
Tuning the selectivity of benzylamine photo-oxidation with different rhodium modes anchored on BiOIO3†
Jiaping
Liu‡
a,
Yan
Wu‡
a,
Qingqing
Chen‡
a,
Rui
Yu
a,
Keqing
Shi
b,
Tao
Jing
 d,
Zhujie
Li
*b,
Zaizhu
Lou
*c and
Gang
Wang
*a
d,
Zhujie
Li
*b,
Zaizhu
Lou
*c and
Gang
Wang
*a
aKey Laboratory of Functional Molecular Solids, Ministry of Education, College of Chemistry and Materials Science, Anhui Normal University, Wuhu, 241002, P. R. China. E-mail: GangWang@ahnu.edu.cn
bAdvanced Technology Research Institute, Beijing Institute of Technology, Jinan, 250300, P. R. China. E-mail: zhujieli89@163.com
cGuangdong Provincial Key Laboratory of Nanophotonic Manipulation, Institute of Nanophotonics, Jinan University, Guangzhou, 511443, P. R. China. E-mail: zzlou@jnu.edu.cn
dCollege of Science, Kaili University, Kaili, 556011, P. R. China
First published on 2nd June 2023
Abstract
Selective benzylamine (BA) oxidation to high value-added imines is of great significance in the chemical industry, but it still remains challenging. In this study, we demonstrate rhodium single atom and nanoparticle modes (Rh1 and RhNP) anchored on BiOIO3 nanosheets as two efficient photocatalysts (Rh1/BOIO and RhNP/BOIO) to achieve the selective BA oxidation reaction. Notably, these two photocatalysts displayed completely opposite selectivity during BA photo-oxidation. Benzonitrile (BN) was the main product over Rh1/BOIO, while RhNP/BOIO preferentially produced N-benzylidenebenzylamine (N-BBA) under the same reaction conditions. To reveal the reason for such differences, we combined density functional theory (DFT) simulation and experiments to investigate the BA oxidation reaction mechanism over these two photocatalysts. As a consequence, it was found that the different active intermediates ˙OH and ˙O2− generated on Rh1 and RhNP would lead to the opposite selectivity of BA photo-oxidation over Rh1/BOIO and RhNP/BOIO, respectively. This study offers a valuable strategy to tune the selectivity of photo-oxidation reactions as required.
 Gang Wang | Gang Wang is currently as Associate Professor at College of Chemistry and Materials Science, Anhui Normal University. He received his PhD in 2015 from State Key Laboratory of Crystal Materials at Shandong University. During 2017–2019, he joined Kyoto University as a postdoc for studying Metal–organic frameworks (MOFs) compounds. His research interests focus on the development of single atom catalysts for solar energy conversion, including CO2 conversion, photosynthesis and overall water splitting. |
1 Introduction
The selective oxidation of amines to imine, amide, and nitrile products is practically significant for feedstock manufacturing in the field of organic synthesis.1–5 However, tuning the selectivity of oxidation products still remained a long-term goal and challenge due to the complicated reaction system and harsh reaction conditions (e.g., high temperature, high pressure, and the use of toxic cyanides) in current amine oxidation reaction systems.6–9 Therefore, it is urgently desirable to develop economical and environmentally friendly catalytic systems toward realizing highly selective aniline oxidation reactions. In recent years, photocatalytic technology has been regarded as one of the promising approaches in organic synthesis owing to its green, cost-effective, and gentle reaction conditions.10–12 Nevertheless, the application of photocatalysts in organic synthesis is still hindered by their inefficient catalytic capacity due to the low separation efficiency of photogenerated charge carriers.Various efforts have been devoted to promoting the charge-separation efficiency of photocatalysts, including metal species loading, which is one of the most effective strategies.13–16 Currently, most of the researches are focused on regulating the patterns of the loaded metal nanoparticles (NPs), such as the particle size, morphology, and their components in photocatalysts. For example, different sizes of Pt nanoparticles were successfully decorated on a TiO2–SiO2 support, leading to the size-dependent activity in photocatalytic CO2 conversion. The charge-transfer efficiency can be promoted by decreasing the size of the Pt NPs.17 Liu and coworkers reported that the existence and absence of Pt NPs on a CdS/Fe2O3 Z-scheme heterojunction exhibited completely different selectivities for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and C–C coupling reactions, respectively. Pt NPs acted as cocatalysts for accelerating the charge separation to facilitate the formation of CN over the CdS/Fe2O3 Z-scheme heterojunction.18 Recently, single-atom photocatalysts (SAPCs), involving the advantages of nearly 100% atom utilization, abundant active sites, and a well-defined coordination structure, have emerged as another encouraging method for promoting the charge-separation efficiency.19–27 For example, a photocatalyst with Ag nanoparticles and in situ-generated Ag single atom (SAAg) on AgF was fabricated to realize selective hydrodehalogenation and dehalogenation–arylation reactions under visible-light irradiation.28 The well-defined coordination structure of SAAg served as the catalytic active center to anchor activated halides for achieving the photocatalytic organic synthesis. Wen and coworkers reported that single-atom Ni on a TiO2 support not only promoted the charge-carrier separation efficiency, but was also favorable to realize the selective sulfonation of enamides into amidosulfones.29 However, there are few reports on the effects on the product selectivity in photocatalytic organic syntheses via anchoring different modes of metal.
N and C–C coupling reactions, respectively. Pt NPs acted as cocatalysts for accelerating the charge separation to facilitate the formation of CN over the CdS/Fe2O3 Z-scheme heterojunction.18 Recently, single-atom photocatalysts (SAPCs), involving the advantages of nearly 100% atom utilization, abundant active sites, and a well-defined coordination structure, have emerged as another encouraging method for promoting the charge-separation efficiency.19–27 For example, a photocatalyst with Ag nanoparticles and in situ-generated Ag single atom (SAAg) on AgF was fabricated to realize selective hydrodehalogenation and dehalogenation–arylation reactions under visible-light irradiation.28 The well-defined coordination structure of SAAg served as the catalytic active center to anchor activated halides for achieving the photocatalytic organic synthesis. Wen and coworkers reported that single-atom Ni on a TiO2 support not only promoted the charge-carrier separation efficiency, but was also favorable to realize the selective sulfonation of enamides into amidosulfones.29 However, there are few reports on the effects on the product selectivity in photocatalytic organic syntheses via anchoring different modes of metal.
In this study, we fabricated two photocatalysts with Rh single atom and nanoparticle modes, respectively, anchored on BiOIO3 nanosheets (Rh1/BOIO and RhNP/BOIO), both of which exhibited highly efficient BA photo-oxidation performances. The difference was that BN was the main product over Rh1/BOIO, while the N-BBA product dominated for RhNP/BOIO under the same reaction conditions. More importantly, the similar phenomena of opposite selectivity also occurred over Rh1/BOIO and RhNP/BOIO with other BA-based compounds as substrates. By combining theoretical calculations and experiments, we could reveal that the Rh–O2 mode in Rh1/BOIO favored combining with ˙OH active intermediates, which were derived from the decomposition of tert-butyl hydroperoxide (t-BuOOH), and then oxidized the PhCHNH intermediate to BN. In the case of RhNP/BOIO, O2 molecules could be generated due to the easy disproportionation of ˙OH radicals on the RhNP mode. After capturing the photogenerated electrons, the O2 molecules became ˙O2− active species and thus could oxidize PhCHNH to PhCHO. Another BA molecule then combined with PhCHO to form the final product of N-BBA.
