS.
- N. Q. Minh, J. Am. Ceram. Soc., 1993, 76, 563–588 CrossRef CAS.
- Y. Arachi, Solid State Ionics, 1999, 121, 133–139 CrossRef CAS.
- O. Posdziech, K. Schwarze and J. Brabandt, Int. J. Hydrogen Energy, 2019, 44, 19089–19101 CrossRef CAS.
- C. Geipel, K. Hauptmeier, K. Herbrig, F. Mittmann, M. Münch, M. Pötschke, L. Reichel, T. Strohbach, T. Seidel, A. Surrey and C. Walter, ECS Trans., 2019, 91, 123–132 CrossRef CAS.
- M. Lang, M. Braig, N. Alqubati and P. Szabo, ECS Trans., 2021, 103, 2041–2051 CrossRef CAS.
- M. Ghatee, M. H. Shariat and J. Irvine, Solid State Ionics, 2009, 180, 57–62 CrossRef CAS.
- T. Politova, Solid State Ionics, 2004, 168, 153–165 CrossRef CAS.
- S. Hussain and L. Yangping, Energy Transition, 2020, 4, 113–126 CrossRef.
- X. Guan, H. Zhou, Y. Wang and J. Zhang, J. Alloys Compd., 2008, 464, 310–316 CrossRef CAS.
- V. V. Kharton, F. M. Figueiredo, L. Navarro, E. N. Naumovich, A. V. Kovalevsky, A. A. Yaremchenko, A. P. Viskup, A. Carneiro, F. M. B. Marques and J. R. Frade, J. Mater. Sci., 2001, 36, 1105–1117 CrossRef CAS.
- Q. Fang, L. Blum and N. H. Menzler, J. Electrochem. Soc., 2015, 162, F907–F912 CrossRef CAS.
- D. Schäfer, Q. Fang, L. Blum and D. Stolten, J. Power Sources, 2019, 433, 126666 CrossRef.
- Q. Fu, J. Schefold, A. Brisse and J. U. Nielsen, Fuel Cells, 2014, 14, 395–402 CrossRef CAS.
- S. D. Ebbesen, J. Høgh, K. A. Nielsen, J. U. Nielsen and M. Mogensen, Int. J. Hydrogen Energy, 2011, 36, 7363–7373 CrossRef CAS.
- J. Mougin, A. Mansuy, A. Chatroux, G. Gousseau, M. Petitjean, M. Reytier and F. Mauvy, Fuel Cells, 2013, 13, 623–630 CrossRef CAS.
- M. Petitjean, M. Reytier, A. Chatroux, L. Bruguière, A. Mansuy, H. Sassoulas, S. Di Iorio, B. Morel and J. Mougin, ECS Trans., 2011, 35, 2905–2913 CrossRef CAS.
- M. Reytier, S. Di Iorio, A. Chatroux, M. Petitjean, J. Cren, M. de Saint Jean, J. Aicart and J. Mougin, Int. J. Hydrogen Energy, 2015, 40, 11370–11377 CrossRef CAS.
- S. H. Jensen, H. Langnickel, N. Hintzen, M. Chen, X. Sun, A. Hauch, G. Butera and L. R. Clausen, J. Energy Storage, 2019, 22, 106–115 CrossRef.
- M. Riedel, M. P. Heddrich, A. Ansar, Q. Fang, L. Blum and K. A. Friedrich, J. Power Sources, 2020, 475, 228682 CrossRef CAS.
- M. Riedel, M. P. Heddrich and K. A. Friedrich, Fuel Cells, 2020, 20, 592–607 CrossRef CAS.
- G. Corre and A. Brisse, ECS Trans., 2015, 68, 3481–3490 CrossRef CAS.
- L. León-Reina, E. R. Losilla, M. Martínez-Lara, S. Bruque and M. A. G. Aranda, J. Mater. Chem., 2004, 14, 1142–1149 RSC.
- T. K. Schultze, J. P. Arnold and S. Grieshammer, ACS Appl. Energy Mater., 2019, 2, 4708–4717 CrossRef CAS.
- S. Nakayama, M. Sakamoto, M. Highchi and K. Kodaira, J. Mater. Sci. Lett., 2000, 19, 91–93 CrossRef CAS.
- S. Nakayama and M. Highchi, J. Mater. Sci. Lett., 2001, 20, 913–915 CrossRef CAS.
- M. Higuchi, H. Katase, K. Kodaira and S. Nakayama, J. Cryst. Growth, 2000, 218, 282–286 CrossRef CAS.
- S. Nakayama, M. Sakamoto, M. Higuchi, K. Kodaira, M. Sato, S. Kakita, T. Suzuki and K. Itoh, J. Eur. Ceram. Soc., 1999, 19, 507–510 CrossRef CAS.
- T. Liao, T. Sasaki and Z. Sun, Phys. Chem. Chem. Phys., 2013, 15, 17553–17559 RSC.
- E. Kendrick, M. S. Islam and P. R. Slater, J. Mater. Chem., 2007, 17, 3104 RSC.
- H. Arikawa, Solid State Ionics, 2000, 136–137, 31–37 CrossRef CAS.
- J. M. Porras-Vázquez, E. R. Losilla, L. León-Reina, D. Marrero-López and M. A. Aranda, J. Am. Ceram. Soc., 2009, 92, 1062–1068 CrossRef.
- N. Liu, M. Shi, C. Wang, Y. P. Yuan, P. Majewski and F. Aldinger, J. Mater. Sci., 2006, 41, 4205–4213 CrossRef CAS.
- J. W. Fergus, J. Power Sources, 2006, 162, 30–40 CrossRef CAS.
- S. Badwal, Solid State Ionics, 2001, 143, 39–46 CrossRef CAS.
- E. Ivers-Tiffée, A. Weber and D. Herbstritt, J. Eur. Ceram. Soc., 2001, 21, 1805–1811 CrossRef.
- S. Badwal, Solid State Ionics, 2000, 136–137, 91–99 CrossRef CAS.
- P. Lacorre, F. Goutenoire, O. Bohnke, R. Retoux and Y. Laligant, Nature, 2000, 404, 856–858 CrossRef CAS.
- L. León-Reina, E. R. Losilla, M. Martínez-Lara, M. C. Martín-Sedeño, S. Bruque, P. Núñez, D. V. Sheptyakov and M. A. G. Aranda, Chem. Mater., 2005, 17, 596–600 CrossRef.
- K. Huang, M. Feng and J. B. Goodenough, J. Am. Ceram. Soc., 1998, 81, 357–362 CrossRef CAS.
- K. Huang, R. S. Tichy and J. B. Goodenough, J. Am. Ceram. Soc., 1998, 81, 2565–2575 CrossRef CAS.
- J. Zhang, C. Lenser, N. Russner, A. Weber, N. H. Menzler and O. Guillon, J. Am. Ceram. Soc., 2023, 106, 93–99 CrossRef CAS.
- T. Jacobsen and M. Mogensen, ECS Trans., 2008, 13, 259–273 CrossRef CAS.
- H. Bouwmeester, H. Kruidhof and A. J. Burggraaf, Solid State Ionics, 1994, 72, 185–194 CrossRef CAS.
- S. B. Adler, J. A. Lane and B. C. H. Steele, J. Electrochem. Soc., 1996, 143, 3554–3564 CrossRef CAS.
- A. V. Nikonov, K. A. Kuterbekov, K. Bekmyrza and N. B. Pavzderin, Eurasian J. Phys. Funct. Mater., 2018, 2, 274–292 CrossRef.
- E. V. Tsipis and V. V. Kharton, J. Solid State Electrochem., 2008, 12, 1039–1060 CrossRef CAS.
- M. Al Daroukh, Solid State Ionics, 2003, 158, 141–150 CrossRef CAS.
- C. Yao, J. Meng, X. Liu, X. Zhang, F. Meng, X. Wu and J. Meng, Electrochim. Acta, 2017, 229, 429–437 CrossRef CAS.
- U. de Haart, Q. Fang, N. H. Menzler and R. Peters, in EFCF 2022 Proceedings of the Conference, vol. 15, pp. 221–230 Search PubMed.
- M. Yuste, J. C. Pérez-Flores, J. R. de Paz, M. T. Azcondo, F. García-Alvarado and U. Amador, Dalton Trans., 2011, 40, 7908–7915 RSC.
- H. Kozuka, K. Ohbayashi and K. Koumoto, Sci. Technol. Adv. Mater., 2015, 16, 26001 CrossRef.
- J. Sunarso, S. S. Hashim, N. Zhu and W. Zhou, Prog. Energy Combust. Sci., 2017, 61, 57–77 CrossRef.
- D. Marinha, J. Hayd, L. Dessemond, E. Ivers-Tiffée and E. Djurado, J. Power Sources, 2011, 196, 5084–5090 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- Y. Song, X. Zhang, K. Xie, G. Wang and X. Bao, Adv. Mater., 2019, 31, e1902033 CrossRef PubMed.
