
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergizing plasmonic Au nanocages with 2D MoS2 nanosheets for significant enhancement in photocatalytic hydrogen evolution†‡
Rui
Peng§
a,
Xiaohan
Ma§
 ab,
Zachary D.
Hood
c,
Abdelaziz
Boulesbaa
d,
Alexander A.
Puretzky
a,
Jianhua
Tong
b and
Zili
Wu
*a
ab,
Zachary D.
Hood
c,
Abdelaziz
Boulesbaa
d,
Alexander A.
Puretzky
a,
Jianhua
Tong
b and
Zili
Wu
*a
aCenter for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA. E-mail: wuz1@ornl.gov
bDepartment of Materials Science and Engineering, Clemson University, Clemson, South Carolina 29634, USA
cApplied Materials Division, Argonne National Laboratory, 9700 S. Cass Avenue, Lemont, Illinois 60439, USA
dDepartment of Chemistry and Biochemistry, California State University Northridge, Northridge, California 91330, USA
First published on 17th July 2023
Abstract
Plasmonic enhancement of photocatalytic hydrogen evolution has been achieved under visible light illumination by integrating strongly plasmonic metal particles such as gold (Au) with semiconducting materials. To understand the effect of plasmon resonance on the photocatalytic hydrogen evolution reaction (HER), in this work, we study the hydrogen evolution reaction (HER) over Au nanocages with systematically tunable localized surface plasmon resonance (LSPR) properties dispersed on an Al2O3 support and covered with multiphasic 2D MoS2 nanosheets. It is interesting to observe that by tuning the LSPR wavelength of Au nanocages, an optimal enhancement in the photocatalytic HER can be obtained over the multiphasic 2D MoS2 nanosheets. In particular, when the LSPR wavelength of Au nanocages is close to the absorption edge of MoS2 nanosheets, a 40-fold increase is observed in the HER rate regarding bare MoS2. Time-resolved transient absorption spectroscopy was conducted to explicitly identify the mechanisms behind the Au/MoS2 system. The results suggest that near field enhancement (NFE) is the dominant LSPR process in this system and a detailed explanation of the working mechanism in this system is proposed. Governed by the NFE process, the energy of the surface plasma is transferred from Au nanocages to MoS2 nanosheets to promote electron–hole excitation in MoS2, and the efficiency reaches the maximum when the LSPR wavelength of Au nanocages matches the MoS2 light absorption edge, resulting in a significantly enhanced photocatalytic hydrogen yield compared to the bare MoS2 nanosheets and Au/MoS2 systems where the LSPR wavelengths of Au nanocages and MoS2 nanosheet absorption edge do not match. The learning from this work provides insights into the design of highly efficient photocatalysts based on plasmonic materials.
Introduction
Earth-abundant transition metal dichalcogenides (TMDs) represent a class of emerging materials for solar energy conversion.1–5 Among these TMDs, MoS2 has received the most intense interest owing to its balanced hydrogen bonding Gibbs free energy on its edge site.6,7 In our recent work,8 we have successfully activated the basal plane in a single-layer MoS2 nanosheet consisting of both 2H (semiconducting) and 1T-like (metallic) phases for photocatalytic hydrogen evolution from water splitting. These multiphasic MoS2 nanosheets possess properties that are favorable for light absorption and hydrogen evolution,9–13 yet their photocatalytic performance is to be further improved.Recently, plasmonic metals have been widely studied as effective materials to boost the overall activity of semiconductor-based photocatalysis, especially for solar hydrogen production.14–18 Photoinduced localized surface plasmon resonance (LSPR) can be activated in plasmonic metal species with irradiation using suitable light sources. The working mechanisms of LSPR-enhanced photocatalysis are quite complicated due to several possible concurrent ones including hot-electron transfer (HET), near-field enhancement (NFE) and plasmon resonant energy transfer (PRET).17–21 Besides, as metal materials, plasmonic metal species can also accept photogenerated electrons from a semiconductor through a classic electron transfer pathway, which makes the process even more complex.
For HET, disequilibrium arrangement of the electrons in the metal nanostructure and surface “hot electrons” can be generated by LSPR. As the metal species are coupled with a semiconductor, LSPR-induced hot electrons can be injected into the conduction band of the semiconductor that is in contact with the metal, which is a nonradiative process.17–21
NFE, however, is a radiative process. The near electromagnetic field generated by the plasmon dipole on a metal surface can significantly enhance the inter-band or other optical transitions in the nearby semiconductor (not necessarily in contact) as the generated field is typically orders of magnitude higher than the incident light.20–22 Therefore, the plasmonic metal is acting like a secondary light source and transferring the energy to the semiconductor. One feature of the radiative process is that the emitted photon will possess the same energy as the absorbed photon.22 Therefore, NFE requires a match of LSPR wavelength to the absorption edge of the semiconductor to maximize the process efficiency.
For PRET, the energy can be transferred from plasmonic metal species to semiconductors to generate excited electrons through nonradiative dipole–dipole coupling between the plasmonic dipole and electron–hole exciton in semiconductors.20,21 Like NFE, no direct contact between the metal and semiconductor is required for PRET, but the energy transfer efficiency highly depends on the spectrum overlap between the plasmonic metal and semiconductor and the special distance between the two parts.18,20–22 Additionally, it has a reverse and competing process called Förster resonant energy transfer (FRET), where energy will transfer from the semiconductor to the plasmonic metal. It becomes stronger when the spatial separation between the metal and semiconductor becomes larger.23
Although the explicit mechanism of charge transport between a plasmonic metal and semiconductor remains controversial, with the strong contribution from plasmonic metal nanostructures, photocatalytic efficiency over the semiconductor species can be drastically enhanced, indicating the positive influence of the plasmonic metals in the photocatalysis process.19,24,25
As a famous plasmonic metal, various plasmonic Au nanostructures, such as Au nanorods,26–29 nanoplates,29,30 nanocubes,29,31,32 nanoantennae,30,33 and nanoparticles16,34–36 have been reported to couple with MoS2 nanosheets to investigate the plasmonic effect on the electronic and/or optical behaviors of MoS2. While the shape effect of plasmonic metal nanostructures on LSPR behaviors has become a hot topic, the LSPR wavelength, another pivotal parameter of plasmonic metal nanostructures, has received much less attention. So far, only limited work has been conducted to reveal how the variation of their various LSPR absorption behaviors impacts the activity of the photocatalyst system.37–40 For example, it has been proposed that PRET may compete with HET when LSPR wavelength is close to the adsorption edge of the semiconductor in an Ag@Cu2O core–shell system and hence suppress the plasmonic enhancement.39 Yue et al.40 reported a Au–Ag/CdS catalyst system, which was composed of CdS as the semiconductor and Au–Ag nanoparticles (NPs) as the plasmonic nanostructure. The Au–Ag NPs were prepared by galvanic replacement. By adjusting the synthesis parameters, the LSPR wavelength of Au–Ag NPs can be finely tuned to a desired frequency. With the help of this feature, they found that excess absorption spectral overlap between the plasmonic metal and semiconductor can lead to a decrease in activity.40 However, a comprehensive understanding of the system was still lacking as they only considered the PRET process of LSPR.
To further understand the plasma-induced photocatalytic enhancement effect, we intentionally designed a series of photocatalysts composed of 2D MoS2 nanosheets loaded with Au nanocages. First reported by Xia's group in 2007,41 Au nanocages are in nature Au shells on the surface of Ag nanocubes. They are similar to the Au–Ag NPs mentioned above as both of them are Au/Ag heterostructures prepared by galvanic replacement and their LSPR wavelength can be finely tuned. Differently, considering the shape effect, Au nanocages can impose a stronger LSPR effect than the sphere-like Au–Ag NPs as Au nanocages have sharper features.22,42 The stronger LSPR effect around their edges and corners22 can lead to a greater impact on photocatalytic performance, making it easier to be observed and analyzed.
In this paper, we aimed to synergize the merits of multiphasic 2D MoS2 and Au nanocages with different LSPR wavelengths and investigate the impact of Au LSPR adsorption behavior on the photocatalytic performance of the system in a more comprehensive and profound way. In this study, the hydrogen evolution reaction (HER) was adopted as a model reaction. It was found that all the Au/MoS2 systems showed better HER performance than bare MoS2 after the loading of Au nanocages, but a particularly large rate enhancement of up to 40 fold (regarding bare MoS2) was observed when the LSPR wavelength of the Au nanocages matched the absorption edge of MoS2. The underlying mechanism was characterized with microscopy and optical and time-resolved transient absorption spectroscopy of the Au nanocages and MoS2 nanosheets.
Experimental
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) in 17.5 mL EG, and (4) 0.3000 g CF3COOAg (>99.99% metal basis, Aldrich) dissolved in 4.18 mL EG (sample concentration 282 mM). Each of these solutions was sufficiently stirred to dissolve the precursors. Next, 0.6 mL of (1) was added to the EG solution at 150 °C. After 4 minutes, 5 mL of (2) was added to the reaction. After another 2 minutes, 12.5 mL of (3) was added to the reaction. After another 2 minutes, 4 mL of (4) was added to the reaction. The reaction was cooled in an ice bath after ∼60 min. The Ag nanocubes were purified by dispersing the products of the reaction in acetone and collecting them by centrifugation at 4900 rpm for 8 min. The Ag nanocubes were washed three times in D.I. H2O (18.2 MΩ cm−1), collected by centrifugation at 14000 rpm for 15 min, and combined in 8 mL of D.I. H2O to create a stock solution of Ag nanocubes.
000 rpm for 20 min. The pellet containing Au nanocages was washed five times by re-dispersing in 25 mL D.I. H2O and centrifuging at 10500 rpm. The as prepared Au nanocages were labeled as Au-460, Au-680 and Au-750 based on their respective LSPR wavelengths.
000) in 17.5 mL EG, and (4) 0.3000 g CF3COOAg (>99.99% metal basis, Aldrich) dissolved in 4.18 mL EG (sample concentration 282 mM). Each of these solutions was sufficiently stirred to dissolve the precursors. Next, 0.6 mL of (1) was added to the EG solution at 150 °C. After 4 minutes, 5 mL of (2) was added to the reaction. After another 2 minutes, 12.5 mL of (3) was added to the reaction. After another 2 minutes, 4 mL of (4) was added to the reaction. The reaction was cooled in an ice bath after ∼60 min. The Ag nanocubes were purified by dispersing the products of the reaction in acetone and collecting them by centrifugation at 4900 rpm for 8 min. The Ag nanocubes were washed three times in D.I. H2O (18.2 MΩ cm−1), collected by centrifugation at 14000 rpm for 15 min, and combined in 8 mL of D.I. H2O to create a stock solution of Ag nanocubes.
000 rpm for 20 min. The pellet containing Au nanocages was washed five times by re-dispersing in 25 mL D.I. H2O and centrifuging at 10500 rpm. The as prepared Au nanocages were labeled as Au-460, Au-680 and Au-750 based on their respective LSPR wavelengths.
Characterization
Powder X-ray diffraction (XRD) patterns of the AMA samples were recorded using a PANalytical X'Pert Pro powder diffractometer equipped with Ni-filtered Cu Kα radiation. Scanning electron microscopy (SEM) images of exfoliated MoS2 nanosheets deposited on a Si wafer were collected with a Zeiss Merlin SEM at 3.0 kV. The transmission electron microscopy (TEM) images of exfoliated MoS2 nanosheets deposited on a lacy carbon grid were recorded on an aberration-corrected FEI Titan S 80-300 TEM/scanning transmission electron microscopy (STEM) operated at 300 kV with a Gatan charge coupled device (CCD) camera. STEM-X-ray energy dispersive spectroscopy (STEM-EDS) was performed on an FEI Talos F200X equipped with an EDS detector (Bruker) operated at 200 kV. Elemental maps were collected with a STEM spot size of 6. Raman and photoluminescence spectra of the exfoliated MoS2 aqueous sample were recorded on an Acton TriVista 555 spectrometer (Princeton Instruments) with laser excitation at 532 nm. Diffuse reflectance spectroscopy (DRS) analysis of the AMA samples was carried out using a Cary 5000 UV-visible spectrophotometer equipped with a praying mantis diffuse reflectance accessory. X-ray photoelectron spectroscopy (XPS) measurements were performed on each powder sample (MoS2–Al2O3) with a Thermo Scientific K-Alpha spectrometer. All spectra were collected using an Al Kα microfused monochromatized source (1486.6 eV) with a step size of 0.1 eV over 50 scans. For all spectra, the spot size was 400 μm and the operating pressure was under 3.0 × 10−7 mbar.Femtosecond transient absorption measurements of the Au/MoS2 deposited on a glass slide were carried out on a home-built pump-probe spectrometer (PPS). A full description of the PPS can be found elsewhere.46 Briefly, the PPS is based on a titanium sapphire (Ti:Sa) oscillator (Micra, Coherent) with its output seeded using a Ti:Sa Coherent Legend amplifier (USP-HE) operating at a 1 kHz repetition rate. The amplifier provides pulses centered at 800 nm, with ∼45 fs duration and 2.2 mJ energy per pulse. The output of the Legend amplifier was divided into two portions: 90% was attenuated to 0.5 mJ and focused on a BBO crystal to generate a 400 nm pump pulse. The second portion (10%) was used to generate a white light continuum (WLC) probe in a 2 mm thick sapphire window. The WLC, which covers the spectral range from 450 nm to 900 nm, was collimated after generation and focused onto the sample using high reflective parabolic mirrors to minimize temporal chirp. After this, the transmitted probe was focused onto a 100-micron core fiber coupled with a spectrometer linear CCD array (USB2000ES, Ocean Optics). The pump passes through a controllable stage-delay and was chopped at 500 Hz frequency to allow the measurement of absorbance change in the transmitted probe between each two successive laser shots. At the sample, the pump and probe spot sizes were 100 and 50 μm, and the pump energy was ∼4 μJ cm−2.
Results and discussion
Structure and morphology
In order to investigate the crystallographic structures of the AMA samples, powder XRD was employed. Fig. 1A shows the XRD patterns of the composite AMA samples along with a pure Al2O3 support material. The XRD patterns of the Au and MoS2 incorporated samples exhibit a pattern similar to that of Al2O3, i.e., intrinsic diffraction peaks of Al2O3 can be observed from all the samples. A few small diffraction peaks falling at 18.1° can be assigned to the presence of Al2O3·3H2O (PDF: 00-001-0259). No peak of Au or MoS2 can be observed due to their low loading on Al2O3. | ||
| Fig. 1 (A) XRD patterns of Al2O3 supported Au/MoS2 samples. (B) Raman spectra of the as-exfoliated MoS2 nanosheets suspended in water (black) and loaded on Al2O3 (red). (C) UV-vis absorption spectra of the as-synthesized three Au nanocages. (D) UV-vis DRS of Au-460, Au-680 and MoS2 nanosheets loaded on Al2O3. | ||
Similar to our previous work,8 Raman spectroscopy is applied to identify the 2D and multiphasic nature of the as-synthesized MoS2 nanosheets. The Raman spectrum of the freshly prepared MoS2 aqueous suspension is shown in Fig. 1B. The typical in-plane mode (E12g) and out-of-plane mode (A1g) peaks of the MoS2 sample can be observed at 388 and 409 cm−1. Compared with those (385 and 410 cm−1) of bulk MoS2, the shift of the two peaks indicates the effective exfoliation of the Li intercalated MoS2 layers and the presence of single-layer MoS2.47 In addition to the E12g and A1g vibration modes, the exfoliated MoS2 nanosheets also present two unique peaks at around 285 (E1g) and 333 cm−1 (J3), both of which originate from the metallic 1T-like phase of MoS2 formed during the Li intercalation.8,48 Specifically, the Raman spectrum of MoS2/Al2O3 is also inspected to see if there is any structure change after the loading on Al2O3. Although the E1g peak becomes less obvious, the existence of J3 still reveals the existence of a 1T-like phase. Meanwhile, although there is a slight peak shift for E12g (from 388 to 386 cm−1) and A1g (from 409 to 407 cm−1), probably caused by the dielectric effect of Al2O3, the separation between these two peaks is almost unchanged after the loading process, suggesting that the MoS2 nanosheets are well dispersed and do not restack on Al2O3.
The AFM image and the layer thickness profiles of the three representative nanosheets displayed in Fig. 2 demonstrate that the majority of the MoS2 nanosheets are about 1 nm thick. This also confirms the successful preparation of single-layer MoS2. Combining the SEM images of these exfoliated nanosheets (shown in Fig. S1 in the ESI‡) and from our previous work on the same MoS2 system,8 it can be concluded that monolayered multiphasic MoS2 nanosheets have been successfully obtained by following our preparation procedures.
 | ||
| Fig. 2 (A) AFM image of the as-exfoliated single-layer and (B) the height profiles of three selected regions indicated in the figure. | ||
The LSPR of the Au nanocages is verified before dispersing onto the Al2O3 support. The Au nanocages are prepared via galvanic replacement between Ag nanocubes and HAuCl4 in an aqueous solution set to 90 °C, similar to our previous reports.44 To systematically investigate the effect of the LSPR on the performance of photocatalytic activity over MoS2 samples for the photocatalytic HER, the LSPR of the Au nanocages was tuned to three different wavelengths: 460, 680, and 750 nm (Fig. 1C) that are off, on, and off the light absorption edge of MoS2. Fig. 1D displays the UV-vis diffuse reflectance spectra (DRS) of the Au nanocages loaded on Al2O3. Only a slight peak shift is observed that does not affect the positions relative to each other. A good match between the absorption edge of MoS2 and the LSPR wavelength of Au-680 can also be observed from the spectra. The as-synthesized Au nanocages have an average size of 48 ± 3.5 nm as measured by transmission electron microscopy (TEM) (Fig. S2‡). When Au nanocages are titrated with different amounts of HAuCl4, their wall thickness changes and pushes the LSPR towards the near-infrared spectral region.
To investigate the heterojunction between gold nanocages and MoS2 nanosheets, we prepared a representative sample for high-angle annular dark-field (HAADF) scanning transmission electron microscopy (STEM) and bright-field STEM (BF-STEM) imaging. Fig. 3 shows the BF- and HAADF-STEM images of the AMA-680 sample as a representative. The BF-STEM images clearly reveal the lattice for the gold nanocage and a honeycomb structure for MoS2. We also investigated the elemental distribution of Au, Mo, S, Ag and Al in the AMA-680 sample by energy-dispersive X-ray spectroscopy (EDS) based on STEM. The STEM-EDS elemental mappings are shown in Fig. S3,‡ where the Au nanocage is clearly seen in contact with MoS2, as expected, on the Al2O3 support. However, it should be noticed that due to the tremendous morphology and size differences between the Au nanocages and MoS2 nanosheets, some of the MoS2 nanosheets are not in contact with Au, which can also be observed from Fig. S3.‡
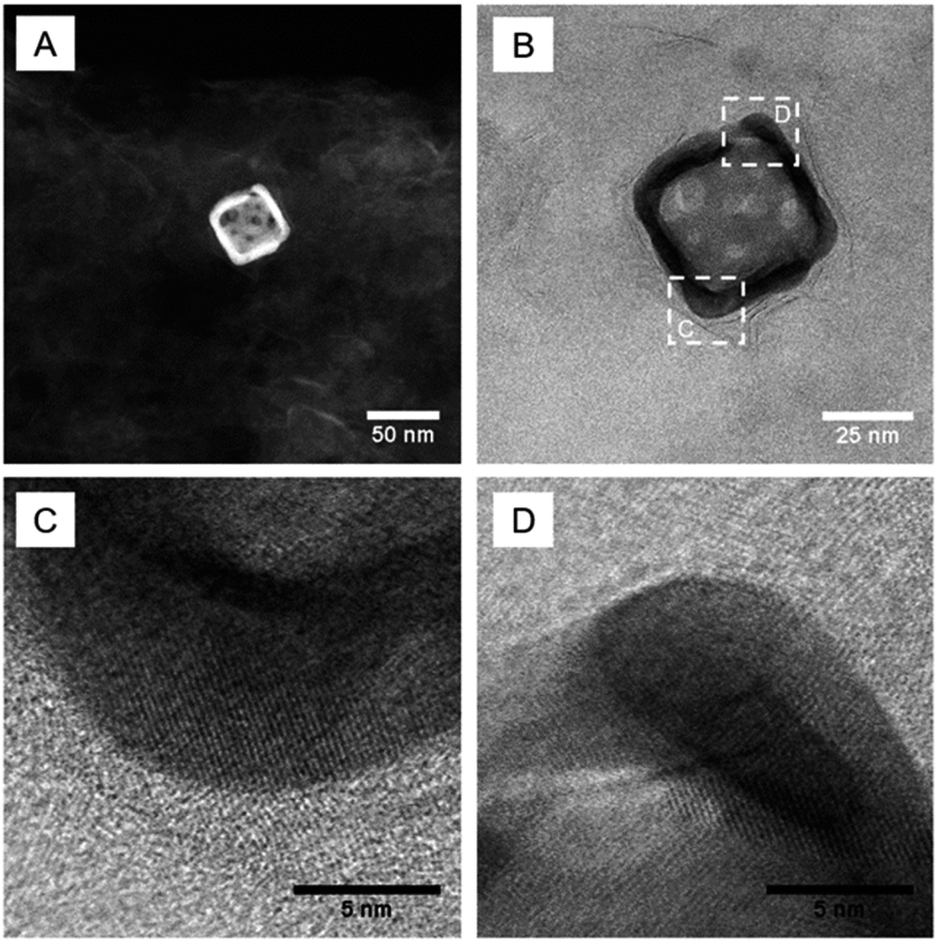 | ||
| Fig. 3 Representative (A) HAADF- and (B–D) BF-STEM images of the AMA-680 sample. | ||
The phase properties of the chemically exfoliated MoS2 are evaluated by X-ray photoelectron spectroscopy (XPS). It must be noted that the thermodynamically stable form of MoS2 is the 2H phase, where each Mo is prismatically coordinated with six S atoms. The 1T-like phase, on the other hand, is composed of octahedrally coordinated Mo with six S atoms. The structure of MoS2 has been previously reported to undergo a structural transformation from the 1T-like into the 2H phase by thermal treatment between room temperature and 300 °C.8 XPS is then employed to further investigate the multiphasic properties of 2D MoS2 in different AMA samples (Fig. 4). The Mo 3d spectra (Fig. 4A) consist of peaks at around 229 and 232 eV, corresponding to Mo4+ 3d5/2 and Mo4+ 3d3/2, respectively. Deconvolution of the Mo 3d spectra reveals a shift in the spectra to higher binding energies (by ∼0.9 eV) when the MoS2 nanosheets are annealed at increased temperatures (e.g., 60 and 120 °C). This shift to higher binding energies can be attributed to the phase transformation from the metallic 1T-like phase into the 2H phase (Table S1‡). The S 2p spectra shown in Fig. 4B display a similar shift to higher binding energies when multiphasic MoS2 is annealed at increased temperatures. The S 2p spectra display doublet peaks, S 2p1/2 and S 2p3/2, appearing at ∼163 and ∼161.9 eV, respectively. Deconvolution of these peaks clearly illustrates the phase shift from the metallic 1T-like phase to the 2H phase when the samples are annealed at 60 and 120 °C. It is worth noting that no peaks are observed from 235 to 240 eV, suggesting that oxidation of Mo4+ to Mo6+ is minimal. The oxidation state and thermal stability of the Au nanocages are also investigated by XPS and TEM imaging after the thermal annealing at 60 and 120 °C. XPS measurements are also performed on the Au 4f region for the different gold nanocages. As shown in Fig. 4C, the Au 4f region shows clear doublet peaks at 84.0 and 87.7 eV, corresponding to Au0 4f5/2 and Au0 4f7/2, respectively. TEM images of the Au nanocages before and after heat treatment at 60 and 120 °C show that the gold nanocages retain their morphology (Fig. S4‡).
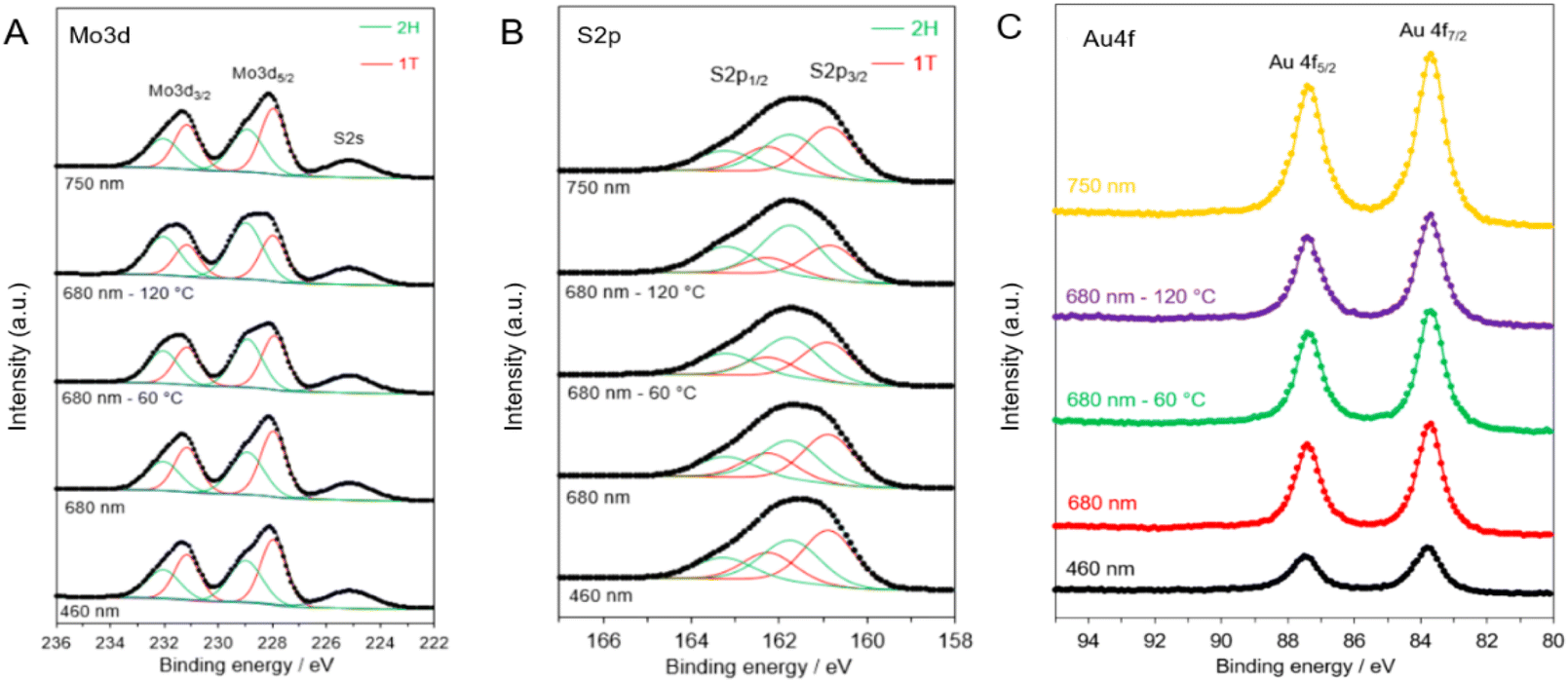 | ||
| Fig. 4 XPS spectra of (A) Mo 3d, (B) S 2p, and (C) Au 4f core level regions of the various AMA samples. | ||
UV-vis DRS is conducted to investigate the optical properties of the Au/MoS2–Al2O3 samples. Fig. S5A‡ displays the spectra of the AMA samples assembled with various Au nanocages. All the spectra in Fig. S5A‡ are similar to each other, a result of the strong light absorption by MoS2. Several absorption bands in the visible light region can be observed in these samples. Two strong absorption peaks that belonged to typical MoS2 are observed in all the samples. The one at ∼420 nm corresponds to convoluted C and D excitonic and the one at ∼620 nm corresponds to A and B excitonic peaks arising from the K point of the Brillouin zone.9 Due to the high intensity of these two peaks, the absorption of Au-460 and Au-680 is eclipsed. However, Au-750 presents an appreciable and broad absorption band at ∼750 nm that can be clearly monitored. We also performed UV-vis DRS on the annealed AMA-680 samples and their spectra are shown in Fig. S5B.‡ It can be seen that all the samples display similar absorption curves indicating the similar chemical composition of the samples after heat treatment.
Photocatalytic performance
In order to investigate the LSPR effect on the photocatalytic HER performance, the activity of the AMA samples is studied under identical experimental conditions. Fig. 5 shows the kinetic plot of the AMA samples in terms of the hydrogen evolution rate normalized to MoS2 weight as a function of reaction time. It can be seen that compared with the pure MoS2 sample, all Au-containing samples exhibit over an order of magnitude enhancement in the hydrogen evolution rate. This phenomenon is thought to be the result of the LSPR effect from Au nanocages. These surface plasmas arising from Au nanocages have been proven to be beneficial factors during the photocatalytic hydrogen evolution reaction.49,50 Interestingly, it can be seen that the AMA-680 sample presents a strikingly higher photocatalytic hydrogen evolution rate compared with the other two AMA samples. As shown in Fig. 5A and C, a ∼40-fold increase in the hydrogen evolution rate is obtained on AMA-680 over the bare MoS2 nanosheets while about a 15-fold increase is found on the other two AMA samples. The superior photocatalytic activity of the AMA-680 sample may be rationalized by the more efficient energy and/or electron transfer between the Au-680 nanocages and the MoS2 nanosheets. A more detailed discussion will be presented in the following part of this paper regarding the mechanism. Fig. 5D shows the cycling stability of AMA-680 in the photocatalytic HER, where above 90% of the activity can be preserved after 5 cycles. This indicates that the Au–MoS2 composite is highly stable under the testing conditions.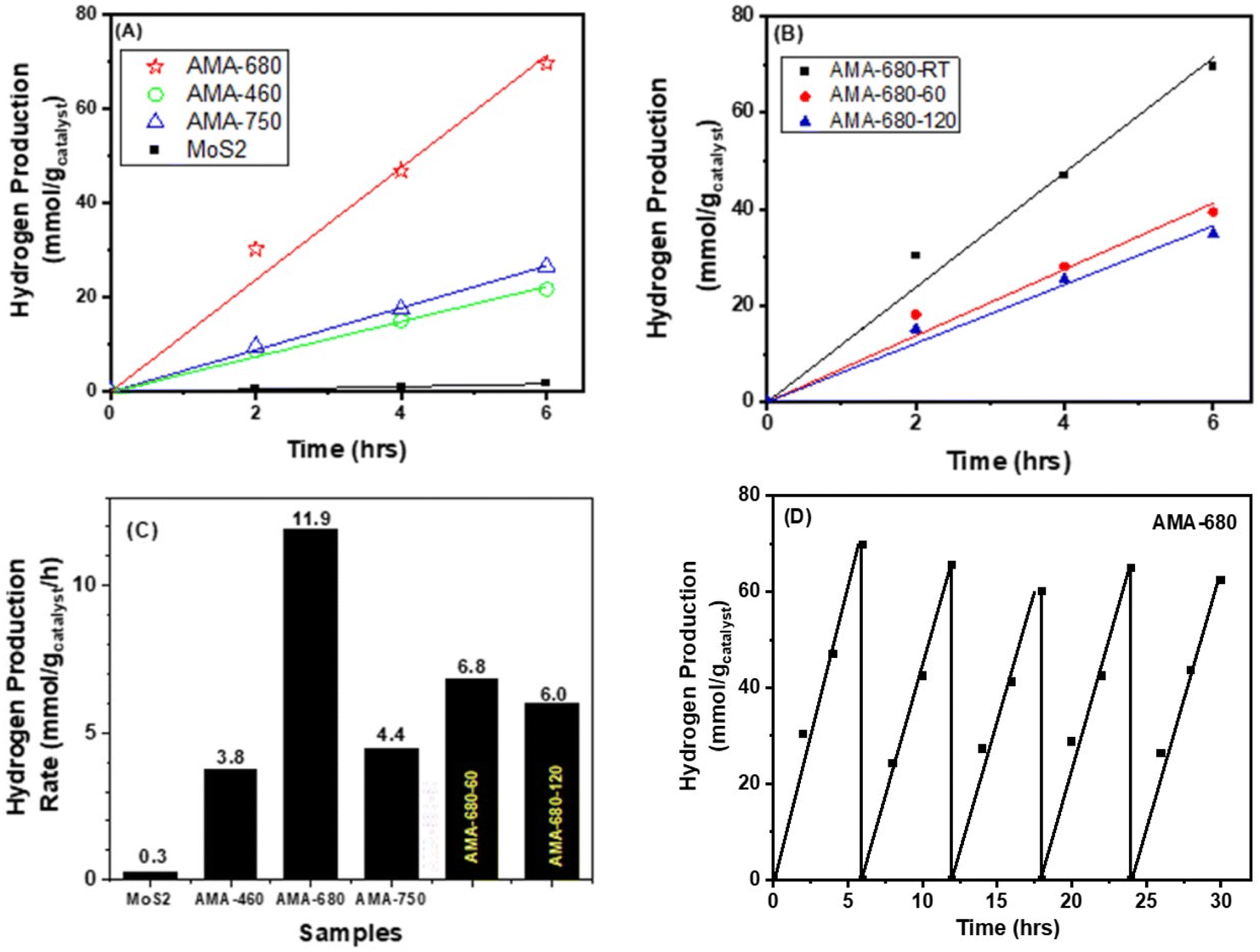 | ||
| Fig. 5 Photocatalytic hydrogen evolution as a function of time over (A) AMA samples with Au nanocages with varying LSPR wavelengths and (B) AMA-680 samples treated at various temperatures. (C) Comparison of the photocatalytic hydrogen production rate over different samples. (D) Cycle stability test result of AMA-680 in photocatalytic hydrogen evolution. | ||
To reveal the role played by the multiphases feature of MoS2 in this reaction, AMA-680 is heated to 60 and 120 °C to vary the ratio of the 1T-like and 2H phases and tested for HER performance. As shown in Fig. 5B, the heat treatment, which leads to a decreased proportion of the 1T-like phase as shown in Table S1,‡ results in a decrease in the HER rate from 11.9 to 6.8 and 6.0 mmol H2 gcatalyst−1 h−1 for AMA-680, -60, and -120, respectively (Fig. 5C). The observation suggests an important role of the 1T-like phase in the HER, consistent with our previous result.8 We previously showed that the photogenerated electrons from the 2H phase of MoS2 had the tendency to diffuse to the 1T-like phase, where the protons were more likely to be reduced into hydrogen. The 1T-like metallic phase of MoS2 is known as a good “electron sink” and exhibits superior electron affinity to the 2H phase MoS2. Hence, the electrons generated, no matter the origin, are inclined to migrate to the 1T-like MoS2 to conduct the HER. But the HER rate of AMA-680-60 and AMA-680-120 is still ∼50% higher than that of the AMA-460 and AMA-750 samples, again pointing to the key role of the match between Au nanocage LSPR wavelength and the MoS2 absorption edge.
Mechanism discussion
In order to unveil the origin of the excited electrons, time-resolved transient absorption spectroscopy (TAS) is carried out to track the destiny of the electrons in the AMA system. In this work, a 400 nm, 45 fs laser pulse is used as the pump to excite the electrons in AMA samples and the time-resolved differential transmission spectra were recorded to track the decay dynamics of the excited electrons. The transient absorption spectra recorded 0.4 ps after excitation are shown in Fig. 6A. They contain a broad feature with a positive sign (pointing up). Within this band, there are two depletion signals (pointing down) at around 660 and 610 nm originating from excitons A and B of MoS2, respectively.51,52 The broad positive band contains two peaks at around 500 (strong) and 700 nm (weak). Considering that the UV-vis spectra of the three samples also showed strong absorption around these two wavelengths, we monitored the three samples at 495 and 730 nm up to a 1 ns time-delay after the probe excitation and plotted their decay dynamics as functions of time in Fig. 6B and C in order to get better signals as well as facilely inspect the electron transfer mechanism under the excitation on and off the LSPR wavelength of Au nanocages. According to previous reports,53,54 excitons in 2D-TMDs decay through three distinct mechanisms: a fast process ascribed to thermalization taking place within the first few picoseconds after excitation, followed by relaxation during the next few picoseconds, and finally a much slower process that can take hundreds of picoseconds or longer assigned to defect-assisted recombination. Based on this, we fitted the dynamics shown in Fig. 6B and C to the following tri-exponential decay function:t1, t2, and t3 (ps) are the time-constants assigned to thermalization, relaxation and defect-assisted recombination processes, respectively, and A1,A2 and A3 (milli optical density, mOD) are their corresponding amplitudes. All three time constants can be affected if there is any electron/energy transfer taking place at the MoS2/Au nanocage interface. However, in this material system, only t1 is the direct indicator of the LSPR effect since the plasmonic energy and/or hot electrons are mostly available during the first few hundreds of femtoseconds after the excitation, which is within the scope of t1. As t2 and t3 represent slower processes, they are good indicators of the classic electron transfer from MoS2 to Au nanocages as this process is not efficient enough to affect t1. The fitted time constants are listed in Table 1 while the corresponding amplitudes are listed in Table S2.‡
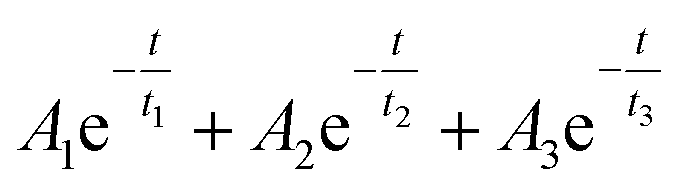
 | ||
| Fig. 6 (A) Transient absorption spectra of the AMA samples and bare MoS2 (recorded at 0.4 ps after excitation at 400 nm) and transient absorption dynamics for AMA samples monitored at (B) 495 nm and (C) 730 nm following pump excitation at 400 nm. Symbols show experimental data, and solid plots show fits. The time-delay axis is shown on a linear scale up to 5 ps and logarithmic scale thereafter. | ||
| Time constant (ps) | Au-680 | MoS2 | AMA-460 | AMA-680 | AMA-750 |
|---|---|---|---|---|---|
| t 1-495 | 2.21 | 0.43 | 0.88 | 0.48 | 0.33 |
| t 1-730 | 5.36 | 0.17 | 0.89 | 1.40 | 0.18 |
| t 2-495 | 3.48 | 18.58 | 30.53 | 22.85 | 9.70 |
| t 2-730 | 5.37 | 8.17 | 30.23 | 26.40 | 0.51 |
| t 3-495 | 7044 | 1011 | 2940 | 1010 | 516 |
| t 3-730 | 2267 | 788 | 2915 | 960 | 99 |
A detailed discussion of the fitting results is given in the ESI.‡ Briefly, based on the fitted time constants, the AMA-460 system is a classic heterojunction photocatalyst system with Au-460 being the metal cocatalyst (as shown in Fig. S6A‡). However, as Au is usually not considered a great cocatalyst for the HER,55 it cannot enhance the activity significantly. Meanwhile, the LSPR effect in AMA-750 just becomes strong enough to marginally overcome the classic electron transfer process (as shown in Fig. S6C‡). Although it converts the system to a plasmonic photocatalyst system, it does not show much enhancement in activity as well due to the overall weak LSPR effect. Unlike AMA-750, AMA-680 is thought to be a LSPR-dominant system (as shown in Fig. S6B‡). Unlike other samples, the decay behavior (depicted using the three time constants in Table 1) of AMA-680 is more analogous to that of Au nanocages, not MoS2, which suggests that it is strongly affected by LSPR. It also exhibits much longer t1 (t1-730, 1.4 ns) around its LSPR wavelength than AMA-750 (t1-730, 0.18 ns) and AMA-460 (t1-730, 0.89 ns). As t1 is the indicator of the LSPR effect, it further confirms that AMA-680 has a stronger LSPR effect than AMA-750 and AMA-460, which can account for its highest photocatalytic activity.
As stated in the introduction, there are potentially several LSPR processes working simultaneously in the plasmonic photocatalyst system, namely HET, NFE and PRET. If we assume that the classic electron transfer efficiency is identical for all three AMA systems (as they are prepared in the same way, the contact between Au and MoS2 should be similar), the LSPR effect in the three systems follows the order AMA-680 > AMA-750 > AMA-460 (expressed in Fig. S6‡ by the weight of arrows) according to the transient absorption results. Importantly, it should be noticed that the approximations of their LSPR wavelength to the absorption edge of MoS2 follow exactly the same order. Based on the introduction part, NFE is the only process whose efficiency relies on the match of LSPR wavelength with the absorption edge of the semiconductor. Therefore, the results strongly show that NFE is the dominant LSPR process in this system. Meanwhile, the HET process was significantly suppressed probably due to the insufficient direct contact between Au nanocages and MoS2 stated above. For the PRET process, although it does not require a direct contact, the bare MoS2 nanosheets around Au nanocages impose a strong FRET effect, nullifying the PRET effect. NFE, however, has no competing process and can affect an area that is 50 nm away22 hence dominating the LSPR process in the AMA-680 system.
Based on the above discussion, a possible mechanism is illustrated for the LSPR promoted photocatalytic hydrogen evolution reaction in the Au/MoS2 composite as shown in Fig. 7. For simplicity, only AMA-680 is presented and the 1T-like MoS2 phase is not shown. Under the illumination of visible light, Au-680 and MoS2 nanosheets will be excited simultaneously. Some electrons located at the conduction band (CB) of MoS2 will migrate to the Fermi level (Ef) of Au-680 by overcoming the Schottky barrier at the interface. Meanwhile, the surface plasma on Au-680 will promote the generation of electron–hole pairs in MoS2 through the NFE process. As the LSPR wavelength of Au-680 significantly matches the absorption edge of MoS2 nanosheets, the ultraefficient NFE process completely dominates AMA-680 and hence, significantly enhances the photocatalytic performance. In our previous study on single layer multiphasic MoS2, we showed that the 1T-like phase of MoS2 is more favorable for electron acceptation and hydrogen evolution. Hence, with the presence of 1T-like MoS2, all the excited electrons in this catalyst system are more inclined and readily transfer to the 1T-like MoS2 region. Through such a synergy between the LSPR effect and the favorable phase structure of MoS2, photon energy can be efficiently utilized for the hydrogen evolution reaction.
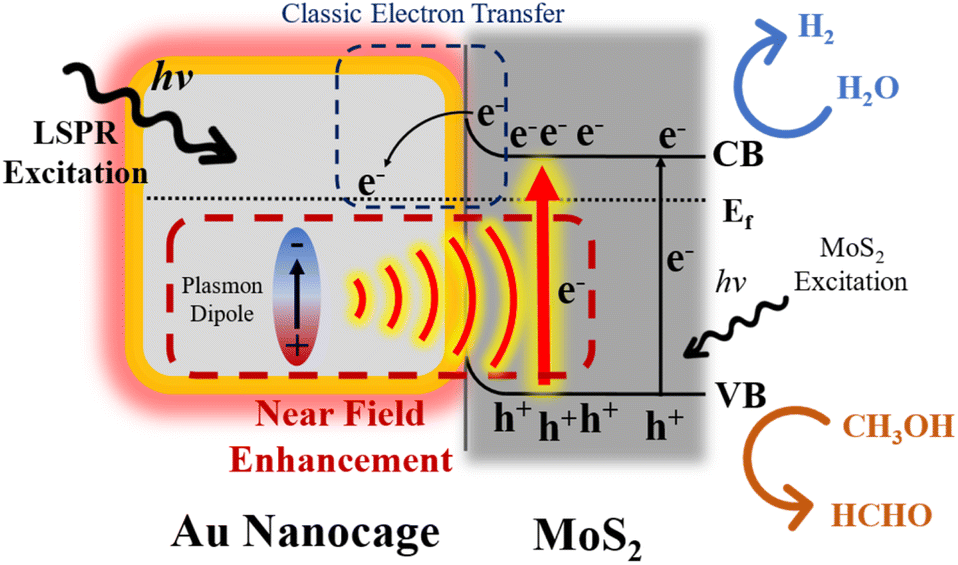 | ||
| Fig. 7 Scheme of the proposed working mechanisms of AMA-680. CB: conduction band; VB: valence band. | ||
Conclusions
We demonstrate that visible-light-driven photocatalytic hydrogen generation can be greatly enhanced by creating heterojunctions between plasmonic Au nanocages and multiphasic single-layer MoS2 nanosheets. We investigate Au nanocages with various LSPR wavelengths in order to understand the effect of the LSPR wavelength in relation to the optical absorption of MoS2 on the photocatalytic hydrogen evolution reaction. The introduction of Au nanocages generally enhances the hydrogen evolution rate of the 2D MoS2 nanosheets. Interestingly, the sample loaded with Au nanocages with the LSPR wavelength at 680 nm displays an impressive 40-fold increase in the HER rate compared to the bare MoS2 nanosheets. The results from time-resolved TAS suggest that the key to this enhancement is related to the ultraefficient energy transfer from Au nanocages to MoS2 through the near field enhancement process when the LSPR wavelength of Au nanocages matches the light absorption edge of MoS2. This finding demonstrates a potential general strategy for designing plasmonic photocatalysts that have high activity under visible light irradiation for solar-to-fuel conversion.Author contributions
R. Peng: investigation, data curation, formal analysis, methodology, writing – original draft; X. Ma: investigation, data curation, formal analysis, writing – review & editing; Z. Hood: data curation, methodology, writing – review and editing; A. Boulesbaa and A. Puretzky: data curation, formal analysis, writing – review & editing; J. Tong: writing – review & editing; Z. Wu: conceptualization, funding acquisition, project administration, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Center for Nanophase Materials Sciences (CNMS), which is a US Department of Energy, Office of Science User Facility at Oak Ridge National laboratory. Z. D. H. was supported by Laboratory Directed Research and Development (LDRD) funding from Argonne National Laboratory, provided by the Director, Office of Science, of the U.S. Department of Energy under Contract No. DE-AC02-06CH11357.Notes and references
- S. Das, M. Kim, J. W. Lee and W. Choi, Crit. Rev. Solid State Mater. Sci., 2014, 39, 231–252 CrossRef CAS.
- M.-R. Gao, Y.-F. Xu, J. Jiang and S.-H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC.
- C. N. R. Rao, H. S. S. Ramakrishna Matte and U. Maitra, Angew. Chem., Int. Ed., 2013, 52, 13162–13185 CrossRef CAS PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- T. Su, Q. Shao, Z. Qin, Z. Guo and Z. Wu, ACS Catal., 2018, 8, 2253–2276 CrossRef CAS.
- P. Afanasiev, C. R. Chim., 2008, 11, 159–182 CrossRef CAS.
- E. Benavente, M. A. Santa Ana, F. Mendizábal and G. González, Coord. Chem. Rev., 2002, 224, 87–109 CrossRef CAS.
- R. Peng, L. Liang, Z. D. Hood, A. Boulesbaa, A. Puretzky, A. V. Ievlev, J. Come, O. S. Ovchinnikova, H. Wang, C. Ma, M. Chi, B. G. Sumpter and Z. Wu, ACS Catal., 2016, 6, 6723–6729 CrossRef CAS.
- G. Eda, H. Yamaguchi, D. Voiry, T. Fujita, M. Chen and M. Chhowalla, Nano Lett., 2011, 11, 5111–5116 CrossRef CAS.
- G. Eda, T. Fujita, H. Yamaguchi, D. Voiry, M. Chen and M. Chhowalla, ACS Nano, 2012, 6, 7311–7317 CrossRef CAS.
- M. Kan, J. Y. Wang, X. W. Li, S. H. Zhang, Y. W. Li, Y. Kawazoe, Q. Sun and P. Jena, J. Phys. Chem. C, 2014, 118, 1515–1522 CrossRef CAS.
- Y.-C. Lin, D. O. Dumcenco, Y.-S. Huang and K. Suenaga, Nat. Nanotechnol., 2014, 9, 391–396 CrossRef CAS PubMed.
- L. Cai, J. He, Q. Liu, T. Yao, L. Chen, W. Yan, F. Hu, Y. Jiang, Y. Zhao, T. Hu, Z. Sun and S. Wei, J. Am. Chem. Soc., 2015, 137, 2622–2627 CrossRef CAS PubMed.
- Y. Tian and T. Tatsuma, J. Am. Chem. Soc., 2005, 127, 7632–7637 CrossRef CAS PubMed.
- K. Awazu, M. Fujimaki, C. Rockstuhl, J. Tominaga, H. Murakami, Y. Ohki, N. Yoshida and T. Watanabe, J. Am. Chem. Soc., 2008, 130, 1676–1680 CrossRef CAS.
- J. L. Yang, Y. L. He, H. Ren, H. L. Zhong, J. S. Lin, W. M. Yang, M. D. Li, Z. L. Yang, H. Zhang, Z. Q. Tian and J. F. Li, ACS Catal., 2021, 11, 5047–5053 CrossRef CAS.
- H. Tada, Nanoscale Adv., 2019, 1, 4238–4245 RSC.
- D. V. Dao, T. T. D. Nguyen, P. Uthirakumar, Y. H. Cho, G. C. Kim, J. K. Yang, D. T. Tran, T. D. Le, H. Choi, H. Y. Kim, Y. T. Yu and I. H. Lee, Appl. Catal., B, 2021, 286, 119947 CrossRef CAS.
- A. Kumar, P. Choudhary, A. Kumar, P. H. C. Camargo and V. Krishnan, Small, 2022, 18, e2101638 CrossRef PubMed.
- X. C. Ma, Y. Dai, L. Yu and B. B. Huang, Light: Sci. Appl., 2016, 5, e16017 CrossRef CAS PubMed.
- Z. Ye, Z. Xu, W. Yue, X. Liu, L. Wang and J. Zhang, Phys. Chem. Chem. Phys., 2023, 2706–2716, 10.1039/d2cp04582f.
- W. R. Erwin, H. F. Zarick, E. M. Talbert and R. Bardhan, Energy Environ. Sci., 2016, 9, 1577–1601 RSC.
- J. T. Li, S. K. Cushing, F. K. Meng, T. R. Senty, A. D. Bristow and N. Q. Wu, Nat. Photonics, 2015, 9, 601–607 CrossRef CAS.
- C. L. Wang and D. Astruc, Chem. Soc. Rev., 2014, 43, 7188–7216 RSC.
- N. L. Reddy, V. N. Rao, M. Vijayakumar, R. Santhosh, S. Anandan, M. Karthik, M. V. Shankar, K. R. Reddy, N. P. Shetti, M. N. Nadagouda and T. M. Aminabhavi, Int. J. Hydrogen Energy, 2019, 44, 10453–10472 CrossRef CAS.
- Y. Shi, J. Wang, C. Wang, T.-T. Zhai, W.-J. Bao, J.-J. Xu, X.-H. Xia and H.-Y. Chen, J. Am. Chem. Soc., 2015, 137, 7365–7370 CrossRef CAS.
- I. Irfan, S. Golovynskyi, O. A. Yeshchenko, M. Bosi, T. Zhou, B. Xue, B. K. Li, J. L. Qu and L. Seravalli, Physica E, 2022, 140, 115213 CrossRef CAS.
- R. Bar-Ziv, P. Ranjan, A. Lavie, A. Jain, S. Garai, A. Bar Hen, R. Popoyitz-Biro, R. Tenne, R. Arenal, A. Ramasubramaniam, L. Lajaunie and M. Bar-Sadan, ACS Appl. Energy Mater., 2019, 2, 6043–6050 CrossRef CAS.
- M. Garai, Z. Y. Zhu, J. Shi, S. S. Li and Q. H. Xu, J. Chem. Phys., 2021, 155, 234201 CrossRef CAS PubMed.
- S. Najmaei, A. Mlayah, A. Arbouet, C. Girard, J. Léotin and J. Lou, ACS Nano, 2014, 8, 12682–12689 CrossRef CAS PubMed.
- J. Li, Z. Chen, H. Yang, Z. Yi, X. Chen, W. Yao, T. Duan, P. Wu, G. Li and Y. Yi, Nanomaterials, 2020, 10, 257 CrossRef CAS PubMed.
- S. H. Wang, Y. Zhang, Y. Y. Zheng, Y. B. Xu, G. D. Yang, S. X. Zhong, Y. L. Zhao and S. Bai, Small, 2023, 19, 2204774 CrossRef CAS.
- Y. Yu, Z. Ji, S. Zu, B. Du, Y. Kang, Z. Li, Z. Zhou, K. Shi and Z. Fang, Adv. Funct. Mater., 2016, 26, 6394–6401 CrossRef CAS.
- X. Yang, W. Liu, M. Xiong, Y. Zhang, T. Liang, J. Yang, M. Xu, J. Ye and H. Chen, J. Mater. Chem. A, 2014, 2, 14798–14806 RSC.
- Y. Kang, S. Najmaei, Z. Liu, Y. Bao, Y. Wang, X. Zhu, N. J. Halas, P. Nordlander, P. M. Ajayan, J. Lou and Z. Fang, Adv. Mater., 2014, 26, 6467–6471 CrossRef CAS PubMed.
- A. Rani, A. S. Patel, A. Chakraborti, K. Singh and P. Sharma, J. Mater. Sci.: Mater. Electron., 2021, 32, 6168–6184 CrossRef CAS.
- E. Thimsen, F. Le Formal, M. Grätzel and S. C. Warren, Nano Lett., 2011, 11, 35–43 CrossRef CAS.
- G. Collins, A. Lonergan, D. McNulty, C. Glynn, D. Buckley, C. Y. Hu and C. O'Dwyer, Adv. Mater. Interfaces, 2020, 7, 1901805 CrossRef CAS.
- J. T. Li, S. K. Cushing, J. Bright, F. K. Meng, T. R. Senty, P. Zheng, A. D. Bristow and N. Q. Wu, ACS Catal., 2013, 3, 47–51 CrossRef CAS.
- X. Yue, J. Hou, Y. Zhang, P. Wu, Y. Guo, S. Peng, Z. Liu and H. Jiang, Dalton Trans., 2020, 49, 7467–7473 RSC.
- S. E. Skrabalak, L. Au, X. D. Li and Y. N. Xia, Nat. Protoc., 2007, 2, 2182–2190 CrossRef CAS PubMed.
- H. M. Zhang, T. Zhu and M. Li, J. Phys. Chem. Lett., 2023, 14, 3853–3860 CrossRef CAS.
- Q. Zhang, W. Li, L. P. Wen, J. Chen and Y. Xia, Chem. –Eur. J., 2010, 16, 10234–10239 CrossRef CAS.
- S. E. Skrabalak, L. Au, X. Li and Y. Xia, Nat. Protoc., 2007, 2, 2182 CrossRef CAS.
- S. E. Skrabalak, J. Chen, Y. Sun, X. Lu, L. Au, C. M. Cobley and Y. Xia, Acc. Chem. Res., 2008, 41, 1587–1595 CrossRef CAS.
- A. Boulesbaa, B. Huang, K. Wang, M.-W. Lin, M. Mahjouri-Samani, C. Rouleau, K. Xiao, M. Yoon, B. Sumpter, A. Puretzky and D. Geohegan, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 115443 CrossRef.
- C. Lee, H. Yan, L. E. Brus, T. F. Heinz, J. Hone and S. Ryu, ACS Nano, 2010, 4, 2695–2700 CrossRef CAS PubMed.
- U. Gupta, B. S. Naidu, U. Maitra, A. Singh, S. N. Shirodkar, U. V. Waghmare and C. N. R. Rao, APL Mater., 2014, 2, 092802 CrossRef.
- S. Mubeen, J. Lee, N. Singh, S. Krämer, G. D. Stucky and M. Moskovits, Nat. Nanotechnol., 2013, 8, 247 CrossRef CAS PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911 CrossRef CAS PubMed.
- Y. Yu, Y. Yu, Y. Cai, W. Li, A. Gurarslan, H. Peelaers, D. E. Aspnes, C. G. Van de Walle, N. V. Nguyen, Y.-W. Zhang and L. Cao, Sci. Rep., 2015, 5, 16996 CrossRef CAS PubMed.
- S. Sim, J. Park, J.-G. Song, C. In, Y.-S. Lee, H. Kim and H. Choi, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 075434 CrossRef.
- Z. E. Eroglu, D. Contreras, P. Bahrami, N. Azam, M. Mahjouri-Samani and A. Boulesbaa, Nanomaterials, 2021, 11, 770 CrossRef CAS PubMed.
- Z. E. Eroglu, O. Comegys, L. S. Quintanar, N. Azam, S. Elafandi, M. Mahjouri-Samani and A. Boulesbaa, Phys. Chem. Chem. Phys., 2020, 22, 17385–17393 RSC.
- C. J. Li, O. J. H. Chai, Q. F. Yao, Z. H. Liu, L. Wang, H. J. Wang and J. P. Xie, Mater. Horiz., 2021, 8, 1657–1682 RSC.
Footnotes |
| † This manuscript has been authored by UT-Battelle, LLC under Contract No. DE-AC05-00OR22725 with the U.S. Department of Energy. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for U.S. Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan). |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta01657a |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |