
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of pore mesostructure on the electrooxidation of glycerol on Pt mesoporous catalysts†
Athira
Anil
a,
Jai
White
b,
Egon
Campos dos Santos
c,
Irina
Terekhina
d,
Mats
Johnsson
d,
Lars G. M.
Pettersson
e,
Ann
Cornell
b and
German
Salazar-Alvarez
 *a
*a
aDepartment of Material Science and Engineering, Ångström Laboratory, Uppsala University, Box 35 751 03, Uppsala, Sweden. E-mail: german.salazar.alvarez@angstrom.uu.se
bDepartment of Chemical Engineering and Technology, KTH Royal Institute of Technology, Stockholm, Sweden
cAdvanced Institute for Materials Research (WPI-AIMR), Tohoku University, Sendai 980-8577, Japan
dDepartment of Materials and Environmental Chemistry, Arrhenius Laboratory, Stockholm University, 106 91 Stockholm, Sweden
eDepartment of Physics, Stockholm University, 106 91 Stockholm, Sweden
First published on 22nd June 2023
Abstract
Glycerol is a renewable chemical that has become widely available and inexpensive owing to the increased production of biodiesel. Noble metal materials are effective catalysts for the production of hydrogen and value-added products through the electrooxidation of glycerol. In this study, we developed three platinum systems with distinct pore mesostructures, e.g., hierarchical pores (HP), cubic pores (CP) and linear pores (LP), all with high electrochemically active surface area (ECSA). The ECSA-normalized GEOR catalytic activity of the systems follows HPC > LPC > CPC > commercial Pt/C. Regarding the oxidation products, we observe glyceric acid as the main three-carbon product (C3), with oxalic acids as the main two-carbon oxidation product. DFT-based theoretical calculations support the glyceraldehyde route going through tartronic acid towards oxalic acid and also help in understanding why the dihydroxyacetone (DHA) route is active despite the absence of DHA amongst the observed oxidation products.
Introduction
The ongoing demand, cost, and low sustainability of traditional fossil fuels have paved the way for the rapid development of biofuels, an alternative renewable energy resource with low greenhouse gas emissions.1–3 Hence, biodiesel, one of the biofuel candidates, has been undergoing rapid study and commercialization in recent years.4–6 This has led to the large-scale accumulation of glycerol, a byproduct of the transesterification reaction for biodiesel synthesis.7 Recent reports have shown that over 2.4 million tons of glycerol is produced every year in the EU, which exceeds the required amount for the entire world.8 Thus, glycerol is an inexpensive,9 non-toxic,10 and renewable three-carbon (C3) waste that must be eliminated or converted. Glycerol can be an ideal source for generating several C1–C3 value-added products, such as dihydroxy acetone (DHA), glyceric acid (GLA), tartronic acid (TTA), oxalic acid (OA), and formic acid (FA) in addition to hydrogen.11 The extensive usage of these products in cosmetic, pharmaceutical, and textile industries has always influenced their high price, which is significantly larger than that of glycerol.12,13Various synthesis and catalysis techniques, such as thermo chemical,14,15 photochemical,16,17 and biocatalytic18–20 techniques, have been used to oxidize glycerol and thus synthesize value-added products. The electrocatalytic conversion of glycerol is an alternative production method that provides a high electrode-to-product efficiency, flexibility of product selectivity, a co-production of H2 at the cathode with a lower operating cell potential compared to water splitting, and moderate reaction conditions.21–27 Several studies have shown that noble metal catalysts, such as Pd, Pt, Au, and Ag, and their alloys are excellent for glycerol electrooxidation reaction (GEOR) with a great advantage of having lower electrode potential and a high fraction percentage of C3 products (where the economic value of C3-chemicals is larger than C1-chemicals).27,28 In addition, the lower onset potential and higher stability over other noble metals make Pt an attractive catalyst.29 In the last decade, many articles have studied the electrooxidation of glycerol with Pt30–36 and Pt-based catalysts.25,37–42
Several experimental reports have emphasized the impact of the high surface area of catalysts for better performance for water splitting and methanol oxidation.43,44 Materials with a high surface area can be achieved either by the reduction of particle size or introducing porosity to the particle morphology. In general, solvothermal, sol–gel, and physical techniques are used for the synthesis of porous materials with high surface area.45–47 Particle aggregation while drop-casting the electrode surface, less uniformity in particle size distribution, and additional requirements for binders, such as Nafion, always impart discrepancies in the electrocatalytic studies.48 Creating a uniform mesoporous thin film on a conducting substrate can limit the differences to a certain level. There are fewer reports on water electrolysis where mesoporous thin films are deposited on conducting electrodes using templates. This technique overcomes the drawbacks of dip coating and provides a homogeneous and reproducible material.49,50
In this study, we created three Pt catalysts with varying porous mesostructures, focusing on GEOR activity: hierarchical pores, cubic pores, and linear pores. The composition of the oxidation products was determined experimentally and theoretically, and the theoretical calculations also provided information on possible pathways and reaction intermediates.
Experimental
Electrodeposition of mesoporous catalysts
A conventional templated electrodeposition route was used for the synthesis of hierarchical, cubic and linear pore catalysts using Pluronic F-127 or phytantriol as porogens. Details are given in the ESI subsections 1.1 and 1.2.† Basically, a template was formed on the surface of polished polycrystalline nickel substrates, and platinum was electrodeposited from a hexachloroplatinic acid aqueous solution and reduced at a potential of −0.25 V vs. Ag/AgCl at room temperature. In the case of the linear pores, the electrodeposition was carried out at 50 °C.Material characterization
The surface morphology and composition of the mesoporous catalysts were studied using a Zeiss LEO 1550 scanning electron microscope (SEM) operated at 15 kV, a Quantera X-ray photoelectron spectroscope (XPS), and a Bruker D5000 X-ray diffractometer under grazing incidence conditions (GI-XRD). A JEOL JEM-2100 transmission electron microscope (TEM) operated at 200 kV was used for high-resolution imaging. The specimens were prepared by detaching the mesoporous catalysts from the substrates using ultrasonication, followed by drop casting on a copper grid (200 mesh). Detailed descriptions of other material characterizations used in this study are given in ESI subsection 1.3.†The small-angle neutron scattering (SANS) pattern, which confirms the micellar formations and the small-angle X-ray scattering (SAXS) pattern representing the phytantriol phases, are shown in ESI Fig. S4a and b,† respectively.
Electrochemical characterization
All electrochemical experiments were performed using a divided cell, with a platinum mesh counter electrode, 0.1 M glycerol in 1 M NaOH as an electrolyte, Nafion 212 membrane as separator and a stirrer bar at 400 rpm. Hg/HgO (1.0 M NaOH) reference electrode with luggin capillary was used for lower IR drop and stability of Hg/HgO at elevated temperatures. The conversion to a reversible hydrogen electrode (RHE) is performed using the following equation: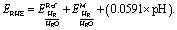
Experimental information regarding the selection of rotation rates and Nafion membrane is further discussed in ESI subsection 1.6 and Fig. S2 and S3.† We evaluated the electrochemical activity of the catalysts using cyclic voltammetry (CV) and potentiostatic studies, such as chronoamperometry (CA) and IR-corrected polarisation curves (ICPCs). Cyclic voltammograms were recorded at 10 mV s−1 in a potential range of 0.10–1.25 V vs. RHE. ICPC studies were conducted from low to high anodic current densities. The chronoamperometric studies were carried out at 0.69 V vs. RHE. Finally, the measured currents from CVs, ICPCs and chronoamperometry were normalized using the electrochemical surface area (ECSA) of the catalysts to obtain the current density curves. Detailed analysis of ECSA of the catalysts and deposited mass of the catalysts are shown in ESI subsection 1.5 and Fig. S1.†
Oxidation product analysis
High-performance liquid chromatography (HPLC) was used to analyse the oxidation product fractions. HPLC measurements were performed using an Agilent 1260 Infinity II isocratic pump, multisampler and multicolumn thermostat with a 1290 Infinity II refractive index detector. The analytical columns, including a Bio-Rad guard column with a standard cartridge holder with a Micro-Guard cation H+ cartridge (4.6 × 30 mm), a Bio-Rad Aminex HPX-87H column, and a Shodex Sugar SH1011 column (8 × 300 mm), were kept in series at a temperature of 30 °C. To obtain the best possible peak separation between glycerol oxidation products, 1 mM H2SO4 and 8 mM H2SO4 were used as mobile phases with a flow rate of 0.25 mL min−1. Calibration curves of glycerol oxidation products are shown in Fig. S6 and S7 of the ESI.†Computational methods
Theoretical calculations were performed following the idea used in our previous studies.51 The Vienna Ab initio Simulation Package (VASP) was used to perform density functional theory (DFT) electronic structure calculations.52 The exchange–correlation functional was approximated as the Bayesian error estimation functional with an additional nonlocal correlation term (BEEF-vdW).53 The calculations were carried out using a (4 × 4 × 4) slab with a vacuum of 20 Å to avoid interactions between periodic images (see ESI Fig. S15†). The free energies of glycerol oxidation intermediates were calculated on Pt (111) and Pt (100) slab surfaces. As indicated in ESI Fig. S16–S24,† elementary oxidation reactions are classified into three groups: deprotonation, hydrolysis, and hydrogen rearrangement. The DFT total energies were corrected using zero-point energies (ZPE) and thermal contributions. The harmonic approximation was employed for the vibrational frequency calculations for all adsorbates and molecules. Henry's law was used to link the gas phase pressure to the aqueous concentration using the NIST-JANAF tables. To account for electrode/electrolyte interface, implicit solvation implemented in VASP was used for the reaction intermediates. The electrochemical environment was modelled based on a computational hydrogen electrode. Further information regarding the computational methodology is provided in ESI Section 4.† An Excel file containing the reaction energetics, including thermodynamics corrections, is provided as ESI.†Results and discussion
Fig. 1 shows the morphology of the catalysts with hierarchical (HPC, Fig. 1a), cubic (CPC, Fig. 1b), and linear pores (LPC, Fig. 1c). The HPC catalysts show a hierarchy in pore sizes with macropores of 322 ± 155 nm diameter and mesopores of 17 ± 7 nm diameter with a wall thickness range of 2–3 nm. CPC comprises a cubic 3-dimensional (3D) network with pores of 5 ± 1 nm, which is consistent with the structure of phytantriol Q224 phase water channels. In the case of LPC, the unsupported platinum nanowires grown on the hexagonal phases of phytantriol were clubbed together to form nanowire bundles with linear pores after removing the template. The LPC showed an average pore size of 34 ± 6 nm. The detailed structural (GIXRD, Fig. S8†) and surface characterizations (XPS, Fig. S9†) of the deposited thin film catalysts can be found in ESI subsections 2.1 and 2.2.† | ||
| Fig. 1 Morphology of mesoporous Pt catalysts: electron microscopy images and pore size distribution plots of pristine (a) hierarchical pores catalyst (HPC), (b) cubic pores catalyst (CPC), and (c) linear pores catalyst (LPC). | ||
Fig. 2a shows the cyclic voltammograms (CV) of the three mesoporous catalysts compared with the commercially used catalyst Pt/C at a temperature of 60 °C (additional CVs carried out at 25 °C are shown in ESI Fig. S14†). The catalyst activity is plotted as specific activity iECSA (normalized by the electrochemical surface area ECSA) against the reversible hydrogen electrode. CV profiles show a clear distinction in the peak current density achieved by each mesostructure with the following current density order: HPC > LPC > CPC > Pt/C. These suggest that the intrinsic activity of mesoporous catalysts is higher than that of the commercial catalyst Pt/C with HPC showing the best performance. Fig. 2b displays the IR-corrected polarisation curves for the three mesoporous catalysts. At a constant potential of 0.7 V, the observed IR-corrected current density is the same as in Fig. 2a, i.e., HPC > LPC > CPC > Pt/C. Similarly, if we consider the operating voltage at a current density of 1.0 mA cmECSA−2 for the catalysts, the HPC has the lowest operating voltage, which is 20 mV, 50 mV, and 200 mV lower than that of LPC, CPC, and Pt/C, respectively. The HPC achieves a higher maximum current density at the lowest operating potential. The Tafel slope of the mesoporous catalysts and Pt/C (ESI Fig. S12†) were calculated using polarisation curves as follows: LPC (84 ± 4.9 mV dec−1), HPC (87 ± 4.2 mV dec−1), CPC (106 ± 4.2 mV dec−1) and Pt/C (145 ± 3.7 mV dec−1). The low onset potential, higher current density and a lower Tafel slope indicate that HPC is the most active catalyst for the GEOR under these conditions.
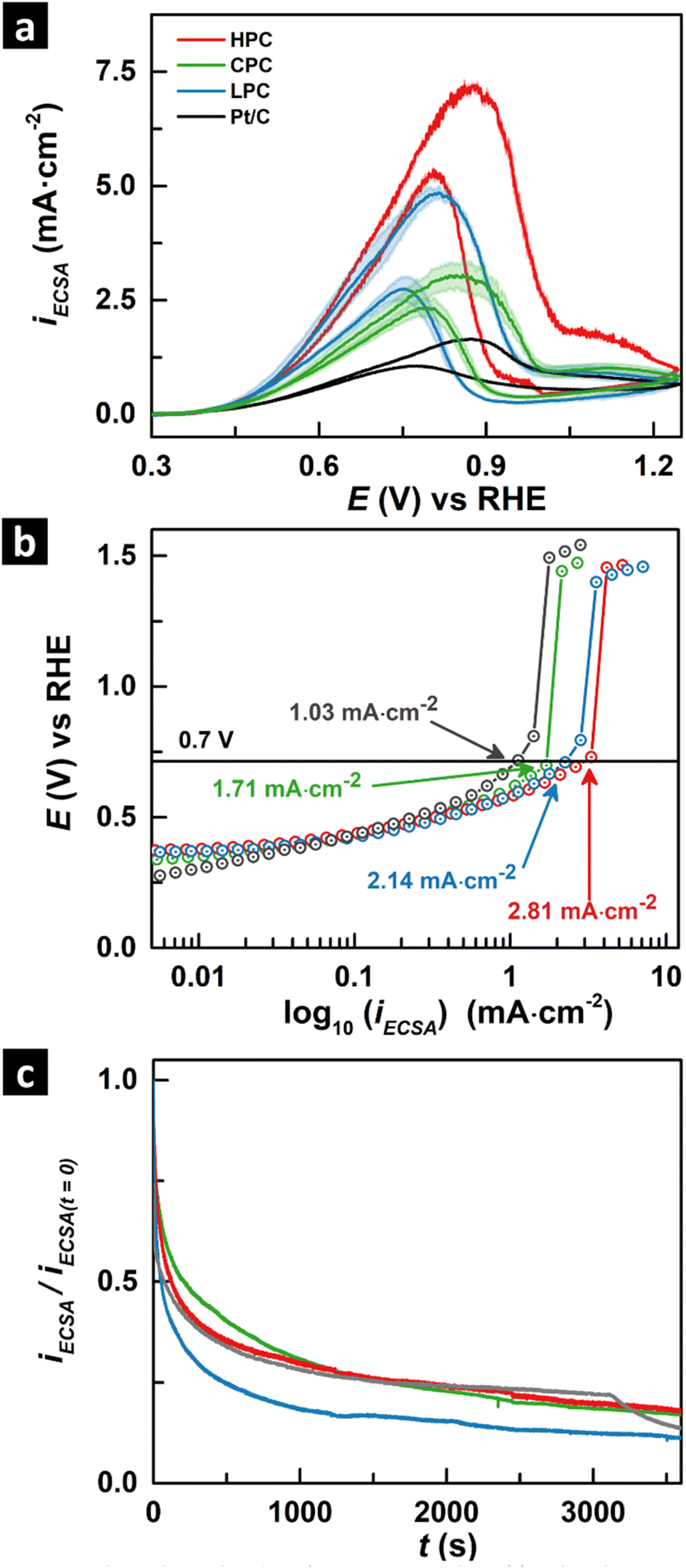 | ||
| Fig. 2 Electrochemical studies of mesoporous catalysts. (a) Cyclic voltammograms, (b) IR-corrected polarisation curves, and (c) normalized chronoamperometric curves (at 0.69 V vs. RHE). All the experiments are carried out in 0.1 M glycerol + 1 M NaOH at scan rate 10 mV s−1 at 60 °C and 400 rpm stirring. The bands in (a) represent the 95% confidence intervals. | ||
Interestingly, although the electrochemical surface area of the catalysts is largest for CPC (≈56 cm2), followed by commercial Pt/C (≈27 cm2) and HPC and LPC with the lowest values (ca. 14 and 11 cm2), the intrinsic activity (ECSA-normalized) follows the pore size with HPC > LPC > CPC. Previous studies have shown that porosity is an important factor and contributes to the ECSA and the mass transport mechanisms affecting the electro-catalyst performance.54,55 According to the Nernst–Planck equation, the mass transport of an electrolyte depends on migration, convection and diffusion.56 In the case of porous films, the majority of the ECSA of the catalyst is internal and exists inside the pores where convection and migration is very limited. Hence, internal diffusion is the main transport mechanism for reactants to access the area within the catalyst and depends strongly on the viscosity of the electrolyte.57 The viscosity of 0.1 M glycerol is 0.58 mPa s, which is higher than that of water (0.46 mPa s) at 60 °C.58,59 The high viscosity of glycerol contributes to the low diffusion rate of glycerol in these 3D nanowire porous structures with small pores (5 ± 1 nm), where the smaller water molecules (≈0.3 nm) can move faster than those of glycerol (≈1 nm) in the 3D cubic porous network, related to the high ECSA with poorer GEOR performance. These results reinforce the idea that designing 3D pore catalysts with a hierarchy in pore sizes is necessary for utilizing the ECSA of the catalysts for GEOR, where the large pores contribute to high diffusivity, whereas the smaller pores result in a larger surface area.
A similar trend of electrochemical performance HPC > LPC > CPC is observed in CVs (ESI Fig. S14†) measured at room temperature (25 °C). The decrease in viscosity causes increased mass transport of glycerol at elevated temperatures, which contributes to higher electrochemical performance at 60 °C (2× that at RT).60,61
ESI Table S1† compares the intrinsic activity of mesoporous catalysts in this study with that of the reported Pt catalysts in the literature under similar reaction conditions.36,38,40,41 The table shows that the mesoporous catalysts shown in this study have better electrochemical performance than reported catalysts, including nanostructured Pt catalysts and carbon-supported Pt catalysts (commercial systems).
Fig. 2c shows the normalized chronoamperometry curves (normalized by the initial current density at t = 0) recorded at 0.69 V vs. RHE of the catalysts in 0.1 M glycerol and 1 M NaOH. All the mesoporous and commercial catalysts show a decreasing current density with time. Similar decays of catalytic currents are reported in the literature.62,63 ESI Fig. S11† presents the chronoamperometric curves at 0.69 V vs. RHE without normalisation. The final current density follows the order HPC > LPC ≈ CPC > Pt/C, suggesting that HPC has a better current density than other catalysts even after 1 h of electrolysis. The catalyst activity is regenerated in HPC (98%), LPC (97%) and CPC (82%) after subsequent CVs in the fresh electrolyte, suggesting that the current decay is attributed to the surface passivation of the catalyst surface by an inactive intermediate rather than morphological changes. Koper et al. discussed the nature of possible inactive intermediates, which can contribute to the blockage of the active sites of the catalyst.64 The oxidation species bound to the catalyst surface by two primary C atoms is referred to as an inactive intermediate candidate that is resistant to further oxidation.
The glycerol oxidation products, as obtained from HPLC, are shown in Fig. 3. Fig. 3a–c shows the concentration normalized by the ECSA, CECSA, for the different catalysts in this study, and Fig. 3d–f shows the fraction percentages (f%) of products formed in different reaction time intervals for HPC, CPC, and LPC, respectively. CECSA and ECSA-normalized glycerol conversion rates of the catalysts follow HPC > LPC > CPC. C3 products contribute to ca. 75% of the total fraction of GEOR products. GLA, formed by primary alcohol oxidation, was observed as the major product in all three mesoporous systems. The highest GLA fraction was observed in the LPC, followed by the CPC and HPC. Apart from GLA, tartronic acid (TTA) and lactic acid (LA) are the other C3 products formed, whereas OA and FA constitute the main C2 and C1 GEOR products. The low stability of glyceraldehyde at pH 14 and the low applied potential for electrolysis likely favoured the highest selectivity of GLA.65 The composition of the oxidation products for the different mesoporous catalysts shows a significant variation in the composition at short times, where HPC and LPC show a higher GLA fraction than CPC, but the relative fractions tend to homogenise after an hour of reaction time. This is likely due to the slower diffusivity of the reactants and products inside the CPC compared to the LPC and HPC. A longer residence time results in products with higher degrees of oxidation. These results agree with the discussion in Fig. 2c, where it is likely that the differences are greater at even shorter times.
 | ||
| Fig. 3 HPLC results of mesoporous catalysts: concentration of products per ECSA (CECSA) as a function of time (a–c) and product fraction percentages (f%) as a function of time (d–f) for HPC (a and d), CPC (b and e), and LPC (c and f). All the experiments are carried out in 0.1 M glycerol + 1 M NaOH at 0.69 V for an electrolysis time of 60 min at 60 °C with stirrer bar of 400 rpm rotational rate in electrolyte. Abbreviations: acetic acid (ACA), formic acid (FA), lactic acid (LA), oxalic acid (OA), tartronic acid (TTA), and glyceric acid (GLA). | ||
The glycerol conversion rate per ECSA of the sample is depicted in ESI Fig. S13.† The conversion follows the order HPC > LPC > CPC. The Faraday efficiency of the HPC, LPC and CPC was 99.6%, 98.9% and 100.5%. The high value of Faraday efficiency might be caused by the changes in the concentration of electrolytes due to electrolyte evaporation, as our experiments were performed at 60 °C. Heterogeneous glycerol oxidation on Pt surfaces could also contribute to higher Faraday efficiency. Table S2 in Section 3 of the ESI† compares the glycerol oxidation products formed by the mesoporous catalysts to the Pt catalysts reported earlier under similar reaction conditions. As mentioned in the previous section, mesoporous catalysts produce GLA as the majority oxidation product, with a fraction of more than 50%. Reported Pt catalysts show a similar distribution with C3 products (GLA or TTA) having a higher fraction over C2/C1 products under similar reaction conditions. Various factors, such as applied voltage and electrolyte concentrations, contribute to variations in glycerol oxidation product distributions.
Fig. 4 shows the predicted GEOR pathway as suggested by both the experimental and computational observations. Fig. S17–S24 in ESI† Section 4 show the possible reactions, intermediates, and potential-determining step (PDS) of reactions occurring in the vicinity of the Pt electrode.
 | ||
| Fig. 4 Representation of calculated glycerol electrooxidation pathway of Pt (111) catalyst in alkaline solution with potential determining step (PDS) values of each oxidation step. The oxidation pathway of each intermediate is shown using different colours as follows: (1) Gly–DHA–HPA (black), (2) Gly–GD (blue), (2′) GD–GLA (blue), (3) GLA–GA (red), (3′) GA–FA (red), (4) GLA–TTA (magenta), (4′) TTA–MLA (magenta), (5) TTA–OA (orange), (6) OA–GA (orange), (7) DHA–PV(green), (7′) PV–LA (green). Here PDS represents the minimum potential require to overcome the energy barrier of the reactions illustrated in Fig. S17–S24.† The PDS values for Pt (100) is shown in Fig. S25.† | ||
The primary alcohol oxidation of glycerol can undergo either pathway 1 (Gly–DHA–HPA) forming dihydroxy acetone (DHA) or hydroxypyruvic acid (HPA) as the primary product or pathways 2 and 2′ (Gly–GD–GLA) forming GLA as the primary oxidation product. The PDS for pathway 2 < pathway 1 suggests that at lower applied potentials, GEOR on Pt catalysts is most likely to result in GLA as the primary alcohol oxidation product rather than HPA. The free energy diagrams of the major pathways shown in ESI Fig. S25† further confirmed this. The Pt (111) and Pt (100) surfaces require a smaller free energy change (ΔG = 0.53 eV) in the GD route to form GLA than following the DHA route to form HPA (ΔG = 0.61 eV). At a higher potential above 0.67 V vs. RHE, GLA can further oxidize to TTA following pathway 4, OA by pathways 4 and 5, glycolic acid (GA) by pathways 4, 5 and 5′, and FA by pathways 4, 5, 5′ and 3′. A further increase in the applied voltage above 0.76 V could produce mesooxalic acid (MLA) through pathways 4 and 4′. The experimentally applied potential of 0.69 V vs. RHE eliminates the possibility of choosing a reaction pathway having a higher PDS value than the applied potential. For example, the PDS of HPA formation is 0.85 V > 0.69 V. The absence of HPA and MLA in the reaction mixture after electrolysis at 0.69 V supports the computational predictions. Hence, the GEOR of Pt at an applied potential of 0.69 V favours reaction pathways 2, 4, 5, and 3′ to form TTA, OA, FA, GA, and GLA as the final product, with Gly–GD–GLA–OA–FA as the major reaction pathway and Gly–DHA–LA as the minor pathway. Note that although the PDS for LA production (0.58 eV) is lower than that of GLA (0.61 eV), the LA pathway is the minor pathway because DHA is unstable under basic conditions and rapidly converts to GD,66,67 which, in turn, rapidly oxidizes to GLA or other C2 or C1 products, thereby disfavouring the production of LA.
ESI Table S3† summarizes the PDS required for the formation of the GEOR product. The PDS of GLA and FA for Pt (111) is 0.69 V and Pt (100) is 0.41 V and 0.43 V, where the difference is only 0.02 V. This suggests that FA formation is unavoidable during GLA synthesis using GEOR. The presence of OA, FA, GA, TTA and GLA and the absence of MLA and HPA in the potential range of 0.67–0.73 V for the reported catalysts in ESI Table S2† are consistent with the PDS results obtained from theoretical calculations in ESI Table S3.† Thus, the computational results obtained strongly support the experimental findings.
Conclusions
In this study, we compared the intrinsic activity of mesoporous Pt catalysts with different pore sizes and structures. Among the catalysts, HPC shows the highest activity in the following order: HPC > LPC > CPC > Pt/C. The advantage of having a hierarchy in pore sizes is likely the combination of large pore sizes that enable fast diffusion, combined with small pores that provide a larger active surface area. In the case of CPC and LPC, the small pores in the structure result in a limited diffusion of glycerol and poor removal of the oxidation products from the surface, resulting in significant deactivation of the catalysts over time. Compared to the commercial catalyst and the literature reports, the mesoporous catalysts presented in this study show the highest intrinsic activity. The ease of being directly used as an electrode for catalysis without further processing provides additional advantages to these catalysts, particularly when compared to particle-based catalysts that require suitable binders, such as Nafion, to form electrode assembly. Furthermore, the GO products of the mesoporous catalysts are similar to reported catalysts under comparable reaction conditions, with GLA as the main product. DFT calculations of the PDS of the elementary steps of the reaction and possible reaction pathways suggested that Gly–GD–GLA–TTA–OA was the dominant pathway at the applied potential, in agreement with the experimental results.Author contributions
Athira Anil: conceptualisation, methodology, investigation and validation, writing the original draft of manuscript. Jai White: investigation and validation, review and editing. Egon Campos dos Santos: computational calculations, investigation and validation, review and editing. Irina Terekhina: TEM characterization, review and editing. Mats Johnsson: supervision, review and editing. Lars Pettersson: supervision, resources, review and editing. Ann Cornell: supervision, resources, review and editing. German Salazar-Alvarez: initial conceptualisation, supervision, project leader, funding and resources, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was funded by the Swedish Foundation for Strategic Research (SSF, Grant No. EM16-0010). AA, ECdS, LGMP and GSA acknowledge SSF for the financial support. The calculations were performed using resources provided by (i) the Swedish National Infrastructure for Computing (SNIC) at the PDC center and (ii) Institute for Materials Research, Tohoku University for the use of MASAMUNE-IMR (Project No. 202208-SCKXX-0212). The authors thank Sugam Kumar of the Bhaba Atomic Research Center (BARC) and Martin Kapuscinski, Uppsala University, for the SANS and SAXS measurements, respectively. We also acknowledge Myfab Uppsala for providing facilities and experimental support. Myfab is funded by the Swedish Research Council (2019-00207) as a national research infrastructure.References
- Economics of Biofuels | US EPA, https://www.epa.gov/environmental-economics/economics-biofuels, (accessed August 9, 2022).
- C. Bessou, F. Ferchaud, B. Gabrielle and B. Mary, in Sustainable Agriculture, Springer Netherlands, Dordrecht, 2009, vol. 2, pp. 365–468 Search PubMed.
- M. H. Hassan and Md. A. Kalam, Procedia Eng., 2013, 56, 39–53 CrossRef.
- V. A. Likhanov and O. P. Lopatin, J. Phys.: Conf. Ser., 2019, 1399, 055016 CrossRef CAS.
- J.-H. Ng, H. K. Ng and S. Gan, Clean Technol. Environ. Policy, 2010, 12, 213–238 CrossRef.
- Y. C. Sharma and B. Singh, Renewable Sustainable Energy Rev., 2009, 13, 1646–1651 CrossRef CAS.
- G. Antolín, F. V. Tinaut, Y. Briceo, V. Castao, C. Pérez and A. I. Ramírez, Bioresour. Technol., 2002, 83, 111–114 CrossRef PubMed.
- M. Pagliaro and M. Rossi, The Future of Glycerol, Royal Society of Chemistry, Cambridge, 2nd edn, 2010 Search PubMed.
- C. A. G. Quispe, C. J. R. Coronado and J. A. Carvalho, Renewable Sustainable Energy Rev., 2013, 27, 475–493 CrossRef CAS.
- Y. Gu and F. Jérôme, Green Chem., 2010, 12, 1127–1138 RSC.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS PubMed.
- J. Kaur, A. K. Sarma, M. K. Jha and P. Gera, Biotechnol. Rep., 2020, 27, e00487 CrossRef.
- C. R. Chilakamarry, A. M. M. Sakinah, A. W. Zularisam and A. Pandey, Syst. Microbiol. Biomanuf., 2021, 1, 378–396 CrossRef CAS.
- B. V. Ayodele, T. A. R. B. T. Abdullah, M. A. Alsaffar, S. I. Mustapa and S. F. Salleh, Int. J. Hydrogen Energy, 2020, 45, 18160–18185 CrossRef CAS.
- P. Kaminski, M. Ziolek and J. A. Van Bokhoven, RSC Adv., 2017, 7, 7801–7819 RSC.
- D. Liu, J. C. Liu, W. Cai, J. Ma, H. Bin Yang, H. Xiao, J. Li, Y. Xiong, Y. Huang and B. Liu, Nat. Commun., 2019, 10, 1–8 CrossRef.
- M. R. Karimi Estahbanati, M. Feilizadeh, F. Attar and M. C. Iliuta, Ind. Eng. Chem. Res., 2020, 59, 22330–22352 CrossRef CAS.
- M. Tudorache, A. Negoi, B. Tudora and V. I. Parvulescu, Appl. Catal., B, 2014, 146, 274–278 CrossRef CAS.
- V. Ripoll, G. De Vicente, B. Morán, A. Rojas, S. Segarra, A. Montesinos, M. Tortajada, D. Ramón, M. Ladero and V. E. Santos, Process Biochem., 2016, 51, 740–748 CrossRef CAS.
- S. N. Suhaimi, L. Y. Phang, T. Maeda, S. Abd-Aziz, M. Wakisaka, Y. Shirai and M. A. Hassan, Braz. J. Microbiol., 2012, 43, 506 CrossRef CAS PubMed.
- C. Coutanceau, S. Baranton and R. S. B. Kouamé, Front. Chem., 2019, 7, 100 CrossRef CAS PubMed.
- M. S. E. Houache, K. Hughes and E. A. Baranova, Sustainable Energy Fuels, 2019, 3, 1892–1915 RSC.
- H. Wan, C. Dai, L. Jin, S. Luo, F. Meng, G. Chen, Y. Duan, C. Liu, Q. Xu, J. Lu and Z. J. Xu, ACS Appl. Mater. Interfaces, 2022, 14, 14293–14301 CrossRef CAS PubMed.
- P. A. Alaba, C. S. Lee, F. Abnisa, M. K. Aroua, P. Cognet, Y. Pérès and W. M. A. Wan Daud, Rev. Chem. Eng., 2021, 37, 779–811 CrossRef CAS.
- Y. Zhou, Y. Shen and J. Piao, ChemElectroChem, 2018, 5, 1636–1643 CrossRef CAS.
- A. M. Verma, L. Laverdure, M. M. Melander and K. Honkala, ACS Catal., 2022, 12, 662–675 CrossRef CAS.
- T. Li and D. A. Harrington, ChemSusChem, 2021, 14, 1472–1495 CrossRef CAS PubMed.
- E. C. dos Santos, R. B. Araujo, M. Valter, G. Salazar-Alvarez, M. Johnsson, M. Bajdich, F. Abild-Pedersen and L. G. M. Pettersson, Electrochim. Acta, 2021, 398, 139283 CrossRef CAS.
- C. Coutanceau, S. Baranton and R. S. B. Kouamé, Front. Chem., 2019, 7, 100 CrossRef CAS PubMed.
- Z. Zhang, L. Xin and W. Li, Appl. Catal., B, 2012, 119–120, 40–48 CrossRef CAS.
- E. Ferreira Frota, V. V. Silva De Barros, B. R. S. De Araújo, Â. G. Purgatto and J. J. Linares, Int. J. Hydrogen Energy, 2017, 42, 23095–23106 CrossRef CAS.
- P. S. Fernández, D. S. Ferreira, C. A. Martins, H. E. Troiani, G. A. Camara and M. E. Martins, Electrochim. Acta, 2013, 98, 25–31 CrossRef.
- Z. Chen, C. Liu, X. Zhao, H. Yan, J. Li, P. Lyu, Y. Du, S. Xi, K. Chi, X. Chi, H. Xu, X. Li, W. Fu, K. Leng, S. J. Pennycook, S. Wang and K. P. Loh, Adv. Mater., 2019, 31, 1804763 CrossRef PubMed.
- Y. Liu, W. Yu, D. Raciti, D. H. Gracias and C. Wang, J. Phys. Chem. C, 2019, 123, 426–432 CrossRef CAS.
- R. M. L. M. Sandrini, J. R. Sempionatto, G. Tremiliosi-Filho, E. Herrero, J. M. Feliu, J. Souza-Garcia and C. A. Angelucci, ChemElectroChem, 2019, 6, 4238–4245 CrossRef CAS.
- J. Han, Y. Kim, D. H. K. Jackson, H. Chang, H. W. Kim, J. Lee, J. R. Kim, Y. Noh, W. B. Kim, K. Y. Lee and H. J. Kim, Appl. Catal., B, 2020, 273, 119037 CrossRef CAS.
- Y. Zhou, Y. Shen, X. Luo, G. Liu and Y. Cao, Nanoscale Adv., 2020, 2, 3423–3430 RSC.
- A. Falase, M. Main, K. Garcia, A. Serov, C. Lau and P. Atanassov, Electrochim. Acta, 2012, 66, 295–301 CrossRef CAS.
- M. S. Ahmad, C. K. Cheng, S. Singh, H. R. Ong, H. Abdullah, C. S. Hong, G. K. Chua and M. R. Khan, J. Nanosci. Nanotechnol., 2020, 20, 5916–5927 CrossRef CAS PubMed.
- L. Y. Jiang, A. J. Wang, X. S. Li, J. Yuan and J. J. Feng, ChemElectroChem, 2017, 4, 2909–2914 CrossRef CAS.
- Q. Sun, F. Gao, Y. Zhang, C. Wang, X. Zhu and Y. Du, J. Colloid Interface Sci., 2019, 556, 441–448 CrossRef CAS PubMed.
- X. Yu, E. C. dos Santos, J. White, G. Salazar-Alvarez, L. G. M. Pettersson, A. Cornell and M. Johnsson, Small, 2021, 17, 2104288 CrossRef CAS PubMed.
- Y. Takasu, T. Kawaguchi, W. Sugimoto and Y. Murakami, Electrochim. Acta, 2003, 48, 3861–3868 CrossRef CAS.
- L. Peng and Z. Wei, Engineering, 2020, 6, 653–679 CrossRef CAS.
- Z. Jiang, Y. Liu, L. Huang, W. H. Gong and P. K. Shen, ACS Sustainable Chem. Eng., 2019, 7, 8109–8116 CrossRef CAS.
- A. Chen and P. Holt-Hindle, Chem. Rev., 2010, 110, 3767–3804 CrossRef CAS PubMed.
- T. Sreethawong, T. Puangpetch, S. Chavadej and S. Yoshikawa, J. Power Sources, 2007, 165, 861–869 CrossRef CAS.
- A. Kaliyaraj Selva Kumar, Y. Zhang, D. Li and R. G. Compton, Electrochem. Commun., 2020, 121, 106867 CrossRef CAS.
- S. Akbar, J. M. Elliott, M. Rittman and A. M. Squires, Adv. Mater., 2013, 25, 1160–1164 CrossRef CAS PubMed.
- K. Eiler, S. Suriñach, J. Sort and E. Pellicer, Appl. Catal., B, 2020, 265, 118597 CrossRef CAS.
- E. Campos dos Santos, R. B. Araujo, M. Valter, G. Salazar-Alvarez, M. Johnsson, M. Bajdich, F. Abild-Pedersen and L. G. M. Pettersson, Electrochim. Acta, 2021, 398, 139283 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Petzold, D. D. Landis, J. K. Nørskov, T. Bligaard and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235149 CrossRef.
- H. Liu, P. He, Z. Li and J. Li, Nanotechnology, 2006, 17, 2167 CrossRef CAS.
- A. Y. Chen, J. W. Wang, Y. Wang, Y. Q. Jia, J. F. Gu, X. F. Xie and D. Pan, Electrochim. Acta, 2015, 153, 552–558 CrossRef CAS.
- T. F. Fuller and J. N. Harb, Electrochemical Engineering, 2018 Search PubMed.
- E. Lind, Influence of mass transport on glycerol electrooxidation on palladium in alkaline media, KTH Royal Institute of Technology, 2022 Search PubMed.
- J. A. Trejo González, M. P. Longinotti and H. R. Corti, J. Chem. Eng. Data, 2011, 56, 1397–1406 CrossRef.
- J. B. Segur and H. E. Oberstar, Ind. Eng. Chem., 1951, 43, 2117–2120 CrossRef CAS.
- T. Zhang, C. Liu, Y. Gu and F. Jérôme, Green Chem., 2021, 23, 7865–7889 RSC.
- J. White, A. Anil, D. Martín-Yerga, G. Salazar-Alvarez, G. Henriksson and A. Cornell, Electrochim. Acta, 2022, 403, 139714 CrossRef CAS.
- M. C. Moraes, G. G. Junco, T. F. M. Moreira, C. J. G. Pinheiro, P. Olivi, D. Profeti and L. P. R. Profeti, J. Environ. Chem. Eng., 2019, 7, 102922 CrossRef CAS.
- P. Lertthahan, S. Yongprapat, A. Therdthianwong and S. Therdthianwong, Int. J. Hydrogen Energy, 2017, 42, 9202–9209 CrossRef CAS.
- A. C. Garcia, M. J. Kolb, C. Van Nierop Y Sanchez, J. Vos, Y. Y. Birdja, Y. Kwon, G. Tremiliosi-Filho and M. T. M. Koper, ACS Catal., 2016, 6, 4491–4500 CrossRef CAS.
- Y. Kwon, K. J. P. Schouten and M. T. M. Koper, ChemCatChem, 2011, 3, 1176–1185 CrossRef CAS.
- B. Katryniok, H. Kimura, E. Skrzyńska, J.-S. Girardon, P. Fongarland, M. Capron, R. Ducoulombier, N. Mimura, S. Paul and F. Dumeignil, Green Chem., 2011, 13, 1960 RSC.
- X. Yu, R. B. Araujo, Z. Qiu, E. Campos Dos Santos, A. Anil, A. Cornell, L. G. M. Pettersson and M. Johnsson, Adv. Energy Mater., 2022, 12, 2103750 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta01738a |
| This journal is © The Royal Society of Chemistry 2023 |