- Y. Li, R. Gemmen and X. Liu, J. Power Sources, 2010, 195, 3345–3358 CrossRef CAS.
- S. P. Jiang, J. Mater. Sci., 2008, 43, 6799–6833 CrossRef CAS.
- S. Carter, Solid State Ionics, 1992, 53–56, 597–605 CrossRef CAS.
- I. Yasuda, Solid State Ionics, 1996, 86–88, 1197–1201 CrossRef CAS.
- J.-H. Kim, R.-H. Song, J.-H. Kim, T.-H. Lim, Y.-K. Sun and D.-R. Shin, J. Solid State Electrochem., 2007, 11, 1385–1390 CrossRef CAS.
- A. Princivalle and E. Djurado, Solid State Ionics, 2008, 179, 1921–1928 CrossRef CAS.
- J. R. Wilson, J. S. Cronin, A. T. Duong, S. Rukes, H.-Y. Chen, K. Thornton, D. R. Mumm and S. Barnett, J. Power Sources, 2010, 195, 1829–1840 CrossRef CAS.
- Z. Liu, X. Zhang, Z. Huang, Z. Zhao, D. Cui and M. Cheng, Int. J. Hydrogen Energy, 2016, 41, 21385–21393 CrossRef CAS.
- A. Barnabé, M. Gaudon, C. Bernard, C. Laberty and B. Durand, Mater. Res. Bull., 2004, 39, 725–735 CrossRef.
- F. Licci, G. Turilli, P. Ferro and A. Ciccarone, J. Am. Ceram. Soc., 2003, 86, 413–419 CrossRef CAS.
- W. Wang, Y. Huang, S. Jung, J. M. Vohs and R. J. Gorte, J. Electrochem. Soc., 2006, 153, A2066 CrossRef CAS.
- J. Schefold, A. Brisse and F. Tietz, J. Electrochem. Soc., 2011, 159, A137–A144 CrossRef.
- A. Petric, Solid State Ionics, 2000, 135, 719–725 CrossRef CAS.
- H. Tu, Y. Takeda, N. Imanishi and O. Yamamoto, Solid State Ionics, 1999, 117, 277–281 CrossRef CAS.
- H. Ullmann, N. Trofimenko, F. Tietz, D. Stöver and A. Ahmad-Khanlou, Solid State Ionics, 2000, 138, 79–90 CrossRef CAS.
- F. Prado, T. Armstrong, A. Caneiro and A. Manthiram, J. Electrochem. Soc., 2001, 148, J7 CrossRef CAS.
- Y. Leng, S. Chan and Q. Liu, Int. J. Hydrogen Energy, 2008, 33, 3808–3817 CrossRef CAS.
- M. Zahid, Proc. Vol., 2005, 2005–2007, 1708–1716 CrossRef.
- S. Jiang, Solid State Ionics, 2002, 146, 1–22 CrossRef CAS.
- R. de Souza, J. Kilner and J. Walker, Mater. Lett., 2000, 43, 43–52 CrossRef CAS.
- S. P. Jiang, Int. J. Hydrogen Energy, 2019, 44, 7448–7493 CrossRef CAS.
- S. T. Aruna, M. Muthuraman and K. C. Patil, J. Mater. Chem., 1997, 7, 2499–2503 RSC.
- F. Tietz, I. Arulraj, M. Zahid and D. Stover, Solid State Ionics, 2006, 177, 1753–1756 CrossRef CAS.
- Y. Ji, J. Kilner and M. Carolan, Solid State Ionics, 2005, 176, 937–943 CrossRef CAS.
- B. Steele, Solid State Ionics, 1996, 86–88, 1223–1234 CrossRef CAS.
- S. Fearn, J. Rossiny, J. A. Kilner and J. Evans, Solid State Ionics, 2012, 211, 51–57 CrossRef CAS.
- A. V. Berenov, A. Atkinson, J. A. Kilner, E. Bucher and W. Sitte, Solid State Ionics, 2010, 181, 819–826 CrossRef CAS.
- V. V. Kharton, E. N. Naumovich, A. A. Vecher and A. V. Nikolaev, J. Solid State Chem., 1995, 120, 128–136 CrossRef CAS.
- A. Egger, E. Bucher, M. Yang and W. Sitte, Solid State Ionics, 2012, 225, 55–60 CrossRef CAS.
- M. Katsuki, Solid State Ionics, 2003, 156, 453–461 CrossRef CAS.
- L. Tai, Solid State Ionics, 1995, 76, 273–283 CrossRef CAS.
- A. Mineshige, J. Izutsu, M. Nakamura, K. Nigaki, J. Abe, M. Kobune, S. Fujii and T. Yazawa, Solid State Ionics, 2005, 176, 1145–1149 CrossRef CAS.
- B. Steele, Solid State Ionics, 1998, 106, 255–261 CrossRef CAS.
- P. Woodward, R.-D. Hoffmann and A. W. Sleight, J. Mater. Res., 1994, 9, 2118–2127 CrossRef CAS.
- G. King and P. M. Woodward, J. Mater. Chem., 2010, 20, 5785 RSC.
- C. Meneghini, S. Ray, F. Liscio, F. Bardelli, S. Mobilio and D. D. Sarma, Phys. Rev. Lett., 2009, 103, 46403 CrossRef CAS PubMed.
- D. Serrate, J. M. de Teresa and M. R. Ibarra, J. Phys.: Condens. Matter, 2007, 19, 23201 CrossRef.
- A. A. Markov, I. A. Leonidov, M. V. Patrakeev, V. L. Kozhevnikov, O. A. Savinskaya, U. V. Ancharova and A. P. Nemudry, Solid State Ionics, 2008, 179, 1050–1053 CrossRef CAS.
- O. Savinskaya and A. P. Nemudry, J. Solid State Electrochem., 2011, 15, 269–275 CrossRef CAS.
- G. Y. Liu, G. H. Rao, X. M. Feng, H. F. Yang, Z. W. Ouyang, W. F. Liu and J. K. Liang, J. Alloys Compd., 2003, 353, 42–47 CrossRef CAS.
- G. Xiao, Q. Liu, X. Dong, K. Huang and F. Chen, J. Power Sources, 2010, 195, 8071–8074 CrossRef CAS.
- L. Zhang, Q. Zhou, Q. He and T. He, J. Power Sources, 2010, 195, 6356–6366 CrossRef CAS.
- Q. Liu, X. Dong, G. Xiao, F. Zhao and F. Chen, Adv. Mater., 2010, 22, 5478–5482 CrossRef CAS PubMed.
- J. Rager, M. Zipperle, A. Sharma and J. L. MacManus-Driscoll, J. Am. Ceram. Soc., 2004, 87, 1330–1335 CrossRef CAS.
- Q. Liu, D. E. Bugaris, G. Xiao, M. Chmara, S. Ma, H.-C. zur Loye, M. D. Amiridis and F. Chen, J. Power Sources, 2011, 196, 9148–9153 CrossRef CAS.
- Y. Teraoka, H. M. Zhang, K. Okamoto and N. Yamazoe, Mater. Res. Bull., 1988, 23, 51–58 CrossRef CAS.
- A. B. Muñoz-García, D. E. Bugaris, M. Pavone, J. P. Hodges, A. Huq, F. Chen, H.-C. zur Loye and E. A. Carter, J. Am. Ceram. Soc., 2012, 134, 6826–6833 CrossRef PubMed.
- B. He, L. Zhao, S. Song, T. Liu, F. Chen and C. Xia, J. Electrochem. Soc., 2012, 159, B619–B626 CrossRef CAS.
- C. Tian, J. Cheng and J. Yang, J. Mater. Sci.: Mater. Electron., 2021, 32, 1258–1264 CrossRef CAS.
- C. Sun, R. Hui and J. Roller, J. Solid State Electrochem., 2010, 14, 1125–1144 CrossRef CAS.
- G. Xiao, Q. Liu, F. Zhao, L. Zhang, C. Xia and F. Chen, J. Electrochem. Soc., 2011, 158, B455 CrossRef CAS.
- D. A. Osinkin, A. V. Khodimchuk, N. M. Porotnikova, N. M. Bogdanovich, A. V. Fetisov and M. V. Ananyev, Energies, 2020, 13, 250 CrossRef CAS.
- M. Sahibzada, Solid State Ionics, 2000, 136–137, 991–996 CrossRef CAS.
- W. Sitte, Solid State Ionics, 2002, 154–155, 517–522 CrossRef CAS.
- K. Huang, M. Feng, J. B. Goodenough and C. Milliken, J. Electrochem. Soc., 1997, 144, 3620–3624 CrossRef CAS.