2 Experimental
2.1. Chemicals
Bismuth nitrate pentahydrate (Bi(NO3)3·5H2O), potassium iodate (KIO3), rhodium(III) chloride (RhCl3·xH2O), ethylene glycol (EG), polyvinylpyrrolidone (PVP, K-30), sodium borohydride (NaBH4), methanol, ethanol, acetone, tert-butyl hydroperoxide (t-BuOOH, 70 wt% in H2O), benzylamine (BA), 4-methylbenzylamine, 4-methoxybenzylamine, 4-fluorobenzylamine and 4-chlorobenzylamine were all obtained from Sinopharm. All the chemicals were analytical grade and used without further treatment for purification. Ultrapure water (18.2 MΩ cm) was purified with a Milli-Q instrument.2.2. Preparation of photocatalysts
Preparation of BiOIO3 (BOIO): BiOIO3 was prepared via a hydrothermal method. First, 8 mmol Bi(NO3)3·5H2O and 8 mmol KIO3 were dissolved in 80 mL H2O. After stirring for 30 min, the mixed suspension was transferred into a 100 mL sealed Teflon-lined autoclave and heated at 150 °C for 5 h. After cooling to room temperature, the obtained sample was washed with water and ethanol several times and dried at 60 °C for 12 h.Preparation of Rh single-atom-loaded BiOIO3 (Rh1/BOIO): first, 0.3 g of the above BOIO was dispersed in 40 mL H2O and 10 mL ethanol mixed solution. Then, 122 μL RhCl3·xH2O solution (0.1 g mL−1) was added into the suspension. After 0.5 h constant stirring, the mixed suspension was irradiated under full light (PLS-SEX300D, 300 W Xenon lamp, Beijing Perfectlight Technology Co., Ltd.) for about 0.5 h. Rh1/BOIO was obtained after washing with water and ethanol several times, and dried at 60 °C for 12 h. Different amounts of Rh1 were also loaded on BOIO surface by controlling the added RhCl3·xH2O. In detail, 61 and 183 μL RhCl3·xH2O solution (0.1 g mL−1) were respectively added into 0.3 g BOIO suspensions to obtain two contrastive samples after light irradiation. The mass fractions of Rh species in these two samples were about 0.34 and 1.04 wt% according to inductively coupled plasma atomic emission spectroscopy (ICP-AES), and denoted as Rh1-0.34 wt% and Rh1-1.04 wt%.
Preparation of Rh nanoparticles-loaded BiOIO3 (RhNP/BOIO): the Rh nanoparticles were synthesized via a chemical reduction method. In detail, 20 mL EG, 418 μL RhCl3·xH2O solution (0.1 g mL−1), and 100 mg PVP were mixed together by constant stirring at 85 °C. After forming a dark mixed solution, the reaction temperature was increased to 115 °C and maintained for 2 h via an oil bath method. Then, NaBH4 was added after the solution cooled to room temperature. The RhNP was obtained after stirring the reaction for 12 h. Before use, RhNP was washed with ethanol and acetone several times. For RhNP/BOIO, 0.3 g BOIO and 2.1 mg of the as-prepared RhNP were added into a mixed solution of 20 mL methanol and 20 mL water. After stirring the reaction for 12 h, RhNP/BOIO was obtained after centrifugation and treatment at 100 °C for 12 h in a vacuum. Different amounts of RhNP were also loaded on the BOIO surface by controlling the added as-prepared RhNP. On the basis of the ICP-AES results, the samples with different RhNP amounts were obtained, and were denoted as RhNP-0.35 wt% and RhNP-1.04 wt%.
2.3. Characterizations
The crystal phases of the samples were analyzed by X-ray diffraction (XRD, D/MAX-2550). The morphologies of the samples were determined by transmission electron microscopy (TEM, JEOL 200CX) and AC HAADF-STEM (JEOL JEM-ARM 200F) with an energy-dispersive X-ray spectrometry system (EDS). Raman spectra and Fourier transform infrared (FT-IR) were performed on a Renshaw in Via Raman spectrometer and Bruker Tensor 22 instrument, respectively. Micromeritics ASAP 2460 adsorption apparatus was used to measure the N2 adsorptions of the samples. UV/vis diffuse reflectance spectra (DRS) of the photocatalysts were recorded on a Hitachi U-3010 spectrophotometer. A Thermo Scientific Plasma Quad inductively coupled plasma atomic emission spectroscopy system (ICP-AES) was used to determine the content of Rh in the samples. The photoluminescence (PL) properties and time-resolved photoluminescence (TRPL) spectra of the samples were measured on fls980 and Horiba Fluomax-4, respectively. X-Ray photoelectron spectroscopy (XPS) tests were performed on a PHI ESCA-5000C electron spectrometer. A gas chromatography-mass spectrometry system (GC-MS, Thermo Scientific Trace 1300) was employed to detect the products. Free radical trapping experiments were conducted using an electron spin resonance (ESR) instrument. For the ˙OH active species, 20 mg sample, 1.0 mL H2O, 50 μL t-BuOOH, and 5, 5-dimethyl-1-pyrroline-N-oxide (DMPO, 0.5 mmol L−1) were mixed together in an argon atmosphere after ultrasonic dispersion. The ESR signals of the samples were obtained after 10 min light irradiation. As for the ˙O2− active species, the test process was similar to that for the ˙OH active species, except for changing the 1.0 mL H2O to 1.0 mL methanol in the mixed suspension. X-Ray absorption fine structure (XAFS) measurements of Rh1/BOIO were conducted at the BL14W1 station in the Shanghai Synchrotron Radiation Facility (SSRF).2.4. Photocatalytic reaction
The photo-oxidation performances over the samples were conducted under full light irradiation. In detail, 20 mg photocatalyst, 1.0 mL t-BuOOH (70 wt%), and 100 μL BA were mixed together after 2 min stirring in a 15 mL thick-walled pressure bottle. Before sealing, Ar gas was injected in to the bottle to remove air. 300 W Xenon lamp (PLS-SEX300D, Beijing Perfectlight Technology Co., Ltd.) was used as the light source for the photocatalytic reactions. After reacting for 6 h, the products were detected via GC-MS. In the scope of the photo-oxidation reactions, the illumination time was increased to 12 h. The conversion rate and selectivity were calculated via the following equations:| Conversion rate (%) = [(C0 − Cr)/C0] × 100% |
| Selectivity rate (%) = [Cp/(C0 − Cr)] × 100% |
2.5. Photoelectrochemical performances
The photoelectrochemical performances of the samples were tested using a three-electrode system in 0.5 M Na2SO4 electrolyte solution. In detail, the sample-modified FTO glasses, platinum foil (1.0 × 1.0 cm2), and Ag/AgCl (saturated KCl) were employed as the working, counter, and reference electrode, respectively. A 300 W Xe lamp was used as the light source and the data were collected on a CHI660E (China) electrochemical station under light illumination and in the dark. During the preparation of the working electrode, 5.0 mg sample, 20 μL of Nafion solution (5.0 wt%), and 1.0 mL H2O were mixed together via ultrasonic dispersion for 1.0 h to form a photocatalyst ink. After that, the photocatalyst-modified FTO glasses were obtained by coating 100 μL ink on FTO glass (1.0 × 1.0 cm2), and dried in the air at room temperature. The same three-electrode system was also used for the Mott–Schottky (M–S) measurements.2.6. Theoretical calculations
Density functional theory (DFT) calculations were performed using the Vienna ab initio Simulation Package (VASP) with the projector augmented-wave potentials.30,31 The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional was employed to describe the exchange-correlation.32,33 The cutoff energy for the plane-wave basis was set to 520 eV for all the calculations. The energy convergence criterion for the self-consistent-field iteration was 10−5 eV, and the atomic positions were fully optimized until all the residual forces were smaller than 0.01 eV Å−1. First, the structure optimization of bulk BiOIO3 was performed with a 7 × 7 × 3 Monkhorst–Pack mesh for the Brillouin zone integration. The optimized bulk BiOIO3 had a cell size of a = 5.71 Å, b = 5.78 Å, c = 11.25 Å with 4 Bi atoms, 4 I atoms, and 16 O atoms in total. Then, the slab model of BiOIO3 was constructed with the IO-terminated (001) surface exposed, with a 15 Å vacuum space along the z-direction included to avoid interlayer interactions. After the optimization of the BiOIO3 slab, four initial configurations of the adsorption structure of a single Rh atom on the BiOIO3 surface were created and optimized. As a result, the four configurations eventually tended to a similar adsorption structure with Rh bonded to two terminal O atoms. Finally, the free energy differences of the t-BuOOH decomposition into O2 were calculated on both the Rh single atom and the Rh nanoparticle (the (111) surface of bulk Rh was created to mimic the Rh nanoparticle). For all the simulations, the DFT-D3 Grimme strategy for dispersion correction of the total energy was used to include the vdW interactions.343 Results and discussion
3.1. Characterization of Rh1/BOIO and RhNP/BOIO
The atomically dispersed Rh was directly anchored on BOIO (Rh1/BOIO) via photodeposition. RhNP/BOIO was obtained after activating the mixed RhNP and BOIO at high temperature in a vacuum (see details in the Experimental part). Rh1/BOIO and RhNP/BOIO were collected as powders for X-ray diffraction (XRD) technique measurements, and both of them could be well matched with the pure BOIO sample (ICSD#262019, Fig. S1†).35–38 Only slightly changes could be detected in the Fourier transform infrared (FT-IR) spectrometry, Raman spectra, and Brunauer–Emmett–Teller (BET) results for the samples, indicating the maintenance of the BOIO crystal structure after the Rh1 and RhNP modes, respectively, were decorated on it (Fig. S2–S4†). Compared with pure BOIO, the slight changes in the Raman, FT-IR, and BET results over Rh1/BOIO and RhNP/BOIO might be attributed to the decreased crystallinity of BOIO after the loading procedure. Next, transmission electron microscopy (TEM) was employed to investigate the morphologies of Rh1/BOIO, RhNP/BOIO and pure BOIO, all of which displayed a random nanosheets morphology (Fig. S5–S7†). Fig. 1a shows that the RhNP were sized about 10 nm and distributed on the surface of BOIO nanosheets. High-resolution TEM (HRTEM) was implemented to obtain more microstructure information of RhNP/BOIO. As presented in Fig. 1b, lattice fringes with inter-fringe distances of 0.282 and 0.225 nm, assigned to the BOIO (200) and Rh (111) lattice planes, respectively, could be observed.39,40 The corresponding energy-dispersive X-ray spectroscopy (EDS) mapping images in Fig. 1c further demonstrated the RhNP mode anchored on the surface of BOIO nanosheets. In the case of Rh1/BOIO, no nanoparticles or clusters could be detected in Fig. 1d. The atomic resolution high-angle annular dark-field scanning TEM (AC HAADF-STEM) images combined with the corresponding EDS mapping images together further confirmed the uniform dispersion of Rh atoms on the BOIO surface (Fig. 1e and f). The content of Rh species was about 0.69 wt% for both Rh1/BOIO and RhNP/BOIO, according to the ICP-AES results.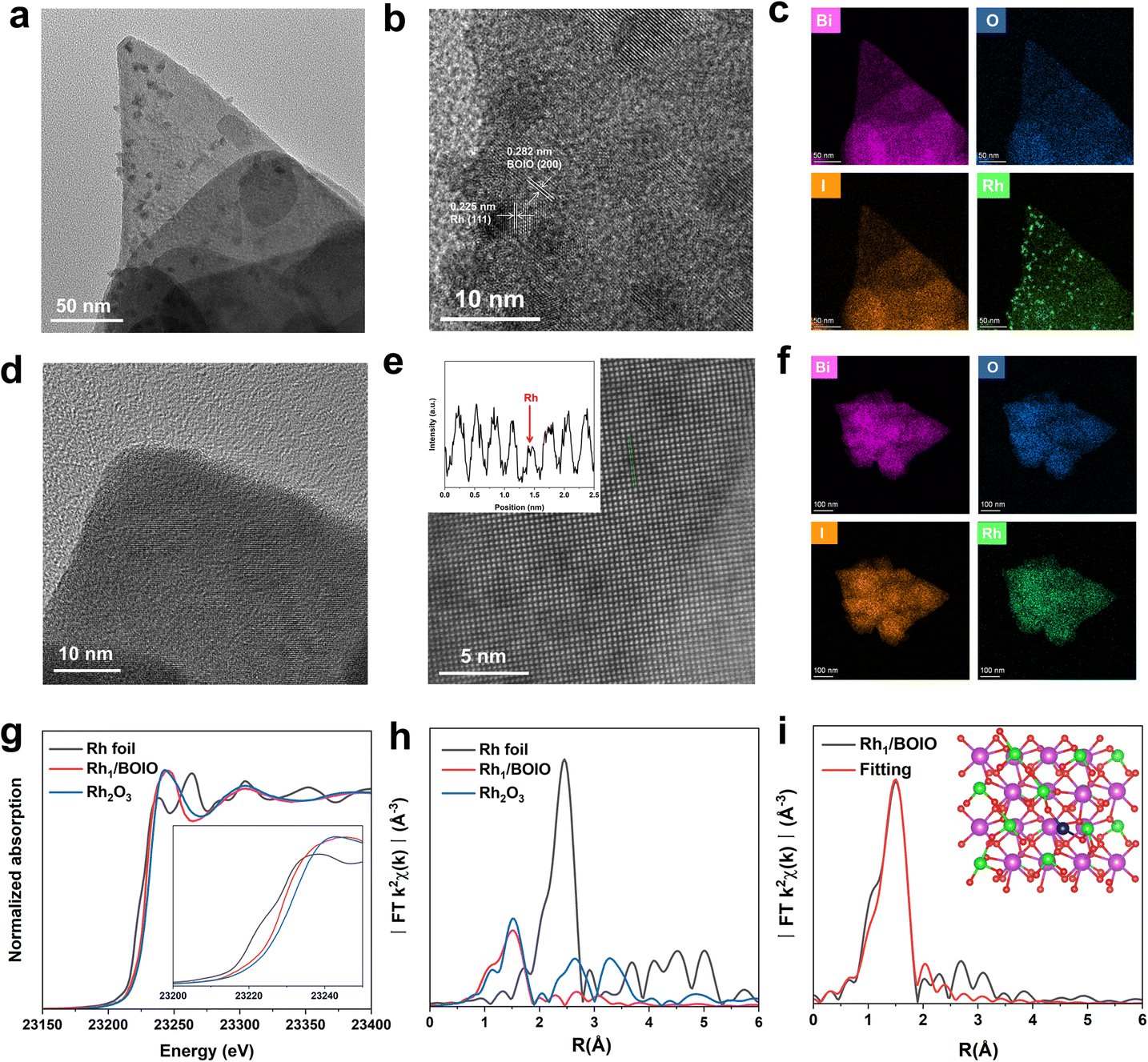 | ||
| Fig. 1 (a) TEM image of RhNP/BOIO. (b) HRTEM image of RhNP/BOIO. (c) The EDS mapping images of RhNP/BOIO. (d) HRTEM image of Rh1/BOIO. (e) Representative AC HAAFD-STEM image of Rh1/BOIO. The inset shows the line intensity profiles taken along the atoms. (f) The EDS mapping of Rh1/BOIO. (g) Rh K-edge XANES spectra of Rh1/BOIO and the reference samples. The inset is the magnified figure. (h) FT-EXAFS spectra of Rh1/BOIO and the reference samples. (i) R-space fitting results. The inset is the optimized structure of Rh1/BOIO, purple ball: Bi, green ball: I, red ball: O, blue ball: Rh. | ||
Moreover, X-ray absorption near-edge structure (XANES) measurements were conducted to investigate the chemical and coordination environment of Rh species in Rh1/BOIO. As shown in Fig. 1g, the near-edge of Rh1/BOIO at the Rh K-edge was between Rh foil and Rh2O3 references, indicating a partial oxidation state of Rh had been formed via the photodeposition method. The Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS, Fig. 1h) spectrum of Rh1/BOIO displayed a prominent peak at about 1.5 Å, which was the first coordination shell of Rh and agreed well with the Rh–O scattering. Obviously, no peak for Rh–Rh coordination could be observed for Rh1/BOIO. In addition, the wavelet transform (WT) EXAFS of Rh1/BOIO presented a maximum peak at about 4.2 Å−1, similar to that for the Rh–O structure in Rh2O3 (Fig. S8†). All the above results suggested the Rh single atom coordinated with an O atom on the surface of Rh1/BOIO. On the basis of the EXAFS fitting results and theoretical simulations (Fig. 1i, S9, S10, and Table S1†), we could confirm the coordination number of Rh was two, indicating the Rh atom anchored on BOIO through a Rh–O2 coordination mode in Rh1/BOIO. By comparing the XPS results of Rh species, a more positive binding energy was also detected in Rh1/BOIO, which was in accordance with the XANES results above. Besides, the metal Rh mode of RhNP/BOIO was confirmed by the XPS measurements (Fig. S11 and S12†).
3.2. Photo-oxidation performances
Based on the well-defined structures of Rh1/BOIO and RhNP/BOIO, we assessed their photo-oxidation activities under full light irradiation. In the model BA photo-oxidation reaction (Fig. 2a and b), Rh1/BOIO displayed a selectivity rate of 86.7% to BN, and completed the reaction without N-BBA product formation after 6 h light irradiation. In sharp contrast, N-BBA was the main product over RhNP/BOIO with a selectivity rate of 82.8% and only 4.6% selectivity to BN. More than a 99% BA conversion rate could be achieved in the photo-oxidation reaction over these two photocatalysts. The as-prepared Rh1/BOIO and RhNP/BOIO exhibited better BA photo-oxidation performances than those in previous reports (Tables S2 and S3†). Comparatively, the selectivity rates of BN and N-BBA products over pure BOIO were much lower than those of Rh1/BOIO and RhNP/BOIO, respectively. In addition, no BN or N-BBA product could be detected in the absence of a photocatalyst or light irradiation, suggesting that the photocatalyst and light irradiation were indispensable for BA photo-oxidation reactions. All these results extensively demonstrated that the different Rh modes on the BOIO surface were able to achieve obviously opposite selectivities during the BA photo-oxidation reaction. To optimize the Rh in Rh1/BOIO and RhNP/BOIO, different amounts of Rh species were loaded on the BOIO surface. As a result, 0.69 wt% was determined to be the optimized Rh loading amount for Rh1/BOIO and RhNP/BOIO (Fig. S13–S15†). Further, cyclic experiments were conducted to evaluate the stability of the Rh1/BOIO and RhNP/BOIO photocatalysts. Fig. 2c and d demonstrate that both of the selectivity rates for BN over Rh1/BOIO and N-BBA over RhNP/BOIO were almost unchanged during the cyclic tests. Next, crystal structure characterizations, including XRD, TEM, and EDS, were carried out to confirm the structural maintenance of the used Rh1/BOIO and RhNP/BOIO (Fig. S16–S18†). In addition, the scope of the photo-oxidation reactions of the BA-based substrates over Rh1/BOIO and RhNP/BOIO, e.g., 4-methylbenzylamine, 4-methoxybenzylamine, 4-fluorobenzylamine, and 4-chlorobenzylamine, also displayed the opposite selectivities. As shown in Fig. 2e and f, R–Bn![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N was the main product over Rh1/BOIO, while the R–BnN–Bn–R product was obtained over RhNP/BOIO (R is CH3, OCH3, F, or Cl), indicating the opposite selectivity of the BA oxidation reaction is applicable to a wider range of substrates over these two photocatalysts.
N was the main product over Rh1/BOIO, while the R–BnN–Bn–R product was obtained over RhNP/BOIO (R is CH3, OCH3, F, or Cl), indicating the opposite selectivity of the BA oxidation reaction is applicable to a wider range of substrates over these two photocatalysts.
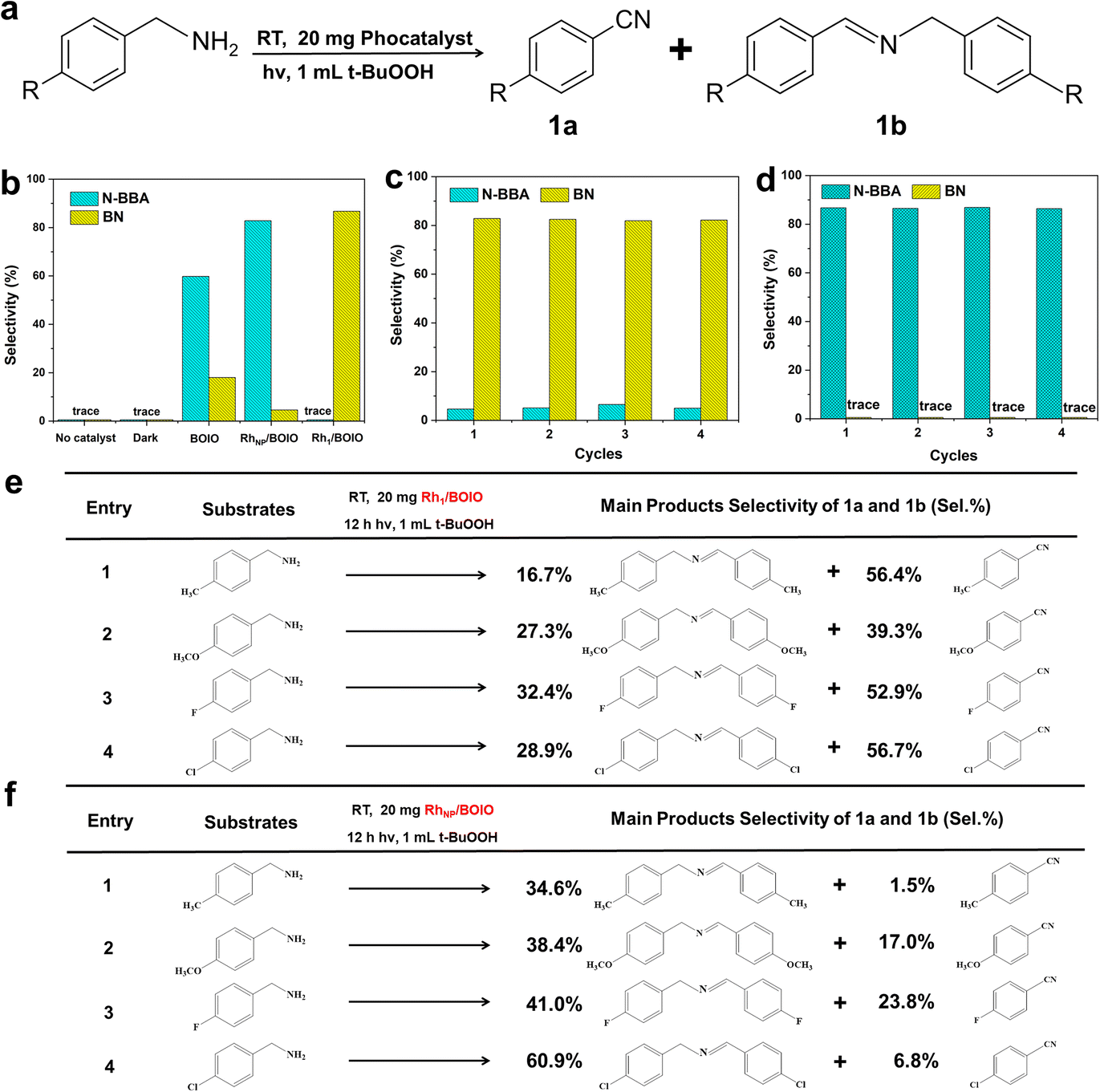 | ||
| Fig. 2 (a) Reaction scheme for the BA photo-oxidation reaction. (b) Selectivity rates of BN and N-BBA after 6 h photo-oxidation reaction over the samples. (c) Stability of Rh1/BOIO. (d) Stability of RhNP/BOIO. (e) Scope of the photo-oxidation reaction over Rh1/BOIO. (f) Scope of the photo-oxidation reaction over RhNP/BOIO. | ||
3.3. Charge-carrier dynamics
The light-harvesting abilities of the samples were characterized by UV/Vis diffuse reflectance spectroscopy (DRS). As displayed in Fig. 3a, Rh1/BOIO displayed stronger visible-light absorption than that of pure BOIO and RhNP/BOIO in the range of 400–800 nm. A slight red-shift of the absorption edges could be observed over Rh1/BOIO and RhNP/BOIO compared with that of BOIO. According to the Kubelka–Munk function and Mott–Schottky results, the bandgaps of pure BOIO and Rh1/BOIO were determined as 3.38 and 3.34 eV, respectively (Fig. 3b, S19 and S20†). The Rh–O2 coordination mode in Rh1/BOIO was able to enhance the light harvesting via the narrower bandgap of BOIO. As for RhNP/BOIO, the promotion of its light-absorption capacity could be mainly attributed to the black color of RhNP. To investigate the dynamic behaviors of the photogenerated carriers, several spectroscopic characterizations were employed. Photoelectrochemical tests were conducted to evaluate the separation efficiency of charge carriers. First, all the samples were evenly coated on the FTO glasses before testing.41 As presented in Fig. 3c, the photocurrent densities of Rh1/BOIO and RhNP/BOIO were much higher than that of pure BOIO. Compared with pure BOIO, Rh1/BOIO possessed the smallest semicircle diameter in the electrochemical impedance spectra (EIS) (Fig. S21†).42 These results suggest that both Rh1 and RhNP were able to effectively facilitate the photogenerated charge transfer. The steady-state PL spectra results of the samples are also given (Fig. S22†). The PL intensity was significantly decreased after Rh1 and RhNP, respectively, were anchored on BOIO, implying that Rh1/BOIO and RhNP/BOIO exhibited lower charge-carrier recombination efficiencies than that of BOIO under light excitation.43,44 Based on the TRPL and fitting results, the lifetimes of BOIO, RhNP/BOIO, and Rh1/BOIO were determined as 1.16, 1.22, and 1.35 ns, respectively (Fig. 3d and Table S4†), which suggests that the lifetimes of the charge carriers were improved after anchoring Rh1 and RhNP on the BOIO surface,45 which corresponds to the steady-state PL spectra results. Taken together, the separation and transmission efficiency of the photogenerated carriers could be improved after respectively anchoring Rh1 and RhNP on the BOIO surface. Despite the separation and transfer efficiency of charge carriers being promoted, the mechanism for the opposite selectivity that occurred over Rh1/BOIO and RhNP/BOIO in the BA photo-oxidation reaction remained unclear.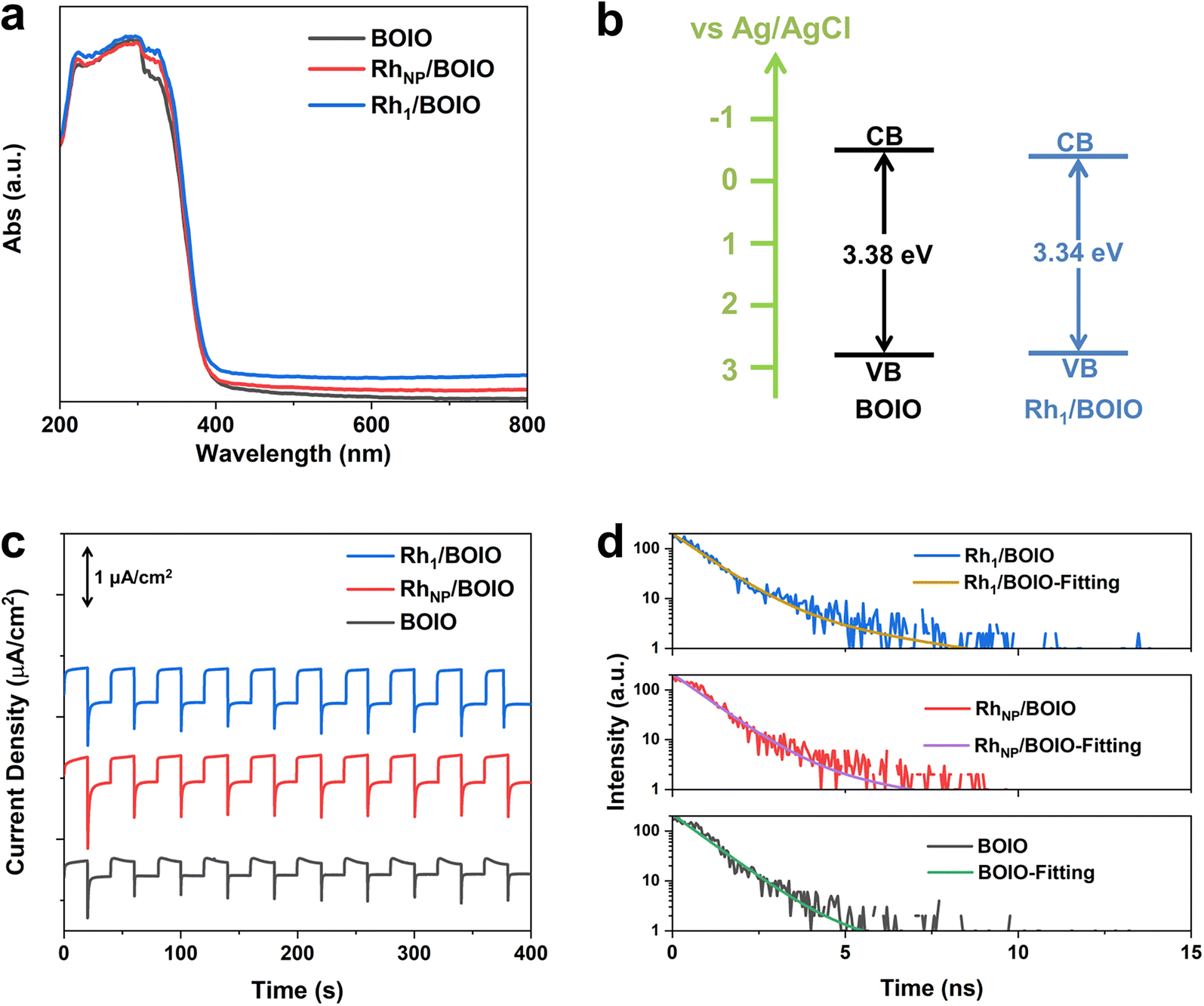 | ||
| Fig. 3 (a) UV/Vis DRS spectra. (b) Band structures of the samples. (c) Photocurrent densities of Rh1/BOIO, RhNP/BOIO, and BOIO. (d) TRPL spectra and fitting results of the samples. | ||
3.4. Mechanism analysis
To elucidate the opposite selectivity mechanism over Rh1/BOIO and RhNP/BOIO, we conducted free radical trapping experiments to explore the active species during the photo-oxidation reaction. As shown in Fig. 3a and b, the characteristic peak intensities of DMPO-˙OH and DMPO-˙O2− species among these samples were totally different. In Rh1/BOIO, DMPO-˙OH were the dominant species under light irradiation, much higher than with RhNP/BOIO and pure BOIO. By contrast, RhNP/BOIO displayed the highest capacity for DMPO-˙O2− species generation among the three samples. Consequently, ˙OH active species could be easily produced on Rh1/BOIO, while ˙O2− were the active species for RhNP/BOIO. Such entirely different active species formation performances were more likely the cause of the opposite selectivity during the BA photo-oxidation reaction over Rh1/BOIO and RhNP/BOIO. Density functional theory (DFT) calculations were performed to further reveal the reaction mechanism. By comparing the density of states between pure BOIO and Rh1/BOIO, an obvious contribution from Rh species appeared near the conduction band minimum of BOIO (Fig. 4c and S23†). Namely, the bandgap of BOIO was narrowed after Rh1 species were anchored on, which was consistent with the band structural analysis in Fig. 3b. Considering the thermodynamic of the active species generation, ˙OH and O2 have been recognized as the main intermediates for decomposing t-BuOOH/OOH−.46,47 Compared to Rh1/BOIO, the free energy changes over RhNP/BOIO were more conductive to generating O2 molecules (Fig. 4d), which implied that the ˙OH could easily undergo coupling with Rh1 and this impedes the disproportionation reaction of two ˙OH. These observations reveal that Rh1 and RhNP in BOIO facilitated the generation of ˙OH and ˙O2− active species, respectively, as well as achieving the opposite selectivity in the BA photo-oxidation reaction.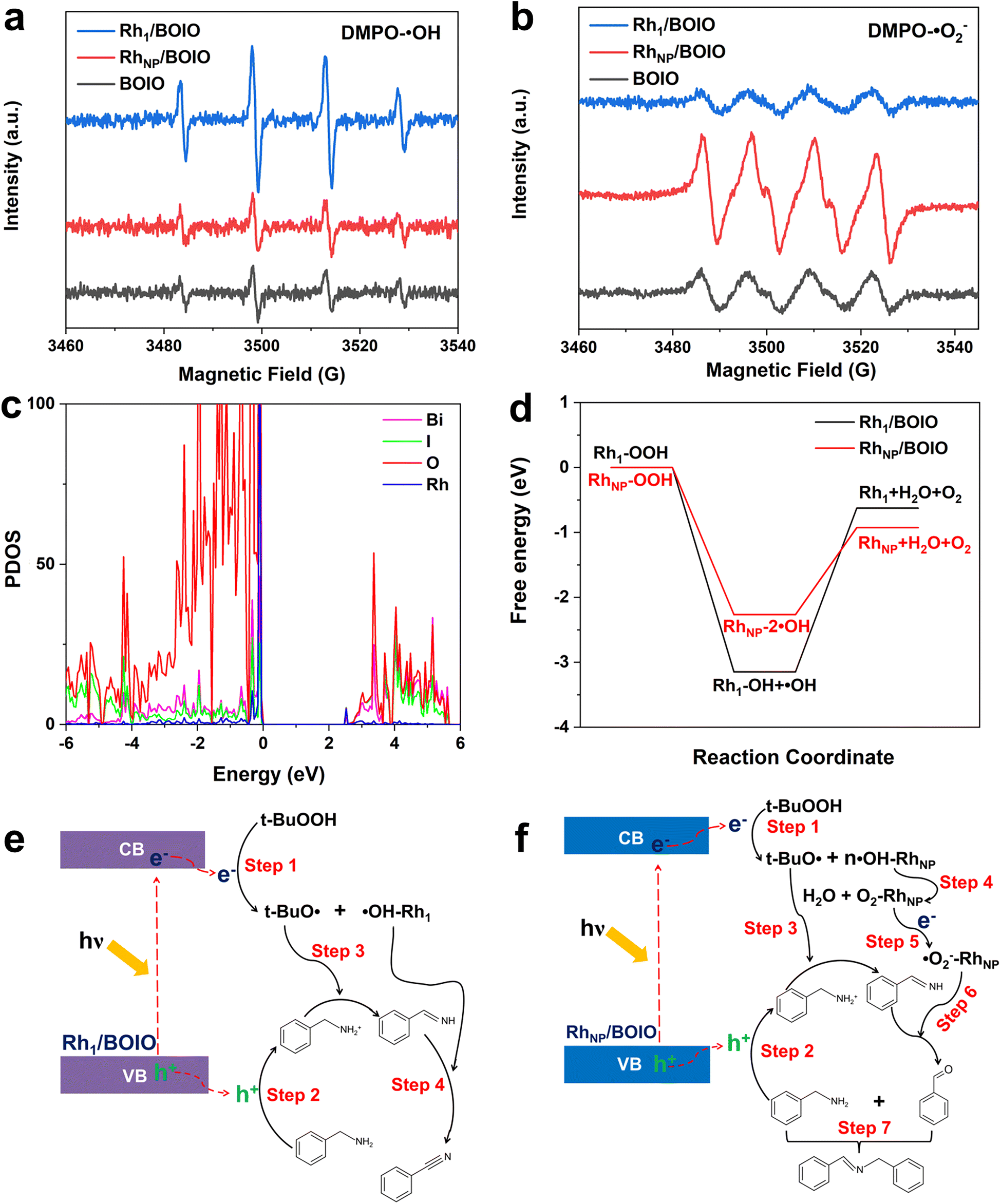 | ||
| Fig. 4 Electron spin resonance (ESR) signals of (a) DMPO-˙OH and (b) DMPO-˙O2− species after 10 min light irradiation over pure BOIO, Rh1/BOIO, and RhNP/BOIO. (c) PDOS results of Rh1/BOIO. (d) Calculated free energy diagram of all the steps for OOH− decomposition to different intermediates on Rh1/BOIO and RhNP/BOIO. Schematic illustration of the possible reaction mechanism for the BA photo-oxidation reaction over (e) Rh1/BOIO and (f) RhNP/BOIO. | ||
On the basis of the above experiments and our theoretical analysis, we propose the possible reaction mechanism over Rh1/BOIO and RhNP/BOIO.48–51 As shown in Fig. 4e, Rh1/BOIO is excited to generate electron–hole pairs during light irradiation. The photogenerated electrons are able to decompose t-BuOOH to t-BuO˙ and ˙OH (Step 1). Besides, BA can be oxidized to PhCHNH intermediate by the photogenerated holes and t-BuO˙ species (Steps 2 and 3). The Rh1 mode of Rh1/BOIO fixes one ˙OH to form ˙OH–Rh1, which not only prevents the disproportionation reaction of two ˙OH to produce O2 and H2O, but also further oxidizes the PhCHNH intermediate to BN product (Step 4). In the case of RhNP/BOIO (Fig. 4f), the RhNP on the BOIO surface prefers to catalyze the disproportionation reaction of ˙OH to generate O2 and H2O, due to the multi-˙OH species adsorbed on RhNP (Step 4). O2 molecules react with photogenerated electrons to generate ˙O2− active species, which then oxidize the PhCHNH to the PhCHO intermediate (Step 6). Another BA molecule couples with PhCHO in the last step to produce N-BBA (Step 7).
4 Conclusions
In summary, two different Rh modes (Rh1 and RhNP) were respectively anchored on BOIO surface to realize a completely opposite selectivity of BA photo-oxidation reaction. The as-prepared Rh1/BOIO exhibited an 86.7% selectivity rate for BN product, while it was 4.6% over RhNP/BOIO. N-BBA was the main product over RhNP/BOIO with an 82.8% selectivity rate, but this could not be detected when using Rh1/BOIO as the photocatalyst under the same reaction conditions. The theoretical simulations and experimental results revealed that the opposite selectivity performances arose from the different active species of ˙OH and ˙O2− generated over Rh1/BOIO and RhNP/BOIO, respectively, during the photocatalytic reaction. Meanwhile, this phenomenon is applicable equally to other types of BA-based substrates, which further confirms that the selective tuning ability can be achieved via anchoring different Rh modes on BOIO. Our work highlights a convenient approach to fabricate catalysts with an ability to tune the product selectivity during photocatalytic reactions.Author contributions
J. L and Y. W. designed, performed the experiments and wrote the paper. G. W. and Z. Lou. supervised the project and revised the manuscript. K. S., T. J. and Z. Li. performed the theoretical calculations. Q. C. and R. Y. analyzed the XAFS results data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22202011, 21971002), the Natural Science Research Project of Higher education in Anhui Province (2022AH020020), and Natural Science Foundation of Shandong Province (ZR2022QB056). The authors thank the BL14W1 station in the Shanghai Synchrotron Radiation Facility (SSRF) for help with characterization.References
- R. D. Patil and M. K. Gupta, Adv. Synth. Catal., 2020, 362, 3987–4009 CrossRef CAS.
- B. Xu, E. M. Hartigan, G. Feula, Z. Huang, J.-P. Lumb and B. A. Arndtsen, Angew. Chem., Int. Ed., 2016, 128, 16034–16038 CrossRef.
- M. Lv, F. Tong, Z. Wang, Y. Liu, P. Wang, H. Cheng, Y. Dai, Z. Zheng and B. Huang, J. Mater. Chem. A, 2022, 10, 19699–19709 RSC.
- M. T. Bender and K. S. Choi, JACS Au, 2022, 2, 1169–1180 CrossRef CAS PubMed.
- J. Qin, Y. Long, F. Sun, P. P. Zhou, W. D. Wang, N. Luo and J. Ma, Angew. Chem., Int. Ed., 2022, 61, e202112907 CAS.
- S. Wang, C. Lai, Y. Zhang, S. Bao, K. Lv and L. Wen, J. Mater. Chem. A, 2022, 10, 20975–20983 RSC.
- R. Ray, A. S. Hazari, G. K. Lahiri and D. Maiti, Chem. Asian. J., 2018, 13, 2138–2148 CrossRef CAS PubMed.
- R. V. Jagadeesh, H. Junge and M. Beller, Nat. Commun., 2014, 5, 4123 CrossRef CAS PubMed.
- Z. Yu, E. R. Waclawik, Z. Wang, X. Gu, Y. Yuan and Z. Zheng, J. Mater. Chem. A, 2017, 5, 4607–4615 RSC.
- Z. Tian, C. Han, Y. Zhao, W. Dai, X. Lian, Y. Wang, Y. Zheng, Y. Shi, X. Pan, Z. Huang, H. Li and W. Chen, Nat. Commun., 2021, 12, 2039 CrossRef CAS PubMed.
- G. Wang, Y. Liu, J. Zhang, Q. Chen, K. Fang and J. Mao, J. Mater. Chem. A, 2022, 10, 21349–21355 RSC.
- F. Wang, X. Han, Z. Jia, Y. Li, T. Zhang, A. Han and J. Liu, Catal. Sci. Technol., 2021, 11, 6947–6951 RSC.
- S. Peiris, J. McMurtrie and H.-Y. Zhu, Catal. Sci. Technol., 2016, 6, 320–338 RSC.
- Y. Li, J. Li and Y. Xu, EnergyChem, 2021, 3, 100047 CrossRef CAS.
- Z. Xue, D. Luan, H. Zhang and X. Lou, Joule, 2022, 6, 92–133 CrossRef CAS.
- L. Tong, L. Ren, A. Fu, D. Wang, L. Liu and J. Ye, Chem. Commun., 2019, 55, 12900–12903 RSC.
- C. Dong, C. Lian, S. Hu, Z. Deng, J. Gong, M. Li, H. Liu, M. Xing and J. Zhang, Nat. Commun., 2018, 9, 1252 CrossRef PubMed.
- X. Liu, D. Dai, Z. Cui, Q. Zhang, X. Gong, Z. Wang, Y. Liu, Z. Zheng, H. Cheng, Y. Dai, B. Huang and P. Wang, ACS Catal., 2022, 12, 12386–12397 CrossRef CAS.
- G. Wang, R. Huang, J. Zhang, J. Mao, D. Wang and Y. Li, Adv. Mater., 2021, 33, 2105904 CrossRef CAS PubMed.
- F. Zhang, J. Ma, Y. Tan, G. Yu, H. Qin, L. Zheng, H. Liu and R. Li, ACS Catal., 2022, 12, 5827–5833 CrossRef CAS.
- Y. Li, Y. Qu and G. Wang, J. Mater. Chem. A, 2022, 10, 5990–5997 RSC.
- G. Wang, Z. Chen, T. Wang, D. Wang and J. Mao, Angew. Chem., Int. Ed., 2022, 61, e202210789 CAS.
- E. Zhao, M. Li, B. Xu, X. L. Wang, Y. Jing, D. Ma, S. Mitchell, J. Perez-Ramirez and Z. Chen, Angew. Chem., Int. Ed., 2022, 61, e202207410 CAS.
- G. Wang, C. He, R. Huang, J. Mao, D. Wang and Y. Li, J. Am. Chem. Soc., 2020, 142, 19339–19345 CrossRef CAS PubMed.
- X. Shi, Y. Huang, Y. Bo, D. Duan, Z. Wang, J. Cao, G. Zhu, W. Ho, L. Wang, T. Huang and Y. Xiong, Angew. Chem., Int. Ed., 2022, 61, e202203063 CAS.
- G. Wang, Y. Wu, Z. Li, Z. Lou, Q. Chen, Y. Li, D. Wang and J. Mao, Angew. Chem., Int. Ed., 2023, 62, e202218460 CAS.
- J. Cai, A. Cao, Z. Wang, S. Lu, Z. Jiang, X. Dong, X. Li and S. Zang, J. Mater. Chem. A, 2021, 9, 13890–13897 RSC.
- W. Wu, E. Cui, Y. Zhang, C. Zhang, F. Zhu, C.-H. Tung and Y. Wang, ACS Catal., 2019, 9, 6335–6341 CrossRef CAS.
- J. Yang, Z. Sun, K. Yan, H. Dong, H. Dong, J. Cui, X. Gong, S. Han, L. Huang and J. Wen, Green Chem., 2021, 23, 2756–2762 RSC.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B, 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B, 1992, 46, 6671–6687 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Chen, H. Huang, L. Ye, T. Zhang, Y. Zhang, X. Han and T. Ma, Adv. Funct. Mater., 2018, 28, 1804284 CrossRef.
- W. Wang, B. Huang, X. Ma, Z. Wang, X. Qin, X. Zhang, Y. Dai and M. H. Whangbo, Chem. - Eur. J., 2013, 19, 14777–14780 CrossRef CAS PubMed.
- F. Chen, H. Huang, C. Zeng, X. Du and Y. Zhang, ACS. Sustain. Chem. Eng., 2017, 5, 7777–7791 CrossRef CAS.
- S. Yu, H. Huang, F. Dong, M. Li, N. Tian, T. Zhang and Y. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 27925–27933 CrossRef CAS PubMed.
- F. Chen, Z. Ma, L. Ye, T. Ma, T. Zhang, Y. Zhang and H. Huang, Adv. Mater., 2020, 32, e1908350 CrossRef PubMed.
- G. Lin, Z. Zhang, Q. Ju, T. Wu, C. U. Segre, W. Chen, H. Peng, H. Zhang, Q. Liu, Z. Liu, Y. Zhang, S. Kong, Y. Mao, W. Zhao, K. Suenaga, F. Huang and J. Wang, Nat. Commun., 2023, 14, 280 CrossRef CAS PubMed.
- M. Ni, Y. Zhu, C. Guo, D. Chen, J. Ning, Y. Zhong and Y. Hu, ACS Catal., 2023, 13, 2502–2512 CrossRef CAS.
- A. Etogo, R. Liu, J. Ren, L. Qi, C. Zheng, J. Ning, Y. Zhong and Y. Hu, J. Mater. Chem. A, 2016, 4, 13242–13250 RSC.
- C. Li, H. Wu, D. Zhu, T. Zhou, M. Yan, G. Chen, J. Sun, G. Dai, F. Ge and H. Dong, Appl. Catal., B, 2021, 297, 120433 CrossRef CAS.
- L. Li, X. Dai, D. Chen, Y. Zeng, Y. Hu and X. Lou, Angew. Chem., Int. Ed., 2022, 61, e202205839 CAS.
- X. Yan, M. Xia, H. Liu, B. Zhang, C. Chang, L. Wang and G. Yang, Nat. Commun., 2023, 14, 1741 CrossRef CAS PubMed.
- J. Van Der Zee, D. P. Barr and R. P. Mason, Free Radicals, 1996, 20, 199–206 CrossRef CAS PubMed.
- M. Zhao, K. Zhang, J. Xu and J. Li, Org. Chem. Front., 2022, 9, 4063–4069 RSC.
- S. Q. Gomes and A. G. Salles, Synth. Commun., 2019, 49, 3389–3399 CrossRef CAS.
- Y. R. Girish, R. Biswas and M. De, Chemistry, 2018, 24, 13871–13878 CrossRef CAS PubMed.
- Y. Fu, M. Zheng, Q. Li, L. Zhang, S. Wang, V. V. Kondratiev and B. Jiang, RSC Adv., 2020, 10, 28059–28065 RSC.
- D. Tantraviwat, A. Nattestad, J. Chen and B. Inceesungvorn, J. Colloid Interface Sci., 2023, 629, 854–863 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta01804k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |