DOI:
10.1039/D3TA01998E
(Paper)
J. Mater. Chem. A, 2023,
11, 15855-15867
Selective radionuclide and heavy metal sorption using functionalised magnetic nanoparticles for environmental remediation†
Received
3rd April 2023
, Accepted 6th June 2023
First published on 6th July 2023
Abstract
In this paper the removal of a wide range of heavy metal ions from different chemical environments has been explored with the use of phosphate-functionalised superparamagnetic iron oxide nanoparticles (SPIONs), specifically magnetite (Fe3O4). These novel complexes have shown a high level of selectivity and increased loading capacity with the ions selected for the study: Na(I), K(I), Cs(I), Ca(II), Cu(II), Co(II), Ni(II), Cd(II), Mg(II), Sr(II), Pb(II), Al(III), Mn(II), Eu(III) and Fe(III). The loading capacities established using these NP-complexes have been shown to be higher than that of conventional surface-ligand systems. The development of these phosphate functionalised complexes, (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4, has successfully shown the feasibility of removing the selected metal ions at pH 1, pH 3 and pH 7. The maximum adsorption capacities of the complexes were tested with single-metal adsorption experiments, showing a degree of selectivity towards all metal ions studied. Multi-metal adsorption experiments were conducted to determine the selectivity of the NP-complexes in the presence of a range of competing ions. These experiments simulated real-world environments that contain the selected heavy metals, which cause great concern for humans and the environment. These experiments allowed for the successful determination of a selectivity series, highlighting the steps in which the various metal ions are removed after sequential sorption experiments. The results that have been presented in this paper highlight the potential use of these magnetic phosphate NP-complexes for selective heavy metal removal from contaminated aqueous wastewaters in the industrial world.
1. Introduction
In recent years, the developments throughout many industrial processes have caused a huge increase in the volume of industrial processes conducted. Many of these processes are inherent producers of aqueous waste streams that require treatment for further disposal. Common examples are galvanisation, mining, energy production and sewage all of which produce enormous volumes of aqueous waste each year. These wastes quite often contain a wide range of contaminants, from non-harmful to very toxic.1 An example is the nuclear industry, where aqueous waste streams that contain radionuclides such as uranium and many toxic heavy metals are being produced.2 Contact of such contaminated waste streams with the environment, through ground water or rivers is to be avoided at all costs, as this would cause harm to both human life and the environment. Not only this, due to new regulatory limits of many of the contaminants found in industrial aqueous waste streams, free release is not feasible until such contaminants have been removed. Therefore, increasing efforts to treat these waste waters are being made which is directly resulting in the development of many novel extraction techniques.3
The development of nanotechnologies has come a long way, where potential solutions for the application of wastewater treatment have become available. The ability to selectively sequester and remove contaminants from aqueous wastes with high loading capacities is of paramount importance to achieve full removal of the contaminants produced in many industries. A recent example of development would be the development of functionalised magnetic nanoparticles, specifically magnetite (Fe3O4). The superparamagnetic behaviour of Fe3O4 magnetic nanoparticles (MNPs) allows for very easy manipulation using an external magnetic field, which is an advantage that many conventional nanomaterials do not have.4 These low cost, non-toxic and easily prepared magnetic nanoparticles are highly biocompatible and have already been widely applied in the biotechnology and biomedical industries. The addition of specific functionalities allows for the fine tuning of the selectivity towards certain elements, therefore allowing full control over the selective removal of a wide range of contaminants. Phosphate functionalised magnetite ((PO)x-Fe3O4) has been shown to have ultra-high loading capacities and a high degree of selectivity towards uranium (U(VI)). Sorption measurements of these nanoparticles have shown them to have loading capacities of 2333 mg U per g Fe (1690 mg U per g Fe3O4 MNP), which is reported to be significantly higher than that in previously reported literature. Even in the presence of competing ions, these nanoparticle complexes have an extremely high selectivity towards U(IV) ions.5 Further developing these NP complexes to allow selectivity towards other elements is a key step in this technology's development, as it would allow the full exploitation of their ability to selectively remove a wide range of contaminant elements from aqueous waste forms. Many waste water streams, in industries such as nuclear and mining, contain a number of contaminants which are highly toxic and hazardous, especially heavy metals, therefore, requiring multiple contaminant removal processes to provide adequate cleanup, where this is currently the case.6,7 The potential development of a cost-effective single technology that could deal with a wide range of contaminant ions present in such wastes, whether in a single or multiple extractions or recycled for repeat extraction cycles, would benefit the water treatment industry greatly. As seen from the literature, phosphate species of many types have been used to selectively remove several contaminant ions from aqueous environments. The removal of contaminants such as Fe(III), Co(II), Ni(II), Pb(II), Cd(II), U(VI) and Pu(III, IV, VI)8–12 have been studied with the use of phosphate bearing substrates, such as nanoparticles, ion exchange resins, thin films and organophosphorus compounds, where phosphate functional groups have been shown to form stable complexes with iron oxide, creating an ideal NP substrate for adsorption of such contaminants.13,14 The formation of stable uranyl–phosphate complexes has been reported previously in the literature.15,16 This, therefore, with some further optimisation, gives a new potential use for (PO)x-Fe3O4 NP complexes that have previously been developed. Due to the numerous environments where these contaminants are found, it is difficult to rely on a single extraction technology. Moreover, many of the environments that require such cleanup are considered extreme environments, which can be highly acidic.17 Therefore, the development of acid resistant NP-complexes is needed. Evidence has shown that such NP-complexes, in the form of (PO)x-SiO2@Fe3O4 NPs, have been successfully manufactured, producing acid resistant NPs, while being able to preserve the surface functionality.18,19
In this work, both single- and multi-metal batch sorption experiments with the use of (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NPs have been conducted to be able to fully determine the selectivity and loading capacity of other elements with these NP–ligand complexes. Single-metal tests identified the selectivity for Na(I), K(I), Cs(I), Ca(II), Cu(II), Co(II), Ni(II), Cd(II), Mg(II), Sr(II), Pb(II), Al(III), Mn(II), Eu(III) and Fe(III). Both (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NP complexes showed selectivity towards all elements selected with rapid kinetics and with significant loading capacities. Multi-metal tests simulating real world environments identified the selectivity of contaminant ions in the presence of competing ions and further determined a selectivity series of elements after repeated extraction cycles. All elements in these tests were selected due to their cause for concern when they are present in natural and wastewater streams. (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NP complexes were successful in the removal of all selected elements, therefore, creating a broad-spectrum adsorbent for the extraction of a wide range of heavy metal and other contaminant ions.
2. Experimental
2.1. Chemicals
1-Octadecene (technical grade, 90%), TritonTM X-100 (laboratory grade), (3-aminopropyl) triethoxysilane (APTES, 99%) and oleic acid (technical grade, 90%) were purchased from Aldrich. Metal salts used in sorption experiments were all purchased from Sigma Aldrich (99.9% trace metal basis). Tetraethyl orthosilicate (TEOS, 98%), aqueous ammonia (28%), absolute ethanol, hexane, and cyclohexane, anhydrous sodium phosphate (99%) and anhydrous potassium phosphate (99%) are of analytical reagent grade. Sodium oleate was purchased from TCI chemicals. All the chemicals were used as-received without further purification. The water used was purified through a Millipore system (18 ΩM).
2.2. Synthesis of 12 nm Fe3O4 and SiO2@Fe3O4 core–shell nanoparticles and surface functionalisation
Highly monodisperse 12 nm magnetite (Fe3O4) nanoparticles were synthesised via a thermal decomposition method prepared according to the literature, producing a two-step reaction.20 Firstly, an iron–oleate complex was prepared at 70 °C and allowed to cool. The iron–oleate complex was then washed several times with ethanol to remove the unwanted organic layer. Secondly, the as-synthesised iron–oleate complex was mixed with oleic acid in 1-octadecene and heated at a controlled heating rate of 3.3 °C per minute up until 320 °C under inert gas (N2). The product was then allowed to cool slowly to room temperature and washed several times with ethanol. The mixture was centrifuged to collect the synthesised oleic acid capped (OA-Fe3O4) NPs, which were finally dispersed in hexane.
A reverse microemulsion coating method was used to manufacture SiO2@Fe3O4 NPs, which were prepared according to the literature.21 A known mass of the as-synthesised OA-Fe3O4 NPs was dispersed in cyclohexane and sonicated for 10 minutes. Triton X-100 was dissolved in cyclohexane and stirred, where the OA-Fe3O4 NPs were added dropwise and left to stir for a further 15 minutes. NH4OH was then added dropwise and stirred for another 15 minutes. Finally, TEOS was added to the sonicated cyclohexane dispersed OA-Fe3O4 NP mixture and stirred for 24 hours. The SiO2@Fe3O4 NPs were washed several times with ethanol and DI water to remove any excess reactants. The NPs were finally dispersed in DI water.
A ligand exchange reaction was conducted with the OA-Fe3O4 NPs, as described in the literature, to graft a phosphate species to the surface.22 A known mass of OA-Fe3O4 NPs was added dropwise to a sodium potassium phosphate buffer produced from Na2HPO4 and KH2PO4. The mixture was stirred for 17 hours at room temperature to allow for complete exchange. The phosphate-functionalised ((PO)x-Fe3O4) NPs were then washed with ethanol several times and dispersed in DI water. Functionalisation of SiO2@Fe3O4 was also conducted by mixing the NPs with APTES and the previously mentioned phosphate buffer and stirring for 4 hours. The final dispersion was then washed and dispersed in DI water (18 ΩM).
2.3. Single-metal batch sorption tests
Metal salts of cationic species of interest (Table 1) were dissolved in deionised water, producing stock solutions. The desired concentration of each metal ion species was 20 mg L−1. All stock solutions with a pH higher than 7 were adjusted to pH 7 using HNO3, whereas stock solutions with pH lower than 7 were adjusted to pH 7 with NaOH. An aliquot of metal ion stock solution (10 mL) was brought into contact with 0.25 mg of the synthesised NP-complexes, (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4, and agitated on an orbital shaker at 150 rpm, room temperature for 1 min, 5 min, 10 min, 30 min, 60 min and 90 min. Upon completion of the reaction time the samples were then analysed using inductively coupled plasma-optical emission spectroscopy (ICP-OES) and energy dispersive X-ray spectroscopy (EDX). To determine the maximum loading capacity of the NP complexes, each sorption study was repeated with a metal ion concentration of 50 mg L−1 and a NP concentration of 0.25 mg. For this study, the NP/metal ion solutions were left to equilibrate on an orbital shaker for 60 min.
Table 1 Contaminant species for sorption experiments
| Species |
Species concentration (mg L−1) |
| Cs(I) |
20 |
| K(I) |
20 |
| Na(I) |
20 |
| Ca(II) |
20 |
| Cd(II) |
20 |
| Co(II) |
20 |
| Cu(II) |
20 |
| Mg(II) |
20 |
| Mn(II) |
20 |
| Mo(II) |
20 |
| Ni(II) |
20 |
| Pb(II) |
20 |
| Sr(II) |
20 |
| Al(III) |
20 |
| Ce(III) |
20 |
| Cr(III) |
20 |
| Eu(III) |
20 |
| Fe(III) |
20 |
| La(III) |
20 |
2.4. Effect of pH on metal ion loading
Further stock solutions of the metal ion species were produced with the same species concentration and pH adjusted to pH 3 (HNO3) and pH 11 (NaOH). Similarly to the above protocol, an aliquot (10 mL) of the pH adjusted metal ion stock solution (20 mg L−1) was brought into contact with 0.25 mg of (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NPs and agitated on an orbital shaker for 1 min, 5 min, 10 min, 30 min, 60 min and 90 min. Due to the adverse effects of solution pH below pH 5 resulting in Fe dissolution, (PO)x-Fe3O4 NPs were only used at pH 7 and pH 11.18 The same goes for (PO)x-SiO2@Fe3O4 NPs, where dissolution of SiO2 is observed at pH > 8; therefore these NPs were used at pH 3. After contact the samples were then analysed using ICP-OES to determine any change in species loading. Previous work studying the dissolution and leaching of OA-Fe3O4, (PO)x-Fe3O4 NPs has shown that under highly acidic conditions (pH 3), these NPs rapidly degrade. However, they have been shown to be highly stable under strongly basic conditions (pH 11). On the other hand, SiO2@Fe3O4 and (PO)x-SiO2@Fe3O4 NPs have shown high stability under strongly acidic conditions, providing evidence that not only the NP structure but also the functionality is stable under highly acidic conditions (see Fig. S2†).18
2.5. Multi-metal batch sorption tests
Multi-species solutions were made with constant metal ion species concentrations, containing groups of contaminants selected from Table 2 to simulate real-world environments. Multi-species stock solutions were made by adding 10 mg L−1 of the selected metal ions and adjusting the pH to 7. An aliquot (10 mL) of the multi-species solution was brought into contact with 0.25 mg of (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NP complexes and agitated for 90 minutes at room temperature. Sequential sorption experiments were also conducted with each selected group to aid in determining a selectivity series of species. ICP-OES measurements were conducted to determine the adsorbed concentration of the adsorbed ions and to determine the concentration of the remaining metal ions in the solution.
Table 2 Simulant compositions for contaminant species adsorption experiments with (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NPs
| Waste Simulant |
Species |
Species concentration (mg L−1) |
| Simulant 1 |
K+ |
10 |
| Na+ |
10 |
| Cs+ |
10 |
| Ca2+ |
10 |
| Mg2+ |
10 |
| Ni2+ |
10 |
| Al3+ |
10 |
| Fe3+ |
10 |
| Simulant 2 |
K+ |
10 |
| Na+ |
10 |
| Ca2+ |
10 |
| Mg2+ |
10 |
| Mn2+ |
10 |
| Eu3+ |
10 |
| Al3+ |
10 |
| Fe3+ |
10 |
| Al3+ |
10 |
| Fe3+ |
10 |
| Simulant 3 |
Co2+ |
10 |
| Cu2+ |
10 |
| Ni2+ |
10 |
| Eu3+ |
10 |
| Fe3+ |
10 |
| Simulant 4 |
Cd2+ |
10 |
| Cu2+ |
10 |
| Pb2+ |
10 |
| Fe3+ |
10 |
| Cr6+ |
10 |
2.6. Instrumentation
OA-Fe3O4 and SiO2–Fe3O4 morphology, phase, size, and shell thickness were analysed to determine the success of the synthesis. The size and morphology were analysed by transmission electron microscopy (TEM) using both JEOL JEM-2100F and JEOL JEM 2100Plus TEMs, operating at 200 keV. Phase determination was carried out using a PANalytical X-ray diffractometer, with a Cu Kα source, where diffraction patterns were acquired in the 2θ range between 20° and 80°. EDX was used to determine the presence of the phosphate functionality and therefore the success of the surface functionalisation of the nanoparticles (NPs). All EDX analyses were conducted using a JEOL JEM-2100F TEM. Concentrations of metal species in aqueous solutions were determined through ICP-OES, using a Scientific iCAP 6000 ICP Spectrometer with a CETAC ASX-520 autosampler.
3. Results
3.1. Fe3O4 and SiO2@Fe3O4 (core–shell) magnetic nanoparticle characterization
The manufacture of magnetite nanoparticles was performed via thermal decomposition using an iron oleate precursor as described above, following an adapted protocol from the literature.20 The manufacture of SiO2@Fe3O4 NP complexes was performed via a reverse oil-in-water microemulsion, following an adapted protocol from the literature.21 The synthesis of highly monodisperse OA-Fe3O4 was successful, showing uniformly shaped NPs. The NP complexes underwent TEM, XRD, ATR-FTIR, DLS, EDX and VSM analyses, to determine the size, morphology, phase purity, surface chemistry and magnetic properties of the NPs. Fe3O4 and SiO2@Fe3O4 NPs were confirmed to have a particle size distribution and hydrodynamic diameter of 12.1 ± 2.1 nm and 12.2 ± 2.2 nm, respectively, whereas, for the SiO2@Fe3O4 NPs these were found to be 31.4 ± 2.3 nm and 34.3 ± 2.1 nm, respectively. (Fig. S1(a) and (b)†). The crystal structure and phase purity of both complexes were confirmed by XRD, where comparison was made with reference patterns of magnetite, maghemite and hematite, confirming that magnetite was the only iron oxide phase present, as well as the presence of amorphous silica (Fig. S1(c) and (d)† for the silica-coated samples, JCPDS card 75-1610). DLS measurements confirmed the absence of aggregation in the OA-Fe3O4 and SiO2@Fe3O4 samples (Fig. S1(e) and (f)†). ATR-FTIR spectroscopy allowed for further surface investigation confirming the presence of the spinel structure of magnetite and oleic acid coverage of OA-Fe3O4 NPs. Signals at 585 and 620 cm−1 corresponding to the Fe–O stretching vibrations and sharp signals located at 2870 and 2925 cm−1 provided evidence of the –CH2– stretching vibrations of the oleic acid chains (Fig. S4(a) and (f)†).23,24 Silanol (Si–OH) bending peaks found at 896 cm−1 and 3690 cm−1 are evidence of the SiO2 presence.25 Also, Si–O–Si stretching vibration peaks can be seen at 615 and 1078 cm−1 (Fig. S4(f)†).25,26 Finally, both OA-Fe3O4 and SiO2@Fe3O4 NP complexes were examined using a vibrating sample magnetometer (VSM) to determine their magnetic behaviour. This analysis confirmed that both complexes, OA-Fe3O4 and SiO2@Fe3O4, exhibited superparamagnetic behaviour at room temperature and at 5 K, with saturated magnetisation values of 61.1 emu g−1 and 30.5 emu g−1, respectively (Fig. S3†).