- Q. Liu, C. Yang, X. Dong and F. Chen, Int. J. Hydrogen Energy, 2010, 35, 10039–10044 CrossRef CAS.
- Y. Wang, T. Liu, S. Fang and F. Chen, J. Power Sources, 2016, 305, 240–248 CrossRef CAS.
- S. Hou and K. Xie, Electrochim. Acta, 2019, 301, 63–68 CrossRef CAS.
- L. Bernadet, C. Moncasi, M. Torrell and A. Tarancón, Int. J. Hydrogen Energy, 2020, 45, 14208–14217 CrossRef CAS.
- A. Maignan, C. Martin, D. Pelloquin, N. Nguyen and B. Raveau, J. Solid State Chem., 1999, 142, 247–260 CrossRef CAS.
- V. Caignaert, I. Mirebeau, F. Bourée, N. Nguyen, A. Ducouret, J.-M. Greneche and B. Raveau, J. Solid State Chem., 1995, 114, 24–35 CrossRef CAS.
- I. O. Troyanchuk, N. V. Kasper, D. D. Khalyavin, H. Szymczak, R. Szymczak and M. Baran, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 2418–2421 CrossRef CAS.
- P. S. Anderson, C. A. Kirk, J. Knudsen, I. M. Reaney and A. R. West, Solid State Sci., 2005, 7, 1149–1156 CrossRef CAS.
- K. Zhang, L. Ge, R. Ran, Z. Shao and S. Liu, Acta Mater., 2008, 56, 4876–4889 CrossRef CAS.
- A. Tarancón, S. J. Skinner, R. J. Chater, F. Hernández-Ramírez and J. A. Kilner, J. Mater. Chem., 2007, 17, 3175 RSC.
- G. Kim, S. Wang, A. J. Jacobson, L. Reimus, P. Brodersen and C. A. Mims, J. Mater. Chem., 2007, 17, 2500 RSC.
- S. Bao, C. Ma, G. Chen, X. Xu, E. Enriquez, C. Chen, Y. Zhang, J. L. Bettis, M.-H. Whangbo, C. Dong and Q. Zhang, Sci. Rep., 2014, 4, 4726 CrossRef PubMed.
- G. Kim, S. Wang, A. Jacobson, L. Reimus, P. Brodersen and C. Mims, Meeting Abstracts, 2006, MA2005-02, p. 836 Search PubMed.
- Y. Wan, Y. Xing, Y. Li, D. Huan and C. Xia, J. Power Sources, 2018, 402, 363–372 CrossRef CAS.
- F. Tietz, Ionics, 1999, 5, 129–139 CrossRef CAS.
- Y. Zhang, L. Shen, Y. Wang, Z. Du, B. Zhang, F. Ciucci and H. Zhao, J. Mater. Chem. A, 2022, 10, 3495–3505 RSC.
- D. Chen, R. Ran, K. Zhang, J. Wang and Z. Shao, J. Power Sources, 2009, 188, 96–105 CrossRef CAS.
- J. Wang, F. Meng, T. Xia, Z. Shi, J. Lian, C. Xu, H. Zhao, J.-M. Bassat and J.-C. Grenier, Int. J. Hydrogen Energy, 2014, 39, 18392–18404 CrossRef CAS.
- G. Kostogloudis and C. Ftikos, Solid State Ionics, 1999, 126, 143–151 CrossRef CAS.
- W. Zhou, R. Ran, Z. Shao, W. Jin and N. Xu, J. Power Sources, 2008, 182, 24–31 CrossRef CAS.
- S. Jiang, W. Zhou, Y. Niu, Z. Zhu and Z. Shao, ChemSusChem, 2012, 5, 2023–2031 CrossRef CAS PubMed.
- Z. Shao and S. M. Haile, in Materials for Sustainable Energy, ed. V. Dusastre, Co-Published with Macmillan Publishers Ltd, UK, 2010, pp. 255–258 Search PubMed.
- E. Boehm, J.-M. Bassat, M. C. Steil, P. Dordor, F. Mauvy and J.-C. Grenier, Solid State Sci., 2003, 5, 973–981 CrossRef CAS.
- C. Munnings, S. Skinner, G. Amow, P. Whitfield and I. Davidson, Solid State Ionics, 2005, 176, 1895–1901 CrossRef CAS.
- A. Flura, S. Dru, C. Nicollet, V. Vibhu, S. Fourcade, E. Lebraud, A. Rougier, J.-M. Bassat and J.-C. Grenier, J. Solid State Chem., 2015, 228, 189–198 CrossRef CAS.
- A. Flura, C. Nicollet, V. Vibhu, B. Zeimetz, A. Rougier, J.-M. Bassat and J.-C. Grenier, J. Electrochem. Soc., 2016, 163, F523–F532 CrossRef CAS.
- S. Saher, J. Song, V. Vibhu, C. Nicollet, A. Flura, J.-M. Bassat and H. J. M. Bouwmeester, J. Mater. Chem. A, 2018, 6, 8331–8339 RSC.
- M. Medarde and J. Rodríguez-Carvajal, Z. Phys. B, 1997, 102, 307–315 CrossRef CAS.
- J. C. Park, D. K. Kim, S. H. Byeon and D. Kim, J. Synchrotron Radiat., 2001, 8, 704–706 CrossRef CAS PubMed.
- Z. Zhang and M. Greenblatt, J. Solid State Chem., 1995, 117, 236–246 CrossRef CAS.
- J. Wu, S. S. Pramana, S. J. Skinner, J. A. Kilner and A. P. Horsfield, J. Mater. Chem. A, 2015, 3, 23760–23767 RSC.
- T. Broux, C. Prestipino, M. Bahout, S. Paofai, E. Elkaïm, V. Vibhu, J.-C. Grenier, A. Rougier, J.-M. Bassat and O. Hernandez, Dalton Trans., 2016, 45, 3024–3033 RSC.
- L. Minervini, R. W. Grimes, J. A. Kilner and K. E. Sickafus, J. Mater. Chem., 2000, 10, 2349–2354 RSC.
- V. V. Kharton, A. P. Viskup, A. V. Kovalevsky, E. N. Naumovich and F. Marques, Solid State Ionics, 2001, 143, 337–353 CrossRef CAS.
- V. V. Kharton, A. P. Viskup, E. N. Naumovich and F. M. B. Marques, J. Mater. Chem., 1999, 9, 2623–2629 RSC.
- J.-M. Bassat, M. Burriel, O. Wahyudi, R. Castaing, M. Ceretti, P. Veber, I. Weill, A. Villesuzanne, J.-C. Grenier, W. Paulus and J. A. Kilner, J. Phys. Chem. C, 2013, 117, 26466–26472 CrossRef CAS.
- M. Burriel, G. Garcia, J. Santiso, J. A. Kilner, R. J. Chater and S. J. Skinner, J. Mater. Chem., 2008, 18, 416–422 RSC.
- E. Boehm, J. Bassat, P. Dordor, F. Mauvy, J. Grenier and P. Stevens, Solid State Ionics, 2005, 176, 2717–2725 CrossRef CAS.
- V. Vibhu, I. C. Vinke, R.-A. Eichel and L. de Haart, J. Power Sources, 2021, 482, 228909 CrossRef CAS.
- L.-P. Sun, Q. Li, H. Zhao, L.-H. Huo and J.-C. Grenier, J. Power Sources, 2008, 183, 43–48 CrossRef CAS.
- A. Chroneos, D. Parfitt, J. A. Kilner and R. W. Grimes, J. Mater. Chem., 2010, 20, 266–270 RSC.
- R. J. Woolley and S. J. Skinner, Solid State Ionics, 2014, 255, 1–5 CrossRef CAS.
- M. Ferkhi, A. Ringuedé, A. Khaled, L. Zerroual and M. Cassir, Electrochim. Acta, 2012, 75, 80–87 CrossRef CAS.
- M. Rieu, R. Sayers, M. A. Laguna-Bercero, S. J. Skinner, P. Lenormand and F. Ansart, J. Electrochem. Soc., 2010, 157, B477 CrossRef CAS.
- S. Skinner, Solid State Ionics, 2000, 135, 709–712 CrossRef CAS.
- F. Mauvy, Solid State Ionics, 2003, 158, 17–28 CrossRef CAS.
- C. Ferchaud, J.-C. Grenier, Y. Zhang-Steenwinkel, M. M. van Tuel, F. P. van Berkel and J.-M. Bassat, J. Power Sources, 2011, 196, 1872–1879 CrossRef CAS.
- A. V. Kovalevsky, V. V. Kharton, A. A. Yaremchenko, Y. V. Pivak, E. N. Naumovich and J. R. Frade, J. Eur. Ceram. Soc., 2007, 27, 4269–4272 CrossRef CAS.
