
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of free volume, hydrogen bonds, and crosslinks on physical aging in polymers of intrinsic microporosity (PIMs)†
Taigyu
Joo
 a,
Katherine
Mizrahi Rodriguez
b,
Hyunhee
Lee
a,
Durga
Acharya
c,
Cara M.
Doherty
c and
Zachary P.
Smith
*a
a,
Katherine
Mizrahi Rodriguez
b,
Hyunhee
Lee
a,
Durga
Acharya
c,
Cara M.
Doherty
c and
Zachary P.
Smith
*a
aDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: zpsmith@mit.edu
bDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
cThe Commonwealth Scientific and Industrial Research Organization (CSIRO) Manufacturing, Private Bag 10, Clayton South 3169, Victoria, Australia
First published on 26th June 2023
Abstract
Physical aging is a slow structural relaxation process characteristic of glassy polymers that results in reduced membrane permeabilities. In this study, PIM-1, the archetypal polymer of intrinsic microporosity (PIM), was post-synthetically modified to introduce components that are known to influence physical aging, such as hydrogen bonds and crosslinks. The effects of physical aging were monitored by permeation and sorption experiments, and structural changes were examined by positron annihilation lifetime spectroscopy (PALS) and other characterization techniques. The results suggest that higher initial fractional free volume is the primary factor contributing to higher rates of physical aging and that the introduction of hydrogen bonds and crosslinks reduces the initial free volume of PIM-1. In contrast, structural factors such as hydrogen bonds and crosslinks were the key factors in determining how permselectivity changed with physical aging. This study provides useful structure–property correlations and design principles related to free volume, hydrogen bonds, and crosslinks on physical aging behavior of microporous polymer membranes.
1. Introduction
Molecular separations play a critical role in a wide variety of chemical processes and engineering applications, making up nearly 50% of total energy consumption in the industrial sector.1 Implementing more energy-efficient separation technologies has the potential to reduce energy consumption and carbon emissions that have reached a new global high in 2022.2 In this regard, the use of membranes is a promising unit operation that could eventually supplement or replace existing energy-intensive industrial processes such as distillation. Membranes have already been explored in various fields, including but not limited to gas separation, water purification, organic solvent nanofiltration, and pharmaceutical production.3–5 In the context of gas separations, membranes have multiple attractive features in addition to energy efficiency, such as modularity, small footprints, the absence of toxic mass separating agents, continuous operation, and potentially lower cost.6,7A major thrust of gas separation membrane research in the last four decades has been the development of new polymeric materials.4 Along these lines, membrane performance is evaluated based on the permeability and selectivity of the polymer, and the Robeson upper bound plots, a comprehensive database of polymers tested for gas separation, are commonly used in the field to assess state-of-the-art performance.8–10 Based on Freeman's theory, the overall separation performance of polymeric materials can be improved by increasing backbone stiffness while simultaneously increasing free volume, and such features are the defining characteristics of polymers of intrinsic microporosity (PIMs).11,12 When this type of polymer is cast from solution, polymer chains pack inefficiently to generate some of the highest free volume structures known within the field of gas separation membranes.13
PIMs are attractive materials in the gas separation field because their micropores allow for high gas sorption capacity while their rigid backbone structures hinder diffusion of larger gas penetrants, making them highly diffusion-selective materials especially when considering their high free volume.14 As a result, PIMs typically have orders-of-magnitude higher permeabilities than conventional polymers that are deployed as commercial gas separation membranes.15,16 Ever since the first reports of the PIM concept,17,18 new categories of these materials have been emerging at a rapid pace (e.g., triptycene, Tröger's base, ROMP, and CANAL-based polymers),19–22 along with new functionalizations (e.g., carboxylic acid, amine, and thioamide),23,24 post-synthetic modifications (e.g., crosslinking, thermal rearrangement, and pyrolysis),25–29 and mixed-matrix concepts (e.g., incorporating nanoparticles, MOFs, GO, CNTs, and PAFs).30–35 PIMs show promise because many compositions surpass the Robeson upper bound, yet they have never been deployed in large-scale industrial settings, in part due to challenges with scaling and stability, including phenomena such as plasticization and physical aging.4,13,36,37
Of the two major stability issues, this paper focuses on physical aging behavior of PIMs. Physical aging is the long timescale structural relaxation process of glassy polymers moving towards an equilibrium state that results in changes in membrane property sets over time. This phenomenon is ubiquitous among all glassy materials.38 Mechanistically, kinetically trapped conformations of polymer segments gradually relax towards equilibrium packing structures, thereby reducing the free volume of the polymer.38 Physical aging obeys an exponential relationship between permeability and free volume, and hence, physical aging has the most pronounced effect on permeability right after a polymer is formed into a film;39 however, this process persists throughout the lifetime of a polymer membrane used in the field and results in membranes “aging out”, requiring operators to discard and replace membrane modules.37 Because stable long-term performance is an important factor in commercial operations, more work is required to understand the underlying mechanisms of this phenomenon.
Some of the seminal studies on physical aging demonstrated that this feature is dependent on the amount of excess free volume and segmental mobility.40,41 Generally, polymers with high free volume are expected to age faster since the amount of excess free volume is the driving force for physical aging,42 while polymers with high glass transition temperatures (Tgs) are considered to be more resistant to physical aging since Tg is correlated with chain mobility.38,43,44 For this reason, various crosslinking methods have been studied to mitigate physical aging by intentionally reducing free volume and forming a network of interchain connections that provide rigidity within the polymer matrix.45–47 In this sense, PIMs have the dual attributes of low segmental mobility and high free volume, the former of which is expected to retard physical aging and the latter of which is expected to accelerate it.
Early PIM research anticipated that these polymers would have high physical aging resistance since PIMs generally lack a detectable Tg by standard techniques and contain permanent micropores arising from their backbone configurations (i.e., configurational free volume) that are theoretically unaffected by physical aging.48,49 In terms of Tg, we note that a study using fast scanning calorimetry has determined a Tg of 715 K for PIM-1, which surpasses its degradation temperature of 673 K in an inert atmosphere.50 However, multiple studies have shown that PIMs exhibit physical aging behavior that is often atypical of traditional polymers. For example, Swaidan et al. observed that TPIM-1, a triptycene-based ladder polymer, aged faster and more extensively than PIM-1, although the backbone of TPIM-1 has fewer flexible dioxane moieties and contains triptycene units that incorporate configurational free volume.48 Recently, Lai et al. reported a stark difference in the aging behavior of two different catalytic arene-norbornene annulation (CANAL) polymers, which are microporous polymers consisting of conformationally restricted fused rings.51 The CANAL polymer with 2D contortions showed a slight gain in selectivity and a larger decrease in permeability with time, while the CANAL polymer with 3D contortions showed a high gain in selectivity and a small decrease in permeability with time.51 The results from such studies suggest that the physical aging characteristics that are accepted for conventional polymers do not always translate to the emerging PIM subclass. Therefore, systematic studies of physical aging in PIM materials are required to glean insights into this important yet poorly understood phenomenon.
Recently, our lab demonstrated a solid-state deprotection strategy for PIMs that resulted in simultaneous enhancements of permeability and selectivity relative to pre-protected PIM analogues.29,52 This method, called free volume manipulation (FVM), uses labile functional groups, such as a tert-butoxycarbonyl group (tBOC), to alter physical packing structures and hence transport properties of the deprotected polymers.29,52 By introducing bulky functional groups to the polymer backbone, the intersegmental distances are intentionally widened when the polymer is processed into a solid-state film. Then, after removing these labile functional groups through simple heat treatment in the solid-state, free volume increases. This method was highly effective for amine-functionalized PIM-1 (PIM-NH2).29Fig. 1 shows the FVM approach for PIM-NH2. In short, the archetypal PIM-1 is converted into its amine-functional counterpart, PIM-NH2, which significantly decreases free volume due to hydrogen bonding.29 Next, the amine groups in PIM-NH2 are protected with tBOC groups to yield PIM-tBOC. PIM-tBOC subsequently undergoes thermal treatment to yield free volume manipulated PIM-NH2 (PIM-NH2-FVM), which has slightly larger average free volume element (FVE) diameters along with the potential for light urea crosslinks.29 This approach results in enhancements of both permeability and selectivity for PIM-NH2.29
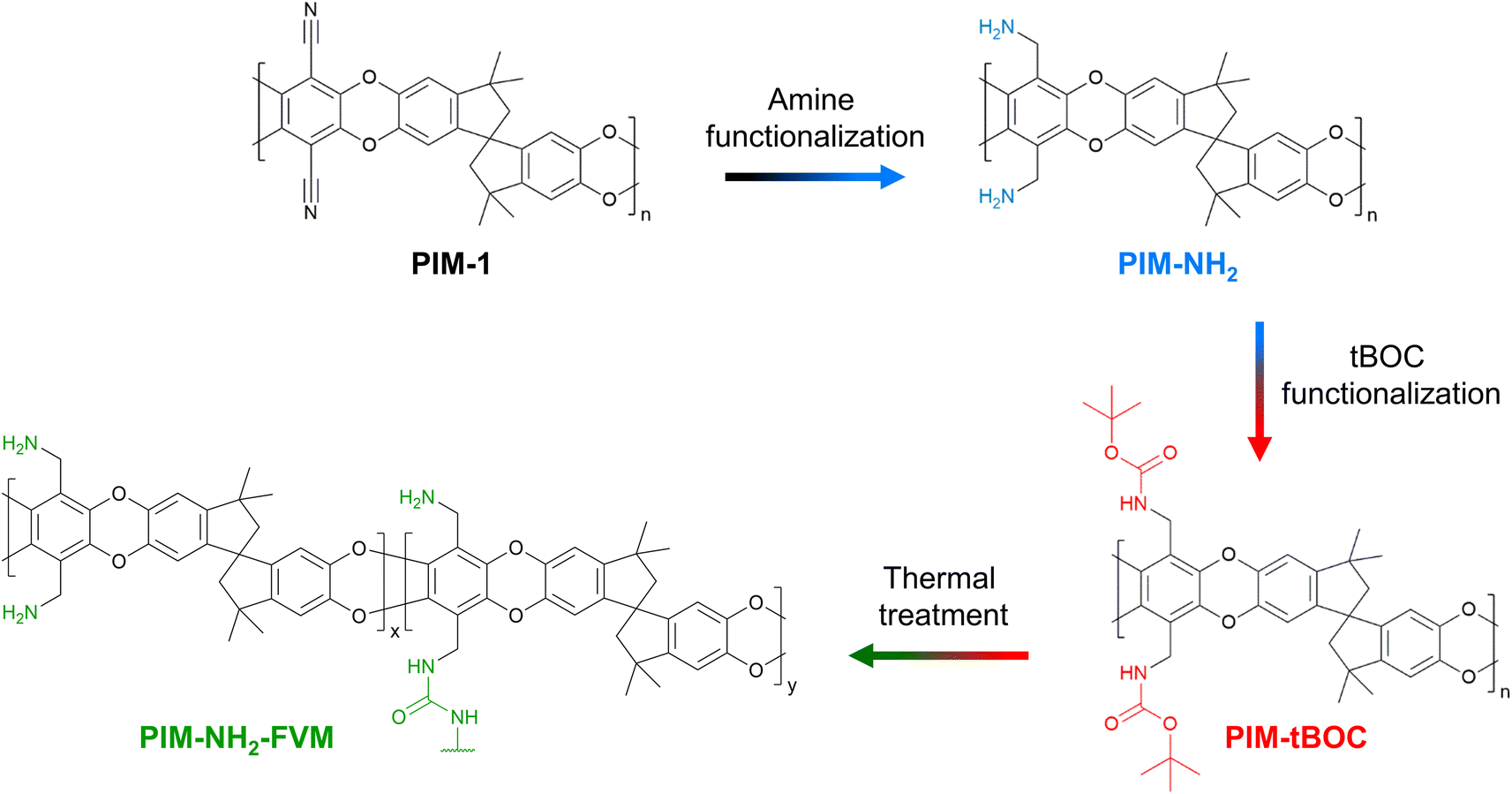 | ||
| Fig. 1 The free volume manipulation (FVM) approach for PIM-NH2 starting with the precursor PIM-1. Note that light crosslinking in PIM-NH2-FVM is represented by a potential urea bond. | ||
In the context of long-term stability, the effect of FVM on physical aging is difficult to predict. FVM increases FVE size and drives the polymer further from its theoretical chain packing equilibrium, which can potentially accelerate the aging rate. However, it also introduces crosslinks that can reduce translational motions of the polymer segments. In previous comparisons of fresh and aged PIM-NH2 and PIM-NH2-FVM permeability data,29 both polymers showed similar permeability reductions, suggesting that FVM may stabilize the polymer matrix from accelerated physical aging despite introducing the additional free volume.
In this study, we investigate this phenomenon in greater depth, studying the influence of amine functionalization and the subsequent free volume manipulation on archetypal PIM-1 films by monitoring pure-gas transport properties over approximately 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 hours using non-plasticizing gases (H2, O2, N2, and CH4). In addition to providing a baseline comparison for the aging behaviors of these post-synthetically modified PIMs, this study aims to bridge the guiding principles already established for conventional polymers on the role of free volume, hydrogen bonds, and crosslinks on physical aging behaviors of microporous polymers. By maintaining identical dioxane backbone structures while modifying hydrogen bonding, crosslinking, and free volume, this set of polymers provides a unique system to directly compare the effect of these features on physical aging.
000 hours using non-plasticizing gases (H2, O2, N2, and CH4). In addition to providing a baseline comparison for the aging behaviors of these post-synthetically modified PIMs, this study aims to bridge the guiding principles already established for conventional polymers on the role of free volume, hydrogen bonds, and crosslinks on physical aging behaviors of microporous polymers. By maintaining identical dioxane backbone structures while modifying hydrogen bonding, crosslinking, and free volume, this set of polymers provides a unique system to directly compare the effect of these features on physical aging.
2. Experimental section
2.1 Materials
PIM-1, PIM-NH2, and PIM-NH2-FVM polymer films with thicknesses of 37.6 ± 0.9, 51.3 ± 1.1, and 51.2 ± 2.0 μm, respectively were prepared as previously reported.29 A detailed procedure is outlined in Section 1 of the ESI.† While thin films generally below 1 μm in thickness are of great interest for commercial gas separation membranes, we consider thick films here for broader comparisons with the open literature and because PIMs have easily observable aging behavior at these length scales.23,29 Physical aging studies of the post-synthetically modified PIMs at industrially relevant thicknesses will be the focus of our future work.2.2 Characterization
Density was measured with a Mettler Toledo density measurement kit (ME-DNY-4) using Archimedes' principle. Room temperature water was used as the reference liquid, and the density for each sample was taken as the average of at least four different film pieces. The error is reported as the standard deviation of the measurements performed. The fractional free volume (FFV) of all samples tested was calculated using the group contribution method:53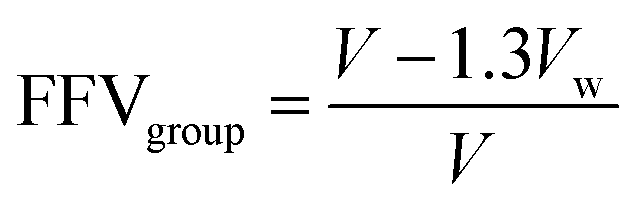 | (1) |
Wide-angle X-ray scattering (WAXS) patterns were collected under 0.08 mbar vacuum using a SAXSLAB machine equipped with a DECTRIS PILATUS3 R 300K detector and Rigaku 002 microfocus X-ray source. All patterns were collected for 1200 s, and the resulting pattern was plotted as intensity I(q) versus scattering wavevector q:
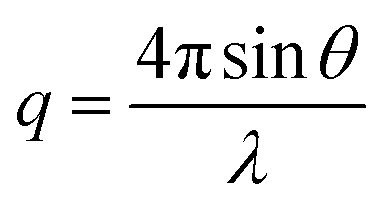 | (2) |
Positron annihilation lifetime spectroscopy (PALS) data were collected using an automated EG&G ORTEC fast–fast coincidence system. A Mylar envelope containing a 22Na radioisotope source was sandwiched between two small stacks of polymer films, each stack approximately 1.5 mm thick. All tests were performed under vacuum (10−6 torr) at room temperature, and a minimum of 5 files with 4.5 × 106 integrated counts were collected for each polymer sample. After each measurement, least squares optimization was performed with the LT v9 program using a four-component (para-positronium, free positron, and ortho-positronium) fitted model.54 In the model, the para-positronium lifetime was fixed to τ1 = 0.125 ns and free positron (τ2) and two ortho-positronium lifetimes (τ3 and τ4) were fitted, indicating bimodal free volume distributions, which typically result in better statistical fitting for high free volume glassy polymers.55 Intensities (Ii) for all of these components were also determined.56 The PAScual software was adapted to calculate pore size distributions,57 and the Tao–Eldrup equation was used to calculate the average FVE size using the obtained ortho-positronium lifetimes:56,58
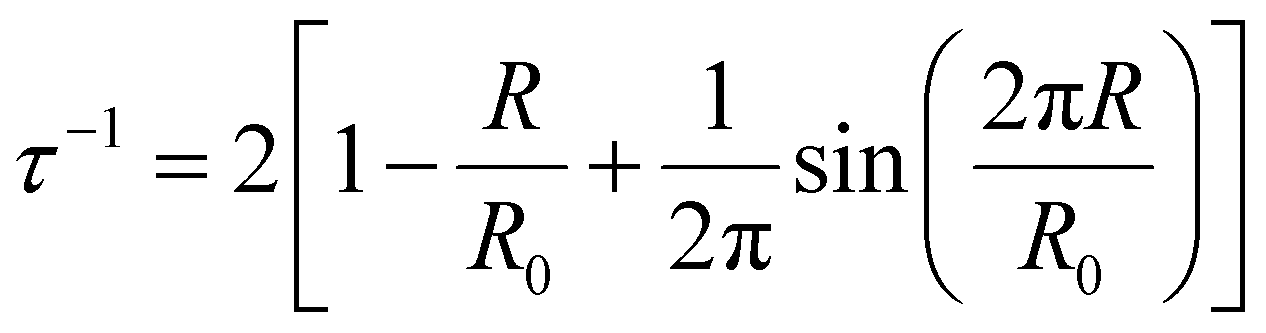 | (3) |
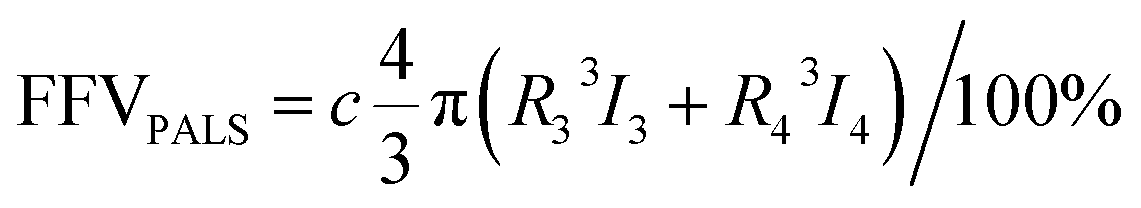 | (4) |
2.3 Pure-gas permeability measurements for physical aging
Before any physical aging experiments, all membrane samples were kept submerged in methanol. At the beginning of the physical aging experiments, the membranes were dried in a vacuum oven at 130 °C for 12 h under dynamic vacuum. PIM-NH2 and PIM-NH2-FVM samples underwent additional thermal treatment, as described in Section 1.3 of the ESI.† The end of the drying or thermal treatment procedure was considered the initial physical aging time (t0). During aging, all films were kept inside a desiccator at ambient temperature (approximately ranging from 20–25 °C).Pure-gas permeabilities of four gases (H2, CH4, N2, and O2) were tested for each aging sample using a constant volume–variable pressure automated permeation testing apparatus (Maxwell Robotics). All samples were masked with epoxy glue on brass supports to ensure no leakage from upstream to downstream. Further details of the testing setup can be found elsewhere.20,29,52 Before any permeation test, the entire apparatus was held under vacuum for 8 h. When changing to a different testing gas, the upstream was flushed with high pressure He, and the entire apparatus was held under vacuum between 0.5 and 1 h depending on the prior gas tested. All permeation tests for physical aging curves were performed at 35 °C with an upstream pressure of 15 psia and a downstream pressure <9.5 torr. For the variable temperature permeation study, the testing conditions were kept similar to those in the previous description, but the tests were performed at 35, 45, 55, and 65 °C.
Permeability (P) was calculated using the following equation:
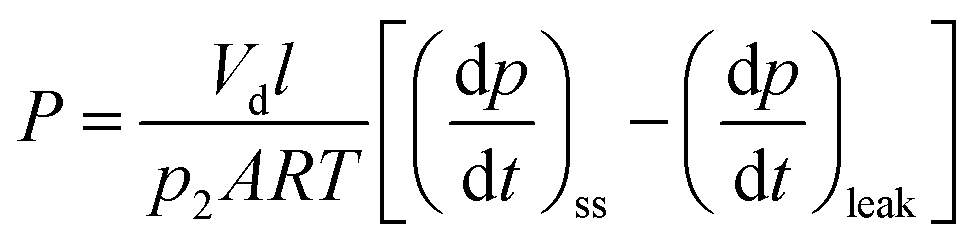 | (5) |
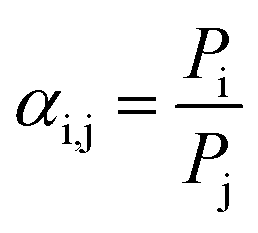 | (6) |
2.4 Pure-gas sorption measurements for physical aging
Similar to permeation tests, all films were kept submerged in methanol until the planned experiments were executed. The membranes were dried and thermally treated right before the sorption tests, marking t0 of the experiments. During aging, all films were kept inside a desiccator at ambient temperature (approximately ranging from 20–25 °C).Pure-gas sorption isotherms of CH4, N2, and O2 were measured at 35 °C using an automated pressure decay sorption testing apparatus (Maxwell Robotics). All connections in the apparatus were sealed using VCR gaskets to limit uncertainty from leaks. For each sorption test, at least 0.1 g of polymer films were loaded into a 3 cm3 sample cell. At the beginning of the sorption measurement, the entire apparatus was degassed under full vacuum for 8 h. When changing to a different testing gas, the apparatus was flushed with high pressure He and held under vacuum for 3 h. The charge volume was held at each testing pressure for 0.3 h before dosing the gas into the sample chamber. Once the testing gas was dosed, the sample chamber was allowed to equilibrate at the testing pressure for 2.5, 2, and 1 h for CH4, N2, and O2, respectively. Each sorption isotherm was collected up to 700 psia and fitted using the dual-mode sorption (DMS) model, which is commonly used to describe sorption in glassy polymers:65
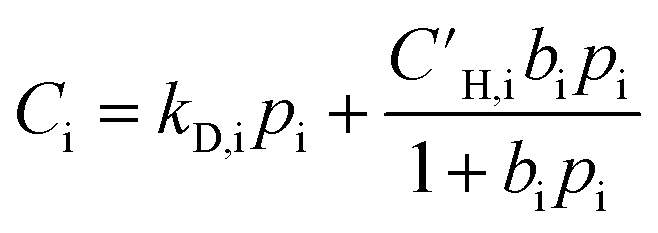 | (7) |
At each testing pressure, the amount of gas sorbed by the sample was calculated by mass balance using the pressure in the charge chamber and the equilibrated pressure after the charge gas was dosed into the sample chamber. The pure-gas sorption coefficients were then calculated by dividing the gas concentration in the polymer by the testing pressure:
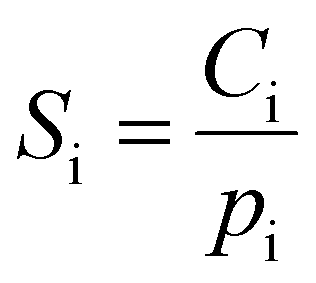 | (8) |
3. Results and discussion
3.1 Physical aging monitored by permeation tests
Physical aging behavior can be affected by multiple factors, such as casting solvent, membrane thickness, previous thermal history, drying conditions, and the environment where samples are stored.44,67 These factors are especially important for samples that have high amounts of non-equilibrium free volume and are hence susceptible to intense aging, such as PIMs. Table 1 shows a summary of the polymers studied and their treatment conditions. All PIM-1 film thicknesses were kept at approximately 40 μm during solution casting, but post-treatments (i.e., amine functionalization and FVM) resulted in slight increases in thickness. The density of PIM-1 in this study was higher compared to the values typically reported in the literature,29,32,68–76 as shown in Table S1.† To ensure internal consistency, the densities of the samples made for this study were used in the FFVgroup calculations, and the batch-to-batch variable for additional samples of PIM-1, PIM-NH2, and PIM-NH2-FVM from our lab is also provided for reference in Table S2.† The FFVgroup of polymers calculated from the group contribution method closely matched what was expected from the pore size investigations from our previous study of these polymers.29 As shown in the table, PIM-1 showed the largest initial FFV, followed by PIM-NH2-FVM and PIM-NH2, respectively. Similarly, the FFVPALS calculated from PALS indicated the largest initial FFV for PIM-1, while PIM-NH2 and PIM-NH2-FVM showed the same initial FFV within the uncertainty of the measurements. As noted in the experimental section, all samples (i.e., PIM-1, PIM-NH2, and PIM-tBOC) were soaked in methanol prior to aging experiments to reset the thermal history of each polymer. At the beginning of the aging experiments, all samples went through the same drying conditions as indicated in Table 1, while PIM-NH2 and PIM-NH2-FVM went through additional post-treatment steps.| Polymer | PIM-1 | PIM-NH2 | PIM-NH2-FVM |
|---|---|---|---|
| a Light urea crosslinks were not considered in the calculation. | |||
| Thickness (μm) | 37.6 ± 0.9 | 51.3 ± 1.1 | 51.2 ± 2.0 |
| Density (g cm−3) | 1.21 ± 0.06 | 1.28 ± 0.02 | 1.25 ± 0.02 |
| FFVgroupa | 0.23 ± 0.04 | 0.16 ± 0.01 | 0.18 ± 0.01 |
| FFVPALS | 0.255 ± 0.005 | 0.215 ± 0.004 | 0.213 ± 0.006 |
| Drying conditions | Vacuum, 130 °C, 12 h | Vacuum, 130 °C, 12 h | Vacuum, 130 °C, 12 h |
| Post-treatment conditions | n/a | Vacuum, 250 °C, 27 h | Vacuum, 250 °C, 27 h |
Fig. 2 depicts a graphical comparison of the pure-gas permeabilities of H2, O2, N2, and CH4 for the three PIM polymers as a function of time to approximately 10000 h. All data points in Fig. 2 are normalized to the first data point for easier comparison of trends among polymers, but comparisons of aging behavior on an absolute scale are shown in Fig. S1† and the tabulated values are in Table S4† for reference. As shown in Table S4,† the permeability and selectivity of fresh films are reasonably similar compared to the previously reported values.29 Given the need to understand uncertainty and reproducibility for membrane-based gas separation,77 at least three total samples from different casting batches were tested up to 1000 h, and the results are shown in Fig. S5 and 6† for comparison. The general trends remained the same within the tested 1000 h, confirming the reproducibility of our results shown in Fig. 2.
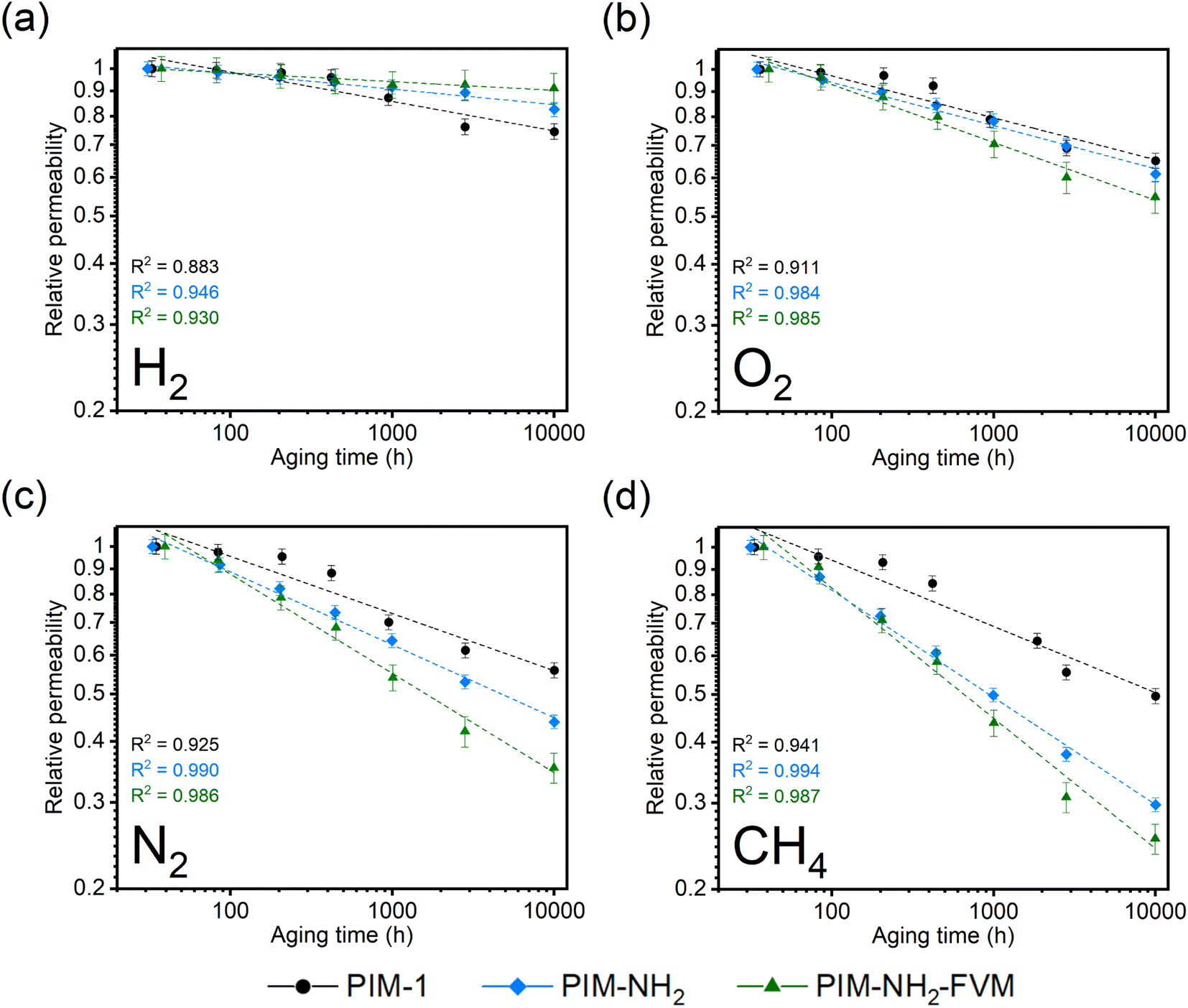 | ||
| Fig. 2 Comparison of aging behavior by tracking pure-gas permeabilities on a normalized scale for (a) H2, (b) O2, (c) N2, and (d) CH4. All data have been normalized to the first data point to show relative permeability. The linear fits of the experimental data on a log–log scale are shown in the dashed lines along with their R2 values. | ||
Physical aging of conventional polymer membranes manifests itself in permeability loss since polymers lose free volume as polymer chains relax toward their equilibrium packing structure.38 As shown in Fig. 2, all three PIM polymers showed this traditionally observed permeability loss with aging time, suggesting that neither the introduction of secondary interactions from amine groups nor the introduction of light crosslinks mitigates free volume reduction. The extent of permeability reduction (CH4 > N2 > O2 > H2) exactly followed the order of kinetic gas diameters (CH4: 3.80 Å; N2: 3.64 Å; O2: 3.46 Å; H2: 2.89 Å), indicating that the dependence of physical aging rate monitored via permeability correlated directly with the sizes of penetrants. Such a dependence is seen in many other studies in the literature,47,78 and it is attributed to the strong effect of free volume loss on diffusivity, as will be further discussed later. Among the three polymers tested, in general, PIM-NH2-FVM showed the largest permeability decrease, resulting in a 75% decrease for the largest gas considered, CH4, followed by PIM-NH2 (70%) and PIM-1 (50%), respectively. Interestingly, the extent of permeability decreases for the polymers considered switched order for the smallest gas considered, H2, as shown in Fig. 2a. In this case, PIM-1 had a 26% decrease from its initial permeability to the final measured permeability at approximately 10000 h, while PIM-NH2 and PIM-NH2-FVM showed a 17% and a 9% decrease, respectively, from their initial hydrogen permeability.
To further elucidate these findings, the permeability data in Fig. 2 have been fitted to the following equation:79,80
| P = P0t−βP | (9) |
000 h, along with high R2 values shown in Fig. 2, indicate that the films are continuously undergoing an aging process and have not yet approached their near-equilibrium state. Additionally, the fits shown in the figure were not constrained to intercept the initial permeability data points because they do not represent the true starting time for physical aging, which is the end of the drying or thermal treatment procedure by our definition. Fig. 3 shows the βP of each polymer as a function of the kinetic diameter squared of gas penetrants. The strong correlations (R2 > 0.968) for all polymers on a semi-log scale suggest that the aging rate is significantly influenced by the sizes of penetrants as previously discussed. The slope obtained by fitting βP against squares of kinetic diameters demonstrates the sensitivity of the free volume change of a polymer to the increasing size of permeating gases. As shown in the inset table in Fig. 3, PIM-NH2-FVM had the steepest slope of 0.199 Å−2, followed by PIM-NH2 (0.137 Å−2) and PIM-1 (0.058 Å−2), which demonstrates that PIM-NH2-FVM has the highest sensitivity to penetrant size when observed physical aging rates are monitored by permeation. PIMs have a characteristic diffusion-selective nature provided by their stiffened polymer backbone structures, so the diffusive barrier is larger for larger gas molecules. Of note, both PIM-NH2 and PIM-NH2-FVM have tighter packing structures induced by hydrogen bonding, and PIM-NH2-FVM also contains light urea crosslinks that preserve the narrow FVE distribution.29 For comparison, the slopes extracted from various aging data of PIM-1 available in the literature are shown in Table S6 and Fig. S4,† with values ranging from 0.039–0.098 Å−2. While some slopes extracted from the literature exhibit higher values compared to that of the PIM-1 film investigated in this study, the highest slope identified in the literature remains smaller than those of both PIM-NH2 and PIM-NH2-FVM. This finding further suggests that the increased diffusion-selective properties from tighter packing structures and crosslinks result in higher sensitivity in observed aging rates for increasing gas sizes.
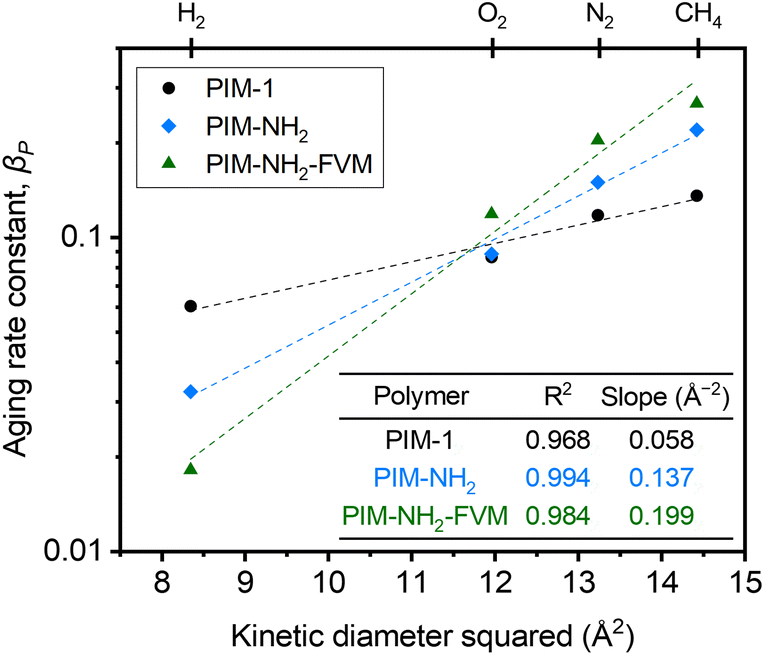 | ||
| Fig. 3 Comparison of permeability aging rate constants (βp) extracted from the experimental permeability data as a function of the kinetic diameter squared of penetrants. The dashed lines are linear fits of βp on a semi-log scale, and the resulting slopes are shown in the inset table. | ||
Regarding the observed change in the order of aging rates when testing H2 for our samples as shown in Fig. 2a, we note that previous studies have proposed that small gases such as H2 may have unique transport behavior relative to larger gases due to small, interconnected void spaces within the membrane matrix.81 We further note that the switch is also readily observed in Fig. 3. The higher relative change in permeability for H2 in PIM-1 compared to PIM-NH2 and PIM-NH2-FVM is simply a result of H2 surveying a broader portion of the free volume distribution in PIM-1 compared to other gas molecules. From a simulation study of PIM-1 by Li et al.,78 the effective FFV of PIM-1 had a strong dependence on the size of the probe molecules surveying its accessible free volume. PIM-1 had an FFV of 0.255 when surveyed with an H2 molecule (Connolly radius of 1.45 Å), while it had an FFV of 0.222 when surveyed with a CO2 molecule (Connolly radius of 1.65 Å).78 In the case of the PIMs studied in this work, PIM-1 had the largest FFV, and the addition of polar functional groups such as amines as well as thermal treatment reduced the amount of free volume inside the polymer matrix for PIM-NH2 and PIM-NH2-FVM, as seen from the FFV values in Table 1. Hence, a more significant effect of aging on the smallest penetrant is observed for PIM-1. Additionally, the stronger influence of free volume loss on diffusivity dominates with increasing gas size for the post-synthetically modified counterparts.
Fig. 4 shows the ideal selectivity of two gas pairs, H2/CH4 and O2/N2, monitored as a function of time. Analogous to the relative permeability aging plots, the selectivities were normalized to the first data point for easier comparison, and the selectivities on an absolute scale are shown in Fig. S2.† The H2/CH4 and O2/N2 selectivities increased for all polymers with aging time. This result is a typical permeability and selectivity trade-off response of physical aging since the reduction of free volume in polymer membranes affects larger molecules more significantly.38 When compared across the samples, PIM-NH2-FVM showed the highest gain in selectivities for both gas pairs throughout the aging period, resulting in a 260% increase for H2/CH4, followed by 180% for PIM-NH2 and 50% for PIM-1. Fig. 5 shows the comparison of selectivity gain and permeability loss with aging compared to other PIMs in the literature that reported aging data with a minimum of 100 days of aging. It is important to note that direct comparisons or correlations cannot be made because all data have different aging times and thicknesses, but a few interesting observations can be gleaned. First, PIMs that have hydrogen bonds without crosslinks (the blue data points in Fig. 5) did not have a particularly distinguishing gain in selectivity or loss in permeability with aging compared to other PIMs. This finding suggests the high sensitivity of the aging rate constant we observed for PIM-NH2 in Fig. 3 may not be a generalizable feature for PIMs with hydrogen bonds. Introducing hydrogen bonds to polymer backbone structures may only tune the free volume structures of PIMs without giving a distinctive aging attribute, at least for non-condensable gases. Further analysis should be made for condensable gases such as CO2 since polar functional groups that induce hydrogen bonds significantly affect gas transport of condensable gases.65 Second, while there are limited aging data for crosslinked PIMs in the literature, crosslinked PIMs generally gained much higher selectivities with aging compared to non-crosslinked PIMs. In fact, the smallest gain in H2/CH4 selectivity among the crosslinked PIMs was 143%, and this was the ninth highest gain among all PIMs considered (N = 58). Given these findings, the high sensitivity of aging rate constants we observed for PIM-NH2-FVM in Fig. 3 may be a generalizable feature of crosslinked PIMs.
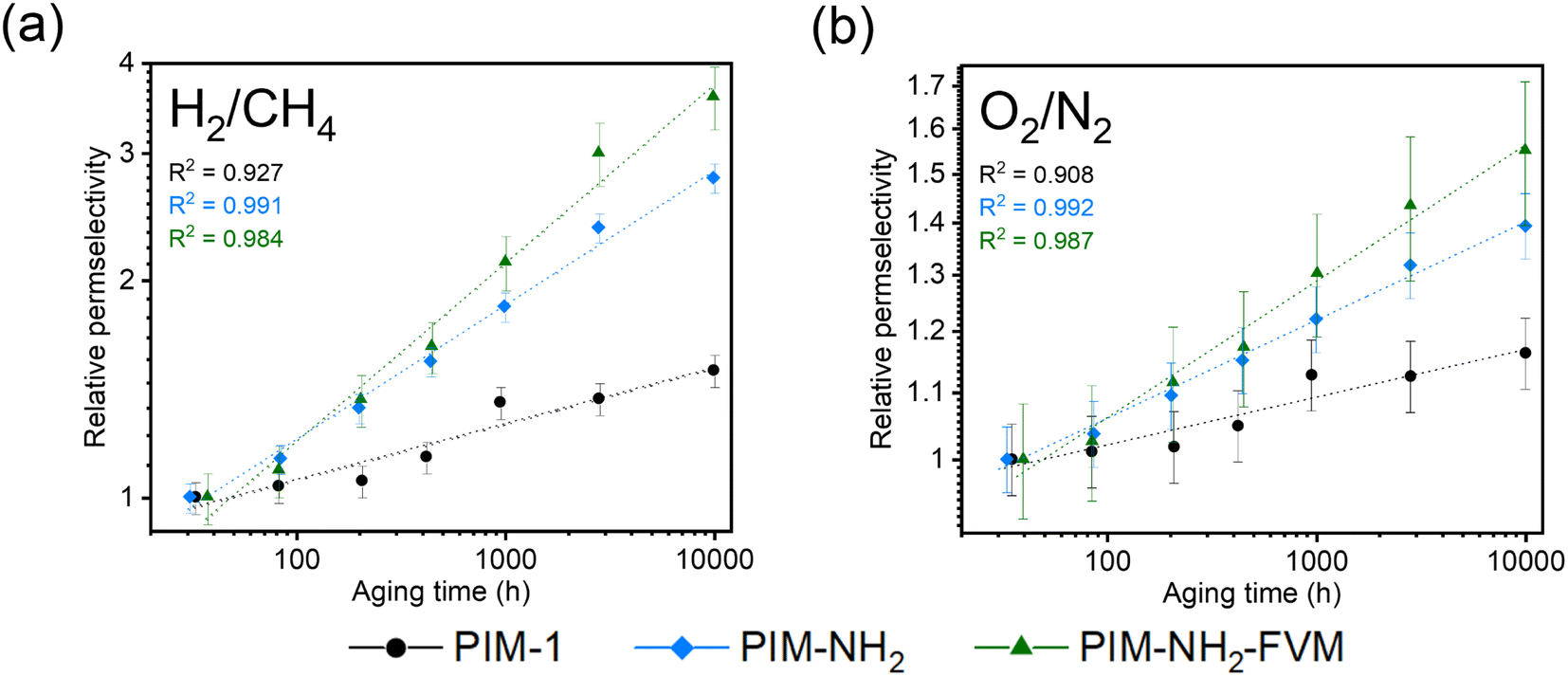 | ||
| Fig. 4 Comparison of selectivity as a function of time on a normalized scale for (a) H2/CH4 and (b) O2/N2. All data have been normalized to the first data point to show relative selectivity. The linear fits of the experimental data on a log–log scale are shown in the dashed lines along with their R2 values. | ||
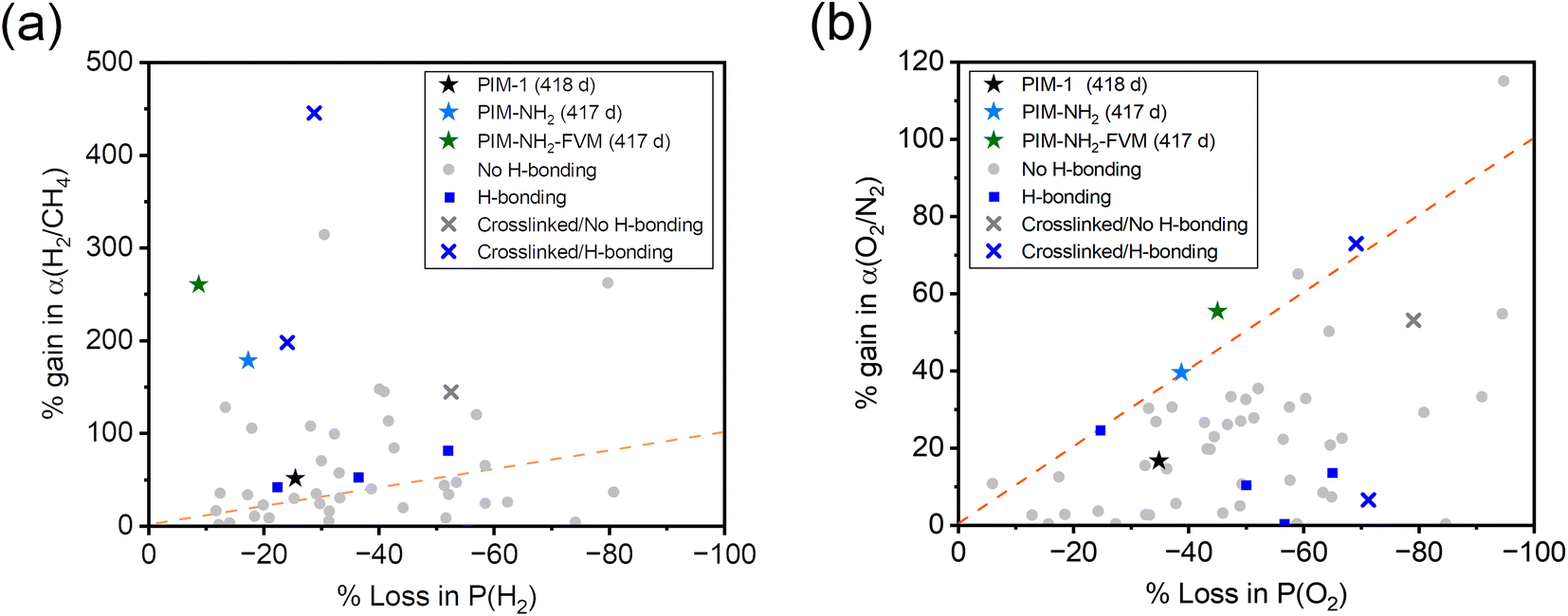 | ||
| Fig. 5 Comparison of long-term physical aging effects on (a) H2/CH4 and (b) O2/N2 separation for various PIMs reported in the literature.13,19,89,94–105 The star symbols are PIMs in this study, and only polymer films with a minimum of 100 days of aging are considered. The dotted orange line is a parity line, representing equal permeability loss and selectivity gain. | ||
What is particularly interesting about PIM-NH2-FVM is that it showed the steepest slope from the origin of the H2/CH4 plot even among the crosslinked PIMs in the literature, highlighting its remarkably large boost in selectivity without losing much permeability to hydrogen. This result suggests that there is unique stability towards smaller gas molecules and increasing molecular screening of large gas molecules with aging. We hypothesize that this observation is a result of the FVM method creating a larger number of small FVE pathways for small gas molecules such as hydrogen. From our previous report on PALS data, applying FVM to PIM-NH2 resulted in a 19% increase in the intensity related to the smaller FVE (I3) along with the narrowing of the FVE distribution,29 indicating an increase in the number of small FVEs and a reduction of larger FVEs, which heavily impacts the diffusion of larger gas molecules. Thus, PIM-NH2-FVM exhibited a significant permeability reduction for larger gas molecules while still permitting rapid and selective diffusion of small gas molecules such as hydrogen.
3.2 Detailed analyses of the physical aging results
Permeability in polymers can be decoupled into an effective diffusion coefficient, Di, and a sorption coefficient, Si, using the sorption–diffusion model:82| Pi = DiSi | (10) |
This model states that permeation of penetrants through a polymer membrane is governed by both thermodynamic factors (i.e., sorption of penetrants into the polymer matrix) and kinetic factors (i.e., diffusion of penetrants through the polymer matrix). By using the ideal gas selectivity equation in eqn (6), diffusion and sorption selectivity can also be decoupled as follows:
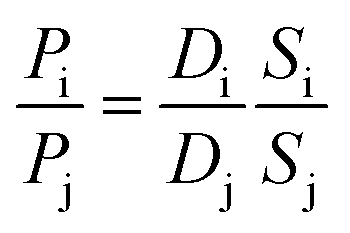 | (11) |
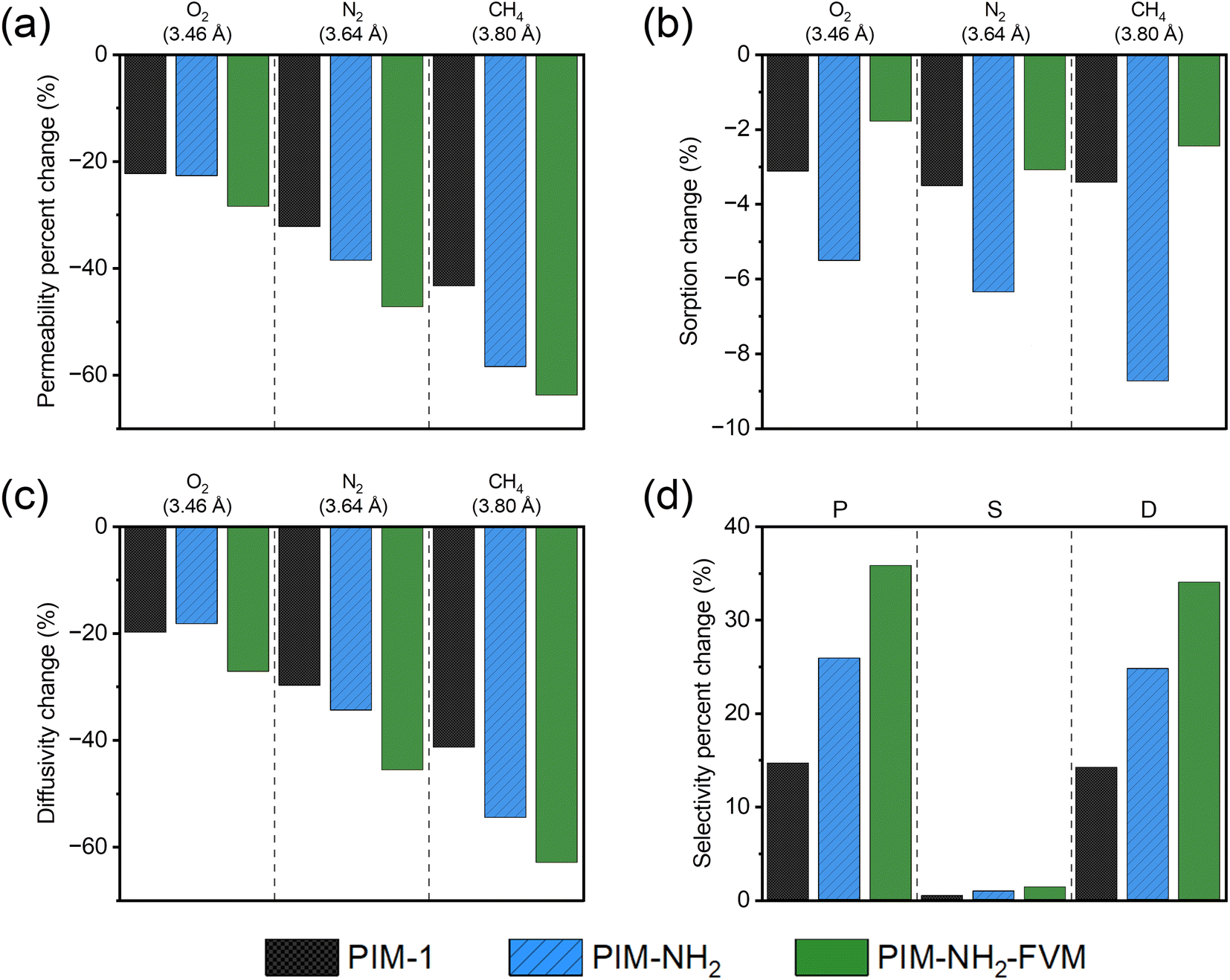 | ||
| Fig. 6 Percent changes in (a) permeability, (b) sorption, (c) diffusivity, and (d) O2/N2 selectivities over approximately 2230 h of physical aging. The gases are ordered from smallest to largest kinetic diameters, and gray dashed lines separate the three tested gases (O2, N2, and CH4) in figures a–c. | ||
As shown in Fig. 6b, the sorption coefficients did not significantly change compared to permeability for all gases and polymers. The changes in sorption coefficients throughout the 2230 h aging period were less than −4% for PIM-1 and PIM-NH2-FVM for all gases. While PIM-NH2 showed a larger decrease among the three polymers, the changes were still small compared to the permeability changes shown in Fig. 6a. As shown in Fig. S8–10,† sorption isotherms did not change significantly even at high pressures for all polymers, and this result demonstrates that a decrease in sorption is not a significant factor regardless of the type of functional groups and additional crosslinks that PIMs contain. Relatively small changes in sorption coefficients have also been observed for CO2 in PIM-1 from a detailed physical aging study by Bernardo et al.,79 even though CO2 has relatively higher sorption compared to the gases tested in this study. Thus, we believe our findings are generalizable to various gases with PIMs.
In contrast to the sorption coefficients, the effective diffusivity decreased significantly with aging, as shown in Fig. 6c. This finding suggests that the mechanism for aging of these PIMs is driven by changes to the packing structure of polymer segments. As expected, changes in both permeability and diffusivity followed identical trends, correlating closely with the size of penetrants, as shown in Fig. 6a and c. The densification of the polymer matrix impacts the diffusion of larger gas penetrants more significantly compared to smaller gas penetrants, and this finding confirms our assertions in Section 3.1 that physical aging monitored by permeability has a large dependence on the kinetic gas diameter due to the diffusion selective nature of PIMs. Hence, the change in diffusion selectivity had a much larger contribution to the permselectivity, as shown in Fig. 6d, and H2/CH4 separation had a much larger gain in selectivity compared to O2/N2 due to the larger difference in penetrant sizes (0.91 Å difference for H2/CH4versus 0.18 Å difference for O2/N2), as shown in Fig. 4.
According to the Brandt model, the activation energy of diffusion, ED, is directly proportional to the square of the penetrant diameter (d2):
| ED = cd2 − f | (12) |
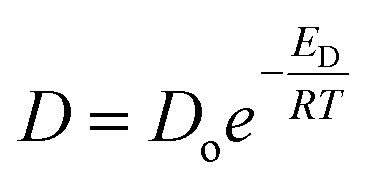 | (13) |
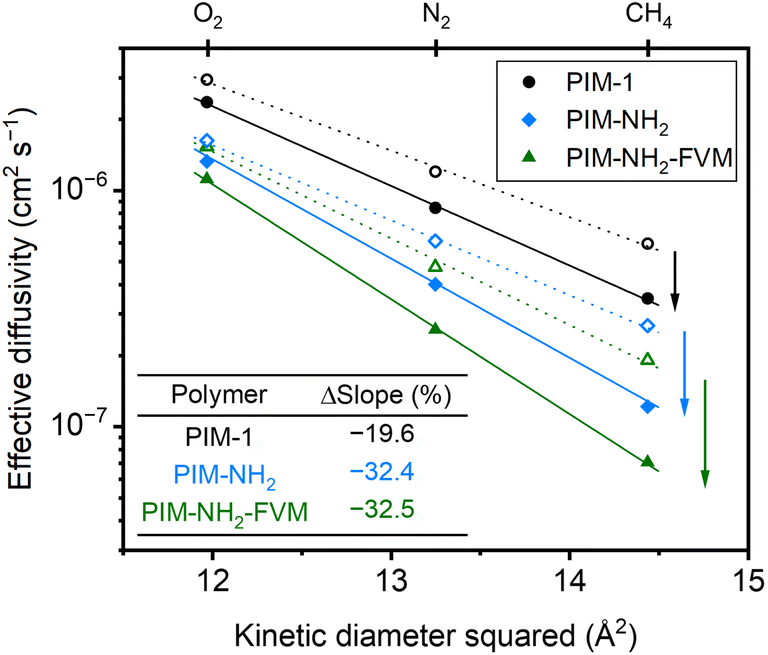 | ||
| Fig. 7 Comparison of effective diffusivity plotted against kinetic diameter squared for fresh and 2230 h aged samples. Unfilled symbols represent fresh samples, and filled symbols represent aged samples. R2 values for all fittings were above 0.997. | ||
To investigate the changes in energetics of gas transport, variable-temperature permeation tests were performed to obtain the activation energy of permeation, Ep:20
| P = P0e−Ep/RT | (14) |
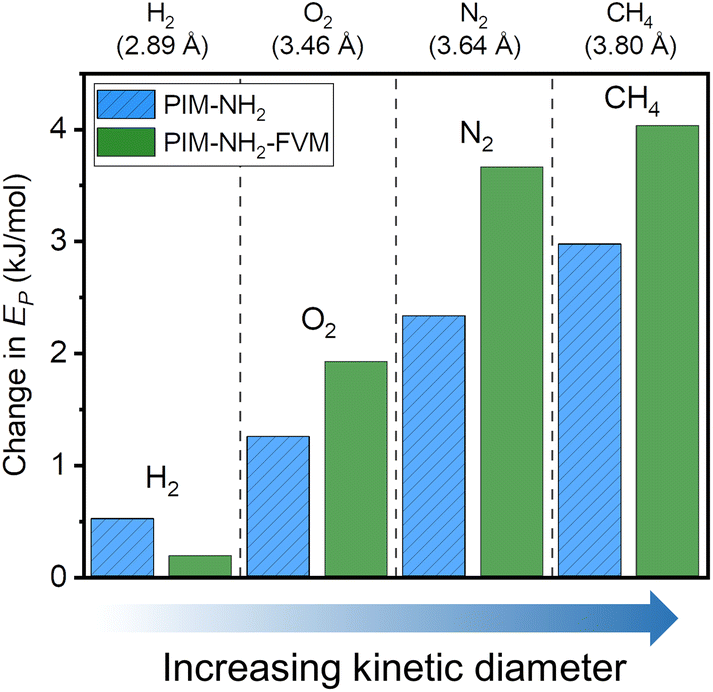 | ||
| Fig. 8 Comparison of changes in activation energy of permeation (Ep) for PIM-NH2 and PIM-NH2-FVM samples upon aging. The samples were aged for 710 h. | ||
3.3 Influence of physical aging on the packing structure and free volume
While permeation is a helpful property to quantify physical aging, permeability reduction is a function of both the initial FFV and the size of the gases, which makes it difficult to directly compare between permeability reduction and structural changes that result from aging. A detailed explanation of this complexity is outlined in Fig. S13† and the surrounding text. In short, if two samples with different initial FFVs have the same amount of free volume reduction, the sample with the smaller initial FFV is likely to have a larger permeability reduction on a normalized scale. In other words, it cannot be simply assumed that PIM-NH2-FVM experiences more free volume contraction with aging since it generally experiences larger permeability reductions. Thus, detailed morphological and free volume analyses have to be made in addition to permeation tests to quantify aging behavior of glassy polymers.The changes in the packing structure of the PIMs considered in this study were investigated using wide-angle X-ray scattering (WAXS). Fig. 9a presents the WAXS patterns for both fresh and aged PIM samples, where the dotted lines indicate the fresh films. All patterns show four distinct peaks, and these peaks clearly resemble the WAXS patterns observed in our previous study.29 A study by McDermott et al. showed that the three peaks in the high-q region, highlighted by the gray background in Fig. 9a, represent characteristic distances of spiro centers on different chains, while the larger peak in the low-q region is closely related to the microporosity of PIMs.85,86 While the locations of the low-q peaks were nearly unchanged for the PIMs studied in this work, the three characteristic peaks in the high-q region showed non-negligible changes, which provides quantitative insights into polymer densification during the aging period. A magnified version of the WAXS patterns in the high-q region is shown in Fig. 9b, and Table S13† presents the d-spacing, which are the average interchain distances that were calculated from these three peaks.
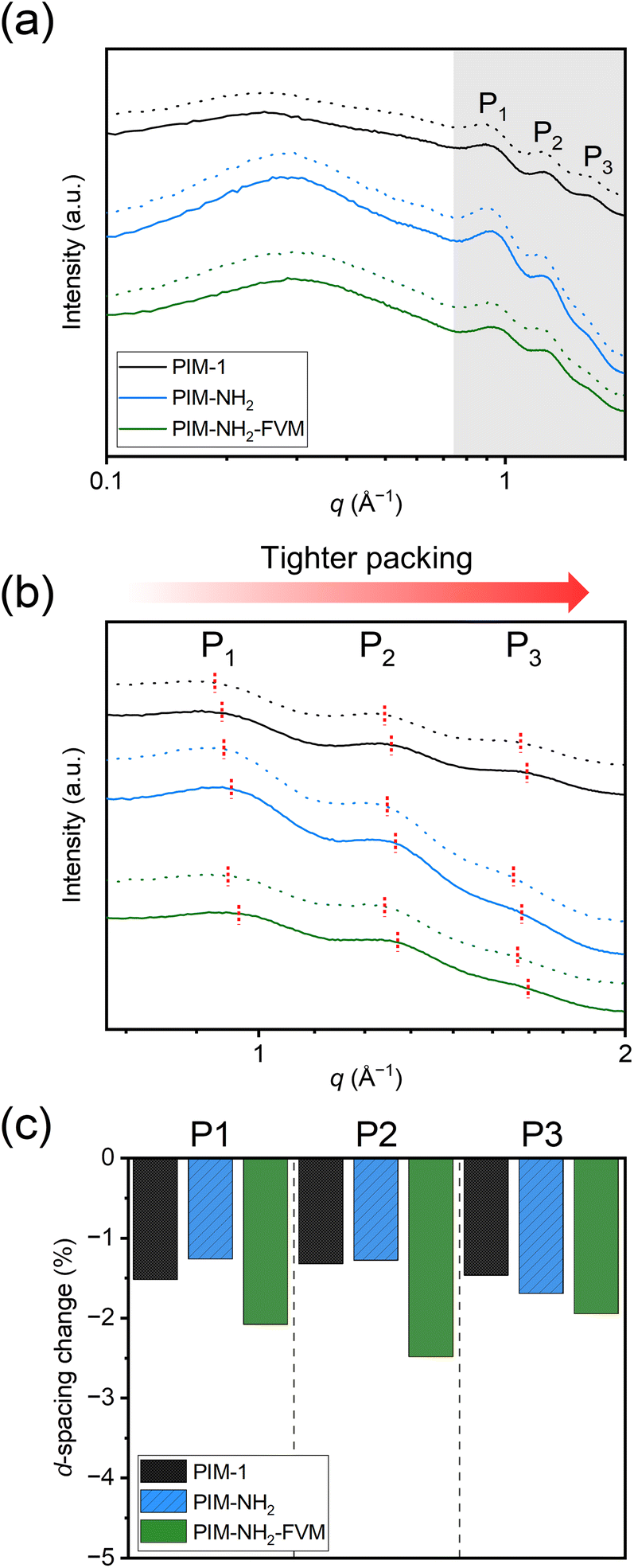 | ||
| Fig. 9 Comparison of wide-angle X-ray scattering patterns on a (a) full scale and (b) magnified scale for PIM-1, PIM-NH2, and PIM-NH2-FVM. The fresh samples are represented by dotted lines, while the samples that have been aged for approximately 1400 h are represented by solid lines. The red dotted lines in (b) represent the peak values used to calculate the d-spacing, and the changes in the d-spacing obtained from the patterns are shown in (c). | ||
As shown in Fig. 9c, the average interchain distances decreased for all polymers with aging. Interestingly, PIM-NH2 had a comparable reduction for all peaks compared to PIM-1, and PIM-NH2-FVM had the largest reduction for all three peaks. While these are very small changes (∼1–2%) and the characteristic spiro center distances do not directly inform the actual free volume sizes in PIMs, we notice a few correlations that agree with the analysis in Fig. 4 and 6. First, the comparable d-spacing reductions for PIM-NH2 and PIM-NH2-FVM with PIM-1 further support our assertion that neither the introduction of secondary interactions from amine groups nor the introduction of light crosslinks mitigates volume contraction. Even if the chain mobilities decrease due to reduced free volume from hydrogen bonding and crosslinks, the polymer packing structures still evolve towards their equilibrium conformations at an appreciable rate within the tested time frame. Thus, permeability reductions were observed for all polymers. Second, the reduction order for peak 3 (P3), which is associated with the smallest d-spacing of around 3.8 Å, correlates with the order of selectivity gains in Fig. 4. This result suggests that d-spacing in the range of gas penetrant size has the largest effect on selectivity, a result that would be consistent with PIMs obeying the sorption–diffusion model, similar to other reports in the literature.24,29,87
Given the thickness of our films for this study, we wanted to investigate ultra-long term aging behavior of our samples. To this end, the free volume changes of the PIM polymers with aging time were investigated using positron annihilation lifetime spectroscopy (PALS), which is able to accurately observe small free volume changes.40,88–92 During a PALS experiment, positrons (e+) are naturally emitted from a radioactive source. These positrons “pick off” electrons in FVEs, forming a transient semi-stable particle ortho-positronium (o-PS) that is subsequently annihilated in the polymer matrix. The lifetimes of the o-PS atoms are correlated with average FVE sizes by using the Tao–Eldrup relationship, and FVE distributions can also be obtained by analyzing the lifetimes and the relative intensities of the model parameters.93 Two o-PS lifetimes (τ3 and τ4) were obtained from PALS for the samples in this work, indicating the presence of a bimodal distribution within the PIM films. The total FFV for a bimodal FVE distribution is the sum of FFV calculated from each bimodal peak as shown in eqn (4). Thus, the amount of free volume contributed by each average FVE diameter (di) can be approximated using the following equation:59,63
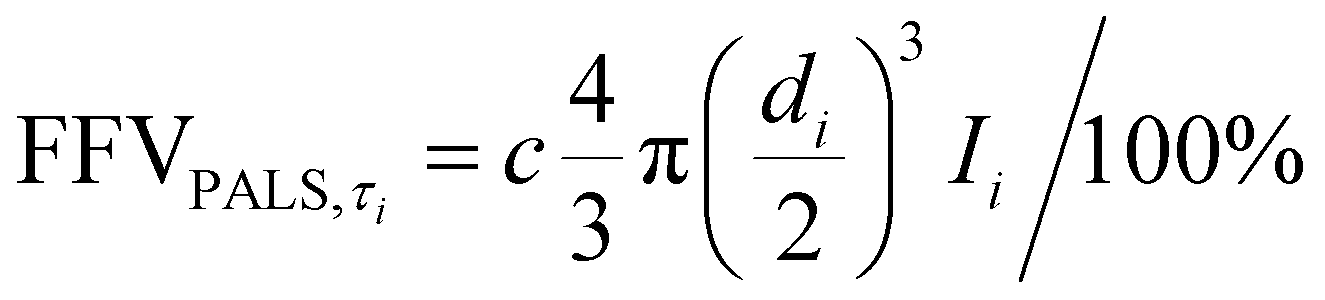 | (15) |
Tables 2 and S14† summarize the PALS parameters obtained for the samples from our previous study29 that have been aged for approximately 20000 h (833 days). Fig. 10a–c show the comparison of the average FVE size distributions (FSDs) obtained for fresh and aged samples. It is noted that our samples have the same chemistry and underwent the same drying conditions as the previous study, but earlier samples had been aged under atmospheric conditions instead of in a desiccator. As shown in Table 2 and Fig. 10a–c, PIM-1 showed the largest average FVE size reduction of −25.6 ± 5.2% and −3.6 ± 0.4% for the small and large FVEs, respectively, compared to PIM-NH2 (−16.6 ± 4.3% and −3.0 ± 0.6%) and PIM-NH2-FVM (−17.5 ± 4.5% and −1.4 ± 0.7%) over this relatively long aging period. The notably larger changes in both FVE sizes for PIM-1 agree with our analysis in Section 3.1 in that PIM-1 had the largest permeability reduction for only H2 but a smaller permeability reduction for larger gases. This interesting finding is related to H2 being able to survey a broader portion of free volume distribution due to its small size. Remarkably, the reduction orders for both d3 and d4 were strongly correlated with the order of the corresponding initial FFVPALS,τi, as shown in Fig. 10d and e. More detailed experiments are required to investigate the correlation between free volume and rates of free volume reduction, but the comparison of FSDs with long aging times suggests that the total extent of structural change is most strongly correlated with the initial amount of free volume and not with amines or light crosslinks. This finding matches known trends for conventional glassy polymers,40,84 suggesting that the same mechanistic and theoretical phenomena for aging apply to PIMs.
000 h (833 days), and the changes are compared with the PALS data from the previous studya
| Aging timeb (h) | PIM-1 | PIM-NH2 | PIM-NH2-FVM | |
|---|---|---|---|---|
| 19170 |
21050 |
19150 |
||
| a Errors were calculated based on the population standard deviations. b The presented time is the aging time at the start of PALS experiments. c PALS data for fresh samples are from a previous study.29 | ||||
| Freshc | d 3 (Å) | 6.4 ± 0.3 | 6.2 ± 0.2 | 6.3 ± 0.2 |
| d 4 (Å) | 11.25 ± 0.04 | 10.13 ± 0.05 | 10.14 ± 0.06 | |
| FFVPALS,τ3 | 0.014 ± 0.002 | 0.012 ± 0.001 | 0.015 ± 0.002 | |
| FFVPALS,τ4 | 0.240 ± 0.005 | 0.203 ± 0.004 | 0.199 ± 0.005 | |
| Aged | d 3 (Å) | 4.76 ± 0.25 | 5.17 ± 0.21 | 5.20 ± 0.23 |
| d 4 (Å) | 10.84 ± 0.03 | 9.83 ± 0.03 | 10.00 ± 0.04 | |
| FFVPALS,τ3 | 0.006 ± 0.001 | 0.007 ± 0.001 | 0.008 ± 0.001 | |
| FFVPALS,τ4 | 0.181 ± 0.003 | 0.199 ± 0.003 | 0.178 ± 0.004 | |
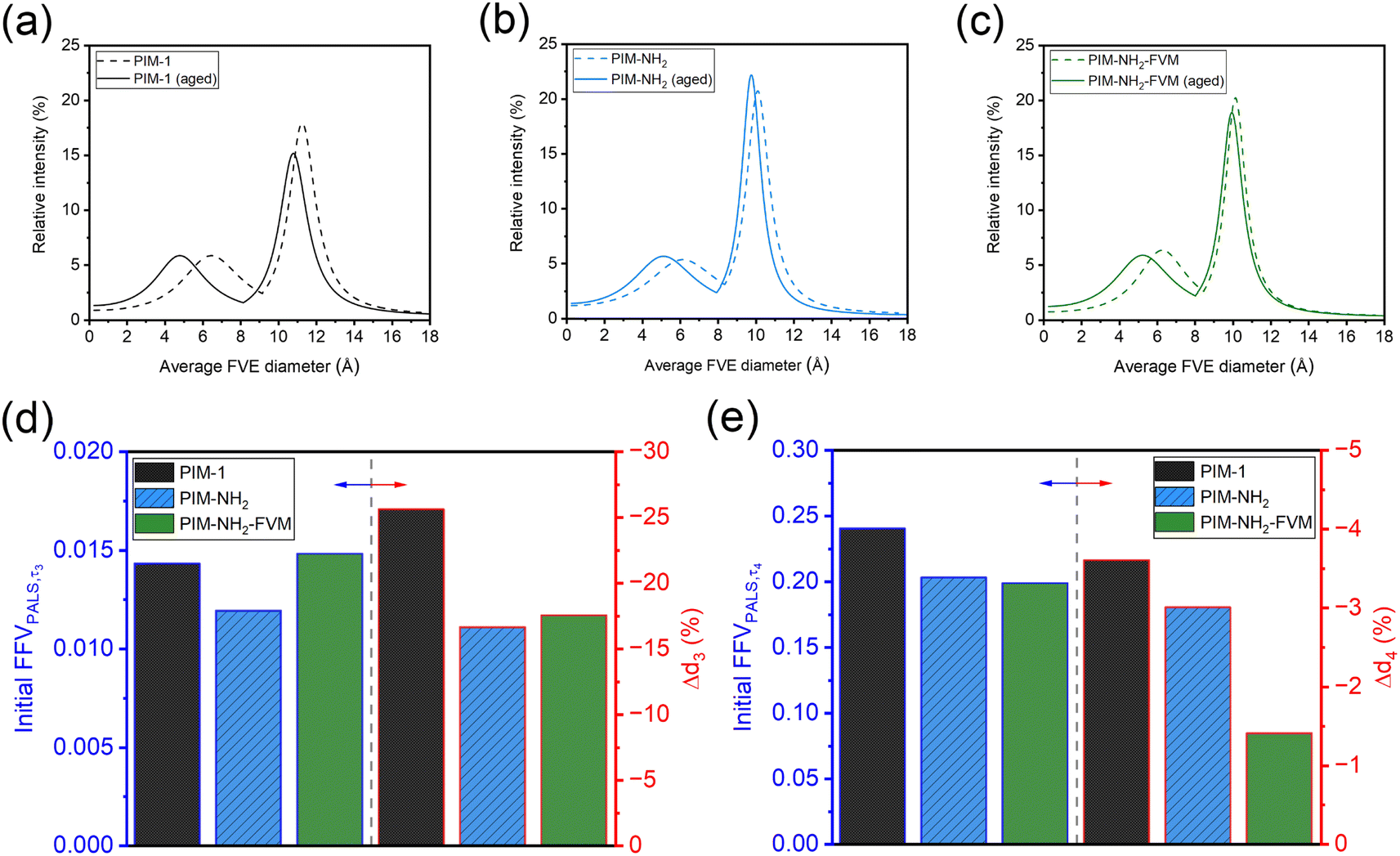 | ||
| Fig. 10 Comparison of change in bimodal FVE size distribution (FSD) for (a) PIM-1 (b) PIM-NH2, and (c) PIM-NH2-FVM. The FVEs of fresh films from the previous study29 are plotted as dotted lines, and samples that have been aged for approximately 20000 h (833 days) are plotted as solid lines. Comparisons of initial FFVPALS,τi for each bimodal peak and changes in average FVE diameters (Δdi) are shown in (d) for the smaller FVE and in (e) for the larger FVE. | ||
4. Conclusions
In this study, physical aging behavior of PIM-1, amine-functionalized PIM-1 (PIM-NH2), and free volume manipulated PIM-NH2 (PIM-NH2-FVM) were systematically investigated via gas transport and structural analyses to closely examine how each modification impacted physical aging. The investigation of free volume size distribution for films aged ∼2 years showed that PIM-1, which had the highest initial free volume compared to its aminated and crosslinked counterparts, had the largest total free volume reduction. This result indicates that initial free volume is the largest driver of free volume relaxation. Gas transport studies revealed that permeability changes from the structural relaxation process are strongly correlated with factors that influence diffusion, such as penetrant size, free volume distribution, and crosslinks. As a result, the aminated PIM-1 derivative with higher initial free volume and light crosslinks showed an accelerated reduction in permeability for larger gases compared to its lower free volume counterpart without crosslinks. Taken together, initial free volume is the primary driving force for reducing free volume, but hydrogen bonds and crosslinks can introduce attributes that strongly impact diffusion of large gas molecules, contributing to improved permselectivity changes in microporous membranes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Separation Science program under Award Number DE-SC0019087 and the Office of Naval Research (ONR) under Award Numbers N00014-20-1-2418 and N00014-21-1-2666. Additionally, support to investigate the free volume size distribution using PALS was provided to CMD through the Veski Inspiring Women Fellowship. The authors would like to thank Pablo Dean for providing densities of the PIM-1 derivatives measured using various buoyant fluids, Dr Stephen DeWitt for his helpful suggestions during the revision process, and Dr Walt Massefski for his help in the characterization of polymers.References
- D. S. Sholl and R. Lively, Nature, 2016, 532, 435–437 CrossRef PubMed.
- International Energy Agency, CO2 Emissions in 2022, 2023 Search PubMed.
- A. W. Mohammad, Y. H. Teow, W. L. Ang, Y. T. Chung, D. L. Oatley-Radcliffe and N. Hilal, Desalination, 2015, 356, 226–254 CrossRef CAS.
- M. Galizia, W. S. Chi, Z. P. Smith, T. C. Merkel, R. W. Baker and B. D. Freeman, Macromolecules, 2017, 50, 7809–7843 CrossRef CAS.
- R. W. Baker, Membrane Technologies and Applications, John Wiley & Sons Ltd, Chichester, 2nd edn, 2004 Search PubMed.
- Y. Wang, X. Ma, B. S. Ghanem, F. Alghunaimi, I. Pinnau and Y. Han, Mater. Today Nano, 2018, 3, 69–95 CrossRef.
- D. F. Sanders, Z. P. Smith, R. Guo, L. M. Robeson, J. E. McGrath, D. R. Paul and B. D. Freeman, Polymer, 2013, 54, 4729–4761 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165–185 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- A. X. Wu, J. A. Drayton and Z. P. Smith, AIChE J., 2019, 65, e16700 CAS.
- B. D. Freeman, Macromolecules, 1999, 32, 375–380 CrossRef CAS.
- P. M. Budd and N. B. McKeown, Polym. Chem., 2010, 1, 63–68 RSC.
- Z. X. Low, P. M. Budd, N. B. McKeown and D. A. Patterson, Chem. Rev., 2018, 118, 5871–5911 CrossRef CAS PubMed.
- P. M. Budd, N. B. McKeown and D. Fritsch, J. Mater. Chem., 2005, 15, 1977–1986 RSC.
- Z. Tong and A. K. Sekizkardes, Membranes, 2021, 11, 156 CrossRef CAS PubMed.
- S. Kim and Y. M. Lee, Prog. Polym. Sci., 2015, 43, 1–32 CrossRef CAS.
- T. M. Long and T. M. Swager, J. Am. Chem. Soc., 2003, 125, 14113–14119 CrossRef CAS PubMed.
- P. M. Budd, E. S. Elabas, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib, C. E. Tattershall and D. Wang, Adv. Mater., 2004, 16, 456–459 CrossRef CAS.
- Y. He, F. M. Benedetti, S. Lin, C. Liu, Y. Zhao, H. Z. Ye, T. Van Voorhis, M. G. De Angelis, T. M. Swager and Z. P. Smith, Adv. Mater., 2019, 31, 1807871 CrossRef PubMed.
- H. W. H. Lai, F. M. Benedetti, Z. Jin, Y. C. Teo, A. X. Wu, M. G. De Angelis, Z. P. Smith and Y. Xia, Macromolecules, 2019, 52, 6294–6302 CrossRef CAS.
- M. Carta, M. Croad, R. Malpass-Evans, J. C. Jansen, P. Bernardo, G. Clarizia, K. Friess, M. Lanč and N. B. McKeown, Adv. Mater., 2014, 26, 3526–3531 CrossRef CAS PubMed.
- I. Rose, C. G. Bezzu, M. Carta, B. Comesanã-Gándara, E. Lasseuguette, M. C. Ferrari, P. Bernardo, G. Clarizia, A. Fuoco, J. C. Jansen, K. E. Hart, T. P. Liyana-Arachchi, C. M. Colina and N. B. McKeown, Nat. Mater., 2017, 16, 932–937 CrossRef CAS PubMed.
- C. R. Mason, L. Maynard-Atem, K. W. J. Heard, B. Satilmis, P. M. Budd, K. Friess, M. Lanč, P. Bernardo, G. Clarizia and J. C. Jansen, Macromolecules, 2014, 47, 1021–1029 CrossRef CAS PubMed.
- K. Mizrahi Rodriguez, A. X. Wu, Q. Qian, G. Han, S. Lin, F. M. Benedetti, H. Lee, W. S. Chi, C. M. Doherty and Z. P. Smith, Macromolecules, 2020, 53, 6220–6234 CrossRef CAS.
- Q. Song, S. Cao, R. H. Pritchard, B. Ghalei, S. A. Al-Muhtaseb, E. M. Terentjev, A. K. Cheetham and E. Sivaniah, Nat. Commun., 2014, 5, 4813 CrossRef PubMed.
- F. Y. Li, Y. Xiao, T. S. Chung and S. Kawi, Macromolecules, 2012, 45, 1427–1437 CrossRef CAS.
- X. Ma, R. Swaidan, B. Teng, H. Tan, O. Salinas, E. Litwiller, Y. Han and I. Pinnau, Carbon, 2013, 62, 88–96 CrossRef CAS.
- H. Shamsipur, B. A. Dawood, P. M. Budd, P. Bernardo, G. Clarizia and J. C. Jansen, Macromolecules, 2014, 47, 5595–5606 CrossRef CAS.
- K. Mizrahi Rodriguez, S. Lin, A. X. Wu, G. Han, J. J. Teesdale, C. M. Doherty and Z. P. Smith, Angew. Chem., Int. Ed., 2021, 60, 6593–6599 CrossRef CAS PubMed.
- J. Ahn, W. J. Chung, I. Pinnau, J. Song, N. Du, G. P. Robertson and M. D. Guiver, J. Membr. Sci., 2010, 346, 280–287 CrossRef CAS.
- A. F. Bushell, M. P. Attfield, C. R. Mason, P. M. Budd, Y. Yampolskii, L. Starannikova, A. Rebrov, F. Bazzarelli, P. Bernardo, J. Carolus Jansen, M. Lanč, K. Friess, V. Shantarovich, V. Gustov and V. Isaeva, J. Membr. Sci., 2013, 427, 48–62 CrossRef CAS.
- M. M. Khan, V. Filiz, G. Bengtson, S. Shishatskiy, M. M. Rahman, J. Lillepaerg and V. Abetz, J. Membr. Sci., 2013, 436, 109–120 CrossRef CAS.
- M. Alberto, R. Bhavsar, J. M. Luque-Alled, A. Vijayaraghavan, P. M. Budd and P. Gorgojo, J. Membr. Sci., 2018, 563, 513–520 CrossRef CAS.
- Q. Song, S. Cao, R. H. Pritchard, H. Qiblawey, E. M. Terentjev, A. K. Cheetham and E. Sivaniah, J. Mater. Chem. A, 2016, 4, 270–279 RSC.
- C. H. Lau, P. T. Nguyen, M. R. Hill, A. W. Thornton, K. Konstas, C. M. Doherty, R. J. Mulder, L. Bourgeois, A. C. Y. Liu, D. J. Sprouster, J. P. Sullivan, T. J. Bastow, A. J. Hill, D. L. Gin and R. D. Noble, Angew. Chem., Int. Ed., 2014, 53, 5322–5326 CrossRef CAS PubMed.
- T. Corrado and R. Guo, Mol. Syst. Des. Eng., 2020, 5, 22–48 RSC.
- R. W. Baker and B. T. Low, Macromolecules, 2014, 47, 6999–7013 CrossRef CAS.
- B. W. Rowe, B. D. Freeman and D. R. Paul, in Membrane Engineering for the Treatment of Gases, ed. E. Drioli and G. Barbieri, Royal Society of Chemistry Books, Cambridge, 1st edn, 2011, vol. 1, pp. 58–83 Search PubMed.
- A. Y. Alentiev and Y. P. Yampolskii, J. Membr. Sci., 2000, 165, 201–216 CrossRef CAS.
- D. Cangialosi, H. Schut, A. Van Veen and S. J. Picken, Macromolecules, 2003, 36, 142–147 CrossRef CAS.
- L. C. E. Struik, Polym. Eng. Sci., 1977, 17, 165–173 CrossRef CAS.
- J. H. Kim, W. J. Koros and D. R. Paul, Polymer, 2006, 47, 3094–3103 CrossRef CAS.
- B. W. Rowe, B. D. Freeman and D. R. Paul, Polymer, 2009, 50, 5565–5575 CrossRef CAS.
- B. W. Rowe, B. D. Freeman and D. R. Paul, Polymer, 2010, 51, 3784–3792 CrossRef CAS.
- M. S. McCaig and D. R. Paul, Polymer, 1999, 40, 7209–7225 CrossRef CAS.
- L. Cui, W. Qiu, D. R. Paul and W. J. Koros, Polymer, 2011, 52, 5528–5537 CrossRef CAS.
- J. H. Kim, W. J. Koros and D. R. Paul, J. Membr. Sci., 2006, 282, 32–43 CrossRef CAS.
- R. Swaidan, B. Ghanem, E. Litwiller and I. Pinnau, Macromolecules, 2015, 48, 6553–6561 CrossRef CAS.
- T. J. Corrado, Z. Huang, D. Huang, N. Wamble, T. Luo and R. Guo, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2022204118 CrossRef CAS PubMed.
- H. Yin, Y. Z. Chua, B. Yang, C. Schick, W. J. Harrison, P. M. Budd, M. Böhning and A. Schönhals, J. Phys. Chem. Lett., 2018, 9, 2003–2008 CrossRef CAS PubMed.
- H. W. H. Lai, F. M. Benedetti, J. M. Ahn, A. M. Robinson, Y. Wang, I. Pinnau, Z. P. Smith and Y. Xia, Science, 2022, 375, 1390–1392 CrossRef CAS PubMed.
- S. Lin, T. Joo, F. M. Benedetti, L. C. Chen, A. X. Wu, K. Mizrahi Rodriguez, Q. Qian, C. M. Doherty and Z. P. Smith, Polymer, 2021, 212, 123121 CrossRef CAS.
- A. X. Wu, S. Lin, K. Mizrahi Rodriguez, F. M. Benedetti, T. Joo, A. F. Grosz, K. R. Storme, N. Roy, D. Syar and Z. P. Smith, J. Membr. Sci., 2021, 636, 119526 CrossRef CAS.
- J. Kansy, Nucl. Instrum. Methods Phys. Res., Sect. A, 1996, 374, 235–244 CrossRef CAS.
- V. P. Shantarovich, I. B. Kevdina, Y. P. Yampolskii and A. Y. Alentiev, Macromolecules, 2000, 33, 7453–7466 CrossRef CAS.
- S. J. Tao, J. Chem. Phys., 1972, 56, 5499 CrossRef CAS.
- C. Pascual-Izarra, A. W. Dong, S. J. Pas, A. J. Hill, B. J. Boyd and C. J. Drummond, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 603, 456–466 CrossRef CAS.
- M. Eldrup, D. Lightbody and J. N. Sherwood, Chem. Phys., 1981, 63, 51–58 CrossRef CAS.
- Y. C. Jean, P. E. Mallon and D. M. Schrader, Principles and Applications of Positron and Positronium Chemistry, World Scientific, River Edge, 2003 Search PubMed.
- Y. Y. Wang, H. Nakanishi, Y. C. Jean and T. C. Sandreczki, J. Polym. Sci., Part B: Polym. Phys., 1990, 28, 1431–1441 CrossRef CAS.
- J. Liu, Q. Deng and Y. C. Jean, Macromolecules, 1993, 26, 7149–7155 CrossRef CAS.
- N. Petzetakis, C. M. Doherty, A. W. Thornton, X. C. Chen, P. Cotanda, A. J. Hill and N. P. Balsara, Nat. Commun., 2015, 6, 7529 CrossRef CAS PubMed.
- Y. C. Jean, J. P. Yuan, J. Liu, Q. Deng and H. Yang, J. Polym. Sci., Part B: Polym. Phys., 1995, 33, 2365–2371 CrossRef CAS.
- J. Liu, Y. C. Jean and H. Yang, Macromolecules, 1995, 28, 5774–5779 CrossRef CAS.
- K. Mizrahi Rodriguez, F. M. Benedetti, N. Roy, A. X. Wu and Z. P. Smith, J. Mater. Chem. A, 2021, 9, 23631–23642 RSC.
- Z. P. Smith, D. F. Sanders, C. P. Ribeiro, R. Guo, B. D. Freeman, D. R. Paul, J. E. McGrath and S. Swinnea, J. Membr. Sci., 2012, 415–416, 558–567 CrossRef CAS.
- R. R. Tiwari, J. Jin, B. D. Freeman and D. R. Paul, J. Membr. Sci., 2017, 537, 362–371 CrossRef CAS.
- Z. G. Wang, X. Liu, D. Wang and J. Jin, Polym. Chem., 2014, 5, 2793–2800 RSC.
- X. Wu, Y. Ren, G. Sui, G. Wang, G. Xu, L. Yang, Y. Wu, G. He, N. Nasir, H. Wu and Z. Jiang, AIChE J., 2020, 66, e16800 CAS.
- W. F. Yong, F. Y. Li, Y. C. Xiao, P. Li, K. P. Pramoda, Y. W. Tong and T. S. Chung, J. Membr. Sci., 2012, 407–408, 47–57 CrossRef CAS.
- L. Hao, P. Li and T. S. Chung, J. Membr. Sci., 2014, 453, 614–623 CrossRef CAS.
- X. M. Wu, Q. G. Zhang, P. J. Lin, Y. Qu, A. M. Zhu and Q. L. Liu, J. Membr. Sci., 2015, 493, 147–155 CrossRef.
- C. Z. Liang, J. T. Liu, J. Y. Lai and T. S. Chung, J. Membr. Sci., 2018, 563, 93–106 CrossRef CAS.
- X. Chen, Z. Zhang, L. Wu, X. Liu, S. Xu, J. E. Efome, X. Zhang and N. Li, ACS Appl. Polym. Mater., 2020, 2, 987–995 CrossRef CAS.
- N. Du, G. P. Robertson, I. Pinnau and M. D. Guiver, Macromolecules, 2010, 43, 8580–8587 CrossRef CAS.
- N. Du, G. P. Robertson, J. Song, I. Pinnau, S. Thomas and M. D. Guiver, Macromolecules, 2008, 41, 9656–9662 CrossRef CAS.
- K. Mizrahi Rodriguez, W. N. Wu, T. Alebrahim, Y. Cao, B. D. Freeman, D. Harrigan, M. Jhalaria, A. Kratochvil, S. Kumar, W. H. Lee, Y. M. Lee, H. Lin, J. M. Richardson, Q. Song, B. Sundell, R. Thür, I. Vankelecom, A. Wang, L. Wang, C. Wiscount and Z. P. Smith, J. Membr. Sci., 2022, 659, 120746 CrossRef CAS.
- F. Y. Li, Y. Xiao, Y. K. Ong, T.-S. Chung, F. Y. Li, T. Chung, Y. K. Ong and Y. Xiao, Adv. Energy Mater., 2012, 2, 1456–1466 CrossRef CAS.
- P. Bernardo, F. Bazzarelli, F. Tasselli, G. Clarizia, C. R. Mason, L. Maynard-Atem, P. M. Budd, M. Lanč, K. Pilnáček, O. Vopička, K. Friess, D. Fritsch, Y. P. Yampolskii, V. Shantarovich and J. C. Jansen, Polymer, 2017, 113, 283–294 CrossRef CAS.
- C. Zhou, T. S. Chung, R. Wang and S. H. Goh, J. Appl. Polym. Sci., 2004, 92, 1758–1764 CrossRef CAS.
- A. Fuoco, C. Rizzuto, E. Tocci, M. Monteleone, E. Esposito, P. M. Budd, M. Carta, B. Comesaña-Gándara, N. B. McKeown and J. C. Jansen, J. Mater. Chem. A, 2019, 7, 20121–20126 RSC.
- J. G. Wijmans and R. W. Baker, J. Membr. Sci., 1995, 107, 1–21 CrossRef CAS.
- W. W. Brandt, J. Phys. Chem., 1959, 63, 1080–1085 CrossRef CAS.
- L. C. E. Struik, Rheol. Acta, 1966, 5, 303–311 CrossRef CAS.
- A. G. McDermott, G. S. Larsen, P. M. Budd, C. M. Colina and J. Runt, Macromolecules, 2011, 44, 14–16 CrossRef CAS.
- A. G. McDermott, P. M. Budd, N. B. McKeown, C. M. Colina and J. Runt, J. Mater. Chem. A, 2014, 2, 11742–11752 RSC.
- X. Ma, R. Swaidan, Y. Belmabkhout, Y. Zhu, E. Litwiller, M. Jouiad, I. Pinnau and Y. Han, Macromolecules, 2012, 45, 3841–3849 CrossRef CAS.
- C. L. Staiger, S. J. Pas, A. J. Hill and C. J. Cornelius, Chem. Mater., 2008, 20, 2606–2608 CrossRef CAS.
- M. Liu, X. Lu, M. D. Nothling, C. M. Doherty, L. Zu, J. N. Hart, P. A. Webley, J. Jin, Q. Fu and G. G. Qiao, ACS Mater. Lett., 2020, 2, 993–998 CrossRef CAS.
- F. Y. Li and T. S. Chung, Int. J. Hydrogen Energy, 2013, 38, 9786–9793 CrossRef CAS.
- C. C. Hu, Y. J. Fu, S. W. Hsiao, K. R. Lee and J. Y. Lai, J. Membr. Sci., 2007, 303, 29–36 CrossRef CAS.
- C. H. Lau, K. Konstas, C. M. Doherty, S. Kanehashi, B. Ozcelik, S. E. Kentish, A. J. Hill and M. R. Hill, Chem. Mater., 2015, 27, 4756–4762 CrossRef CAS.
- A. J. Hill, in Polymer Characterization Techniques and Their Application to Blends, ed. G. P. Simon, American Chemical Society, Washington D.C., 2003, pp. 401–435 Search PubMed.
- B. Comesaña-Gándara, J. Chen, C. G. Bezzu, M. Carta, I. Rose, M. C. Ferrari, E. Esposito, A. Fuoco, J. C. Jansen and N. B. McKeown, Energy Environ. Sci., 2019, 12, 2733–2740 RSC.
- F. Li, C. Zhang and Y. Weng, Macromol. Chem. Phys., 2019, 220, 1900047 CrossRef.
- Y. Wang, B. S. Ghanem, Y. Han and I. Pinnau, Polymer, 2020, 201, 122619 CrossRef CAS.
- X. Chen, Y. Fan, L. Wu, L. Zhang, D. Guan, C. Ma and N. Li, Nat. Commun., 2021, 12, 6140 CrossRef CAS PubMed.
- Z. Zhu, J. Zhu, J. Li and X. Ma, Macromolecules, 2020, 53, 1573–1584 CrossRef CAS.
- R. Malpass-Evans, I. Rose, A. Fuoco, P. Bernardo, G. Clarizia, N. B. McKeown, J. C. Jansen and M. Carta, Membranes, 2020, 10, 62 CrossRef CAS PubMed.
- S. L. Li, Z. Zhu, J. Li, Y. Hu and X. Ma, Polymer, 2020, 193, 122369 CrossRef CAS.
- F. Alghunaimi, B. Ghanem, N. Alaslai, M. Mukaddam and I. Pinnau, J. Membr. Sci., 2016, 520, 240–246 CrossRef CAS.
- M. Longo, M. P. De Santo, E. Esposito, A. Fuoco, M. Monteleone, L. Giorno, B. Comesaña-Gándara, J. Chen, C. G. Bezzu, M. Carta, I. Rose, N. B. McKeown and J. C. Jansen, Ind. Eng. Chem. Res., 2020, 59, 5381–5391 CrossRef CAS.
- R. Williams, L. A. Burt, E. Esposito, J. C. Jansen, E. Tocci, C. Rizzuto, M. Lanč, M. Carta and N. B. McKeown, J. Mater. Chem. A, 2018, 6, 5661–5667 RSC.
- X. Ma and I. Pinnau, Macromolecules, 2018, 51, 1069–1076 CrossRef CAS.
- B. Shrimant, S. V. Shaligram, U. K. Kharul and P. P. Wadgaonkar, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 16–24 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta01680c |
| This journal is © The Royal Society of Chemistry 2023 |