
Thermocatalytic and photocatalytic chemoselective reduction of cinnamaldehyde to cinnamyl alcohol and hydrocinnamaldehyde over Ru@ZnO/CN†
Arzoo
Chauhan‡
 a,
Rajat
Ghalta‡
a,
Rajaram
Bal
b and
Rajendra
Srivastava
*a
a,
Rajat
Ghalta‡
a,
Rajaram
Bal
b and
Rajendra
Srivastava
*a
aCatalysis Research Laboratory, Department of Chemistry, Indian Institute of Technology Ropar, Rupnagar, Punjab-140001, India. E-mail: rajendra@iitrpr.ac.in; Tel: +91-1881-232064
bNanocatalysis Area Conversion and Catalysis Division, CSIR-Indian Institute of Petroleum, Dehradun-248005, India
First published on 3rd May 2023
Abstract
The selective hydrogenation of α–β unsaturated carbonyl compounds requires a catalyst with a suitable combination of the support and active sites to activate a specific functional group. In this work, ZnO and g-C3N4(CN) nanocomposite (ZnO/CN) supported Ru catalysts were synthesized for thermal and photochemical selective hydrogenation of cinnamaldehyde (CAL). Under thermal conditions, formic acid (FA) was employed as a hydrogenating agent in water, and 85% cinnamyl alcohol (COL) selectivity was achieved with nearly complete CAL conversion. The acidity of ZnO activated the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of CAL, the basicity of CN facilitated the adsorption of FA, and the decorated Ru assisted the FA to H2 formation leading to the selective production of COL. A FT-IR study confirmed the effective adsorption of CAL through CO, yielding the selective formation of COL. In contrast, under photochemical conditions, hydrocinnamaldehyde (HCAL) was the selective hydrogenation product that was formed due to the efficient migration of charge carriers at the interface of the Z-scheme heterojunction of CN and ZnO. The synergistic effects at the interface were crucial for the charge transfer mechanism, enhancing the charge carriers' lifetime and enabling excellent charge separation under photocatalytic conditions. Detailed characterization and control reactions were performed to establish the structure–activity relationship and to conclude the plausible reaction mechanism under both conditions.
O group of CAL, the basicity of CN facilitated the adsorption of FA, and the decorated Ru assisted the FA to H2 formation leading to the selective production of COL. A FT-IR study confirmed the effective adsorption of CAL through CO, yielding the selective formation of COL. In contrast, under photochemical conditions, hydrocinnamaldehyde (HCAL) was the selective hydrogenation product that was formed due to the efficient migration of charge carriers at the interface of the Z-scheme heterojunction of CN and ZnO. The synergistic effects at the interface were crucial for the charge transfer mechanism, enhancing the charge carriers' lifetime and enabling excellent charge separation under photocatalytic conditions. Detailed characterization and control reactions were performed to establish the structure–activity relationship and to conclude the plausible reaction mechanism under both conditions.
Introduction
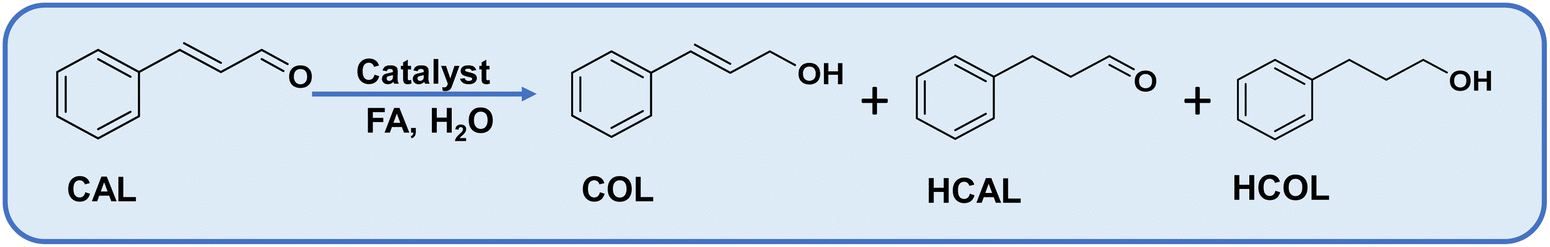
The selective reduction of α–β unsaturated carbonyl compounds is attractive to researchers and industrialists.1 Cinnamaldehyde (CAL), a typical example of α–β unsaturated carbonyl compounds, has gained interest over the last two decades due to the vast demand for its hydrogenated products, cinnamyl alcohol (COL) and hydrocinnamaldehyde (HCAL), due to their applications in pharmaceutical, cosmetics, flavorings, and fine chemicals industries.2,3 The selective synthesis of COL is challenging because the adsorption and reduction of CC are kinetically and thermodynamically favourable over CO.4 Uncontrolled hydrogenation produces the fully hydrogenated product hydrocinnamyl alcohol (HCOL) (Scheme 1).5 The selective synthesis of COL has been achieved with a few heterogeneous catalysts, which are mainly Ir and Pt-based and are non-preferable for industrial exploration due to their high cost.6,7
 | ||
| Scheme 1 The possible products formed in the selective hydrogenation of cinnamaldehyde. | ||
A low-cost Ru-based catalyst has shown promising activity for the selective hydrogenation of α–β unsaturated aldehydes to α–β unsaturated alcohols.8–10 Research has been conducted to increase the catalytic efficiency of Ru by modulating the nature of the support, hydrogenating agents, and introducing a second metal with Ru, solvents, and reaction conditions.11–13 The introduction of a second metal is witnessed to improve the efficiency but inevitably adds to the cost.14 Good metal–support interaction improves the metal dispersion, stability, and selectivity, owing to more efficient catalysis. N-containing carbon supports such as C3N4 (CN) exhibit considerable basicity due to high N content, which holds the metal nanoparticles, preventing the deactivation of the catalyst.15,16 The incorporation of metal oxides assists in establishing the interaction between the carbonyl group of the substrate and the catalyst surface through effective adsorption of CO, resulting in selective hydrogenation due to the Lewis acidic character of metal oxides.17 Primarily, molecular hydrogen is used as a hydrogenating agent for α–β unsaturated compounds.18–23 However, the process becomes more challenging when H2 is substituted by indirect hydrogenating agents such as formic acid, as maintaining alcohol selectivity is difficult.24 We have recently developed metal-free and metal-based catalysts for utilizing formic acid as an H2 and formylation source to form selective products.25–28 In addition to the catalyst design, the solvent governs the activity and product selectivity. Water is preferable in developing sustainable catalytic protocols. Due to its dipolar nature, water is effective in hydrogenating CO over CC in unsaturated substrates.29,30 Moreover, the water molecules undergo a hydrogen exchange with the formic acid and, thus, accelerate the reaction kinetics.31
Besides thermocatalytic exploration, cinnamaldehyde reduction can also be explored under photocatalytic conditions with different light sources.32 Generally, COL is detected as the primary product, and very few reports suggest the formation of HCAL as a major product under photocatalytic conditions.33 Most of the photocatalytic processes are based on noble-metal catalysts. A bimetallic catalyst composed of Au and Pd decorated on an acidic support offered good selectivity for HCAL (91%).34 A fully hydrogenated product, hydrocinnamyl alcohol (HCOL), was observed as the final product over a transition metal catalyst.35,36 Thus, designing an economic catalyst without compromising the HCAL selectivity needs to be developed.
Herein, a Ru decorated ZnO/g-C3N4 catalyst is synthesized for the selective hydrogenation of cinnamaldehyde under thermocatalytic and photocatalytic conditions. Under thermal conditions, COL was obtained with 85% selectivity in water with formic acid as a hydrogen source. To the best of our search, it will be the first report wherein formic acid has been utilized as a hydrogen donor for the selective hydrogenation of cinnamaldehyde in water. Interestingly, ∼100% selective production of HCAL was achieved under photocatalytic conditions (visible light). The interface formation between g-C3N4 (CN) and ZnO was key in the charge transfer mechanism, enhancing the charge carriers' lifetime and resulting in excellent charge separation under photocatalytic conditions. Two different products by switching thermocatalytic to photocatalytic conditions make this study exciting and would attract significant attention from researchers in diverse areas.