3.2. Surface functionalisation of Fe3O4 and SiO2@Fe3O4 nanoparticles
Successful surface functionalisation of OA-Fe3O4 and SiO2@Fe3O4 was achieved through a ligand exchange reaction and a two-step ligand reaction, respectively. This was shown by characteristic peaks of the symmetric and asymmetric –CH2– stretching vibrations of the oleic acid chains at 2870 and 2925 cm−1 of the OA-Fe3O4 NPs (green curve, Fig. S4a†), being replaced with a large broad peak at 3300 cm−1 (red curve, Fig. S4a†). A broad peak present between 900 and 1250 cm−1 indicates the success of the ligand exchange reaction. The broad peak between 900 and 1250 cm−1 (Fig. S4a,† red curve) contains the characteristic peaks that are representative of vibrational stretching wavelengths of typical phosphate species. These phosphate peaks are found at 940, 995 and 1078 cm−1, typical of P–O–Fe, P–O and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations, respectively.27 The combination of the peaks at 1039 and 1078 cm−1 has been observed previously in the literature and suggests the presence of bidentate phosphate complexes on the iron oxide surface.28 An interesting peak at 1150 cm−1 is the contribution of a H2PO4 species containing PO and two –OH groups.29,30 The peaks located at 1640 and 3300 cm−1 are characteristic of H2O bending and stretching modes. With all peaks between 2800 and 3200 cm−1 absent, this confirms the complete replacement of the oleic acid capping with phosphate molecules. The EDX analysis (Fig. S4(b)†) further confirms the presence of phosphate with an intense peak at 2.01 keV. TEM micrographs (Fig. S4(c), (d), (h) and (i)†) show the (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NPs, with successful coating of the Fe3O4 NPs with SiO2 observed. Additionally, zeta potential measurement (Fig. S4(e) and (j)†) show evidence of the phosphate functionality. With zeta potentials of −42 mV and −30 mV, respectively, this shows a negative surface charge, suggesting that the phosphate coated NPs are deprotonated. The functionalisation of SiO2@Fe3O4 NPs to (PO)x-SiO2@Fe3O4 NPs was conducted through a two-step reaction, as the ligand exchange reaction was not feasible; therefore an aminosilane branching ligand, (3-aminopropyl) triethoxysilane (APTES), was used. In the FTIR data the characteristic peaks of the phosphate species are still present at 810, 990, and 1075 cm−1, representative of P–O–Fe, P–O and PO stretching vibrations, respectively (red curve, Fig. S4f†).27 Again, the peak at 1160 cm−1 is evidence of the PO groups characteristic of the H2PO4 group.29 Similarly the peaks at 1645 and 3300 cm−1 are representative of the H2O bending and stretching modes. The characteristic peaks of the APTES branching ligand are observed to be present in the FTIR spectrum. The peaks present at 1403 cm−1 represent the deformation mode of the Si–CH2 group. This peak along with the shallow Si–O–Si scissoring absorption mode peak at 550 cm−1 suggests the complete functionalisation of APTES on the SiO2 surface.31 Furthermore, it can be observed that the deformation modes of the amine group are present at 1480 and 1540 cm−1. The stretching modes of the –CH2– contained within the APTES chain are found at 2870 and 2975 cm−1, which are semi-masked by the broad Si–OH and H2O bending and stretching modes.32 Finally, the symmetric and asymmetric stretching modes of –NH2 can be seen at 3295 and 3370 cm−1.33 These data suggest the successful grafting of the APTES branching ligand and the further grafting of the phosphate species. Again, the EDX analysis shows the presence of phosphate with an intense peak at 2.01 keV (Fig. S4g†). (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NP complexes remained stable in suspension for over a year, where no sedimentation or aggregation was detected. VSM analysis was further conducted with (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NP complexes to investigate any deviations in magnetic properties due to the phosphate capping. Similarly to OA-Fe3O4 and SiO2@Fe3O4 NPs (Fig. S3†), these NPs exhibited superparamagnetic behaviour at room temperature and at 5 K, with saturated magnetisation values of 63.7 emu g−1 and 29.1 emu g−1, respectively, showing no change in magnetic saturation (Fig. S3†).
O stretching vibrations, respectively.27 The combination of the peaks at 1039 and 1078 cm−1 has been observed previously in the literature and suggests the presence of bidentate phosphate complexes on the iron oxide surface.28 An interesting peak at 1150 cm−1 is the contribution of a H2PO4 species containing PO and two –OH groups.29,30 The peaks located at 1640 and 3300 cm−1 are characteristic of H2O bending and stretching modes. With all peaks between 2800 and 3200 cm−1 absent, this confirms the complete replacement of the oleic acid capping with phosphate molecules. The EDX analysis (Fig. S4(b)†) further confirms the presence of phosphate with an intense peak at 2.01 keV. TEM micrographs (Fig. S4(c), (d), (h) and (i)†) show the (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NPs, with successful coating of the Fe3O4 NPs with SiO2 observed. Additionally, zeta potential measurement (Fig. S4(e) and (j)†) show evidence of the phosphate functionality. With zeta potentials of −42 mV and −30 mV, respectively, this shows a negative surface charge, suggesting that the phosphate coated NPs are deprotonated. The functionalisation of SiO2@Fe3O4 NPs to (PO)x-SiO2@Fe3O4 NPs was conducted through a two-step reaction, as the ligand exchange reaction was not feasible; therefore an aminosilane branching ligand, (3-aminopropyl) triethoxysilane (APTES), was used. In the FTIR data the characteristic peaks of the phosphate species are still present at 810, 990, and 1075 cm−1, representative of P–O–Fe, P–O and PO stretching vibrations, respectively (red curve, Fig. S4f†).27 Again, the peak at 1160 cm−1 is evidence of the PO groups characteristic of the H2PO4 group.29 Similarly the peaks at 1645 and 3300 cm−1 are representative of the H2O bending and stretching modes. The characteristic peaks of the APTES branching ligand are observed to be present in the FTIR spectrum. The peaks present at 1403 cm−1 represent the deformation mode of the Si–CH2 group. This peak along with the shallow Si–O–Si scissoring absorption mode peak at 550 cm−1 suggests the complete functionalisation of APTES on the SiO2 surface.31 Furthermore, it can be observed that the deformation modes of the amine group are present at 1480 and 1540 cm−1. The stretching modes of the –CH2– contained within the APTES chain are found at 2870 and 2975 cm−1, which are semi-masked by the broad Si–OH and H2O bending and stretching modes.32 Finally, the symmetric and asymmetric stretching modes of –NH2 can be seen at 3295 and 3370 cm−1.33 These data suggest the successful grafting of the APTES branching ligand and the further grafting of the phosphate species. Again, the EDX analysis shows the presence of phosphate with an intense peak at 2.01 keV (Fig. S4g†). (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NP complexes remained stable in suspension for over a year, where no sedimentation or aggregation was detected. VSM analysis was further conducted with (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NP complexes to investigate any deviations in magnetic properties due to the phosphate capping. Similarly to OA-Fe3O4 and SiO2@Fe3O4 NPs (Fig. S3†), these NPs exhibited superparamagnetic behaviour at room temperature and at 5 K, with saturated magnetisation values of 63.7 emu g−1 and 29.1 emu g−1, respectively, showing no change in magnetic saturation (Fig. S3†).
3.3. Single-metal batch sorption tests
Single-metal sorption experiments were conducted to investigate the potential selectivity and extraction capabilities of (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NP complexes towards a wide range of heavy metals and common contaminants in natural and waste waters without competing ions (Table 1). Each NP complex was separately brought into contact with aqueous solutions of single-metal ion species and analysed over time (1–90 min), with repeats of at least three times per adsorption experiment. The extraction percentages of each metal ion species tested can be found in the ESI (Table S2†). Both NP complexes showed affinity towards all metal ion species selected for this study. Fig. 1(a) and (b) show that the metal ion extraction exceeds 80% for Eu(III), Fe(III) and La(III) over the 90-minute period. All metal ions were extracted above 50% except for Mo(II) and Pb(II) with the (PO)x-Fe3O4 NPs and Ca(II), Cd(II) and Pb(II) with the (PO)x-SiO2@Fe3O4 NPs (Fig. 1(c) and (d)). As expected, with an increase in time, there is an increase in extraction efficiency.
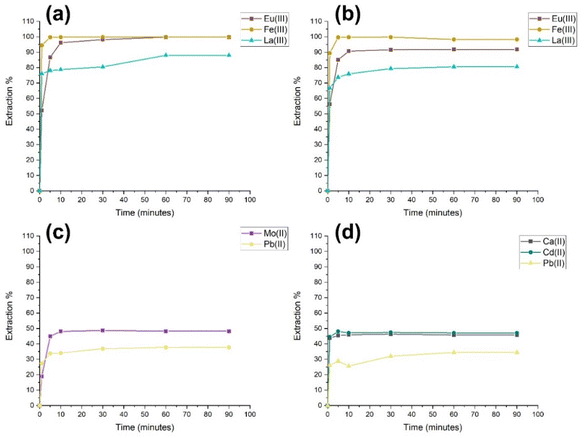 |
| | Fig. 1 Extraction of all metal ion species above 80% (a) Eu(III), Fe(III) and La(III) with (PO)x-Fe3O4 NPs; (b) Eu(III), Fe(III) and La(III) with (PO)x-SiO2@Fe3O4 NPs; extraction below 50% (c) Mo(II) and Pb(II) with (PO)x-Fe3O4 NPs; (d) Ca(II), Cd(II) and Pb(II) with (PO)x-SiO2@Fe3O4 NPs at pH 7. | |
From these data, the maximum loading capacities were calculated (see Table S3†). A minor difference between the loading capacity of (PO)x-Fe3O4 compared to (PO)x-SiO2@Fe3O4 NPs is always seen. However, as this difference is found to be generally consistent, these were considered to be within the errors in NP surface calculations whilst normalising for sorption studies. However, one example of a significant difference was found in the extraction percentage of Mo(II) with (PO)x-@Fe3O4 compared to that with (PO)x-SiO2@Fe3O4, where it can be seen that the functionalisation with (PO)x-SiO2 affects the sorption capacity of (PO)x-SiO2@Fe3O4 by 200 mg metal ion per g of MNP, possibly related to the formation of Fe–Mo species. Several reports of redox reactions between complexes of molybdenum and iron have been found in the literature, where the reduction of Mo(III) by Fe(III) and Mo(VI) by Fe(II), as well as the formation of Fe–Mo complexes have been observed.34–36 It has also been reported that Fe-based and Mo-based redox proxies, specifically interactions between these species have been reported in marine sediments.37 These interactions between Fe in the core particles and the Mo species in solution might explain the significant difference in Mo loading found for the (PO)x-Fe3O4 NPs compared to that for (PO)x-SiO2@Fe3O4 NPs, due to inaccessibility of the Fe3O4 core.
Although previous work on U(VI) adsorption with similar NP complexes, which are known to have an extreme degree of selectivity towards U(VI), shows a high loading capacity, these complexes have shown a significant degree of affinity towards the metal ions selected, where adsorbed concentrations for (PO)x-Fe3O4 NPs and (PO)x-SiO2@Fe3O4 NPs show adsorption feasibility for each metal ion selected in this study.
3.4. Effect of pH on metal ion loading
Batch-wise sorption experiments were conducted on solutions containing the single contaminant species in Table 1 after pH adjustments to pH 3 and pH 11. All extraction data for each species towards (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NP showed a significant change in loading (Table S3†). Increasing the [H+] concentration (pH 3) caused the extraction efficiency of (PO)x-SiO2@Fe3O4 NPs to reduce slightly, showing a degree of suppression of loading by less than 10%. However, the extraction percentage of Cs(II), Co(II), Pb(II) and Sr(II) dropped significantly, decreasing to 46, 46, 28 and 48%, respectively from 55, 70, 35 and 67%. Fig. 2(b) shows the total species removed from solution to be below 50%, where the number of species below 50% removal has increased. Interestingly, increasing the [OH−] concentration (pH 11) caused instead a significant increase in the removal of all metal ions by the (PO)x-Fe3O4 NPs, as shown in Fig. 1(a) where almost all species were removed over 90%, resulting in enhanced removal compared to that at pH 7. In general, all removal percentages increased, resulting in nearly all contaminants being completely removed from solution, with the exception of Na(I), Ca(II), Sr(II) and Cr(III), which showed removal percentages of 69, 70, 70 and 86% respectively. Fig. 2b shows the species removed above 95%, indicating an enhanced removal of all species tested.
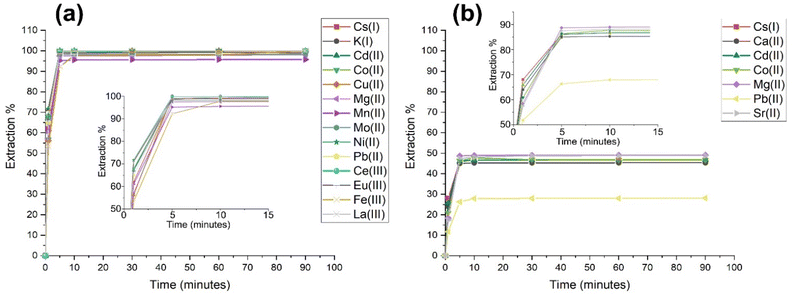 |
| | Fig. 2 Total removal from solution of (a) (PO)x-Fe3O4 NPs with increasing [OH−] above 95% (pH 11); and extraction percentages of (b) (PO)x-SiO2@Fe3O4 NPs with increasing [H+] below 50% (pH 3). | |
3.5. Multi-metal batch sorption tests
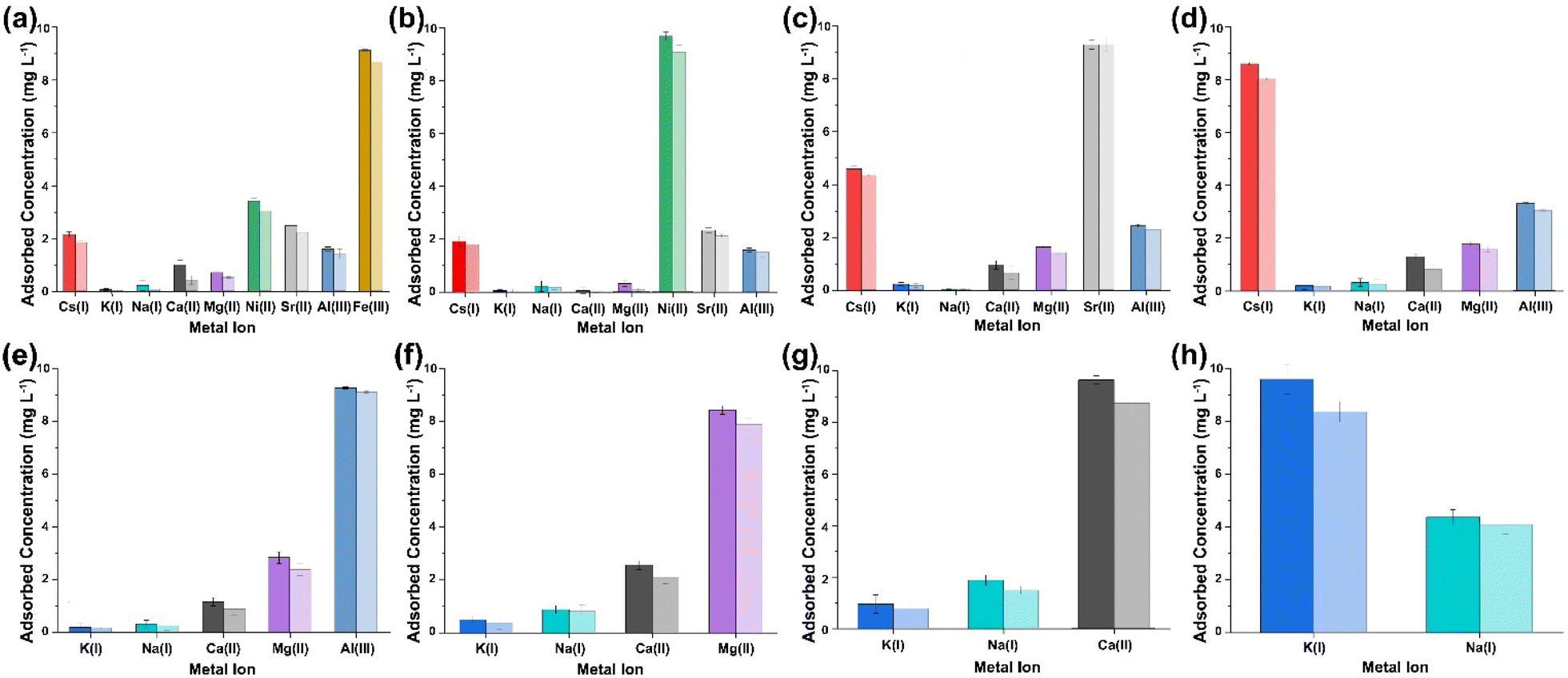
Aqueous solutions containing contaminant species (Table 1) were brought into contact with (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 in a batchwise system where the metal ion concentrations were constant at 10 mg L−1 for each metal ion in solution at pH 7. The metal ions contained within each solution were selected to simulate real-world contaminated aqueous waste streams that are produced by a range of industrial processes, to assess the selectivity of the NP complexes while in the presence of several competing ions. Four simulant wastewater solutions were produced to reflect contaminants contained in (i) spent nuclear fuel ponds, (ii) post uranium mining waters, (iii) reactor coolant loop water, and (iv) heavy metal containing sewage waste, here labelled, ‘simulant 1’, ‘simulant 2’, ‘simulant 3’ and ‘simulant 4’, respectively (Table 2). It was found that with every multi-metal solution (simulant 1, 2, 3 and 4), Fe(III) constantly showed the highest recovery percentage with both NP complexes, no matter what competing ions were present with recovery percentages exceeding 95%. Moreover, excluding some cases of potential multisorption, the NP complexes were observed to extract one element at a time, allowing for a selectivity series to be produced (Fig. 3–6). Through sequential sorption tests, selectivity series were obtained for each simulant composition, with a different selectivity series for each simulant:
| Simulant 1: Fe(III) > Ni(II) > Sr(II) > Cs(I) > Al(III) > Mg(II) > Ca(II)> K(I) > Na(I) |
| Simulant 2: Fe(III) > Eu(III) > Al(III) > Mg(II) > Ca(II) > Mn(II) > K(I) > Na(I) |
| Simulant 3: Fe(III) > Eu(III) > Cs(I) > Cu(II) > Ni(II) > Co(II) > Ca(II) |
| Simulant 4: Fe(III) > Pb(II) > Cd(II) > Cu(II) > Cr(III) |
It can be seen from Fig. 3–6, that co-extraction of all metal ions present in solution is observed with every sorption cycle. However, there is an observed suppression of loading capacity for all metal ions except the metal ion being selectively extracted, when compared to the loading capacities of the single ion experiments. On average, the competing ions in the initial sorption tests for each simulant had extraction percentages lower than 20%, as further evidence of this suppression. However, in some cases, multi-sorption of additional ions exceeded 50%. It is currently difficult to pin-point the specific mechanism(s) that these systems of metal ions follow due to the lack of modelling and key experimental data (such as electron energy loss spectroscopy (EELS) and X-ray absorption near edge structure (XANES) data) – even for single ion systems. With these speciation data it would be possible to calculate the binding energies and edge energies which would in turn allow us to fully explore the thermodynamics of the sorption of the multi-metal systems.
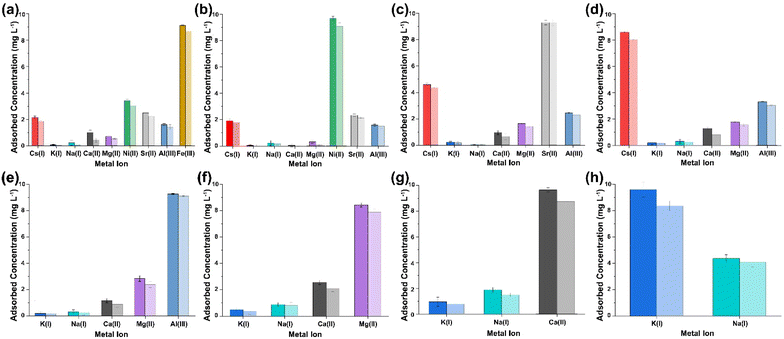 |
| | Fig. 3 Multi ICP-OES results obtained from the analysis of the supernatant collected after sequential multi-metal adsorption experiments with simulant 1 at pH 7 (a–h) [dark shaded colours represent (PO)x-Fe3O4 NPs and light shaded colours represent (PO)x-SiO2@Fe3O4 NPs]. | |
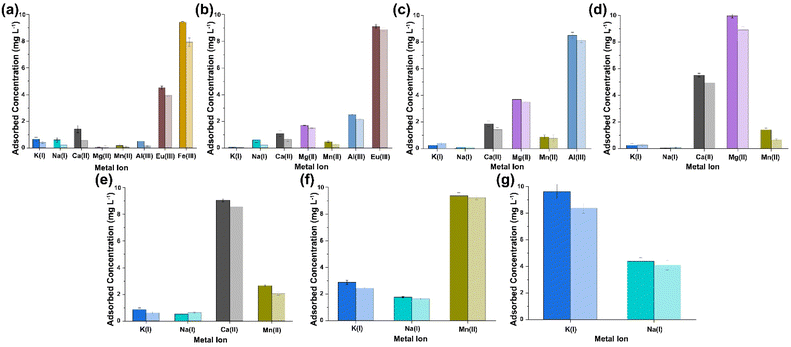 |
| | Fig. 4 ICP-OES results obtained from the analysis of the supernatant collected after sequential multi-metal adsorption experiments with simulant 2 at pH 7 (a–g) [dark shaded colours represent (PO)x-Fe3O4 NPs and light shaded colours represent (PO)x-SiO2@Fe3O4 NPs]. | |
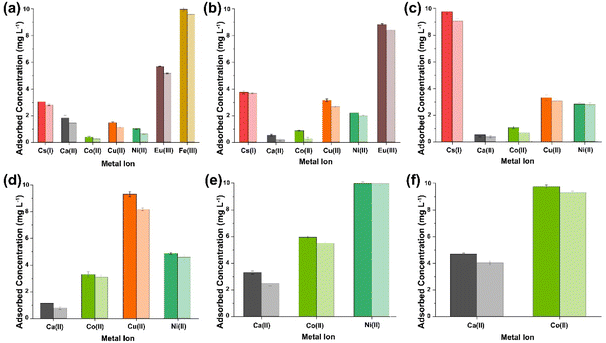 |
| | Fig. 5 ICP-OES results obtained from the analysis of the supernatant collected after sequential multi-metal adsorption experiments with simulant 3 at pH 7 (a–f) [dark shaded colours represent (PO)x-Fe3O4 NPs and light shaded colours represent (PO)x-SiO2@Fe3O4 NPs]. | |
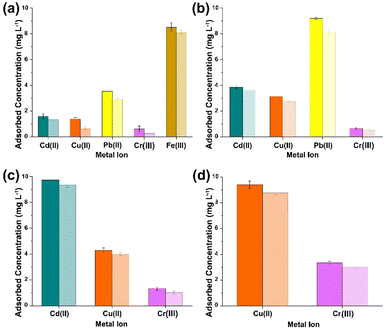 |
| | Fig. 6 ICP-OES results obtained from the analysis of the supernatant collected after sequential multi-metal adsorption experiments with simulant 4 at pH 7 (a–d) [dark shaded colours represent (PO)x-Fe3O4 NPs and light shaded colours represent (PO)x-SiO2@Fe3O4 NPs]. | |
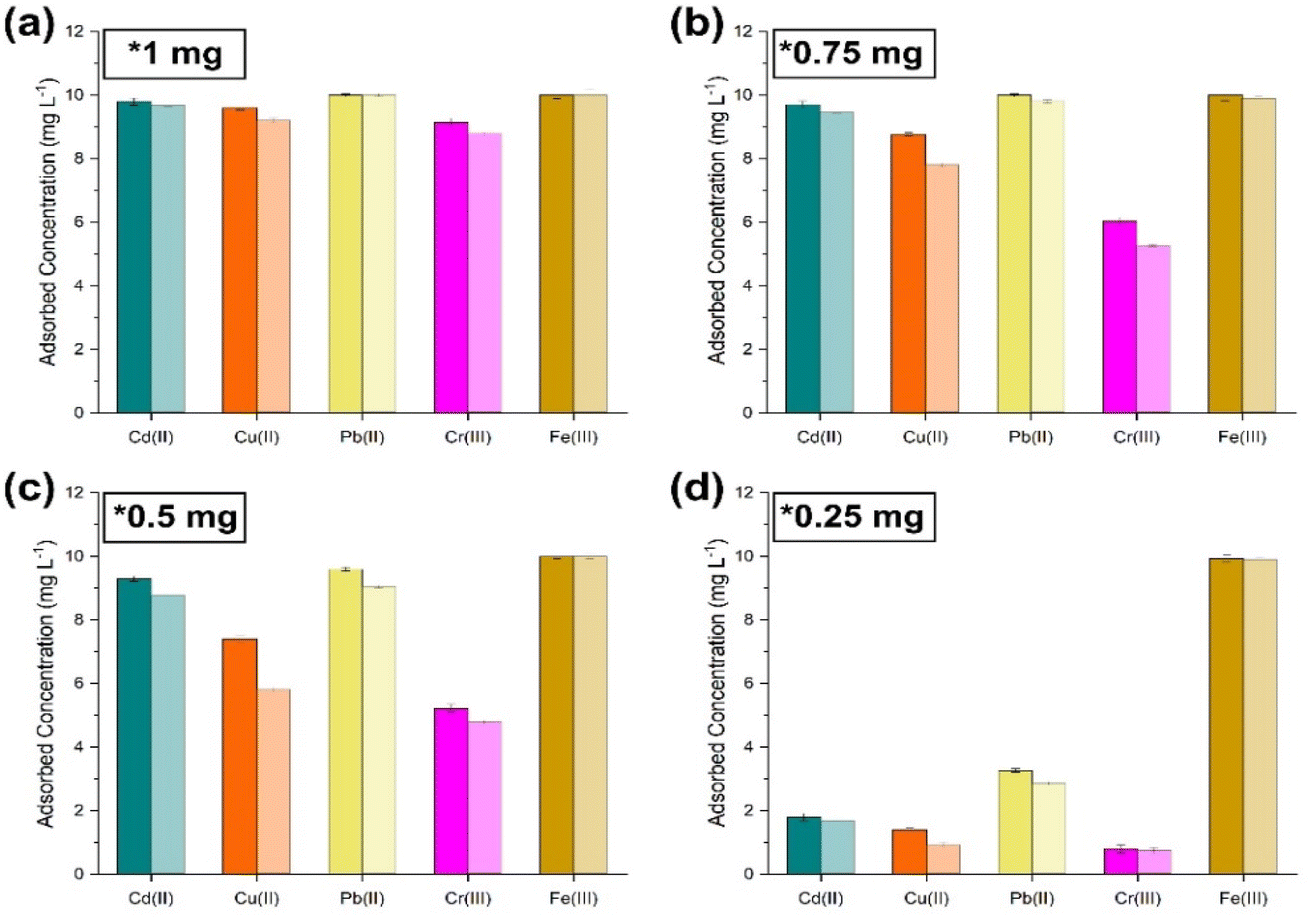
To fully understand the limit of this observed suppression of extraction of the metal ions in solution, the concentrations of NPs added were increased until all metal ions were entirely co-extracted in a single sorption cycle. Fig. 7–10 show the adsorbed concentrations of each metal ion present in the simulants with increasing NP concentrations, where it is observed that at 1 mg of NPs, all metal ions were co-extracted in one sorption cycle due to an increased number of phosphate binding sites.
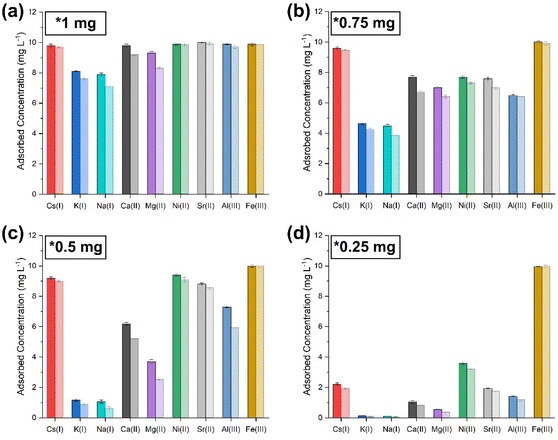 |
| | Fig. 7 Co-extraction limit tests carried out by sequentially lowering the amount of sorbent ((PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NPs) with simulant 1. (a) 1 mg; (b) 0.75 mg; (c) 0.5 mg; (d) 0.25 mg [dark shaded colours represent (PO)x-Fe3O4 NPs and light colour represents (PO)x-SiO2@Fe3O4 NPs]. | |
4. Discussion
Previously, the average Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P ratio for similar 12 nm phosphate-NP complexes was found to be 3.5 ± 2.1 Fe atoms per P atom,38 compared to the estimated Fe:P ratio of a simple phosphate monolayer of 20:1.27 These data show that the NPs exhibit an increased moiety that is 3.5 times the estimated monolayer and therefore points towards a possible multilayer structure on the surface. This can potentially be confirmed by the presence of both protonated phosphates and polyphosphate species signals in the ATR-FTIR analysis carried out to identify the presence of phosphate species (Fig. S4(a) and (f)†). Reported signals at 950 and 1200 cm−1 suggest the presence of a monoprotonated binuclear phosphate structure on the surface ((FeO)2(PO)(OH)).27 The signal present at 1600 cm−1 corresponds to a bihydroxlyated phosphate complex at the P–O peak position.27 The signal at 1160 cm−1 is possibly due to deprotonation of the phosphate complex (H2PO4) at the P–O band, where there is a slight shift in the peak position.39 Finally, the signals at 1350 and 1220 cm−1 are typical of polyphosphate chains.40,41 Therefore these ATR-FTIR findings support the presence of a wide range of phosphate-based species on the surface of the Fe3O4 and SiO2@Fe3O4 NPs. This increased Fe:P ratio has allowed us to increase the loading capacities of these NP complexes due to the higher number of M+ binding sites.
:P ratio for similar 12 nm phosphate-NP complexes was found to be 3.5 ± 2.1 Fe atoms per P atom,38 compared to the estimated Fe:P ratio of a simple phosphate monolayer of 20:1.27 These data show that the NPs exhibit an increased moiety that is 3.5 times the estimated monolayer and therefore points towards a possible multilayer structure on the surface. This can potentially be confirmed by the presence of both protonated phosphates and polyphosphate species signals in the ATR-FTIR analysis carried out to identify the presence of phosphate species (Fig. S4(a) and (f)†). Reported signals at 950 and 1200 cm−1 suggest the presence of a monoprotonated binuclear phosphate structure on the surface ((FeO)2(PO)(OH)).27 The signal present at 1600 cm−1 corresponds to a bihydroxlyated phosphate complex at the P–O peak position.27 The signal at 1160 cm−1 is possibly due to deprotonation of the phosphate complex (H2PO4) at the P–O band, where there is a slight shift in the peak position.39 Finally, the signals at 1350 and 1220 cm−1 are typical of polyphosphate chains.40,41 Therefore these ATR-FTIR findings support the presence of a wide range of phosphate-based species on the surface of the Fe3O4 and SiO2@Fe3O4 NPs. This increased Fe:P ratio has allowed us to increase the loading capacities of these NP complexes due to the higher number of M+ binding sites.
During the single-metal sorption experiments all metal ions selected for testing were shown to have a significant degree of extraction with both (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NP complexation. As all of the metal ions chosen were cationic, these results would be expected due to the anionic nature of the phosphate species (PO4−) functionalising the surface of the NPs. Another expected result was the complete extraction of Fe(III) ions from solutions, as Fe(III) has been reported to be in high concentrations in phosphate-rich soils, where it is found bonded to the phosphates.42 In this work, Fe(III) extraction consistently exceeds 95% extraction with both NP complexes presented. This suggests that phosphates have a high affinity towards Fe(III), similar to that of U(VI).43 As Fe(III) ions are prevalent throughout all of the scenarios presented here and indeed in other contaminated wastewaters, this result is highly promising. Both Eu(III) and La(III) were also highly extracted, exceeding 80% extraction for both NP complexes. This is again expected as phosphate affinity towards Eu(III) and La(III) has been reported previously.44,45 Al(III), Ce(III) and Cr(III) on the other hand showed a slightly lower extraction percentage (∼70%, Table S1†), in agreement with previous studies.46–48 In contrast, ions such as Ca(II), Cd(II), and Pb(II) showed a significant reduction in the extraction percentage, where Ca(II) and Cd(II) are below 50% for (PO)x-SiO2@Fe3O4 NP studies (45.75 and 47.10%, respectively) and Pb(II) is below 50% for both (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NP studies (37,74 and 34.45%, respectively).
With pH-dependent single-metal sorption studies, it was found that when the [H+] concentration is increased, i.e., decreasing the pH, the extraction percentages are reduced slightly. This shows evidence of a degree of suppression when it comes to metal ion sorption onto the (PO)x-SiO2@Fe3O4 NP surface. The (PO)x-SiO2@Fe3O4 NP complex tested in this work showed a tolerance to low pH environments, with no significant reduction in extraction efficiencies, where extraction percentages were reduced by less than 5% on average. However, at this pH Cs(II), Co(II), Pb(II) and Sr(II) extraction percentages were shown to be reduced by 16, 33, 18 and 27%, respectively. Optimal extraction environments for these metal ions are known to be above pH 6, where studies conducted previously show their reduced effectiveness below this pH value.9,49–51 Overall there was little effect when decreasing the pH on the sorption and loading capacities of the NP complexes. Due to the observed rapid dissolution of (PO)x-Fe3O4 NPs at this pH, they were not tested and therefore only tested in high pH environments (pH 11).52 On the other hand, due to the deterioration of SiO2 at pH above 8, these NPs were also excluded from the study.53 At pH 11, a significant increase in total removal for all metal ions in solution was observed. The (PO)x-Fe3O4 NPs showed a total removal above 90% for all metal ions selected for this study. The only metal ions with an extraction value below 90% were Na(I), Ca(II), Sr(II) and Cr(III), where they were removed by 69, 70, 70 and 86%, respectively. The enhanced removal percentages were deduced to be because of a high level of chemical precipitation occurring in such basic environments. The metal ions in Table 1 have been previously reported to undergo chemical precipitation at pH values about pH 8 and therefore this is an expected result.54,55 Overall, this study shows that sorption was able to be achieved at pH 3 with a significant extraction percentage and potentially at pH 11 with a large portion of removal being from chemical precipitation.
Fig. 3–6 show that the presence of several competing metal ions causes a large degree of competition towards the phosphate binding sites. It can be seen that Fe(III) ions are consistently the most extracted metal ions from all simulants tested. This is to be expected due to the affinity of Fe towards P, as reported previously.42 Excluding the small degree of multi-metal sorption shown most clearly in Fig. 4 for a single sorption experiment, there seems to be only one metal ion extracted at any one time. This suppression of other metal ions could be due to the fact that the fast-sorbed metal ion layer is blocking access to the phosphate binding sites, therefore reducing the number of metal ions extracted at once. Indeed, when reducing the concentration of metal ions in solution multi-metal sorption can be seen to increase, providing evidence that co-extraction of metals can be achieved when sufficient binding sites are available. There is an observed difference between the loading capacity of (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NPs, where a small reduction in loading capacity can be seen for (PO)x-SiO2@Fe3O4 NPs, with the reduction being on average less than 5%. This small decrease corresponds with the error in accurately calculating the approximate surface area of the NP complexes when normalising the surface area for both NP complexes and therefore the difference was assumed to be negligible at pH 7. From Fig. 7–10, with an increase in the NP concentration in all solutions for each simulant it can be observed that with a single sorption cycle it is possible to achieve co-extraction of all metal ions using 1 mg of NPs. This is due to the significant increase in phosphate binding sites available for the metal ions in solution, essentially reducing the competition for binding sites. Specificity on why these metal ions follow the selectivity series has not been determined mainly due to the lack of literature found simulating these specific systems of metal ions, to our knowledge. As there are several adsorption mechanisms that these sorption processes can follow, such as chemical adsorption, physical adsorption and isothermal adsorption, it is difficult to determine the specific route the metal ions take when being sorbed in these systems. To fully explore the thermodynamics of the remarkable sorption seen in this work, additional experimental work (such as EELS and/or XANES analysis of speciation and coordination) could provide valuable insights and enable the conduct of further modelling on these types of systems. This will be the subject of future studies. Overall, the NP complexes have shown great affinity towards all metal ions selected and have achieved the sorption of single metal ions at the concentrations tested.
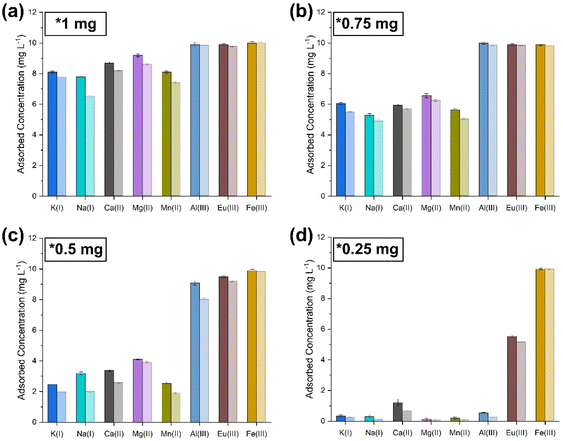 |
| | Fig. 8 Co-extraction limit tests carried out by sequentially lowering the amount of sorbent ((PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NPs) with simulant 2. (a) 1 mg; (b) 0.75 mg; (c) 0.5 mg; (d) 0.25 mg [dark shaded colours represent (PO)x-Fe3O4 NPs and light colour represents (PO)x-SiO2@Fe3O4 NPs]. | |
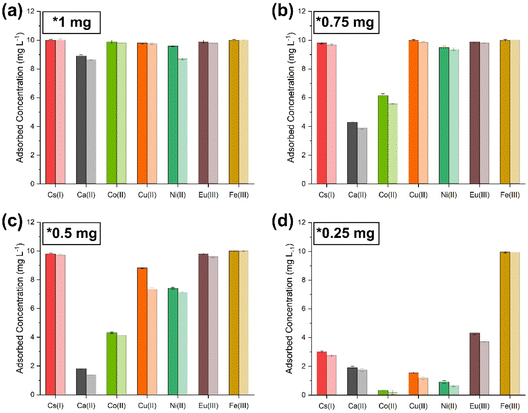 |
| | Fig. 9 Co-extraction limit tests carried out by sequentially lowering the amount of sorbent ((PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NPs) with simulant 3. (a) 1 mg; (b) 0.75 mg; (c) 0.5 mg; (d) 0.25 mg [dark shaded colours represent (PO)x-Fe3O4 NPs and light colour represents (PO)x-SiO2@Fe3O4 NPs]. | |
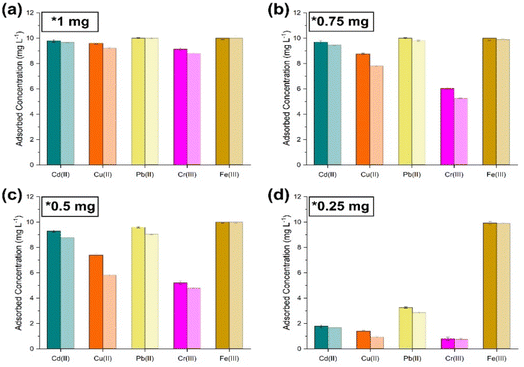 |
| | Fig. 10 Co-extraction limit tests carried out by sequentially lowering the amount of sorbent ((PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NPs) with simulant 4. (a) 1 mg; (b) 0.75 mg; (c) 0.5 mg; (d) 0.25 mg [dark shaded colours represent (PO)x-Fe3O4 NPs and light colour represents (PO)x-SiO2@Fe3O4 NPs]. | |
5. Conclusion
In conclusion, we have developed a potential broad-spectrum adsorbent for use in a range of chemical environments, showing high affinity and extremely high loading capacity towards Na(I), K(I), Cs(I), Ca(II), Cu(II), Co(II), Ni(II), Cd(II), Mg(II), Sr(II), Pb(II), Al(III), Mn(II), Eu(III) and Fe(III). By selecting different environments from real-world scenarios, such as nuclear fuel cooling ponds, mining wastewaters, coolant loops and sewage waters, the NP complexes were tested and analysed for their sorption capacity in the presence of competing ions. After sequential sorption tests with the same solutions, a selectivity series for each simulated environment was listed, therefore creating a portfolio of metal ions that the complexes are selective towards. Importantly, the metal ions were selected based on their potential harm to surrounding water systems, due to their toxicity to both the environment and humans. The pH study allowed the determination of the feasibility of the use of these NPs complexes for heavy metal removal in different chemical or industrial environments. We have demonstrated high efficiency at pH 7, pH 3 and pH 1 of the (PO)x-Fe3O4 and (PO)x-SiO2@Fe3O4 NP complexes, hence opening up the possibility of their use for both alkaline and acidic scenarios.
Conflicts of interest
The authors declare that there are no conflicts of interest regarding this work. This paper is dedicated to the memory of our dear colleague and friend Luc Vandeperre. We are grateful to have had the opportunity to know and work with him.
Acknowledgements
S. A. is a member of the EPSRC Centre for Doctoral Training in Nuclear Energy (EP/L015900/1). M. P. R. is grateful to the Royal Academy of Engineering and the Armourers and Brasier's Company for additional support. We acknowledge the technical assistance of Dr Mahmound Ardakani (TEM), Mr Richard Sweeney (XRD), Dr Irena Nevjestic (Magnetic Characterisation), and Mr Benjamin Chan (ICP-OES). Electron microscopy was carried out in the Harvey Flowers Microscopy suite, and magnetic characterisation in the SPIN-LAB (EP/P030548/1) at Imperial College London.
References
- E. Iakovleva and M. Sillanpää, The use of low-cost adsorbents for wastewater purification in mining industries, Environ. Sci. Pollut. Res., 2013, 20, 7878–7899 CrossRef CAS PubMed.
- A. Vazquez-Ortega, N. Perdrial, E. Reinoso-Maset, P. A. O'Day and J. Chorover, Phosphate Controls Uranium Release from Acidic Waste-Weathered Hanford Sediments, J. Hazard. Mater., 2021, 416, 126240 CrossRef CAS PubMed.
- J. Yang, B. Hou, J. Wang, B. Tian, J. Bi, N. Wang, X. Li and X. Huang, Nanomaterials for the Removal of Heavy Metals from Wastewater, Nanomaterials, 2019, 9, 424 CrossRef CAS PubMed.
- S. W. Wu and C. Z. Jiang, Designed synthesis and surface engineering strategies of magnetic iron oxide nanoparticles for biomedical applications, Nanoscale, 2016, 8, 19421–19474 RSC.
- E. Cali, J. Qi, O. Preedy, S. Chen, D. Boldrin, W. R. Branford, L. Vandeperre and M. P. Ryan, Functionalised magnetic nanoparticles for uranium adsorption with ultra-high capacity and selectivity, J. Mater. Chem. A, 2018, 6, 3063–3073 RSC.
- I. Mantis, D. Voutsa and C. Samara, Assessment of the environmental hazard from municipal and industrial wastewater treatment sludge by employing chemical and biological methods, Ecotoxicol. Environ. Saf., 2005, 62, 397–407 CrossRef CAS PubMed.
- N. K. Srivastava and C. B. Majumder, Novel biofiltration methods for the treatment of heavy metals from industrial wastewater, J. Hazard. Mater., 2008, 151, 1–8 CrossRef CAS PubMed.
- B. Gajda and M. B. Bogacki, The Effect of Tributyl Phosphate on the Extraction of Nickel(ii) and Cobalt(II), Physicochem. Probl. Miner. Process., 2007, 41, 145–152 CAS.
- R. He, W. Li, D. Deng, W. Chen, H. Li, C. Wei and Y. Tang, Efficient removal of lead from highly acidic wastewater by periodic ion imprinted mesoporous SBA-15 organosilica combining metal coordination and co-condensation, J. Mater. Chem. A, 2015, 3, 9789–9798 RSC.
- M. Mouflih, A. Aklil, N. Jahroud, M. Gourai and S. Sebti, Removal of lead from aqueous solutions by natural phosphate, Hydrometallurgy, 2006, 81, 219–225 CrossRef CAS.
- H. T. Hahn, The Mechanism of Uranium Extraction by Tributyl Phosphate, J. Am. Chem. Soc., 1957, 79, 4625–4629 CrossRef CAS.
- M. H. Taha, A. M. Masoud, Y. M. Khawassek, A. E. M. Hussein, H. F. Aly and E. Guibal, Cadmium and iron removal from phosphoric acid using commercial resins for purification purpose, Environ. Sci. Pollut. Res., 2020, 27, 31278–31288 CrossRef CAS PubMed.
- L. Fang, L. Huang, P. E. Holm, X. Yang, H. C. B. Hansen and D. Wang, Facile upscaled synthesis of layered iron oxide nanosheets and their application in phosphate removal, J. Mater. Chem. A, 2015, 3, 7505–7512 RSC.
- S. S. Lee, W. Li, C. Kim, M. Cho, B. J. Lafferty and J. D. Fortner, Surface functionalized manganese ferrite nanocrystals for enhanced uranium sorption and separation in water, J. Mater. Chem. A, 2015, 3, 21930–21939 RSC.
- K. Kazy, S. F. Dsouza and P. Sar, Uranium and thorium sequestration by a Pseudomonas sp.: mechanism and chemical characterization, J. Hazard. Mater., 2009, 163, 65–72 CrossRef PubMed.
- M. Sutton and S. R. Burastero, Uranium(iv) solubility and speciation in simulated elemental human biological fluids, Chemosphere, 2007, 70, 263–269 CrossRef PubMed.
- P. Rudnicki, Z. Hubicki and D. Kołodyńska, Evaluation of heavy metal ions removal from acidic waste water streams, Chem. Eng. J., 2014, 252, 362–373 CrossRef CAS.
- S. Aberdeen, C. An Hur, E. Cali, L. Vandeperre and M. P. Ryan, Acid resistant functionalised magnetic nanoparticles for radionuclide and heavy metal adsorption, J. Colloid Interface Sci., 2022, 608, 1728–1738 CrossRef CAS PubMed.
- I. I. Slowing, J. L. Vivero-Escoto, B. G. Trewyn and V. S.-Y. Lin, Mesoporous silica nanoparticles: structural design and applications, J. Mater. Chem. A, 2010, 20, 7924–7937 RSC.
- J. Park, K. An, Y. Hwang, J. E. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Ultra-large-scale syntheses of monodisperse nanocrystals, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- H. L. Ding, Y. X. Zhang, S. Wang, J. M. Xu, S. C. Xu and G. H. Li, Fe3O4@SiO2 core/shell nanoparticles: the silica coating regulations with a single core for different core sizes and shell thicknesses, Chem. Mater., 2012, 24, 4572–4580 CrossRef CAS.
- Y. Zhao, J. Li, L. Zhao, S. Zhang, Y. Huang, X. Wu and X. Wang, Synthesis of amidoxime-functionalized Fe3O4@SiO2 core–shell magnetic microspheres for highly efficient sorption of U(IV), Chem. Eng. J., 2014, 235, 275–283 CrossRef CAS.
- L. Zhang, R. He and H. C. Gu, Oleic acid coating on the monodisperse magnetite nanoparticles, Appl. Surf. Sci., 2006, 253, 2611–2617 CrossRef CAS.
- M. Klokkenburg, J. Hilhorst and B. H. Erné, Surface analysis of magnetite nanoparticles in cyclohexane solutions of oleic acid and oleylamine, Vib. Spectrosc., 2007, 43, 243–248 CrossRef CAS.
- K. Yoshinaga, H. Yoshida, Y. Yamamoto, K. Takakura and M. Komatsu, A convenient determination of surface hydroxyl group on silica gel by conversion of silanol hydrogen to dimethylsilyl group with diffuse reflectance FTIR spectroscopy, J. Colloid Interface Sci., 1992, 153, 207–211 CrossRef CAS.
- B. McCool, L. Murphy and C. P. Tripp, A simple FTIR technique for estimating the surface area of silica powders and films, J. Colloid Interface Sci., 2006, 295, 294–298 CrossRef CAS PubMed.
- T. J. Daou, S. Begin-Colin, J. M. Grenèche, F. Thomas, A. Derory, P. Bernhardt, P. Legaré, G. Pourroy, J. M. Grenche, F. Thomas, A. Derory, P. Bernhardt, P. Legar, G. Pourroy and P. Legare, Phosphate Adsorption Properties of Magnetite-Based Nanoparticles, Chem. Mater., 2007, 19, 4494–4505 CrossRef CAS.
- A. Zach-Maor, R. Semiat and H. Shemer, Adsorption–desorption mechanism of phosphate by immobilized nano-sized magnetite layer: interface and bulk interactions, J. Colloid Interface Sci., 2011, 363, 608–614 CrossRef CAS PubMed.
- E. J. Elzinga and D. L. Sparks, Phosphate adsorption onto hematite: an in situ ATR-FTIR investigation of the effects of pH and loading level on the mode of phosphate surface complexation, J. Colloid Interface Sci., 2007, 308, 53–70 CrossRef CAS PubMed.
- C. Sun, D. Xu and D. Xue,
In situ FTIR-ATR observation of structural dynamics of H2PO4 in precrystallisation solution, Mater. Res. Innovations, 2014, 18, 370–375 CrossRef CAS.
- N. Majoul, S. Aouida and B. Bessaïs, Progress of porous silicon APTES-functionalization by FTIR investigations, Appl. Surf. Sci., 2015, 331, 388–391 CrossRef CAS.
- V. C. Karade, A. Sharma, R. P. Dhavale, R. P. Dhavale, S. R. Shingte, P. S. Patil, J. H. Kim, D. R. T. Zahn, A. D. Chougale, G. Salvan and P. B. Patil, APTES monolayer coverage on self-assembled magnetic nanospheres for controlled release of anticancer drug Nintedanib, Sci. Rep., 2021, 11, 1–12 CrossRef PubMed.
- R. A. Bini, R. F. C. Marques, F. J. Santos, J. A. Chaker and M. Jafelicci, Synthesis and functionalization of magnetite nanoparticles with different amino-functional alkoxysilanes, J. Magn. Magn. Mater., 2012, 324, 534–539 CrossRef CAS.
- H. Diebler and C. Millant, Redox reactions between
complexes of molybdenum and iron, Polyhedron, 1986, 5, 539–545 CrossRef CAS.
- A. M. Hutchings, A. Basu, A. J. Dickson and A. V. Turchyn, Molybdenum geochemistry in salt marsh pond sediments, Geochim. Cosmochim. Acta, 2020, 284, 75–91 CrossRef CAS.
- A. Gaur, M. Stehle, K. V. Raun, J. Thrane, A. D. Jensen, J. D. Grunwaldt and M. Høj, Structural dynamics of an iron molybdate catalyst under redox cycling conditions studied with: in situ multi edge XAS and XRD, Phys. Chem. Chem. Phys., 2020, 22, 11713–11723 RSC.
- S. Eroglu, F. Scholz, R. Salvatteci, C. Siebert, R. Schneider and M. Frank, The impact of postdepositional alteration on iron- and molybdenum-based redox proxies, Geology, 2021, 49, 1411–1415 CrossRef CAS.
- Y. Mao and Q. Yue, Kinetic Modeling of Phosphate Adsorption by Preformed and in situ formed Hydrous Ferric Oxides at Circumneutral pH, Sci. Rep., 2016, 6, 35292 CrossRef CAS PubMed.
- M. Tejedor-Tejedor and M. Anderson, The protonation of phosphate on the surface of goethite as studied by CIR-FTIR and electrophoretic mobility, Langmuir, 1990, 6, 602–611 CrossRef CAS.
- A. Khoshmanesh, P. L. M. Cook and B. R. Wood, Quantitative determination of polyphosphate in sediments using Attenuated Total Reflectance-Fourier Transform Infrared (ATR-FTIR) spectroscopy and partial least squares regression, Analyst, 2012, 137, 3704 RSC.
- Z. He, C. W. Honeycutt, B. Xing, R. W. McDowell, P. J. Pellechia and T. Zhang, Solid-State Fourier Transform Infrared and 31P Nuclear Magnetic Resonance Spectral Features of Phosphate Compounds, Soil Sci., 2007, 7, 501–515 CrossRef.
- E. Herndon, L. Kinsman-Costello, N. Di Domenico, K. Duroe, M. Barczok, C. Smith and S. D. Wullschleger, Iron and iron-bound phosphate accumulate in surface soils of ice-wedge polygons in arctic tundra, Environ. Sci.: Processes Impacts, 2020, 22, 1475–1490 RSC.
- N. Dacheux, N. Clavier, A. C. Robisson, O. Terra, F. Audubert, J. É. Lartigue and C. Guy, Immobilisation of actinides in phosphate matrices, C. R. Chim., 2004, 7, 1141–1152 CrossRef CAS.
- D. Copetti, K. Finsterle, L. Marziali, F. Stefani, G. Tartari, G. Douglas, K. Reitzel, B. M. Spears, I. J. Winfield, G. Crosa, P. D'Haese, S. Yasseri and M. Lürling, Eutrophication management in surface waters using lanthanum modified bentonite: a review, Water Res., 2015, 97, 162–174 CrossRef PubMed.
- S. Y. Huang, M. Qian and V. C. Pierre, A Combination of Factors: tuning the Affinity of Europium Receptors for Phosphate in Water, Inorg. Chem., 2019, 58, 16087–16099 CrossRef CAS PubMed.
-
P. H. Hsu, Interaction between aluminum and phosphate in aqueous solution, in Trace Inorganics In Water, 1968, pp. 115–127 Search PubMed.
- P. Puszrai, E. Toth-Szeles, D. Horvath, A. Toth, A. Kukovexz and Z. Konya, A simple method to control the formation of cerium phosphate architectures, CrystEngComm, 2015, 17, 8477–8485 RSC.
- G. P. Haight, F. Smentowski, M. Rose and C. Heller, Reactions of Chromium(VI) with Phosphorus(III)1 and Phosphorus(I). II. Ethyl-Substituted Phosphorus(III) Compounds, J. Am. Chem. Soc., 1968, 90, 6325–6328 CrossRef CAS.
- F. Giannakopoulou, C. Haidouti, A. Chronopoulou and D. Gasparatos, Sorption behavior of cesium on various soils under different pH levels, J. Hazard. Mater., 2007, 149, 553–556 CrossRef CAS PubMed.
- S. S. Al-Shahrani, Treatment of wastewater contaminated with cobalt using Saudi activated bentonite, Alexandria Eng. J., 2014, 53, 205–211 CrossRef.
- I. Smičiklas, I. Coha, M. Jović, M. Nodilo, M. Šljivić-Ivanović, S. Smiljanić and Ž. Grahek, Efficient separation of strontium radionuclides from high-salinity wastewater by zeolite 4A synthesized from Bayer process liquids, Sci. Rep., 2021, 11, 1–14 CrossRef PubMed.
- A. E. Chastukhin, A. D. Izotov, I. G. Gorichev and A. M. Kutepov, Analysis of Fe2O3 and Fe3O4 Dissolution Kinetics in Terms of the Chain Mechanism Model, Theor. Found. Chem. Eng., 2003, 37, 398–406 CrossRef CAS.
- Y. Niibori, M. Kunita, O. Tochiyama and T. Chida, Dissolution Rates of Amorphous Silica in Highly Alkaline Solution, J. Nucl. Sci. Technol., 2000, 37, 349–357 CrossRef CAS.
- A. Pohl, Removal of Heavy Metal Ions from Water and Wastewaters by Sulfur-Containing Precipitation Agents, Water, Air, Soil Pollut., 2020, 231, 503 CrossRef CAS.
- M. M. Matlock, B. S. Howerton and D. A. Atwood, Chemical precipitation of heavy metals from acid mine drainage, Water Res., 2002, 36, 4757–4764 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterisation of the as-synthesised 12 nm Fe3O4 NPs including TEM micrographs, size-distribution, DLS measurements, VSM hysteresis loop and XRD data. See DOI: https://doi.org/10.1039/d3ta01998e |
| ‡ Deceased. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Eleonora
Cali
ac,
Luc
Vandeperre‡
a and
Mary. P.
Ryan
*ab
a,
Eleonora
Cali
ac,
Luc
Vandeperre‡
a and
Mary. P.
Ryan
*ab