- V. Vibhu, J.-M. Bassat, A. Flura, C. Nicollet, J.-C. Grenier and A. Rougier, ECS Trans., 2015, 68, 825–835 CrossRef CAS.
- C. Nicollet, A. Flura, V. Vibhu, A. Rougier, J.-M. Bassat and J.-C. Grenier, Int. J. Hydrogen Energy, 2016, 41, 15538–15544 CrossRef CAS.
- G. Amow, I. Davidson and S. Skinner, Solid State Ionics, 2006, 177, 1205–1210 CrossRef CAS.
- G. Amow and S. J. Skinner, J. Solid State Electrochem., 2006, 10, 538–546 CrossRef CAS.
- G. Amow, Proc. Vol., 2005, 2005–2007, 1745–1750 CrossRef.
- J. M. Bassat, C. Allançon, P. Odier, J. P. Loup, M. D. Carvalho and A. Wattiaux, Eur. J. Solid State Inorg. Chem., 1998, 35, 173–188 CrossRef CAS.
- E. N. Naumovich and V. V. Kharton, J. Mol. Struct.: THEOCHEM, 2010, 946, 57–64 CrossRef CAS.
- V. Vibhu, I. C. Vinke, R.-A. Eichel, J.-M. Bassat and L. de Haart, J. Power Sources, 2019, 444, 227292 CrossRef CAS.
- C. Berger, A. Egger, R. Merkle, E. Bucher, B. Stuhlhofer, N. Schrödl, J. Lammer, C. Gspan, G. Logvenov, J. Maier and W. Sitte, J. Electrochem. Soc., 2019, 166, F1088–F1095 CrossRef CAS.
- T. Inprasit, S. Wongkasemjit, S. J. Skinner, M. Burriel and P. Limthongkul, RSC Adv., 2015, 5, 2486–2492 RSC.
- J. Kilner, Solid State Ionics, 2002, 154–155, 523–527 CrossRef CAS.
- I. D. Unachukwu, V. Vibhu, I. C. Vinke, R.-A. Eichel and L. G. J. de Haart, Energies, 2022, 15, 2136 CrossRef CAS.
- V. Dusastre and J. A. Kilner, Solid State Ionics, 1999, 126, 163–174 CrossRef CAS.
- V. Vibhu, A. Rougier, C. Nicollet, A. Flura, J.-C. Grenier and J.-M. Bassat, Solid State Ionics, 2015, 278, 32–37 CrossRef CAS.
- V. Vibhu, A. Flura, A. Rougier, C. Nicollet, S. Fourcade, T. Hungria, J.-C. Grenier and J.-M. Bassat, J. Energy Chem., 2020, 46, 62–70 CrossRef.
- V. Vibhu, M. R. Suchomel, N. Penin, F. Weill, J.-C. Grenier, J.-M. Bassat and A. Rougier, Dalton Trans., 2018, 48, 266–277 RSC.
- J.-M. Bassat, V. Vibhu, C. Nicollet, A. Flura, S. Fourcade, J.-C. Grenier and A. Rougier, ECS Trans., 2017, 78, 655–665 CrossRef CAS.
- R. J. Woolley and S. J. Skinner, J. Power Sources, 2013, 243, 790–795 CrossRef CAS.
- T. Ramos, J. Hjelm and M. Mogensen, J. Electrochem. Soc., 2011, 158, B814 CrossRef CAS.
- M. Brown, S. Primdahl and M. Mogensen, J. Electrochem. Soc., 2000, 147, 475 CrossRef CAS.
- R. Barfod, M. Mogensen, T. Klemensø, A. Hagen, Y.-L. Liu and P. Vang Hendriksen, J. Electrochem. Soc., 2007, 154, B371 CrossRef CAS.
- V. Haanappel, J. Mertens, D. Rutenbeck, C. Tropartz, W. Herzhof, D. Sebold and F. Tietz, J. Power Sources, 2005, 141, 216–226 CrossRef CAS.
- V. A. C. Haanappel, J. Mertens and A. Mai, J. Fuel Cell Sci. Technol., 2006, 3, 263–270 CrossRef CAS.
- J. Mertens, V. A. C. Haanappel, C. Tropartz, W. Herzhof and H. P. Buchkremer, J. Fuel Cell Sci. Technol., 2006, 3, 125–130 CrossRef CAS.
- M. Mogensen, Solid State Ionics, 1996, 86–88, 1151–1160 CrossRef CAS.
- M. Jørgensen, Solid State Ionics, 2001, 139, 1–11 CrossRef.
- M. Sase, J. Suzuki, K. Yashiro, T. Otake, A. Kaimai, T. Kawada, J. Mizusaki and H. Yugami, Solid State Ionics, 2006, 177, 1961–1964 CrossRef CAS.
- D. Beckel, U. Muecke, T. Gyger, G. Florey, A. Infortuna and L. Gauckler, Solid State Ionics, 2007, 178, 407–415 CrossRef CAS.
- V. Haanappel, D. Rutenbeck, A. Mai, S. Uhlenbruck, D. Sebold, H. Wesemeyer, B. Röwekamp, C. Tropartz and F. Tietz, J. Power Sources, 2004, 130, 119–128 CrossRef CAS.
- A. Mai, V. A. Haanappel, S. Uhlenbruck, F. Tietz and D. Stöver, Solid State Ionics, 2005, 176, 1341–1350 CrossRef CAS.
- N. H. Menzler and V. A. Haanappel, J. Power Sources, 2010, 195, 5340–5343 CrossRef CAS.
- S. Uhlenbruck, N. Jordan, D. Sebold, H. P. Buchkremer, V. Haanappel and D. Stöver, Thin Solid Films, 2007, 515, 4053–4060 CrossRef CAS.
- M. Sogaard, P. Hendriksen, M. Mogensen, F. Poulsen and E. Skou, Solid State Ionics, 2006, 177, 3285–3296 CrossRef CAS.
- F. Han, R. Mücke, T. van Gestel, A. Leonide, N. H. Menzler, H. P. Buchkremer and D. Stöver, J. Power Sources, 2012, 218, 157–162 CrossRef CAS.
- N. H. Menzler, D. Sebold, Y. J. Sohn and S. Zischke, J. Power Sources, 2020, 478, 228770 CrossRef CAS.
- S. E. Wolf, V. Vibhu, E. Tröster, I. C. Vinke, R.-A. Eichel and L. G. J. de Haart, Energies, 2022, 15, 5449 CrossRef CAS.
- D. Marinha, L. Dessemond, J. S. Cronin, J. R. Wilson, S. A. Barnett and E. Djurado, Chem. Mater., 2011, 23, 5340–5348 CrossRef CAS.
- D. Marinha, L. Dessemond and E. Djurado, J. Power Sources, 2012, 197, 80–87 CrossRef CAS.
- O. Celikbilek, C.-A. Thieu, F. Agnese, E. Calì, C. Lenser, N. H. Menzler, J.-W. Son, S. J. Skinner and E. Djurado, J. Mater. Chem. A, 2019, 7, 25102–25111 RSC.
- R. K. Sharma, M. Burriel, L. Dessemond, V. Martin, J.-M. Bassat and E. Djurado, J. Power Sources, 2016, 316, 17–28 CrossRef CAS.
- B. Joshi, E. Samuel, Y. Kim, A. L. Yarin, M. T. Swihart and S. S. Yoon, Adv. Funct. Mater., 2021, 31, 2008181 CrossRef CAS.
- D. Marinha, L. Dessemond and E. Djurado, ECS Trans., 2010, 28, 93–103 CrossRef CAS.
- R. Sayers, M. Rieu, P. Lenormand, F. Ansart, J. A. Kilner and S. J. Skinner, Solid State Ionics, 2011, 192, 531–534 CrossRef CAS.
- N. Hildenbrand, P. Nammensma, D. H. Blank, H. J. Bouwmeester and B. A. Boukamp, J. Power Sources, 2013, 238, 442–453 CrossRef CAS.
- M. Zhi, S. Lee, N. Miller, N. H. Menzler and N. Wu, Energy Environ. Sci., 2012, 5, 7066 RSC.
- B. Zhang, Z. Leng, Y. Ling, H. Bai, S. Li, J. Zhou and S. Wang, Crystals, 2022, 12, 1624 CrossRef CAS.
- S. T. Aruna, L. S. Balaji, S. S. Kumar and B. S. Prakash, Renewable Sustainable Energy Rev., 2017, 67, 673–682 CrossRef CAS.
- J. Nowotny, Science of Ceramic Interfaces II, Elsevier, Burlington, 1995 Search PubMed.
- W. Lee, J. W. Han, Y. Chen, Z. Cai and B. Yildiz, J. Am. Chem. Soc., 2013, 135, 7909–7925 CrossRef CAS PubMed.
- K. Szot and W. Speier, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 5909–5926 CrossRef CAS.
- Y. Chen, W. Jung, Z. Cai, J. J. Kim, H. L. Tuller and B. Yildiz, Energy Environ. Sci., 2012, 5, 7979 RSC.
- H. Yokokawa, N. Sakai, T. Kawada and M. Dokiya, J. Electrochem. Soc., 1991, 138, 2719–2727 CrossRef CAS.
- H. Yokokawa, Solid State Ionics, 1996, 86–88, 1161–1165 CrossRef CAS.
- A. Mitterdorfer, Solid State Ionics, 1998, 111, 185–218 CrossRef CAS.
- C. Brugnoni, Solid State Ionics, 1995, 76, 177–182 CrossRef CAS.
- M. Mori, Solid State Ionics, 1999, 123, 113–119 CrossRef CAS.
- H. Türk, F.-P. Schmidt, T. Götsch, F. Girgsdies, A. Hammud, D. Ivanov, I. C. Vinke, L. de Haart, R.-A. Eichel, K. Reuter, R. Schlögl, A. Knop-Gericke, C. Scheurer and T. Lunkenbein, Adv. Mater. Interfaces, 2021, 8, 2100967 CrossRef.
- Developments in Solid Oxide Fuel Cells and Lithium Ion Batteries. Proceedings of the 106th Annual Meeting of the American Ceramic Society, Indianapolis, Indiana, USA (2004), American Ceramic Society, Westerville, Ohio, 2005, vol. 161 Search PubMed.
- M. Backhaus-Ricoult, K. Adib, T. S. Clair, B. Luerssen, L. Gregoratti and A. Barinov, Solid State Ionics, 2008, 179, 891–895 CrossRef CAS.
- M. Backhaus-Ricoult, Solid State Ionics, 2006, 177, 2195–2200 CrossRef CAS.
- Y. Chen, Y. Fan, S. Lee, G. Hackett, H. Abernathy, K. Gerdes and X. Song, J. Power Sources, 2019, 438, 227043 CrossRef CAS.
- S. He, K. Chen, M. Saunders, J. Li, C. Q. Cui and S. P. Jiang, J. Electrochem. Soc., 2017, 164, F1437–F1447 CrossRef CAS.
- S. He, K. Chen, M. Saunders, Z. Quadir, S. Tao, J. T. Irvine, C. Q. Cui and S. P. Jiang, Solid State Ionics, 2018, 325, 176–188 CrossRef CAS.
- H. Türk, T. Götsch, F.-P. Schmidt, A. Hammud, D. Ivanov, L. G. J. de Haart, I. C. Vinke, R.-A. Eichel, R. Schlögl, K. Reuter, A. Knop-Gericke, T. Lunkenbein and C. Scheurer, ChemCatChem, 2022, 14, e202200300 CrossRef.
- K. Chen and S. P. Jiang, Int. J. Hydrogen Energy, 2011, 36, 10541–10549 CrossRef CAS.
- A. V. Virkar, Int. J. Hydrogen Energy, 2010, 35, 9527–9543 CrossRef CAS.
- J. R. Mawdsley, J. David Carter, A. Jeremy Kropf, B. Yildiz and V. A. Maroni, Int. J. Hydrogen Energy, 2009, 34, 4198–4207 CrossRef CAS.
- A. Momma, T. Kato, Y. Kaga and S. Nagata, J. Ceram. Soc. Jpn., 1997, 105, 369–373 CrossRef CAS.
- V. Brichzin, Solid State Ionics, 2002, 152–153, 499–507 CrossRef CAS.
- J. Mizusaki, T. Saito and H. Tagawa, J. Electrochem. Soc., 1996, 143, 3065–3073 CrossRef CAS.
- R. Knibbe, M. L. Traulsen, A. Hauch, S. D. Ebbesen and M. Mogensen, J. Electrochem. Soc., 2010, 157, B1209 CrossRef CAS.
- J. Kim, H.-I. Ji, H. P. Dasari, D. Shin, H. Song, J.-H. Lee, B.-K. Kim, H.-J. Je, H.-W. Lee and K. J. Yoon, Int. J. Hydrogen Energy, 2013, 38, 1225–1235 CrossRef CAS.
- M. Keane, M. K. Mahapatra, A. Verma and P. Singh, Int. J. Hydrogen Energy, 2012, 37, 16776–16785 CrossRef CAS.
- P. Hjalmarsson, X. Sun, Y.-L. Liu and M. Chen, J. Power Sources, 2014, 262, 316–322 CrossRef CAS.
- S. J. Kim and G. M. Choi, Solid State Ionics, 2014, 262, 303–306 CrossRef CAS.
- Z. Pan, Q. Liu, M. Ni, R. Lyu, P. Li and S. H. Chan, Int. J. Hydrogen Energy, 2018, 43, 5437–5450 CrossRef CAS.
- N. Li, M. Keane, M. K. Mahapatra and P. Singh, Int. J. Hydrogen Energy, 2013, 38, 6298–6303 CrossRef CAS.
- K. Chen, N. Ai and S. P. Jiang, Electrochem. Commun., 2012, 19, 119–122 CrossRef CAS.
- H. Yokokawa, N. Sakai, T. Horita, K. Yamaji, M. E. Brito and H. Kishimoto, J. Alloys Compd., 2008, 452, 41–47 CrossRef CAS.
- J. Druce, H. Téllez, M. Burriel, M. D. Sharp, L. J. Fawcett, S. N. Cook, D. S. McPhail, T. Ishihara, H. H. Brongersma and J. A. Kilner, Energy Environ. Sci., 2014, 7, 3593–3599 RSC.
- H. Téllez, J. Druce, Y.-W. Ju, J. Kilner and T. Ishihara, Int. J. Hydrogen Energy, 2014, 39, 20856–20863 CrossRef.
- H. Téllez, J. Druce, J. A. Kilner and T. Ishihara, Faraday Discuss., 2015, 182, 145–157 RSC.
- B. Wei, M. Schroeder and M. Martin, ACS Appl. Mater. Interfaces, 2018, 10, 8621–8629 CrossRef CAS PubMed.
- B. Philippeau, F. Mauvy, C. Mazataud, S. Fourcade and J.-C. Grenier, Solid State Ionics, 2013, 249–250, 17–25 CrossRef CAS.
- A. Montenegro-Hernández, J. Vega-Castillo, L. Mogni and A. Caneiro, Int. J. Hydrogen Energy, 2011, 36, 15704–15714 CrossRef.
- A. M. Hernández, L. Mogni and A. Caneiro, Int. J. Hydrogen Energy, 2010, 35, 6031–6036 CrossRef.
- R. Sayers, J. Liu, B. Rustumji and S. J. Skinner, Fuel Cells, 2008, 8, 338–343 CrossRef CAS.
- V. Vibhu, A. Rougier, C. Nicollet, A. Flura, S. Fourcade, N. Penin, J.-C. Grenier and J.-M. Bassat, J. Power Sources, 2016, 317, 184–193 CrossRef CAS.
- S. Badwal, R. Deller, K. Foger, Y. Ramprakash and J. Zhang, Solid State Ionics, 1997, 99, 297–310 CrossRef CAS.
- S. Taniguchi, M. Kadowaki, H. Kawamura, T. Yasuo, Y. Akiyama, Y. Miyake and T. Saitoh, J. Power Sources, 1995, 55, 73–79 CrossRef CAS.
- S. C. Paulson and V. I. Birss, J. Electrochem. Soc., 2004, 151, A1961 CrossRef CAS.
- K. Schiemann, V. Vibhu, S. Yildiz, I. C. Vinke, R.-A. Eichel and L. de Haart, ECS Trans., 2017, 78, 1027–1034 CrossRef CAS.
- L. G. J. de Haart, J. Mougin, O. Posdziech, J. Kiviaho and N. H. Menzler, Fuel Cells, 2009, 9, 794–804 CrossRef CAS.
- L. Blum, Q. Fang, S. M. Groß-Barsnick, L. de Haart, J. Malzbender, N. H. Menzler and W. J. Quadakkers, Int. J. Hydrogen Energy, 2020, 45, 8955–8964 CrossRef CAS.
- N. H. Menzler, D. Sebold and Q. Fang, Meeting Abstracts, 2015, MA2015-03, p. 25 Search PubMed.
- M. Kornely, A. Neumann, N. H. Menzler, A. Weber and E. Ivers-Tiffée, ECS Trans., 2011, 35, 2009–2017 CrossRef CAS.
- Q. Fang, N. H. Menzler and L. Blum, J. Electrochem. Soc., 2021, 168, 104505 CrossRef CAS.
- S. P. Jiang, J. P. Zhang, L. Apateanu and K. Foger, J. Electrochem. Soc., 2000, 147, 4013 CrossRef CAS.
- T. Horita, Y. Xiong, M. Yoshinaga, H. Kishimoto, K. Yamaji, M. E. Brito and H. Yokokawa, Electrochem. Solid-State Lett., 2009, 12, B146 CrossRef CAS.
- K. Chen, J. Hyodo, A. Dodd, N. Ai, T. Ishihara, L. Jian and S. P. Jiang, Faraday Discuss., 2015, 182, 457–476 RSC.
- S. P. Jiang and X. Chen, Int. J. Hydrogen Energy, 2014, 39, 505–531 CrossRef CAS.
- B. Wei, K. Chen, L. Zhao, Z. Lü and S. P. Jiang, Phys. Chem. Chem. Phys., 2015, 17, 1601–1609 RSC.
- V. I. Sharma and B. Yildiz, J. Electrochem. Soc., 2010, 157, B441 CrossRef CAS.
- P. Qiu, J. Lin, L. Lei, Z. Yuan, L. Jia, J. Li and F. Chen, ACS Appl. Energy Mater., 2019, 2, 7619–7627 CrossRef CAS.
- B. Wei, K. Chen, C. C. Wang, Z. Lü and S. P. Jiang, Electrochim. Acta, 2015, 174, 327–331 CrossRef CAS.
- N. Schrödl, A. Egger, C. Gspan, T. Höschen, F. Hofer and W. Sitte, Solid State Ionics, 2018, 322, 44–53 CrossRef.
- N. Schrödl, A. Egger, J. Lammer, F. Hofer and W. Sitte, J. Electrochem. Soc., 2021, 168, 14509 CrossRef.
- S.-N. Lee, A. Atkinson and J. A. Kilner, ECS Trans., 2013, 57, 605–613 CrossRef.
- K. J. Lee, J. H. Chung, M. J. Lee and H. J. Hwang, J. Korean Ceram. Soc., 2019, 56, 160–166 CrossRef CAS.
- J. Schuler, H. Lübbe, A. Hessler-Wyser and J. van Herle, J. Power Sources, 2012, 213, 223–228 CrossRef.
- M. A. Laguna-Bercero, J. Power Sources, 2012, 203, 4–16 CrossRef CAS.
- M. Ni, M. K. H. Leung and D. Y. C. Leung, Chem. Eng. Technol., 2006, 29, 636–642 CrossRef CAS.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- F. Thaler, Q. Fang, U. de Haart, L. G. J. de Haart, R. Peters and L. Blum, ECS Trans., 2021, 103, 363–374 CrossRef.
- K. Zhao, Q. Bkour, X. Hou, S. W. Kang, J. C. Park, M. G. Norton, J.-I. Yang and S. Ha, Chem. Eng. J., 2018, 336, 20–27 CrossRef CAS.
- S.-D. Kim, D.-W. Seo, A. K. Dorai and S.-K. Woo, Int. J. Hydrogen Energy, 2013, 38, 6569–6576 CrossRef CAS.
- A. Hauch, S. D. Ebbesen, S. H. Jensen and M. Mogensen, J. Electrochem. Soc., 2008, 155, B1184–B1193 CrossRef CAS.
- M. H. Pihlatie, A. Kaiser, M. Mogensen and M. Chen, Solid State Ionics, 2011, 189, 82–90 CrossRef CAS.
- M. A. Laguna-Bercero, A. R. Hanifi, L. Menand, N. K. Sandhu, N. E. Anderson, T. H. Etsell and P. Sarkar, Electrochim. Acta, 2018, 268, 195–201 CrossRef CAS.
- L. Mingyi, Y. Bo, X. Jingming and C. Jing, Int. J. Hydrogen Energy, 2010, 35, 2670–2674 CrossRef.
- C. Neofytidis, E. Ioannidou, L. Sygellou, M. Kollia and D. K. Niakolas, J. Catal., 2019, 373, 260–275 CrossRef CAS.
- S. P. Jiang and S. H. Chan, J. Mater. Sci., 2004, 39, 4405–4439 CrossRef CAS.
- D. A. Osinkin, D. I. Bronin, S. M. Beresnev, N. M. Bogdanovich, V. D. Zhuravlev, G. K. Vdovin and T. A. Demyanenko, J. Solid State Electrochem., 2014, 18, 149–156 CrossRef CAS.
- S.-Y. Park, C. W. Na, J. H. Ahn, U.-J. Yun, T.-H. Lim, R.-H. Song, D.-R. Shin and J.-H. Lee, J. Power Sources, 2012, 218, 119–127 CrossRef CAS.
- V. Gil, J. Tartaj and C. Moure, Ceram. Int., 2009, 35, 839–846 CrossRef CAS.
- S. Aruna, M. Muthuraman and K. Patil, Solid State Ionics, 1998, 111, 45–51 CrossRef CAS.
- E. Drożdż, J. Wyrwa and M. Rękas, Ionics, 2013, 19, 1733–1743 CrossRef.
- A. K. Soman, P. Kuppusami and A. M. Rabel, 2017, 17, pp. 224–236.
- S. P. Patil, L. D. Jadhav and M. Chourashiya, Open Ceram., 2022, 9, 100230 CrossRef CAS.
- A. Brisse, J. Schefold and M. Zahid, Int. J. Hydrogen Energy, 2008, 33, 5375–5382 CrossRef CAS.
- C. Xi, J. Sang, A. Wu, J. Yang, X. Qi, W. Guan, J. Wang and S. C. Singhal, Int. J. Hydrogen Energy, 2022, 47, 10166–10174 CrossRef CAS.
- Z. Zhan, W. Kobsiriphat, J. R. Wilson, M. Pillai, I. Kim and S. A. Barnett, Energy Fuels, 2009, 23, 3089–3096 CrossRef CAS.
- Y. Tao, S. D. Ebbesen and M. B. Mogensen, J. Power Sources, 2016, 328, 452–462 CrossRef CAS.
- M. Zheng, S. Wang, Y. Yang and C. Xia, J. Mater. Chem. A, 2018, 6, 2721–2729 RSC.
- V. Singh, H. Muroyama, T. Matsui and K. Eguchi, ECS Trans., 2014, 64, 53–64 CrossRef CAS.
- I. D. Unachukwu, V. Vibhu, I. C. Vinke, R.-A. Eichel and L. de Haart, J. Power Sources, 2023, 556, 232436 CrossRef CAS.
- V. Vibhu, I. C. Vinke, F. Zaravelis, S. G. Neophytides, D. K. Niakolas, R.-A. Eichel and L. G. J. de Haart, Energies, 2022, 15, 2726 CrossRef CAS.
- J. Zhou, Z. Ma, L. Zhang, C. Liu, J. Pu, X. Chen, Y. Zheng and S. H. Chan, Int. J. Hydrogen Energy, 2019, 44, 28939–28946 CrossRef CAS.
- Z. Ma, Y. Li, Y. Zheng, W. Li, X. Chen, X. Sun, X. Chen and J. Zhou, Ceram. Int., 2021, 47, 23350–23361 CrossRef CAS.
- C. Jin, C. Yang, F. Zhao, D. Cui and F. Chen, Int. J. Hydrogen Energy, 2011, 36, 3340–3346 CrossRef CAS.
- R. Xing, Y. Wang, S. Liu and C. Jin, J. Power Sources, 2012, 208, 276–281 CrossRef CAS.
- S. Chen, K. Xie, D. Dong, H. Li, Q. Qin, Y. Zhang and Y. Wu, J. Power Sources, 2015, 274, 718–729 CrossRef CAS.
- S. Tao and J. T. S. Irvine, J. Electrochem. Soc., 2004, 151, A252 CrossRef CAS.
- Y. Song, Q. Zhong and W. Tan, Int. J. Hydrogen Energy, 2014, 39, 13694–13700 CrossRef CAS.
- K.-T. Wu and T. Ishihara, ECS Trans., 2021, 103, 561–567 CrossRef.
- T. Ishihara, S. Wang and K.-T. Wu, Solid State Ionics, 2017, 299, 60–63 CrossRef CAS.
- F. Tietz, I. A. Raj, M. Zahid, A. Mai and D. Stöver, Prog. Solid State Chem., 2007, 35, 539–543 CrossRef CAS.
- Y. S. Chung, T. Kim, T. H. Shin, H. Yoon, S. Park, N. M. Sammes, W. B. Kim and J. S. Chung, J. Mater. Chem. A, 2017, 5, 6437–6446 RSC.
- F. Y. Farsani, M. Jafari, E. Shahsavari and H. Salamati, Int. J. Hydrogen Energy, 2020, 45, 8915–8929 CrossRef.
- X. Peng, Y. Tian, Y. Liu, W. Wang, L. Jia, J. Pu, B. Chi and J. Li, J. CO2 Util., 2020, 36, 18–24 CrossRef CAS.
- F. B. Wede, Master thesis, TU Graz, 2013.
- S. Wang and T. Ishihara, ECS Trans., 2013, 57, 3171–3176 CrossRef.
- Q. Qin, K. Xie, H. Wei, W. Qi, J. Cui and Y. Wu, RSC Adv., 2014, 4, 38474–38483 RSC.
- O. Marina, Solid State Ionics, 2002, 149, 21–28 CrossRef CAS.
- Q. X. Fu, F. Tietz and D. Stöver, J. Electrochem. Soc., 2006, 153, D74 CrossRef CAS.
- D. Neagu and J. T. S. Irvine, Chem. Mater., 2010, 22, 5042–5053 CrossRef CAS.
- S. Li, Y. Li, Y. Gan, K. Xie and G. Meng, J. Power Sources, 2012, 218, 244–249 CrossRef CAS.
- Y. Li, J. Zhou, D. Dong, Y. Wang, J. Z. Jiang, H. Xiang and K. Xie, Phys. Chem. Chem. Phys., 2012, 14, 15547–15553 RSC.
- S. Paydar, O. Korjus, M. Maide, I. Kivi, E. Lust and G. Nurk, ECS Trans., 2021, 103, 1971–1979 CrossRef CAS.
- O. A. Marina, L. R. Pederson, M. C. Williams, G. W. Coffey, K. D. Meinhardt, C. D. Nguyen and E. C. Thomsen, J. Electrochem. Soc., 2007, 154, B452 CrossRef CAS.
- W. Qi, Y. Gan, D. Yin, Z. Li, G. Wu, K. Xie and Y. Wu, J. Mater. Chem. A, 2014, 2, 6904–6915 RSC.
- S. E. Wolf, V. Vibhu, C. L. Coll, N. Eyckeler, I. C. Vinke, R.-A. Eichel and L. de Haart, ECS Trans., 2023, 111, 2119–2130 CrossRef.
- S. Zhang, K. Zhu, X. Hu, R. Peng and C. Xia, J. Mater. Chem. A, 2021, 9, 24336–24347 RSC.
- Y. Jiang, Y. Yang, C. Xia and H. J. M. Bouwmeester, J. Mater. Chem. A, 2019, 7, 22939–22949 RSC.
- Y. Wang, T. Liu, M. Li, C. Xia, B. Zhou and F. Chen, J. Mater. Chem. A, 2016, 4, 14163–14169 RSC.
- Y. Li, B. Hu, C. Xia, W. Q. Xu, J. P. Lemmon and F. Chen, J. Mater. Chem. A, 2017, 5, 20833–20842 RSC.
- B.-W. Zhang, M.-N. Zhu, M.-R. Gao, X. Xi, N. Duan, Z. Chen, R.-F. Feng, H. Zeng and J.-L. Luo, Nat. Commun., 2022, 13, 4618 CrossRef CAS PubMed.
- C. Li, Y. Deng, L. Yang, B. Liu, D. Yan, L. Fan, J. Li and L. Jia, Adv. Powder Mater., 2023, 2, 100133 CrossRef.
- K. Kamlungsua and P.-C. Su, Electrochim. Acta, 2020, 355, 136670 CrossRef CAS.
- N. Dai, Z. Wang, T. Jiang, J. Feng, W. Sun, J. Qiao, D. Rooney and K. Sun, J. Power Sources, 2014, 268, 176–182 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- T. Xia, N. Lin, H. Zhao, L. Huo, J. Wang and J.-C. Grenier, J. Power Sources, 2009, 192, 291–296 CrossRef CAS.
- R. Lan, P. I. Cowin, S. Sengodan and S. Tao, Sci. Rep., 2016, 6, 31839 CrossRef CAS PubMed.
- B. Ge, J. T. Ma, D. Ai, C. Deng, X. Lin and J. Xu, Electrochim. Acta, 2015, 151, 437–446 CrossRef CAS.
- B. Ge, J. Sheng, Y. Zhuang, J. Ma, Z. Yang, C. Li and S. Peng, Mater. Res. Express, 2020, 7, 56302 CrossRef CAS.
- S. Tao, J. Canales-Vázquez and J. T. S. Irvine, Chem. Mater., 2004, 16, 2309–2316 CrossRef CAS.
- R. Fu, P. Jiang, H. Xu, B. Niu, F. Jiang, L. Yang, T. Feng and T. He, Int. J. Hydrogen Energy, 2019, 44, 31394–31405 CrossRef CAS.
- J. Zhou, Mater. Lett., 2019, 257, 126758 CrossRef CAS.
- B. S. Teketel, B. A. Beshiwork, D. Tian, S. Zhu, H. G. Desta, K. Kashif, Y. Chen and B. Lin, Catalysts, 2022, 12, 488 CrossRef CAS.
- S. Sengodan, S. Choi, A. Jun, T. H. Shin, Y.-W. Ju, H. Y. Jeong, J. Shin, J. T. S. Irvine and G. Kim, Nat. Mater., 2015, 14, 205–209 CrossRef CAS PubMed.
- H. Ding, Z. Tao, S. Liu and J. Zhang, Sci. Rep., 2015, 5, 18129 CrossRef CAS PubMed.
- R. M. Clemmer and S. F. Corbin, Solid State Ionics, 2009, 180, 721–730 CrossRef CAS.
- S. K. Pratihar, A. Dassharma and H. S. Maiti, J. Mater. Sci., 2007, 42, 7220–7226 CrossRef CAS.
- A. R. Hanifi, M. A. Laguna-Bercero, N. K. Sandhu, T. H. Etsell and P. Sarkar, Sci. Rep., 2016, 6, 27359 CrossRef CAS PubMed.
- M. B. Mogensen, M. Chen, H. L. Frandsen, C. Graves, A. Hauch, P. V. Hendriksen, T. Jacobsen, S. H. Jensen, T. L. Skafte and X. Sun, Fuel Cells, 2021, 21, 415–429 CAS.
- F. Monaco, M. Hubert, J. Vulliet, J. P. Ouweltjes, D. Montinaro, P. Cloetens, P. Piccardo, F. Lefebvre-Joud and J. Laurencin, J. Electrochem. Soc., 2019, 166, F1229–F1242 CrossRef CAS.
- A. Hauch, K. Brodersen, M. Chen and M. B. Mogensen, Solid State Ionics, 2016, 293, 27–36 CrossRef CAS.
- A. Hauch, PhD thesis, Risoe National Lab., DTU, Roskilde, Denmark, 2007.
- A. Hagen, R. Barfod, P. V. Hendriksen, Y.-L. Liu and S. Ramousse, J. Electrochem. Soc., 2006, 153(6), A1165–A1171 CrossRef CAS.
- A. Hauch, S. D. Ebbesen, S. H. Jensen and M. Mogensen, J. Mater. Chem., 2008, 18(20), 2331–2340 RSC.
- M. Trini, A. Hauch, S. de Angelis, X. Tong, P. V. Hendriksen and M. Chen, J. Power Sources, 2020, 450, 227599 CrossRef CAS.
- S. He and S. P. Jiang, Prog. Mater. Sci., 2021, 31, 341–372 CAS.
- Q. Fang, L. Blum, N. H. Menzler and D. Stolten, ECS Trans., 2017, 78, 2885–2893 CrossRef CAS.
- M. Trini, P. S. Jørgensen, A. Hauch, J. J. Bentzen, P. V. Hendriksen and M. Chen, J. Electrochem. Soc., 2019, 166, F158–F167 CrossRef CAS.
- S. D. Ebbesen, X. Sun and M. B. Mogensen, Faraday Discuss., 2015, 182, 393–422 RSC.
- Y. Wang, W. Li, L. Ma, W. Li and X. Liu, J. Mater. Sci. Technol., 2020, 55, 35–55 CrossRef CAS.
- P. Moçoteguy and A. Brisse, Int. J. Hydrogen Energy, 2013, 38, 15887–15902 CrossRef.
- X. Sun, Y. Liu, P. V. Hendriksen and M. Chen, J. Power Sources, 2021, 506, 230136 CrossRef CAS.
- C. E. Frey, Q. Fang, D. Sebold, L. Blum and N. H. Menzler, J. Electrochem. Soc., 2018, 165, F357–F364 CrossRef CAS.
- F. Tietz, D. Sebold, A. Brisse and J. Schefold, J. Power Sources, 2013, 223, 129–135 CrossRef CAS.
- Q. Fang, L. Blum, N. H. Menzler and D. Stolten, ECS Trans., 2017, 78, 2885–2893 CrossRef CAS.
- J. Uecker, I. D. Unachukwu, V. Vibhu, I. C. Vinke, R.-A. Eichel and L. de Haart, Electrochim. Acta, 2023, 452, 142320 CrossRef CAS.
- Y. Song, Z. Zhou, X. Zhang, Y. Zhou, H. Gong, H. Lv, Q. Liu, G. Wang and X. Bao, J. Mater. Chem. A, 2018, 6, 13661–13667 RSC.
- J. Schefold, A. Brisse, A. Surrey and C. Walter, Int. J. Hydrogen Energy, 2020, 45, 5143–5154 CrossRef CAS.
- C. K. Lim, Q. Liu, J. Zhou, Q. Sun and S. H. Chan, Fuel Cells, 2017, 17, 464–472 CrossRef CAS.
- D. Schäfer, L. Queda, V. Nischwitz, Q. Fang and L. Blum, Processes, 2022, 10, 598 CrossRef.
- D. Schäfer, T. Janßen, Q. Fang, F. Merten and L. Blum, Energies, 2021, 14, 544 CrossRef.
- W. Z. Zhu and S. C. Deevi, Mater. Sci. Eng., A, 2003, 348, 227–243 CrossRef.
- S. Fontana, R. Amendola, S. Chevalier, P. Piccardo, G. Caboche, M. Viviani, R. Molins and M. Sennour, J. Power Sources, 2007, 171, 652–662 CrossRef CAS.
- K. Huang and S. C. Singhal, J. Power Sources, 2013, 237, 84–97 CrossRef CAS.
- S. Geng, J. Zhu and Z. Lu, Solid State Ionics, 2006, 177, 559–568 CrossRef CAS.
- B. Königshofer, M. Höber, G. Nusev, P. Boškoski, C. Hochenauer and V. Subotić, J. Power Sources, 2022, 523, 230982 CrossRef.
- R. Sachitanand, J.-E. Svensson and J. Froitzheim, Oxid. Met., 2015, 84, 241–257 CrossRef CAS.
- M. Stanislowski, E. Wessel, K. Hilpert, T. Markus and L. Singheiser, J. Electrochem. Soc., 2007, 154, A295 CrossRef CAS.
- S. Fontana, S. Chevalier and G. Caboche, Oxid. Met., 2012, 78, 307–328 CrossRef CAS.
- B. Talic, H. Falk-Windisch, V. Venkatachalam, P. V. Hendriksen, K. Wiik and H. L. Lein, J. Power Sources, 2017, 354, 57–67 CrossRef CAS.
- R. Vaßen, N. Grünwald, D. Marcano, N. H. Menzler, R. Mücke, D. Sebold, Y. J. Sohn and O. Guillon, Surf. Coat. Technol., 2016, 291, 115–122 CrossRef.
- N. Grünwald, D. Sebold, Y. J. Sohn, N. H. Menzler and R. Vaßen, J. Power Sources, 2017, 363, 185–192 CrossRef.
- M. Kornely, N. H. Menzler, A. Weber and E. Iver-Tiffée, Meeting Abstracts, 2012, MA2012-02, p. 1972 Search PubMed.
- M. Bianco, J. Tallgren, J.-E. Hong, S. Yang, O. Himanen, J. Mikkola, J. van Herle and R. Steinberger-Wilckens, J. Power Sources, 2019, 437, 226900 CrossRef CAS.
- S. Frangini, A. Masi, L. Della Seta, M. Bianco and J. van Herle, J. Electrochem. Soc., 2018, 165, F97–F104 CrossRef CAS.
- M. M. Del Juez Lorenzo, V. Kolarik, V. Kuchenreuther-Hummel, M. Pötschke and D. Schimanke, Oxid. Met., 2017, 88, 279–290 CrossRef.
- C.-H. Park and K.-H. Baik, Met. Mater. Int., 2014, 20, 63–67 CrossRef CAS.
- N. H. Menzler, D. Sebold and O. Guillon, J. Power Sources, 2018, 374, 69–76 CrossRef CAS.
- J. Malzbender, P. Batfalsky, R. Vaßen, V. Shemet and F. Tietz, J. Power Sources, 2012, 201, 196–203 CrossRef CAS.
- N. Grünwald, P. Lhuissier, L. Salvo, J. Villanova, N. H. Menzler, O. Guillon, C. L. Martin and R. Vaßen, J. Mater. Sci., 2020, 55, 12725–12736 CrossRef.
- N. Grünwald, Y. J. Sohn, X. Yin, N. H. Menzler, O. Guillon and R. Vaßen, J. Eur. Ceram. Soc., 2019, 39, 449–460 CrossRef.
- J. Zurek, N. Margaritis, D. Naumenko, N. H. Menzler and W. J. Quadakkers, Oxid. Met., 2019, 92, 353–377 CrossRef CAS.
- K. Sick, N. Grigorev, N. H. Menzler and O. Guillon, in Proceeding of the 42nd International Conference on Advanced Ceramics and Composites, ed. J. Salem, D. Koch, P. Mechnich, M. Kusnezoff, N. Bansal, J. LaSalvia, P. Balaya, Z. Fu and T. Ohji, John Wiley & Sons, Inc, Hoboken, NJ, USA, 2019, pp. 99–111 Search PubMed.
- Z. Yang, G. Xia, P. Singh and J. W. Stevenson, J. Power Sources, 2006, 155, 246–252 CrossRef CAS.
- J. Kupecki, R. Kluczowski, D. Papurello, A. Lanzini, M. Kawalec, M. Krauz and M. Santarelli, Int. J. Hydrogen Energy, 2019, 44, 19405–19411 CrossRef CAS.
- D. Yan, C. Zhang, L. Liang, K. Li, L. Jia, J. Pu, L. Jian, X. Li and T. Zhang, Appl. Energy, 2016, 175, 414–420 CrossRef.
- W. Wu, G. L. Wang, W. B. Guan, Y. F. Zhen and W. G. Wang, Fuel Cells, 2013, 13(5), 743–750 CAS.
- M. C. Tucker, L. Cheng and L. C. DeJonghe, J. Power Sources, 2011, 196, 8313–8322 CrossRef CAS.
- X. Li, S. M. Groß-Barsnick, T. Koppitz, S. Baumann, W. A. Meulenberg and G. Natour, J. Eur. Ceram. Soc., 2022, 42, 2879–2891 CrossRef CAS.
- Y. Mizutani, M. Tamura, M. Kawai and O. Yamamoto, Solid State Ionics, 1994, 72, 271–275 CrossRef CAS.
- A. Hammouche, E. Siebert and A. Hammou, Mater. Res. Bull., 1989, 24, 367–380 CrossRef CAS.
- J. Xiang, Z.-G. Liu, J.-H. Ouyang and F.-Y. Yan, J. Power Sources, 2014, 251, 305–310 CrossRef CAS.
- L. Blum, L. de Haart, J. Malzbender, N. H. Menzler, J. Remmel and R. Steinberger-Wilckens, J. Power Sources, 2013, 241, 477–485 CrossRef CAS.
- F. Tietz, A. Schmidt and M. Zahid, J. Solid State Chem., 2004, 177, 745–751 CrossRef CAS.
- M.-F. Han, X. Du and Z. Liu, ECS Trans., 2009, 25, 1379–1386 CrossRef CAS.
- X. Montero, N. Jordán, J. Pirón-Abellán, F. Tietz, D. Stöver, M. Cassir and I. Villarreal, J. Electrochem. Soc., 2009, 156, B188 CrossRef CAS.
- X. Montero, F. Tietz, D. Sebold, H. P. Buchkremer, A. Ringuede, M. Cassir, A. Laresgoiti and I. Villarreal, J. Power Sources, 2008, 184, 172–179 CrossRef CAS.
- C. Yang, A. Coffin and F. Chen, Int. J. Hydrogen Energy, 2010, 35, 3221–3226 CrossRef CAS.
- M. Liang, B. Yu, M. Wen, J. Chen, J. Xu and Y. Zhai, J. Power Sources, 2009, 190, 341–345 CrossRef CAS.
- Y. Tao, S. D. Ebbesen and M. B. Mogensen, J. Electrochem. Soc., 2014, 161, F337–F343 CrossRef CAS.
- Y. Li, Z. Zhan and C. Xia, Catal. Sci. Technol., 2018, 8, 980–984 RSC.
- V. Vibhu, I. C. Vinke, R.-A. Eichel and L. G. J. de Haart, ECS Trans., 2021, 103, 1505–1515 CrossRef CAS.
- A. Usenka, V. Pankov, V. Vibhu, A. Flura, J.-C. Grenier and J.-M. Bassat, ECS Trans., 2019, 91, 1341–1353 CrossRef CAS.
- V. Vibhu, I. C. Vinke, R.-A. Eichel, J.-M. Bassat and L. de Haart, ECS Trans., 2019, 91, 1327–1339 CrossRef CAS.
- T. Ogier, F. Mauvy, J.-M. Bassat, J. Laurencin, J. Mougin and J.-C. Grenier, Int. J. Hydrogen Energy, 2015, 40, 15885–15892 CrossRef CAS.
Footnote |
| † These authors contributed equally to the publication. |
| This journal is © The Royal Society of Chemistry 2023 |