Experimental
Preparation of ZnO(X)/CN nanocomposites
The detailed synthesis procedures of ZnO and CN, and their properties are provided in the ESI.† 1 g of CN was dispersed in 100 mL of ethanol and stirred for 30 minutes. A specific quantity of zinc acetate (1, 1.5, and 2 g) was added and stirred until the complete evaporation of the ethanol at 80 °C. The dried materials were calcined at 400 °C for 3 h, and the resultant materials are denoted as ZnO(X)/CN (where X = 1, 1.5, and 2 g of Zn(OAc)2·2H2O).Preparation of (Y%) Ru@ZnO(X)/CN nanocomposites
The ZnO(X)/CN composites were dispersed in 50 mL of ethanol and subjected to sonication for 30 minutes. Subsequently, predetermined quantities of RuCl3·xH2O were dissolved in 10 mL of ethanol and added dropwise to the ZnO(X)/CN solution under continuous stirring. The resulting solution was stirred at 80 °C until the complete evaporation of ethanol. The materials were then dried at 75 °C in an oven overnight. The synthesized materials were reduced in a tube furnace at 300 °C for 3 h with a ramp rate of 5 °C min−1, in a 10% H2/Ar atmosphere to yield Y% Ru@ZnO(X)/CN (where Y = 0.5, 1, 1.5, 2, and 3 w% Ru).Catalytic reactions
Elaborated catalytic protocols are presented in the ESI.† The photocatalytic setup used in this study is shown in Fig. S1.† The details of quantification, gas analysis, quantum yield calculations, etc., are provided in the ESI.†Results and discussion
The synthesis of the Ru@ZnO/CN catalyst was carried out in three steps. Initially, urea was calcined to prepare CN. In the second step, a specific amount of zinc salt was impregnated into CN. By calcination, ZnO/CN composites with different amounts of ZnO were prepared. Desired quantities of Ru salts were impregnated into the composites, followed by a reduction in a tube furnace in a H2/Ar atmosphere to yield Ru@ZnO/CN. The step-wise synthesis procedure is presented in Scheme 2. | ||
| Scheme 2 Schematic presentation for the synthesis of Ru@ZnO/CN nanocomposites. | ||
Physicochemical characterization
The degree of purity of the materials was evaluated using Powder X-ray Diffraction (XRD) analysis. The diffraction pattern exhibited characteristic peaks corresponding to the (100), (002), and (101) crystallographic planes of ZnO (JPCDS card number 36-1451), which were observed at 2θ values of 31.8°, 34.4°, and 36.3°, respectively (Fig. 1a).37 The sharp peaks indicate the crystalline and hexagonal (wurtzite) structure of ZnO.38 The intensity of the planes (100) and (101) is relatively higher than that of (002), suggesting the dominance of rod-like ZnO over filament-like ZnO,37,38 but the (002) plane confirms the co-existence of both the phases, confirmed by the SEM images (discussed later).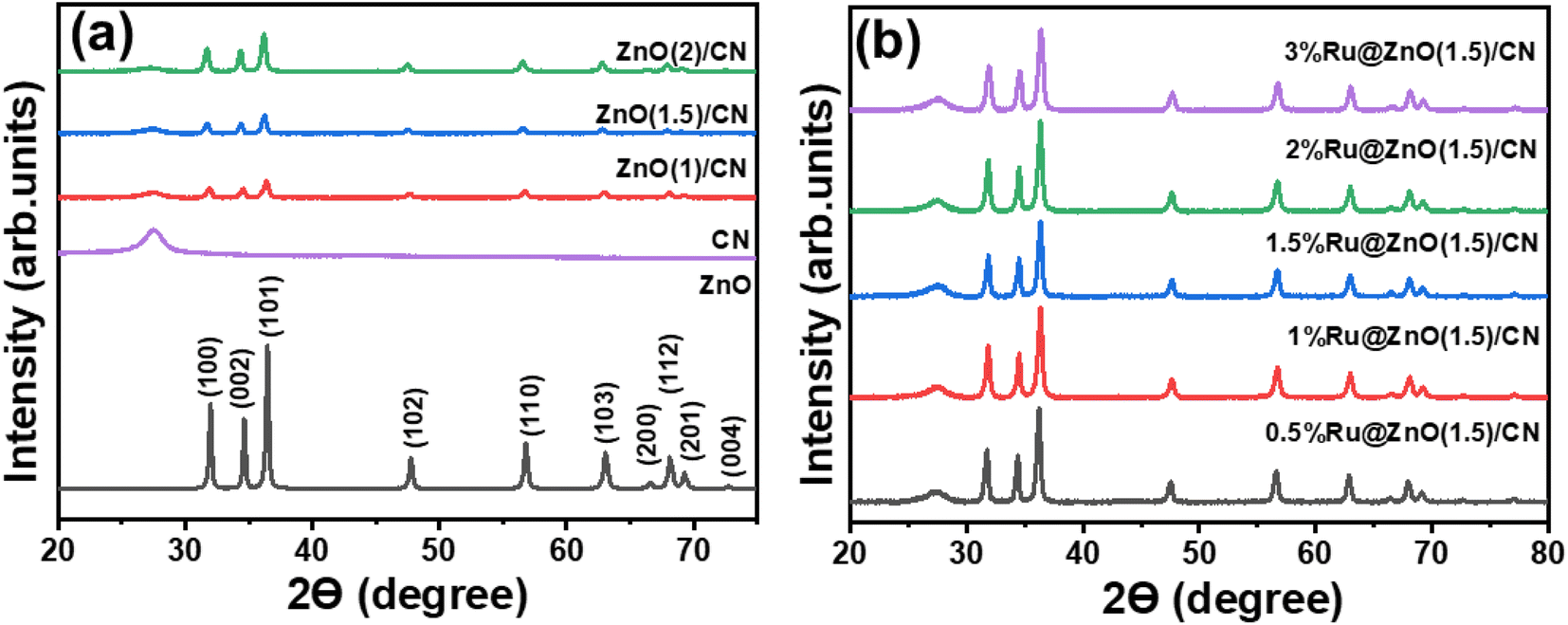 | ||
| Fig. 1 XRD patterns of (a) ZnO and ZnO(X)/CN nanocomposites, and (b) (Y%) Ru@ZnO(X) CN catalysts. | ||
All the ZnO/CN nanocomposites have a distinctive peak at 27.4°, attributed to the (002) plane of graphitic carbon of CN (Fig. 1a).39 Decrease in the intensity of (002) was observed with increasing ZnO content (Fig. 1a). It indicates the successful formation of the ZnO/CN composites where (002) plane intensity varies with the amount of CN in nanocomposites. Fig. 1b shows the XRD patterns of the Ru-decorated ZnO/CN composites. For better presentation, a plot keeping the intensity at the same scale on the Y-axis for all the catalysts is provided (Fig. S2a†), indicating no change in the intensity of CN with increasing Ru content. No peaks corresponding to Ru NPs were observed. For an accurate determination of the existence of Ru NPs, a 10% Ru/SS catalyst was prepared by loading 10 weight % of Ru NPs on silica spheres. The material was reduced employing the same reduction conditions as were used for the preparation of Ru-decorated ZnO(1.5)/CN catalysts. The XRD pattern of 10% Ru/SS shows intense peaks ascribed to different planes of the Ru NPs (Fig. S2b†) corresponding to the JCPDS card number 6-663. Comparing the XRD patterns of 10% Ru/SS and Ru-decorated ZnO(1.5)/CN, no peak corresponding to Ru NPs was observed in Ru-decorated ZnO(1.5)/CN catalysts. No diffraction peak corresponding to Ru NPs in the catalysts synthesized in this study is attributed to lower % Ru loading, small size, and high dispersion of Ru NPs.
The surface morphology of the catalysts was examined by scanning electron microscopy (SEM), and an irregular morphology was observed in the SEM image of ZnO at low magnification. On higher magnification, a mixture of rod and filament-like ZnO was observed with rods in excess, as suggested by the XRD analysis (Fig. S3a and b†).38 The SEM images of ZnO/CN composites are provided in Fig. S3c–e,† and the SEM images of 1% Ru@ZnO(1.5)/NC and 3% Ru@ZnO(1.5)/NC are provided in Fig. S3f and g.† The FESEM images of the ZnO(1.5)/CN composite show rods of ZnO distended outwards from the sheets of CN, which proposes the 3-dimensional growth of ZnO over the sheet-like morphology of CN, confirming the stacked interlinked network at the interface of the two materials (Fig. 3c and d). The HR-TEM images of 1% Ru@ZnO(1.5)/NC confirm the ZnO and CN phases. ZnO nanorods are visibly protruding outwards from the nanosheets of CN, forming an interface between the two materials (Fig. 3e and f). The (002) plane of hexagonal wurtzite ZnO was identified as the source of lattice fringes with a d-spacing of 0.261 nm (Fig. 3g).40 In Fig. 2h, the distant lattice fringes, at 0.200 and 0.253 nm, are associated with the (101) plane of metallic Ru NPs and (011) of ZnO, which suggests the successful Ru(0) incorporation in the 1% Ru@ZnO(1.5)/NC catalyst.41,42 Similar trends are observed for 3% Ru@ZnO(1.5)/NC (Fig. S3h–j†). The atomic % and metal dispersion of 1% Ru@ZnO(1.5)/NC and 3% Ru@ZnO(1.5)/NC catalysts were analyzed by EDS and elemental mapping (Fig. S4 and S5†). The elemental mapping demonstrates that Ru is homogeneously dispersed on the ZnO/CN nanocomposite.
 | ||
| Fig. 2 FESEM images of (a) CN, (b) ZnO, and (c) the ZnO(1.5)/CN nanocomposite, (d)–(e) HRTEM image of the 1% Ru@ZnO(1.5)/CN nanocomposite, (f) magnified image of a portion of the image (e) showing the interface between ZnO and CN, (g) image showing fringes of ZnO from a selected domain, and (h) image showing fringes of ZnO and Ru. | ||
X-ray photoelectron spectroscopy analysis was conducted for the 1% Ru@ZnO(1.5)/CN and 3% Ru@ZnO(1.5)/CN to examine the chemical states of the elements. The catalyst comprises Ru, Zn, C, N, and O, specified by the surface survey scan of 1% Ru@ZnO(1.5)/CN (Fig. 3a). The Zn XPS spectrum is composed of two peaks, at binding energies of 1044.8 and 1021.7 eV, assigned to the Zn 2p1/2 and Zn 2p3/2 peaks of Zn2+, respectively.43 The O 1s exhibits four peaks at binding energies 533.0, 531.4, 531.7, and 530.4 eV associated with Ru–O, defects present in the framework, O–H groups attached to the CN sheets, and the lattice oxygen (Zn–O), respectively.44 N 1s was deconvoluted into three peaks, 400.8 eV (NHx, amine functional groups), 399.8 (NC3, tertiary nitrogen), and 398.5 eV (pyridinic N).45 The dominance of the pyridinic N is in accordance with the urea-derived pristine CN. The high-resolution spectrum for C 1s is composed of three peaks. The peaks at 287.9, 284.8, and 284.1 are ascribed to sp2 hybridized N–C–N carbon, (C)3–N bonded carbon, and C–C bonded carbon.46 The peak at 284.47 is due to the overlapping of Ru 3d and C 1s and the high intensity is due to the merger of Ru and C signals.45 Furthermore, the greater peak intensity of C 1s in 3% Ru@ZnO(1.5)/CN (Fig. S6e†) than that of 1% Ru@ZnO(1.5)/CN confirms this observation. Fig. 3e shows the deconvoluted peaks for Ru 3d and C 1s. For the sake of clarity and better understanding, Ru 3p was considered for the further confirmation of chemical states (Fig. 3f), which also consist of an intense peak at 475.2 for Zn LMM47 (refer to surface scan, Fig. 3a) due to which Ru 3p3/2 and Ru 3p1/2 were deconvoluted separately (Fig. 3g and h). Fig. 3g shows two deconvoluted peaks of Ru 3p3/2 at 461.9 and 464.4 eV corresponding to Ru(0) and Ru(+4). Similarly, Fig. 3h shows peaks at 483.6 and 486.3 eV for Ru 3p1/2 corresponding to Ru(0) and Ru(+4), respectively.48,49 The XPS analysis plots of 3% Ru@ZnO(1.5)/CN are provided in Fig. S6 in the ESI.† Similar results were obtained except for more intensified peaks corresponding to Ru due to the higher % loading. The atomic % of the elements based on XPS is provided in Table S1.† Bulk elemental analysis (using microwave plasma-atomic emission spectroscopy) was conducted to determine the Ru NP concentration, which was estimated to be 0.94% and 2.72% for 1% Ru@ZnO(1.5)/CN and 3% Ru@ZnO(1.5)/CN, respectively.
 | ||
| Fig. 3 (a) XPS surface survey of 1% Ru@ZnO(1.5)/CN, (b) Zn 2p, (c) O 1s, (d) N 1s, (e) Ru 3d and C 1s, (f) Ru 3p and Zn LMM, (g) Ru 3p3/2, and (h) Ru 3p1/2. | ||
The BET analysis was conducted for all the synthesized catalysts to compare the effect of composite formation and metal loading on the surface properties. The adsorption isotherms for all the ZnO/CN composites and Ru decorated composite show a type II isotherm with H3 hysteresis loops, indicating the formation of interparticle mesopores in the nanocomposites (Fig. S7†).50 Moreover, with the introduction of ZnO into the CN, the surface area of the composite increased from 45 to 65 m2 g−1 (Table S2†), which could be due to the thermal exfoliation and fragmentation of CN sheets, as indicated by SEM images. The metal loading marginally increased the surface area because Ru NP sites provided additional sites for N2-adsorption.51,52 Table S2† summarises all synthesized catalysts' surface area and total pore volume.
