
Enhancing the cycling stability of MgH2 using nitrogen modified titanate†
 *ad
*ad
Abstract
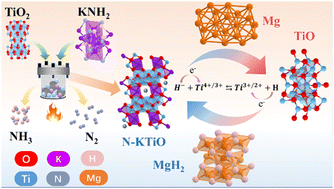
By introducing nitrogen anions, we obtained a N-doped K2Ti6O13 (N-KTiO) catalyst with a considerably reduced band gap. The experimental results showed that N-KTiO could improve the de/re-hydrogenation kinetics and long-term cycling stability of the Mg/MgH2 system. In particular, the peak temperature of MgH2 with a 5 wt% N-KTiO catalyst was ca. 45 °C and 80 °C lower than those of TiO2 catalysed and pure ones, respectively. After full dehydrogenation, 4.6 wt% H2 was absorbed in 1 hour, even under room temperature. Moreover, after cycling for two hundred times at 300 °C, the H2 capacity remained at 6.8 wt% with a retention as high as 94.4%, which is significantly better than those of the reported MgH2-TiO2 composites. Further investigation reveals that band structure changes of the catalyst may affect the catalytic effects, and the reversible changes in the valence state of the titanium species in catalysts are intrinsically associated with the enhanced kinetic properties and stability. This research may offer novel insights into developing effective catalysts for light metal hydrogen storage materials.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