For calculating the compositions of the ZnO/CN nanocomposites, the zinc source (Zn(OAc)2)·2H2O was subjected to TGA. 1/5 mass of ZnO was obtained from Zn(OAc)2·2H2O (Fig. S8a†). A sharp weight loss near 300 °C was observed in the TGA of Zn(OAc)2·2H2O. Above 300 °C, Zn(OAc)2·2H2O got converted into ZnO, and no significant weight loss was observed above 400 °C. Therefore, 400 °C was the best calcination temperature for preparing ZnO. The ZnO showed good stability in the thermogram with ∼2% weight loss up to 800 °C (Fig. S8b†). In contrast, CN showed 100% weight loss below 700 °C (Fig. S8a†). The composites show different % of weight loss up to 700 °C, which depends on the composition of the nanocomposite (Fig. S8b†). Furthermore, all the catalysts were thermally stable up to 400 °C, which signifies that catalysts are not prone to decomposition at the reaction temperature adopted in this study.
CO2-TPD was conducted to estimate the basicity of the catalysts imparted by CN predominantly. The basicity is helpful for the adsorption and activation of FA. A peak associated with the moderate basic sites in the temperature range of 400 to 500 °C was observed in all the ZnO(X)/CN catalysts. The peak intensity was directly proportional to the CN content in the catalyst (Fig. S9a†). Similarly, NH3-TPD was undertaken to evaluate the acidic sites due to ZnO, which is required for the adsorption and activation of the CO group of CAL. The acidity increased with the increasing amount of ZnO in the composite catalysts. ZnO(1)/CN had the lowest acidity, whereas ZnO(2)/CN had the highest value of acidity (Fig. S9b†).
Optical characterization
The optical properties of CN, ZnO, and their composite were examined through solid-state diffuse reflectance ultraviolet-visible spectroscopy (DRUV-vis). The aforementioned catalysts exhibit an absorbed edge in the visible area of the electromagnetic spectrum (Fig. 4a). ZnO showed a band edge in the UV domain, while the CN adsorption edge was in the visible domain. Upon the formation of the composite of these two materials, a red shift was observed. The increased Ru content in the catalyst redshifted the band edge.53 The bandgap of the synthesized catalysts was determined through Tauc plots (Fig. S10†), which employed the formula (αhν)1/r = β(hν − Eg). The said formula comprises various parameters, including the absorption coefficient (α), photon energy (hν), bandgap (Eg), band tailing parameter (β), and power factor (r).54,55 It is well established in the literature that both catalysts execute direct transition with an r value of ½.56 Table S3† lists the band gaps of all the synthesized catalysts. For a comparative purpose, the band gap of the composite was also calculated. The reduction in the band gap of the composite is attributed to the decoration of Ru NPs, which possess a low Fermi energy. It facilitated the transfer of photogenerated electrons in the conduction band of the Ru NPs, leading to improved charge separation and a decreased band gap.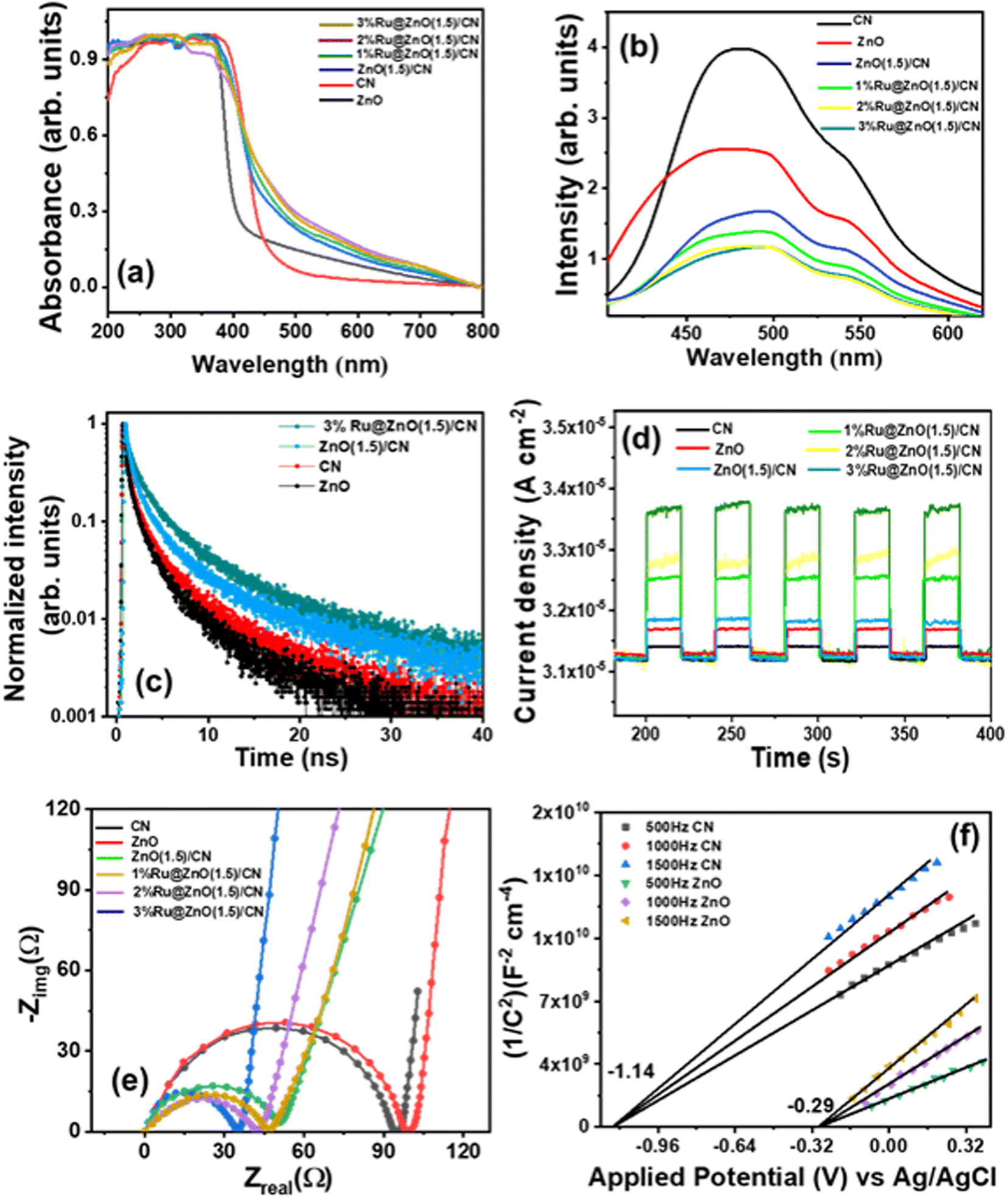 | ||
| Fig. 4 (a) DRUV-vis spectra, (b) photoluminescence spectra, (c) photoluminescence decay, (d) transient photocurrent (i–t) response, (e) electrochemical impedances of various photocatalysts and (f) M–S plots for CN and ZnO. | ||
A photoluminescence (PL) experiment was performed to evaluate the charge separation efficiency (Fig. 4b). CN exhibited a broad and most intense emission covering the visible region with a peak maximum of 480 nm. ZnO showed a lower intense emission spectrum than CN. The heterojunction of CN and ZnO separates the charge carriers more efficiently; therefore, less intense emission spectra were observed.55 Moreover, the Ru-decorated nanocomposite showed relatively lower emission with a broader peak in the visible region. The Ru NPs, with their higher work function and low Fermi level, effectively captured the photogenerated electrons from the semiconductor, resulting in efficient separation of charge carriers. This process leads to a reduction in the intensity of the photoluminescence (PL) emission peak. The Ru NPs also increased the lifetime of charge carriers by reducing their recombination rate, potentially leading to enhanced photocatalytic efficiency. The luminescence decay characteristics of unmodified CN, ZnO, and their composite were examined with the Time-Correlated Single Photon Counting (TCSPC) technique. The decay curves were fitted using a mathematical equation  that accounts for the contribution of discrete emissive species.45 The pre-exponential factors and the time of excited-state luminescence decay were determined for each component. The average decay time, 〈τ〉, was calculated using
that accounts for the contribution of discrete emissive species.45 The pre-exponential factors and the time of excited-state luminescence decay were determined for each component. The average decay time, 〈τ〉, was calculated using  , and the fractional contribution for each decay was obtained using
, and the fractional contribution for each decay was obtained using  .52 A three-exponential fit was used to model the decay curve. The result showed that the nanocomposite had a higher average decay lifetime of 3.2 ns than the individual components, and the introduction of Ru NPs further raised the decay lifetime to 4.11 ns (Fig. 4c). The TCSPC analysis confirmed that the nanocomposite had a better decay lifetime than pristine CN and ZnO, and introducing Ru NPs enhanced the charge retention capability by accommodating photogenerated electrons. The details of the investigation and calculated parameters are provided in Table S4.†
.52 A three-exponential fit was used to model the decay curve. The result showed that the nanocomposite had a higher average decay lifetime of 3.2 ns than the individual components, and the introduction of Ru NPs further raised the decay lifetime to 4.11 ns (Fig. 4c). The TCSPC analysis confirmed that the nanocomposite had a better decay lifetime than pristine CN and ZnO, and introducing Ru NPs enhanced the charge retention capability by accommodating photogenerated electrons. The details of the investigation and calculated parameters are provided in Table S4.†
Photoelectrochemical characterization
The photoelectrochemical performance of the materials was assessed by conducting various electrochemical analyses, including linear sweep voltammetry (LSV), on–off photocurrent (i–t), and electrochemical impedance (EIS). These techniques were used to evaluate the stability and lifetime of the generated charge carriers in CN, ZnO, and their heterojunction with varying amounts of Ru NPs. Additionally, Mott–Schottky (M–S) plots were constructed at different frequencies (500 Hz, 1000 Hz, and 1500 Hz) to calculate the flat band potential of CN and ZnO. LSV experiments were conducted under dark and illuminated conditions in the persistence of having a survey for their charge separation and migration proficiencies (Fig. S11†). It was observed that the current density increased significantly with increasing potential under both dark and illuminated conditions. Without light irradiation, the current density values for CN and ZnO were 1.8 × 10−3 A cm−2 and 2.1 × 10−3 A cm−2, respectively, at 1.7 V vs. Ag/AgCl. Upon exposure to light, these values increased to 2.9 × 10−3 A cm−2 and 3.1 × 10−3 A cm−2, respectively, indicating light-induced photoactivity in both cases.57,58 The heterojunction catalysts transport the charge carriers to the surface more efficiently, gradually increasing the current density to 2.5 × 10−3 A cm−2 and 3.7 × 10−3 A cm−2 at 1.7 V vs. Ag/AgCl under dark and light conditions. In the Ru NP decorated catalyst, the metal NPs more efficiently channel the charge; therefore, it has the highest current density of 3.1 × 10−3 A cm−2 at 1.7 V vs. Ag/AgCl in light.The transient current response of all photocatalysts was recorded with an on–off photocurrent experiment (i–t) (Fig. 4d). The semiconductor material exhibited an increased current response upon irradiation and promptly returned to its initial value upon turning off the light source. The analysis of the current response was repeated for multiple test cycles, and a consistent response was observed in each cycle, indicating stable performance. CN showed the least response among all catalysts because of easy charge recombination. ZnO possessed a moderately higher response for the test cycle evidencing the slow recombination. The CN/ZnO heterojunction showed a relatively higher response than bare CN and ZnO because, in the heterojunction, the electrons and holes were separated in different parts of the junction, enhancing its catalytic activity. The Ru NP decorated CN/ZnO catalysts exhibited a higher photocurrent response than the bare ZnO(1.5)/CN. The rise in the Ru content in the heterojunction resulted in an increase in the current response because Ru NPs aided in the effective migration of charge carriers.57 The heterojunction catalyst with 3% Ru@ZnO(1.5)/CN showed the highest current response, which can be attributed to its higher conductivity. The relationship between carrier concentration (N) and electrical conductivity (σ) can be expressed using the equation σ = Neμ, where “e” is the charge and μ denotes carrier mobility.45 Therefore, in 3% Ru@ZnO(1.5)/CN, a significantly higher charge carrier generation and migration was observed during the light illumination. In conclusion, Ru NPs promoted the mobility of the produced charge carriers and improved the catalyst's performance.
EIS was performed for all the photocatalysts under light illumination, and the resulting Nyquist plots (Fig. 4e) showed semicircles, the size of which correlates directly with the charge transfer rate.59 The diameter of the semicircle reflects the interfacial charge transfer resistance (Rct) present at the electrode–electrolyte interface. A reduction in the diameter of the semicircle signifies a decline in interfacial charge transfer resistance, thereby indicating improved charge transfer for the migration of charge carriers. The Rct values were determined for CN, ZnO, ZnO/CN, and Ru-decorated ZnO/CN. The largest semicircle diameter (Rct) was obtained for ZnO, whereas CN has a comparably smaller arc diameter, indicating better charge migration. The heterojunction showed a lower semicircle arc compared to ZnO and CN. The introduction of Ru NPs facilitated the charge carriers' migration, resulting in a reduction of the arc diameter with an increase in the Ru content. Among the various photocatalysts tested, 3% Ru@ZnO(1.5)/CN demonstrated the smallest Rct value, indicating its superior photoactivity. The Ru NPs expedited the proficient migration of charge carriers by stimulating the charge transfer mechanism.
Band structure evaluation
Mott–Schottky analysis was performed to find the band edges of the photocatalysts (Fig. 4f). The equation 1/C2 = 2 [V − Vfb − (kbT/e)]/(εε0eA2Nd), where C denotes capacitance, V stands for applied potential, Vfb presents flat band potential, k denotes the Boltzmann constant, T denotes temperature, e is the charge (1.602 × 10−19 C), ε0 is the vacuum permittivity (8.85 × 10−14 F cm−2), εr is the dielectric constant of the photocatalyst, and Nd stands for donor density, was applied, and the plots were constructed for 1/C2vs. applied potential. The M–S plots for CN and ZnO exhibited a positive slope, implying n-type semiconductors.60 The flat band potentials (Efb) for CN and ZnO were determined from the x-intercepts of the Mott–Schottky plots and were found to be −1.14 V vs. Ag/AgCl and −0.29 V vs. Ag/AgCl, respectively (Fig. 4f). In n-type semiconductors, the flat band potential lies below the conduction band (CB) edge. The Efb values were employed to establish the position of the conduction band edge (Ecb) by using the relation Ecb = Efb − 0.1 V.61 The equation (ENHE = EAg/AgCl + 0.196) was applied to standardize the potential values.62 The calculated values of Ecb for CN and ZnO were −1.04 and −0.19 V vs. NHE (pH = 7). Using the ECB, the valence band potential (EVB) was obtained by applying the relation EVB = ECB + Eg. CN possesses a valence band edge at 1.79 V vs. NHE, whereas ZnO has an Evb potential of 2.84 V vs. NHE.The complete band structures of CN, ZnO, and ZnO(1.5)/CN were explored using valence band X-ray photoelectron spectroscopy (VB-XPS), ultraviolet photoelectron spectroscopy (UPS), and diffuse reflectance UV-visible (DRUV-vis) spectroscopy. The VB-XPS spectra of CN and ZnO (Fig. 5a and c) revealed that their valence band maxima (VBM) were positioned at 1.71 eV and 2.30 eV, respectively, with respect to their Fermi levels (Ef).63 The Fermi level of both CN and ZnO was linked to their work function Φ, which is the energy difference between the Fermi level and vacuum. The UPS spectra (Fig. 5b and d) provided further insight into the work function Φ values for CN and ZnO, which were calculated to be 4.51 eV and 5.1 eV, respectively, using the relation Φ = 21.22 − ESE cutoff + Ecutoff.45,64 By combining the data of VB-XPS, DRUV-vis, and UPS spectra, the calculated conduction band (CB) and valence band (VB) values of CN were 6.22 eV and 3.40 eV, respectively, with respect to vacuum.64 The CB and VB values of ZnO were 7.40 eV and 4.27 eV, respectively (Fig. 5e). The band edge values are expressed in Evac and can be changed to ENHE through the addition of −4.44 ± 0.02 eV.65 The band edges of CN and ZnO (Fig. 5e) were observed to align well with the band structure estimated from the Mott–Schottky plots, with a slight shift.
 | ||
| Fig. 5 (a) VB-XPS spectrum of CN, (b) UPS spectrum of CN, (c) VB-XPS spectrum of ZnO, (d) UPS spectrum of ZnO, and (e) band structures of photocatalysts of this study. | ||
The Fermi level is the maximum energy level at which an electron can exist. When CN and ZnO are combined in a composite, the Fermi level of CN (4.51 eV vs. vacuum) declined, and ZnO (5.1 eV vs. vacuum) rose till it attained a new equilibrium at 4.63 eV vs. vacuum observed in the UPS spectrum of ZnO(1.5)/CN (Fig. S12a†). Therefore, the nanocomposite had a newly formed Fermi level at 4.63 eV vs. vacuum (Fig. 5e). It is thoroughly described in the literature that Ru has a Fermi level of 4.7 eV vs. vacuum, which is lower than the calculated Fermi level of the nanocomposite (Fig. 5e).66–68 Therefore, the decoration of Ru NPs in the ZnO(1.5)/CN nanocomposite leads to the transfer of electrons over Ru NPs with marginal rearrangement in the Fermi level (discussed later). The UPS spectrum of 3% Ru@ ZnO(1.5)/CN showed that the material had a Fermi level at 4.66 eV vs. vacuum (Fig. S12b†), which lies between the Ru and ZnO(1.5)/CN nanocomposite Fermi levels (Fig. 5e).
Catalytic activity
Thermocatalytic performance for selective hydrogenation of cinnamaldehyde with formic acid
Catalysts were employed for the hydrogenation of cinnamaldehyde (CAL) to cinnamyl alcohol (COL) as a selective product using formic acid as a hydrogen donor in water. Cinnamaldehyde can undergo hydrogenation at two functional sites, (i) CO and (ii) CC. The hydrogenation of the former gives COL, whereas the hydrogenation of the latter forms hydrocinnamaldehyde (HCAL). The complete reduction product is hydrocinnamyl alcohol (HCOL) (Scheme 1). A combination of the support and active metal sites is obligatory for the adsorption of the cinnamaldehyde through a specific mode, dispersion of active sites for the effective dissociation of FA to H2 and CO2. In consideration, CN was chosen as a basic support for the adsorption of FA and dispersion of metal NPs, and metal oxides as an additional active support to tune the surface properties of the catalyst.69 Amphoteric oxides are beneficial for the adsorption of α–β unsaturated substrates through the carbonyl group and obtaining α–β unsaturated alcohols selectively, as evidenced by previous reports.70 Due to these reasons, various amphoteric metal oxides were explored for this transformation (Table S5†). Among them, ZnO was superior, yielding 17% CAL conversion with 100% COL selectivity. The pronounced selectivity for COL is attributed to the electropositive nature of Zn2+, which assists in the adsorption and activating of the carbonyl group. CN alone offered only 6% conversion, but combining ZnO/CN elevated the catalytic activity (Table 1, compare entries 2 and 4–6). Different contents of Ru were decorated over ZnO/CN composites to obtain the maximum COL yield. Two control catalysts, 1% Ru@ZnO and 1% Ru@CN, were synthesized to elucidate the role of ZnO and CN separately. The former offered 40% conversion with 89% COL selectivity, whereas the latter afforded exceptionally high activity with 100% conversion and selectivity for the completely reduced product, HCOL (Table 1, entries 7 and 8). It can be inferred that in order to acquire improved catalytic performance, a combination of ZnO and CN is obligatory. The highest COL selectivity (85%) was achieved over 1% Ru@ZnO(1.5)/CN (Table 1, entry 10). On further increasing the Ru % to 1.5%, the COL selectivity decreased at the expense of the wholly reduced product HCOL. It suggests that a lower content of Ru was suitable for the selective reduction of CAL to COL. Moreover, when the same catalyst was employed for hydrogenation with molecular H2, 100% COL selectivity was obtained, but CAL conversion was only 30%. The results suggest that the reaction was limited due to the mass transfer of H2 to the active sites, while the in situ FA dissociation generated H2 that was efficiently picked by the neighboring Ru sites and provided Ru–H species for the reduction of CAL to COL. The gas generated during the reaction was collected and subjected to gas chromatography (GC) using a gas-tight syringe and monitored using a TCD detector. The chromatogram shows the formation of H2 and CO2 (Fig. S13†). For comparison, four control catalysts based on cheap transition metals, such as Co and Ni, were prepared (please see XRD Fig. S2c†). Noble metals such as Pd were not chosen as their cost is much higher than Ru. The control catalysts were synthesized by following the synthesis protocol of this study. The catalysts were evaluated under optimized reaction conditions (Table S6†). Based on the catalytic activity data, it can be concluded that cheaper transition metal catalysts were not suitable for this reaction. The cheapest noble metal Ru was the best choice for such a selective catalytic transformation.
|
|
||||||
|---|---|---|---|---|---|---|
| EN | Catalyst | Conversion (%) | Selectivity (%) | TOF (min−1) | ||
| CAL | COL | HCAL | HCOL | |||
| a Reaction conditions: CAL (0.5 mmol), FA (4.5 mmol), catalyst (20 mg), water (3 mL), temperature (140 °C), and time (6 h). b Average of three measurements presented as an integer. c Reaction conducted with 10 bar H2 instead of FA. d TOF is calculated considering the whole mass of the catalyst. e TOF is calculated considering the Ru content. | ||||||
| 1 | None | 2 | 0 | 100 | 0 | — |
| 2 | CN | 6 | 0 | 100 | 0 | 1.4 |
| 3 | ZnO | 15 | 100 | 0 | 0 | 18.3 |
| 4 | ZnO(1)/CN | 20 | 60 | 40 | 0 | 4.5 |
| 5 | ZnO(1.5)/CN | 18 | 65 | 35 | 0 | 3.9 |
| 6 | ZnO(2)/CN | 16 | 67 | 33 | 0 | 0.7 |
| 7 | 1% Ru@ZnO | 40 | 89 | 0 | 11 | 8.1 e(1015) |
| 8 | 1% Ru@CN | 100 | 0 | 0 | 100 | 23 e(2538) |
| 9 | 0.5% Ru@ZnO(1.5)/CN | 61 | 73 | 0 | 27 | 13.4 e(3096) |
| 10 | 1% Ru@ZnO(1.5)/CN | 96 | 85 | 0 | 15 | 40 e(2436) |
| 11 | 1.5% Ru@ZnO(1.5)/CN | 100 | 77 | 0 | 23 | 42 e(1685) |
| 12 | 1% Ru@ZnO(1.5)/CN | 30 | 100 | 0 | 0 | 12.6 e(761) |
The involvement of water in the hydrogen-exchange pathway with formic acid is already documented, which suggests that it is a suitable solvent for the selective formation of COL.25,31 Furthermore, water also assists in the effective dissociation of FA by the exchange mechanism, already proposed in our previous report.25 Two control reactions for the formic acid-mediated hydrogenation were conducted to confirm the exchange pathway between water and formic acid: first using H2O as a solvent and the other based on isotope labelling in which D2O was used instead of H2O. Both reactions were conducted under optimized conditions. The reaction mixtures were then subjected to GC-MS analysis. The mass spectra of the products formed in both cases are provided in Fig. S14.† In H2O, cinnamyl alcohol (C9H10O) was the established product with a molecular mass of 134 g mol−1, consistent with the analyzed mass spectrum (B). Three noticeable fragments, in this case, are 105, 115, and 134 (highlighted in yellow), with the base peak 92. However, in the D2O mediated reaction, deuterated cinnamyl alcohol (C9H9DO) with a molecular mass of 135 (A) was formed instead of cinnamyl alcohol (C9H10O). An increment of 1 was observed in the three fragments, which appeared at 106, 116, and 135 with the same base peak of 92. It confirms that an exchange reaction occurred between formic acid and H2O, which facilitated formic acid to dissociate into the H2 and CO2 (confirmed by GC analysis, Fig. S13†). When D2O was used, the exchange of “D” from D2O occurred with HCOOH and formed HCOOD, which decomposed to HD and CO2. The generated HD reacted with cinnamaldehyde to produce deuterated cinnamyl alcohol (C9H9DO). Hence, the molecular mass of deuterated cinnamyl alcohol increased by “1” when the aldehydic group of cinnamaldehyde was hydrogenated in the presence of formic acid and D2O. A GC-MS chromatogram of the reaction mixture recovered after 3 h in thermal hydrogenation of CAL shows the formation of COL and HCOL (Fig. S15†). 1H NMR of COL in the reaction mixture of thermal hydrogenation of CAL is provided in Fig. S16.†
All the parameters influencing the catalytic activity were varied to optimize the reaction conditions. Initially, the temperature was varied from 120 °C to 160 °C. Only 18% CAL conversion with 90% COL selectivity was obtained at 120 °C. On conducting the reaction at an elevated temperature (160 °C), almost 100% conversion was achieved, but instead of COL, HCOL was selectively obtained. So, the temperature (140 °C) was the optimum temperature, yielding 85% COL with 96% conversion (Fig. 6a). The role of FA concentration was also explored (Fig. 6b). 4.5 mmol of FA was sufficient for achieving the highest yield of COL (85%), whereas further increasing the FA, the COL selectivity was reduced. Similarly, the reaction time and catalyst amount were optimized. The best COL yield was achieved in 6 h with 20 mg catalyst (Fig. 6c and d). To our knowledge, this is the first report where FA is employed as a hydrogen source for the selective reduction of CAL to COL over a heterogeneous catalyst. Therefore, providing comparative activity data for this catalytic protocol is difficult.
 | ||
| Fig. 6 Variation of (a) reaction temperature (°C) [reaction conditions: CAL (0.5 mmol), FA (4.5 mmol), catalyst (20 mg), water (3 mL), and time (6 h)], (b) formic acid concentration (mmol) [reaction conditions: CAL (0.5 mmol), catalyst (20 mg), water (3 mL), time (6 h), and temperature (140 °C)], (c) time (h) [reaction conditions: CAL (0.5 mmol), catalyst (20 mg), water (3 mL), temperature (140 °C), and FA (4.5 mmol)] and (d) catalyst amount (mg) [reaction conditions: CAL (0.5 mmol), water (3 mL), time (6 h) temperature (140 °C), and FA (4.5 mmol)] for selective hydrogenation of CAL over the 1% Ru@ZnO(1.5)/CN catalyst. | ||
The kinetics of the reaction was investigated by conducting experiments at 140 °C for different durations. The CAL conversion was assessed for various order equations, and a first-order rate equation provided a linear graph. The rate constant (k) at 140 °C was 4.33 × 10−5 s−1, deduced from the slope of the linear plot. The experiment was conducted at different temperatures (130–150 °C), resulting in an increased reaction rate and rate constant with a temperature rise. The rate constant values were 4.33 × 10−5 s−1 at 130 °C and 7.26 × 10−5 s−1 at 150 °C. The Arrhenius equation was used to compute the Ea, and a linear plot of ln[k] vs. 1/T was obtained with an R2 value of 0.99 (Fig. 7b). The resulting straight-line plot ascertained the Ea as 82.44 K J mol−1.
 | ||
Fig. 7 (a) Kinetic plots of thermocatalytic CAL hydrogenation at various temperatures (130, 140 and 150 °C) using 1% Ru@ZnO(1.5)/CN, (b) ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k vs. 1/T plots for Ea calculation of CAL hydrogenation, (c) kinetic plots of photocatalytic CAL hydrogenation at different light intensities (270, 590, 925, and 1210 W cm−2) using 3% Ru@ZnO(1.5)/CN, and (d) dependence of the rate constant k on light intensity at ambient temperature for CAL hydrogenation over 3% Ru@ZnO(1.5)/CN. k vs. 1/T plots for Ea calculation of CAL hydrogenation, (c) kinetic plots of photocatalytic CAL hydrogenation at different light intensities (270, 590, 925, and 1210 W cm−2) using 3% Ru@ZnO(1.5)/CN, and (d) dependence of the rate constant k on light intensity at ambient temperature for CAL hydrogenation over 3% Ru@ZnO(1.5)/CN. | ||
Plausible reaction mechanism
The selective production of a particular product requires a specific reaction site to be adsorbed and activated on a catalyst surface. Cinnamaldehyde is a tricky substrate with two functional adsorption sites, a double bond in conjugation with an aldehyde group. Numerous research reports have documented and illustrated the higher feasibility of the hydrogenation of the conjugated double bond over the carbonyl group due to favorable reaction kinetics and thermodynamics.1,71,72 An additional energy is required to activate the carbonyl group, and the surface energetics of the catalyst plays a key role. The large size of the benzene ring occupies a large space and creates a steric hindrance for the adsorption of CC, and promotes the adsorption of CO on the catalyst surface. Such promotional adsorption resulted in the higher selectivity of the hydrogenation of CO.73,74 A temperature-dependent adsorption study was conducted using an FT-IR study. For this purpose, 5 μL of cinnamaldehyde was adsorbed on 50 mg of catalyst at different temperatures ranging from 25 °C to 130 °C. The physisorbed substrate was removed by flowing Ar for 5 min. The peak at 1675 cm−1 arose due to the carbonyl stretching of the cinnamaldehyde at 25 °C (Fig. S17†). With the increase in the temperature, a gradual red shift is observed from 1675 to 1656 cm−1. The red shift indicates that with the rise in temperature, the cinnamaldehyde chemisorbed over the surface of the catalyst through the CO group, preferably. Therefore, under the thermal conditions, the COL was obtained as the particular product, which suggests that a specific temperature was required to reach the threshold barrier for the activation of the α–β unsaturated carbonyl group. Moreover, incorporating a suitable amount of Lewis acidic ZnO assists in interacting electronegative O2− of the CO group of the CAL with the Zn2+ on the catalyst's surface.75,76 The adsorption and activation of FA were achieved by combining basic CN and Ru NPs, which preferentially dissociate FA into H2 + CO2 in water through the metal-formato complex.21,22 The generated H2 was dissociatively adsorbed on the Ru NPs resulting in the formation of Ru–H.26 The Ru adsorbed H was transferred to the CO of CAL to yield COL as the selective product. A schematic presentation for the thermo-catalytic plausible reaction mechanism for FA-assisted CAL hydrogenation to COL is provided in Scheme 3.
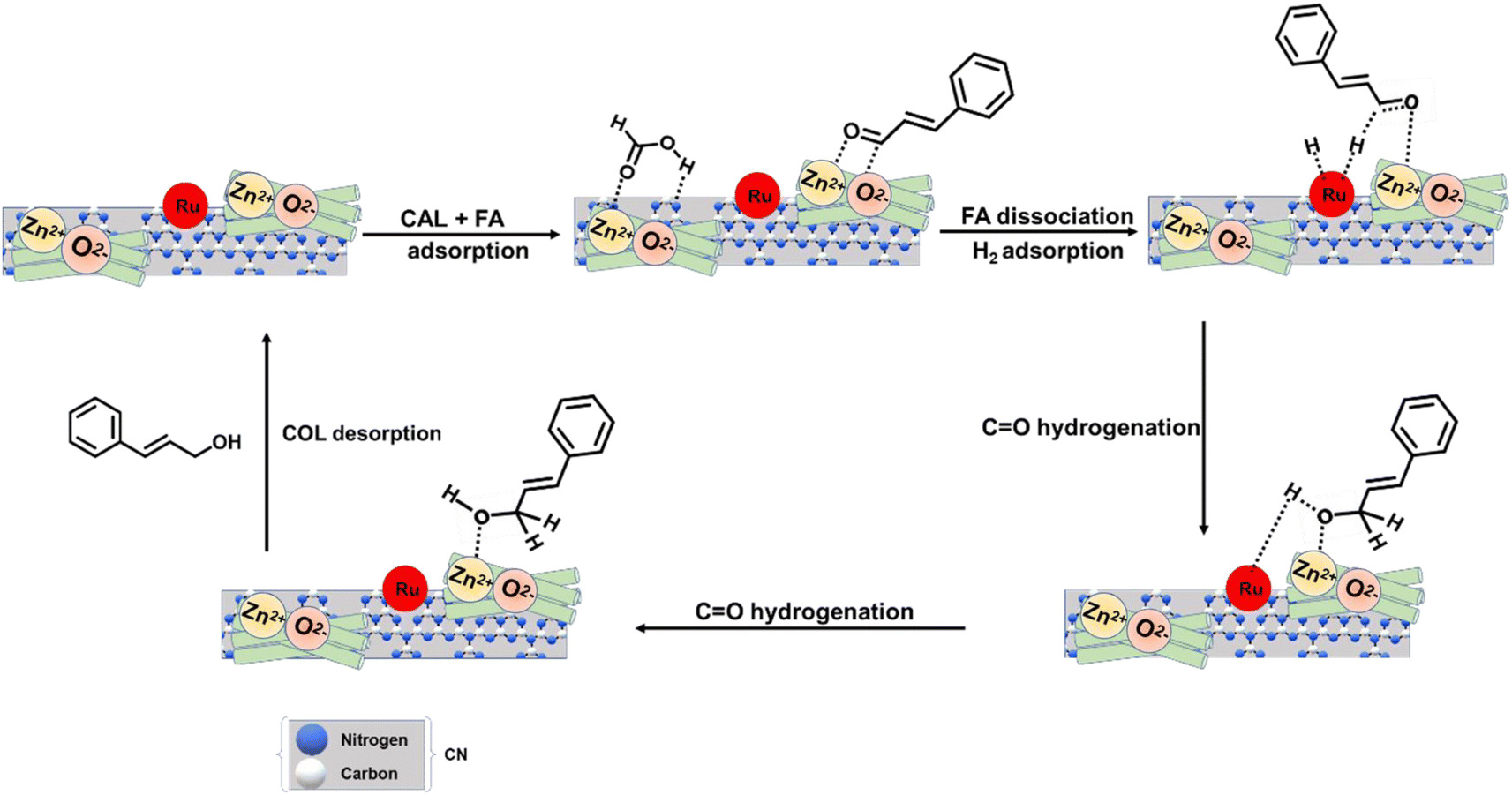 | ||
| Scheme 3 Schematic presentation for the thermo-catalytic plausible reaction mechanism for FA-assisted CAL hydrogenation over the 1% Ru@ZnO(1.5)/CN catalyst. | ||
Photocatalytic activity of Ru@ZnO(1.5)/CN for selective hydrogenation of cinnamaldehyde
After successfully optimizing the thermocatalytic reaction conditions, it was interesting to explore the photochemical activity of the 1% Ru@ZnO(1.5)/CN catalyst because all three components of the catalyst, namely (i) Ru NPs, (ii) ZnO, and (iii) CN, are photoactive specifically in the visible region. When selective hydrogenation of CAL was targeted employing the 1% Ru@ZnO(1.5)/CN catalyst with molecular H2 and IPA as a solvent, 18% conversion was observed, but HCAL was the selective product. The result motivated us to further explore the catalyst to yield HCAL with promising selectivity. To serve this purpose, various catalysts with different %Ru loadings were synthesized. The complete conversion and selectivity were obtained with the 4% Ru@ZnO(1.5)/CN catalyst, but considering a low Ru content, 3% Ru@ZnO(1.5)/CN was chosen as the best catalyst, which is also suggested by the apparent quantum yield (Table 2). Cheaper transition metal decorated catalysts such as 3% Co@ZnO(1.5)/CN and 3%Ni@ZnO(1.5)/CN showed inferior activity to 3% Ru@ZnO(1.5)/CN (Table S6†), suggesting that the cheapest Noble metal Ru was the best choice for such a selective catalytic transformation under photocatalytic conditions. To realize the role of Ru NPs in this reduction, a control reaction was carried out with the heterojunction of CN and ZnO without Ru NPs. Only a 7% conversion was obtained in this case (Table 2, entry 3), which confirms the importance of Ru NPs in reducing CAL. Two control reactions were conducted over ZnO and CN to compare the activity of the heterojunction with individual photocatalysts. Over ZnO, the least activity was found with a 2% CAL conversion. Similarly, the CN photocatalyst afforded only 3% CAL conversion. An efficient charge separation (discussed above) in the heterojunction (ZnO(1.5)/CN) compared to individual CN and ZnO afforded higher activity. Furthermore, to understand the structure–activity relationship and mechanism, various photochemical characterization techniques were performed. In our search, only one report was found for photocatalytic reduction of CAL to HCAL with molecular H2, where an Au–Pd/ultrathin SnNb2O6 nanosheet catalyst afforded 99.8% conversion and 91.0% selectivity for HCAL.34 The present catalyst is cheaper and simple to synthesize and affords better selectivity for HCAL. A GC-MS chromatograph and 1H NMR of the reaction mixture recovered after 5.5 h in photocatalytic hydrogenation of CAL are provided in Fig. S18 and S19.†|
|
||||
|---|---|---|---|---|
| EN | Catalyst | Conversion (%) | Selectivity (%) | AQY |
| a Reaction conditions: substrate (0.5 mmol), catalyst amount (20 mg), IPA (5 mL), 150 W LED, room temperature, H2 (2 bar), and time (5 h). b Average of three measurements is presented as an integer. c Without H2. d Dark. e Sunlight (4 h reaction time and 10 am–2 pm). | ||||
| 1 | ZnO | 2 | >99 | 13.9 × 10−3 |
| 2 | CN | 3 | >99 | 18.8 × 10−3 |
| 3 | ZnO(1.5)/CN | 7 | >99 | 43.5 × 10−3 |
| 4 | 1% Ru@CN | 13 | >99 | 80.8 × 10−3 |
| 5 | 1% Ru@ZnO | 10 | >99 | 68.4 × 10−3 |
| 2 | 1% Ru@ZnO(1.5)/CN | 18 | >99 | 111.1 × 10−3 |
| 3 | 2% Ru@ZnO(1.5)/CN | 32 | >99 | 194.3 × 10−3 |
| 4 | 3% Ru@ZnO(1.5)/CN | 96 | >99 | 580.2 × 10−3 |
| 5 | 4% Ru@ZnO(1.5)/CN | 99 | >99 | 595.8 × 10−3 |
| 6c | 3% Ru@ZnO(1.5)/CN | 12 | >95 | 72.5 × 10−3 |
| 7d | 3% Ru@ZnO(1.5)/CN | — | — | 6 × 10−3 |
| 8e | 3% Ru@ZnO(1.5)/CN | 99 | >99 | 931.4 × 10−3 |

The spectral response of 3% Ru@ZnO(1.5)/CN was analyzed by photocatalytic reactions with violet, blue, green, red, and white 9 W LED bulbs under the optimized reaction conditions. The emission peak maxima for violet, blue, green, and red LEDs were observed in the visible region. The effect of the spectral response on the photocatalytic CAL conversion was studied, and the results demonstrate that the catalyst's activity followed the order: violet > blue > white > green > red, which correlates with the catalyst's light absorption properties (Fig. S20†).
The photocatalytic reduction of CAL was carried out at room temperature, and only the first-order rate equation resulted in a linear graph for CAL conversion (Fig. 7c). The rate constant was calculated to be 6.25 × 10−5 s−1 from the straight-line plot for first-order kinetics under photocatalytic conditions. Since the reaction is mediated by the photocatalyst, the kinetics depend on the light source's power. Thus, ln [CAL] vs. time plots were obtained for reactions at different light intensities. The kinetic profiles revealed that the rate constant decreased from 6.25 × 10−5 to 1.23 × 10−5 s−1 as the light intensity was reduced from 1210 to 270 W m−2. A linear relationship between the rate constant and light intensity with an R2 value of 0.98 was detected (Fig. 7d), indicating that the reaction kinetics were directly proportional to light intensity.
Photocatalytic reaction mechanism
First, it is important to recognize the charge carrier's interaction with the interface at the heterojunction photocatalyst to understand the possible mechanism. The Fermi level values for CN and ZnO are 4.51 eV and 5.1 eV vs. vacuum. The Fermi level of ZnO is lower than that of CN. When CN and ZnO are in direct contact as heterojunctions, there is an electron transfer from CN to ZnO until Fermi level equilibration at 4.63 eV vs. vacuum is attained, resulting in the generation of an electric field over the interface of both materials. It leads to upward band bending in CN and downward in ZnO towards the interface (Fig. 8 a(i) and (ii)). Once the Fermi levels are equilibrated, the charge transfer channel at the CN and ZnO interface is disrupted. Upon light irradiation, charge carriers are generated over the CB and VB of CN and ZnO, and two possible charge migration mechanisms could follow, namely, Type II heterojunction and Z-scheme heterojunction. In Type II, holes from the VB of ZnO are transferred to the VB of CN, while the electron from the CB of CN goes to the CB of ZnO, leading to photocatalytic oxidation at the VB of CN and reduction at the CB of ZnO. In the Z scheme, the electron from the CB of ZnO and holes from the VB of CN are transferred to the materials' interfaces, leading to recombination. In this case, oxidation occurs at the VB of ZnO, and reduction occurs at the CB of CN. To conclude the exact mechanism, some control experiments were performed. The NBT experiment shows the capability of the photocatalyst to generate the superoxide radical from molecular oxygen, for which the reduction potential of −0.33 V vs. NHE is required (Fig. 8b). The CB of ZnO is below this reduction potential; hence it is incapable of superoxide formation (Fig. 8b). A similar result was obtained in the NBT test (Fig. S21a†). A small decrease in the concentration of NBT was observed when the ZnO catalyst was employed. But the CB position of CN is more negative than the reduction potential required for superoxide formation. A good amount of superoxide was formed at CN. In the case of the composite, there are two possibilities: (i) reduction occurs at the CB of ZnO (type II heterojunction) and (ii) reduction occurs at the CB of CN (Z scheme heterojunction). According to the NBT experiment, the heterojunction efficiently generates the superoxide (Fig. S21a†), which confirms that the reduction in the heterojunction occurred at the CB of CN. It confirms that the ZnO/CN heterojunction is a Z scheme heterojunction (Fig. 8b). | ||
| Fig. 8 (a) Schematic band structure of CN and ZnO (i) before and (ii) after coming into contact, the charge transfer in the presence of light in (iii) the binary CN/ZnO heterojunction and (iv) the ternary Ru@ZnO/CN heterojunction, (b) possible mechanisms for the spatial separation in (i) the traditional type-II heterojunction and (ii) the Z-scheme heterojunction system, and (c) overall photocatalytic mechanism for CAL hydrogenation to HCAL over the 3% Ru@ZnO(1.5)/CN catalyst. | ||
To scrutinize the ability of the photocatalyst to form OH radicals, a terephthalic acid test was executed as a control experiment, utilizing PL spectroscopy. The generation of OH necessitates an oxidation potential of 2.8 vs. NHE. However, the VB position of CN is less negative than this oxidation potential, resulting in its incapacity to produce the OH radical. Nonetheless, the VB of ZnO is more positive than the oxidation potential of OH radical formation; therefore, ZnO can proficiently produce OH radicals, as validated by the terephthalic acid test (Fig. S21b†). When a heterojunction is formed, two possibilities exist: oxidation can occur at the VB of CN (type II heterojunction) or oxidation can occur at the VB of ZnO (Z-scheme heterojunction). The terephthalic acid test evinced that the heterojunction effectively generated OH radicals and manifested robust PL spectrum emission, evincing that oxidation in the heterojunction occurred at the VB of ZnO. It verifies that the heterojunction is a Z-scheme heterojunction (Fig. 7b). Electrons from the CB of ZnO and holes from the VB of CN move to the interface and recombine (Fig. 8(iii)). The photogenerated electron perseveres in the CB of CN and is transported to the Ru NPs owing to the low Fermi level of Ru (Fig. 8(iv)).
Control experiments were carried out to elucidate the role of charge carriers in photocatalytic reduction using the Ru@ZnO/CN heterojunction. Initially, CAL reduction was executed in the presence of electron scavengers, CCl4, and formic acid (Fig. S22a†). A marked drop in CAL conversion was noted in both scenarios, implying the significance of electrons in photocatalytic reduction. Moreover, IPA served as a hole scavenger, solely offering photogenerated electrons for reduction. To scrutinize the impact of isopropyl alcohol (IPA) on the photocatalytic reduction process, a control experiment was carried out by substituting IPA with acetonitrile (ACN), which proved to be ineffective (Fig. S22b†). To further amplify the effectiveness of the photocatalytic reduction reaction, a strategic approach involving the addition of external hole scavengers to the ACN solvent was employed. In one control experiment, triethanolamine (TEA) was added as an external additive to scavenge holes, resulting in a reduction of CAL. The CAL conversion was markedly improved with increasing amounts of TEA, from 100 μL to 200 μL (Fig. S22b†). In this instance, the reduction of CAL is solely attributed to the presence of H2, in the absence of IPA. Therefore, the overall photocatalytic reduction process is mainly driven by molecular hydrogen, with IPA's minimal involvement in the form of transfer hydrogenation. Nonetheless, IPA plays a critical role in quenching the photogenerated holes and enhancing the availability of photogenerated electrons, thus enabling Ru NPs' efficient photocatalytic reduction. Another control experiment involved adding IPA to the ACN solvent, which made the catalyst activity in a mixture of these two solvents, thus demonstrating IPA's role as a hole scavenger (Fig. S22c†). As the amount of IPA was increased, CAL conversion also increased. Consequently, effective quenching of photogenerated holes is an indispensable prerequisite for a successful photocatalytic reduction process, as it enables the availability of electrons, ultimately enhancing the production of HCAL.
The Ru nanoparticles (NPs) in the Ru@ZnO/CN heterojunction accept and trap the electrons produced by ZnO/CN under illumination, thereby promoting the desorption of hydrogen molecules (as discussed subsequently). To test this hypothesis, a non-photocatalytic material comprising Ru NPs supported on SBA-15 was utilized. A reaction using 3% Ru@SBA-15 under 150 W LED irradiation at room temperature resulted in negligible CAL conversion. The absence of electrons on the Ru NPs at room temperature prevented the desorption of H2 molecules. Thus, the photocatalyst absorbs light and generates electron–hole pairs, with IPA serving as a hole scavenger and the electrons accumulating on the Ru NP sites enabling successful desorption of hydrogen and reducing the double bond of CAL.
The photoactive catalyst (3% Ru@ZnO(1.5)/CN) induces the separation of charges within the ZnO/CN composite upon exposure to light. The resulting photogenerated electrons originating from the conduction band of ZnO and holes from the valence band of CN are recombined at the interface of the two materials. Subsequently, the electrons from the conduction band of CN are transferred to the surface of Ru NPs due to their higher work function and lower Fermi energy relative to the ZnO/CN composite. Furthermore, the externally supplied H2 adsorbed on the surface of the Ru NPs. The surface electrons on the Ru NPs efficiently dissociate and desorb the adsorbed H2, thereby promoting the reduction of the CC bond of CAL adsorbed on the catalyst surface, ultimately resulting in the selective production of HCAL (Fig. 8c). Additionally, the holes present in the valence band of ZnO participate in the oxidation of isopropyl alcohol (IPA), leading to the formation of acetone (Fig. 8c).
Stability of the catalyst under thermal and photochemical conditions
The recycling experiments for 1% Ru@ZnO(1.5)/CN and 3% Ru@ZnO(1.5)/CN were performed under thermal and photocatalytic conditions, respectively. The catalysts were subjected to centrifugation, followed by washing with ethanol and drying in an oven before being reused for consecutive runs under optimized conditions for a shorter duration. In the case of thermal recyclability, the 1% Ru@ZnO(1.5)/CN catalyst exhibited a conversion rate of 49%, after five consecutive cycles starting from 55%. To confirm the heterogeneous nature of the catalyst, hot filtration tests were conducted, and the reaction was prolonged after removing the catalyst at half the reaction time. There was no significant increase in catalytic activity after the removal of the catalyst, indicating that the process was indeed mediated by the heterogeneous catalyst (Fig. S23a and b†). Similarly, the 3% Ru@ZnO(1.5)/CN catalyst showed good stability under photocatalytic recycling experiments, with no substantial decrease in activity. The spent 3% Ru@ZnO(1.5)/CN catalyst was characterized by DRUV-vis, and no changes were observed in the absorbed edge (Fig. S24b†). Both the spent catalysts were characterized by XRD, XPS, SEM, and TEM techniques, which indicated no visible changes in the phase, elemental composition and oxidation states, morphology, and characteristics of both catalysts (Fig. S23c–g and S24c–g†).Overall, under thermal conditions, the role of ZnO is to adsorb and activate the –CO group of cinnamaldehyde. CN adsorbs formic acid due to the basic nature of the CN support. The role of Ru is to assist the effective dissociation of HCOOH into H2 + CO2 and dissociatively adsorb the generated H2. The independent roles of all the counterparts of the catalyst were determined by various control experiments. The catalytic activity of ZnO, CN, and Ru was independently evaluated, and the following results were obtained. ZnO afforded 17% CAL conversion and 100% COL selectivity. The pronounced selectivity for COL is attributed to the electropositive nature of Zn2+, which assisted in the adsorption and activating of the carbonyl group. CN alone offered only 6% conversion, but combining ZnO/CN elevated the catalytic activity. This observation suggests that formic acid activation was required along with CAL activation. CN being a basic support provided adsorption sites to FA, which were further enhanced by Ru NPs. Ru NPs dissociate FA into H2 + CO2 following the decarboxylation route (also confirmed by GC analysis), and the H2 generated was dissociatively adsorbed on Ru NPs. The dissociatively adsorbed –H hydrogenated the cinnamaldehyde. Moreover, CN also assisted the dispersion of the Ru NPs by providing a surface. Hence, it can be inferred that an optimum balance of Ru NPs, ZnO, and CN was essential to acquire efficient catalytic performance.
Under photocatalytic conditions, ZnO plays the role of an oxidative photocatalyst, whereas CN plays the role of a reductive photocatalyst because of their respective band edge positions. Combining both materials, a Z-scheme heterojunction photocatalyst was prepared (proved through control experiments) with enhanced charge separation compared to the individual components. Furthermore, due to the lower Fermi level of Ru, Ru accepted electrons generated by the ZnO/CN Z-scheme heterojunction and helped in the efficient dissociation of adsorbed H2 over Ru NPs. Hence, Ru assisted in photo-generated electron-mediated dissociation of H2 for the efficient hydrogenation of CAL.
Conclusions
A single catalyst was developed for the selective reduction of cinnamaldehyde to cinnamyl alcohol under thermal conditions using formic acid in water and cinnamaldehyde reduction to hydrocinnamaldehyde under photocatalytic conditions in IPA involving H2. The interface between the CN and ZnO components of the catalyst support was thoroughly examined under thermal and photocatalytic conditions. 1% Ru@ZnO(1.5)/CN yielded cinnamyl alcohol with 85% selectivity due to the successful activation of the CO group of CAL and FA through the optimum acidity of ZnO and the basicity of CN, respectively. The temperature-dependence selective activation and adsorption of cinnamaldehyde were confirmed through adsorption studies at different temperatures, employing FT-IR analysis. The FA was adsorbed over basic CN, and decorated Ru NPs facilitated the dissociation of formic acid into H2 + CO2 in water and formation of Ru–H. The Ru-adsorbed –H was transferred to the CO of cinnamaldehyde to yield COL as the selective product. Under photochemical conditions, the catalyst with an increased %Ru loading, i.e., 3% Ru@ZnO(1.5)/CN, selectively yielded hydrocinnamaldehyde as the selective product (>99%) with (>99%) cinnamaldehyde conversion. Various spectroscopic techniques and control experiments demonstrated the efficient role of the ZnO/CN heterojunction in facilitating better charge separation than individual entities. The photocatalyst followed the Z-scheme pathways for the selective reduction of cinnamaldehyde to hydrocinnamaldehyde. The electrons from the CB of CN are transferred to the surface of Ru NPs, which assisted in the effective dissociation and desorption of the adsorbed H2, reducing the CC of the cinnamaldehyde and yielding hydrocinnamaldehyde as the selective product. The surface–activity relationship established through extensive catalyst characterization under thermal and photocatalytic conditions was consistent with the proposed reaction mechanisms. The efficient recyclability and thermal and photostability of materials are other advantages of this work. The ability to selectively synthesize two products using a single catalyst under different experimental setups is of significant academic and industrial interest.
Author contributions
The work was planned by RS, AC, and RG. Experimental work, in-house characterization, and data presentation were done by AC. Photochemical characterization and data collection were conducted by RG. TEM investigations were conducted by RB. The manuscript was written by AC, RG, and RS.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
RS thanks IIT Ropar for the faculty and Innovation award. AC (09/1005(0033)/2020-EMR-I) and RG (09/1005(0031)/2020-EMR-I) are grateful to CSIR for the fellowship. The authors are grateful to the Advanced Material Research Centre (AMRC), IIT Mandi, for XPS characterization support, and the Time-Resolved Fluorescence Spectroscopy and Microscopy Facility at SAIF, IIT Bombay, for TCSPC analysis.Notes and references
- X. Wang, X. Liang, P. Geng and Q. Li, ACS Catal., 2020, 10, 2395–2412 CrossRef CAS.
- H. G. Manyar, B. Yang, H. Daly, H. Moor, S. McMonagle, Y. Tao, G. D. Yadav, A. Goguet, P. Hu and C. Hardacre, ChemCatChem, 2013, 5, 506–512 CrossRef CAS.
- X. Lan and T. Wang, ACS Catal., 2020, 10, 2764–2790 CrossRef CAS.
- M. Luneau, J. S. Lim, D. A. Patel, E. C. H. Sykes, C. M. Friend and P. Sautet, Chem. Rev., 2020, 120, 12834–12872 CrossRef CAS PubMed.
- R. Geng, H. Jia, Y. Xie, D. Pan, F. Yu and B. Fan, Microporous Mesoporous Mater., 2022, 338, 111968 CrossRef CAS.
- X. Gao, H. Dai, L. Peng, D. Lu, X. Wan, C. Zhou, J. Zheng, Y. Dai, H. Wang and Y. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 2516–2524 CrossRef CAS PubMed.
- J. BREEN, Appl. Catal., A, 2004, 268, 267–274 CrossRef CAS.
- H. Shen, H. Tang, H. Yan, W. Han, Y. Li and J. Ni, RSC Adv., 2014, 4, 30180–30185 RSC.
- T. Wu, H. Meng and R. Dang, Inorg. Chem. Front., 2021, 8, 4712–4719 RSC.
- C. M. Cova, A. Zuliani, M. J. Muñoz-Batista and R. Luque, Green Chem., 2019, 21, 4712–4722 RSC.
- J. Zhu, X. Ding, D. Li, M. Dou, M. Lu, Y. Li and F. Luo, ACS Appl. Mater. Interfaces, 2019, 11, 16443–16451 CrossRef CAS PubMed.
- K. N. Patil, P. Manikanta, P. M. Srinivasappa, A. H. Jadhav and B. M. Nagaraja, J. Environ. Chem. Eng., 2023, 11, 109168 CrossRef CAS.
- Y. Bonita, V. Jain, F. Geng, T. P. O'Connell, N. X. Ramos, N. Rai and J. C. Hicks, Appl. Catal., B, 2020, 277, 119272 CrossRef CAS.
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 683–733 CrossRef CAS PubMed.
- X. Liu, P. Su, Y. Chen, B. Zhu, S. Zhang and W. Huang, New J. Chem., 2018, 42, 9449–9454 RSC.
- H. Mao, S. Peng, H. Yu, J. Chen, S. Zhao and F. Huo, J. Mater. Chem. A, 2014, 2, 5847 RSC.
- B. Bachiller-Baeza, I. Rodríguez-Ramos and A. Guerrero-Ruiz, Appl. Catal., A, 2001, 205, 227–237 CrossRef CAS.
- N. Mahata, F. Gonçalves, M. F. R. Pereira and J. L. Figueiredo, Appl. Catal., A, 2008, 339, 159–168 CrossRef CAS.
- Y. Li, P.-F. Zhu and R.-X. Zhou, Appl. Surf. Sci., 2008, 254, 2609–2614 CrossRef CAS.
- H. Vu, F. Goncalves, R. Philippe, E. Lamouroux, M. Corrias, Y. Kihn, D. Plee, P. Kalck and P. Serp, J. Catal., 2006, 240, 18–22 CrossRef CAS.
- W. Feng, H. Dong, L. Niu, X. Wen, L. Huo and G. Bai, J. Mater. Chem. A, 2015, 3, 19807–19814 RSC.
- Q. Liu, Y. Li, Y. Fan, C.-Y. Su and G. Li, J. Mater. Chem. A, 2020, 8, 11442–11447 RSC.
- Z. Konuspayeva, G. Berhault, P. Afanasiev, T.-S. Nguyen, S. Giorgio and L. Piccolo, J. Mater. Chem. A, 2017, 5, 17360–17367 RSC.
- M. Butt, X. Feng, Y. Yamamoto, A. I. Almansour, N. Arumugam, R. S. Kumar and M. Bao, Asian J. Org. Chem., 2017, 6, 867–872 CrossRef CAS.
- A. Chauhan, A. Banerjee, A. K. Kar and R. Srivastava, ChemSusChem, 2022, 15, e202201560 CAS.
- A. Chauhan, A. K. Kar and R. Srivastava, Appl. Catal., A, 2022, 636, 118580 CrossRef CAS.
- A. Kumar Kar and R. Srivastava, ChemCatChem, 2021, 13, 3174–3183 CrossRef CAS.
- A. K. Kar and R. Srivastava, ACS Sustainable Chem. Eng., 2019, 7, 13136–13147 CrossRef CAS.
- Y. Dai, X. Gao, X. Chu, C. Jiang, Y. Yao, Z. Guo, C. Zhou, C. Wang, H. Wang and Y. Yang, J. Catal., 2018, 364, 192–203 CrossRef CAS.
- Y. Dai, X. Chu, J. Gu, X. Gao, M. Xu, D. Lu, X. Wan, W. Qi, B. Zhang and Y. Yang, Appl. Catal., A, 2019, 582, 117098 CrossRef CAS.
- N. Akiya and P. E. Savage, AIChE J., 1998, 44, 405–415 CrossRef CAS.
- C.-H. Hao, X.-N. Guo, Y.-T. Pan, S. Chen, Z.-F. Jiao, H. Yang and X.-Y. Guo, J. Am. Chem. Soc., 2016, 138, 9361–9364 CrossRef CAS PubMed.
- R. Xie, G. Fan, Q. Ma, L. Yang and F. Li, J. Mater. Chem. A, 2014, 2, 7880 RSC.
- H. Wang, Y. Shi, Z. Wang, Y. Song, M. Shen, B. Guo and L. Wu, J. Catal., 2021, 396, 374–386 CrossRef CAS.
- K. Wu, B. Zhao, C. Yang, Q. Wang, W. Liu and H. Zhou, J. Energy Chem., 2020, 43, 16–23 CrossRef.
- M. G. Prakash, R. Mahalakshmy, K. R. Krishnamurthy and B. Viswanathan, Catal. Sci. Technol., 2015, 5, 3313–3321 RSC.
- A. Khorsand Zak, R. Razali, W. H. B. Abd Majid and M. Darroudi, Int. J. Nanomed., 2011, 1399 CrossRef CAS PubMed.
- H. Lei, R. Nie, G. Wu and Z. Hou, Fuel, 2015, 154, 161–166 CrossRef CAS.
- A. Kumar, P. Kumar, C. Joshi, M. Manchanda, R. Boukherroub and S. Jain, Nanomaterials, 2016, 6, 59 CrossRef PubMed.
- X. Liu, L. Ye, S. Liu, Y. Li and X. Ji, Sci. Rep., 2016, 6, 38474 CrossRef CAS PubMed.
- S. Ghosh and B. R. Jagirdar, ChemCatChem, 2018, 10, 3086–3095 CrossRef CAS.
- F. Ye, H. Liu, J. Yang, H. Cao and J. Yang, Dalton Trans., 2013, 42, 12309 RSC.
- Y.-C. Liang and C.-C. Wang, RSC Adv., 2018, 8, 5063–5070 RSC.
- H. Wang, X. Li, Q. Ruan and J. Tang, Nanoscale, 2020, 12, 12329–12335 RSC.
- R. Ghalta and R. Srivastava, Catal. Sci. Technol., 2023, 13, 1541–1557 RSC.
- J. Büker, X. Huang, J. Bitzer, W. Kleist, M. Muhler and B. Peng, ACS Catal., 2021, 11, 7863–7875 CrossRef.
- L. S. Dake, D. R. Baer and J. M. Zachara, Surf. Interface Anal., 1989, 14, 71–75 CrossRef CAS.
- C. L. Bianchi, V. Ragaini and M. G. Cattania, Mater. Chem. Phys., 1991, 29, 297–306 CrossRef CAS.
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- M. O. Fuentez-Torres, F. Ortiz-Chi, C. G. Espinosa-González, M. Aleman, A. Cervantes-Uribe, J. G. Torres-Torres, M. K. Kesarla, V. Collins-Martínez, S. Godavarthi and L. Martínez-Gómez, Top. Catal., 2021, 64, 65–72 CrossRef CAS.
- X. Su, A. Vinu, S. S. Aldeyab and L. Zhong, Catal. Lett., 2015, 145, 1388–1395 CrossRef CAS.
- R. Ghalta and R. Srivastava, Sustainable Energy Fuels, 2023, 7, 1707–1723 RSC.
- J. Ning, Int. J. Electrochem. Sci., 2022, 220855 CrossRef CAS.
- R. Ghalta, A. K. Kar and R. Srivastava, Chem. – Asian J., 2021, 16, 3790–3803 CrossRef CAS PubMed.
- G. S. More, N. Kushwaha, R. Bal and R. Srivastava, J. Colloid Interface Sci., 2022, 619, 14–27 CrossRef CAS PubMed.
- G. Algara-Siller, N. Severin, S. Y. Chong, T. Björkman, R. G. Palgrave, A. Laybourn, M. Antonietti, Y. Z. Khimyak, A. V. Krasheninnikov, J. P. Rabe, U. Kaiser, A. I. Cooper, A. Thomas and M. J. Bojdys, Angew. Chem., Int. Ed., 2014, 53, 7450–7455 CrossRef CAS PubMed.
- C. Mahala, M. D. Sharma and M. Basu, ACS Appl. Nano Mater., 2020, 3, 1999–2007 CrossRef CAS.
- Z. Wu, X. Chen, X. Liu, X. Yang and Y. Yang, Nanoscale Res. Lett., 2019, 14, 147 CrossRef PubMed.
- H. Yang, Z. Jin, H. Hu, G. Lu and Y. Bi, Catalysts, 2017, 7, 99 CrossRef.
- C. Liu, Y. Qiu, F. Wang, K. Wang, Q. Liang and Z. Chen, Adv. Mater. Interfaces, 2017, 4, 1700681 CrossRef.
- A. Ishikawa, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2002, 124, 13547–13553 CrossRef CAS PubMed.
- S. Hoang, S. Guo, N. T. Hahn, A. J. Bard and C. B. Mullins, Nano Lett., 2012, 12, 26–32 CrossRef CAS PubMed.
- R. Pandiyan, N. Delegan, A. Dirany, P. Drogui and M. A. El Khakani, J. Phys. Chem. C, 2016, 120, 631–638 CrossRef CAS.
- I. Shown, H.-C. Hsu, Y.-C. Chang, C.-H. Lin, P. K. Roy, A. Ganguly, C.-H. Wang, J.-K. Chang, C.-I. Wu, L.-C. Chen and K.-H. Chen, Nano Lett., 2014, 14, 6097–6103 CrossRef CAS PubMed.
- R. Beranek, Adv. Phys. Chem., 2011, 2011, 1–20 CrossRef.
- F. Hayashi, Y. Toda, Y. Kanie, M. Kitano, Y. Inoue, T. Yokoyama, M. Hara and H. Hosono, Chem. Sci., 2013, 4, 3124 RSC.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S.-W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- H. B. Michaelson, J. Appl. Phys., 1977, 48, 4729–4733 CrossRef CAS.
- F. Su, S. C. Mathew, G. Lipner, X. Fu, M. Antonietti, S. Blechert and X. Wang, J. Am. Chem. Soc., 2010, 132, 16299–16301 CrossRef CAS PubMed.
- Y. Zhang, S. Zhang, X. Pan, M. Bao, J. Huang and W. Shen, Catal. Lett., 2017, 147, 102–109 CrossRef CAS.
- A. B. da Silva, E. Jordão, M. J. Mendes and P. Fouilloux, Appl. Catal., A, 1997, 148, 253–264 CrossRef CAS.
- H. Chen, D. A. Cullen and J. Z. Larese, J. Phys. Chem. C, 2015, 119, 28885–28894 CrossRef CAS.
- H. Li, H. Yang and H. Li, J. Catal., 2007, 251, 233–238 CrossRef CAS.
- J. Zhaoa, V. Malgrasb, J. Nac, R. Lianga, Y. Caia, Y. Kangb, A. A. Alshehrie, K. A. Alzahranie, Y. G. Alghamdie, T. Asahid, D. Zhanga, B. Jiangb, H. Lia and Y. Yamauchic, Chem. Eng. J., 2020, 398(1–6), 125564 CrossRef.
- M. Consonni, D. Jokic, D. Yu Murzin and R. Touroude, J. Catal., 1999, 188, 165–175 CrossRef CAS.
- E. V. Ramos-Fernández, J. Ruiz-Martínez, J. C. Serrano-Ruiz, J. Silvestre-Albero, A. Sepúlveda-Escribano and F. Rodríguez-Reinoso, Appl. Catal., A, 2011, 402, 50–58 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: XRD patterns, XPS spectra of the 3% Ru@ZnO/CN catalyst, SEM images of all the materials, TEM images of the 3% Ru@ZnO/CN catalyst, N2-adsorption/desorption isotherms, EDS and elemental mapping, TGA profiles of composites, CO2 & NH3 temperature-programmed desorption profiles, Tauc plots of all synthesized photocatalysts, LSV spectra in the dark and light, UPS spectra of 3% CN(1.5)/ZnO and Ru@CN(1.5)/ZnO, catalytic activity data of Ni and Co decorated catalysts, GC chromatogram of H2 + CO2, GC-Mass spectra recorded to establish the deuterium exchange, 1H NMR spectra of the crude reaction mixture showing the formation of COL and HCAL, temperature-dependent FT-IR spectra of CAL adsorbed on the catalyst, comparative absorption spectra of NBT and fluorescence spectra of THA solution and characterization of the spent catalyst under thermal and photochemical conditions. See DOI: https://doi.org/10.1039/d3ta02000b |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |