
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh performance N-doped carbon nanosheet/MnO2 cathode derived from bacterial cellulose for aqueous Zn-ion batteries†
Wenhai
Wang
ab,
Ashley P.
Black
b,
Cheng
Liu
b,
Vlad
Martin-Diaconescu
c,
Laura
Simonelli
 c and
Dino
Tonti
*b
c and
Dino
Tonti
*b
aInstitute of Clean Energy and Advanced Nanocatalysis (iClean), School of Chemistry and Chemical Engineering, Anhui University of Technology, Maanshan 243002, China
bInstitut de Ciència de Materials de Barcelona, Consejo Superior de Investigaciones Científicas (ICMAB-CSIC), Campus UAB Bellaterra, Barcelona, Spain. E-mail: dino.t@csic.es
cALBA Synchrotron Light Source, 08290 Cerdanyola del Vallès, Barcelona, Spain
First published on 27th July 2023
Abstract
Rechargeable aqueous Zn-ion batteries (ZIBs) have obtained extensive attention owing to their high safety, low-cost, environmental friendliness and high energy density. Nevertheless, developing suitable cathode materials remains challenging due to requirements for appropriate microstructure. We present a porous N-doped carbon nanosheet/MnO2 (NCS/MnO2) derived from bacterial cellulose (BC) by a simple route. BC chunks were soaked in urea solution and then carbonized under Ar flow at 900 °C. N-doped carbon nanosheets were obtained and MnO2 was added by reaction with NaMnO4. Benefiting from both the conductivity and porosity of the NCS support, the NCS/MnO2 composite delivers a high capacity and long cycling stability (114 mA h g−1 at 2 A g−1 after 1800 cycles). The electrode reaction mechanism was further investigated and the MnO2 dissolution/deposition mechanism was confirmed, with a critical role of zinc sulfate hydroxide (ZSH) to assist the deposition of MnO2.
Introduction
Due to the limited amount of fossil energy and the climate problems, the demand to explore renewable energy systems becomes more urgent. To favor their exploitation enormous efforts have been devoted to developing suitable energy storage systems. Li-ion batteries are considered as one of the most convincing commercial options, however the safety problems related to organic electrolyte and limited lithium abundance restrict their development for large-scale application.1 Recently, rechargeable aqueous zinc-ion batteries (ZIBs) with nearly neutral electrolytes have obtained enormous interest.2,3 Indeed, metal zinc possesses a striking theoretical capacity (820 mA h g−1), low reduction potential (−0.76 vs. SHE), excellent stability in water and large abundance.4 Moreover, aqueous electrolytes are safer and cheaper than organic electrolyte,5 and present much higher ionic conductivity.6Despite aqueous ZIBs present important advantages, there are still many challenges for developing satisfactory cathodes. Various cathode materials (such as manganese-based oxides, Prussian blue analogues, vanadium based oxides and organic electrodes) have been explored for aqueous ZIBs.7,8 Among them, MnO2 has gained much interest, because of considerable capacity, decent voltage, low-cost and rich abundance.9–11 However, MnO2 exhibits poor electrical conductivity, seriously undermining the electrochemical performance of ZIBs.12,13 To handle this problem, conductive polymers and carbon materials (graphene, CNT, N-doped carbon) have been integrated with MnO2.14–17 However, conductive polymers tend to degrade by swelling and shrink during cycling, which negatively affects the lifetime.18,19 Instead, CNT and graphene are relatively high-cost, which restricts their commercial applications. N-doped carbon has emerged as an effective candidate thanks to lower cost, good electrical conductivity and good electrochemical properties.20 In many reports, metal–organic frameworks (MOFs) were utilized as the sources of N-doped carbon.17,21–23 Nevertheless MOFs are often difficult to prepare at a large-scale, and suffer poor chemical stability.24–26 It is more appealing to fabricate N-doped carbons from biomass, which is usually facile, low-cost, ecofriendly and easy to scale up. Bacterial cellulose (BC) as a novel biomass material, typically produced from Acetobacter xylinum,27,28 owns outstanding characteristics of purity, 3D porous structure and high water-absorbing capacity,29–31 which make it employed in many fields such as food packing, biomedical field, water treatment, electrochemical energy storage and conversion.32–34 BC has been exploited to develop N-doped carbons, by polymerization of polypyrrole, polyaniline, polyacrylonitrile on BC35–37 or high temperature ammonia treatment38 to introduce the nitrogen source. These ways are time-consuming, complicated or risky, thus their economic viability remains limited.
Here we report a simple, green and scalable fabrication of porous N-doped carbon/MnO2 derived from BC, which is used as the cathode for ZIBs. In this process, BC was soaked in a urea solution to obtain the nitrogen source and then after carbonization porous N-doped carbon with an unexpected nanosheet morphology (NCS) was obtained. The MnO2 deposition was achieved by a reaction between NCS and NaMnO4. The ability of BC to acquire the nitrogen source by absorbing urea solution avoids more complex or hazardous routes. Meanwhile, the decomposition of absorbed urea on BC can create high porosity for NCS during the process of pyrolysis. Due to the resulting good conductive and high porosity NCS carbon, the resulting NCS/MnO2 composite derived from BC shows a large capacity and long-term cyclability (114 mA h g−1 at 2 A g−1 after 1800 cycles). Furthermore, the electrochemical mechanism of NCS/MnO2 is further scrutinized by multiple analytical methods. These results indicate that the mechanism of NCS/MnO2 involves MnO2 dissolution/deposition.
Experimental section
Materials
Bacterial cellulose (BC, Q-Phil Products International), urea (CH4N2O, Thermo Scientific), sodium permanganate solution (NaMnO4, 40 wt% in H2O, Sigma-Aldrich), commercial manganese dioxide (MnO2, Sigma-Aldrich), zinc sulfate heptahydrate (ZnSO4·7H2O, Labkem), manganese sulfate monohydrate (MnSO4·H2O, Labkem), 1-methyl-2-pyrrolidone (C5H9NO, Sigma-Aldrich), polyvinylidene fluoride (PVDF, Sigma-Aldrich), carbon black (Super P, Timcal), carbon paper (Freudenberg, H2315, 210 μm thickness), Zn foil (Advent Research Materials Ltd, 0.125 mm thickness), glass fibre filter (PRAT DUMAS, 270 μm thickness).Preparation of materials
Bacterial cellulose cubes (approx. size 15 × 15 × 15 mm3) was purified by washing in Milli-Q water as described previously.39,40 Then individual BC cubes were pressed by a ∼150 g Teflon cylinder for 10 minutes to remove most water. The pressed BC was soaked in urea solution (0.1 g mL−1) with stirring for 2 h. Then the soaked BC was extracted and dried in oven at 60 °C for 24 h. The dried BC was carbonized in a tubular furnace under an Ar flow of 100 mL min−1 with a ramp of 10 °C min−1 to 900 °C and kept there for 1 h.N-doped carbon/MnO2 preparation: 24 mg N-doped carbon was added in 35 mL solution containing 0.28 g NaMnO4 and stirred for 4 h at room temperature. Then the product was washed by Milli-Q water for several times and dried at 60 °C for 24 h. Note that in spite of the large NaMnO4 excess, according to thermogravimetric analysis MnO2 is ∼42% of the composite mass (see Results and discussion), which strongly suggests that in our conditions the reaction between NaMnO4 and NCS is self-limited.
Carbon-w was produced from simply carbonizing purified BC in Ar atmosphere at 900 °C for 1 h, as in our previous article.39 C/MnO2 is then obtained by reaction between carbon-w and NaMnO4, as described for N-doped carbon/MnO2.
Characterization
FEI Quanta 200 FEG-ESEM equipment (15 kV voltage) was performed to obtain Scanning Electron Microscopy (SEM) images. JEOL JEM1210 TEM (voltage 120 kV) and JEM-2011 TEM (voltage 200 kV) were carried out to obtain Transmission electron microscopy (TEM) images. Scanning transmission electron microscopy was performed at a Tecnai G2 F20 at 200 kV. Fourier transform infrared (FTIR) spectra of samples were conducted by a Jasco 4700 Spectrophotometer. Siemens D-5000 diffractometer with Cu Kα radiation was used to collect X-ray diffraction (XRD) patterns. Raman spectra of samples were recorded by a Renishaw InVia Raman Microscope with 633 nm laser. Thermogravimetric analysis (TGA) was done by a NETZSCH STA 449 F1 Jupiter equipment under air atmosphere with 10 °C min−1. N2 adsorption/desorption measurements were probed by Micromeritics ASAP 2020 equipment. X-Ray Photoelectron Spectroscopy (XPS) measurements were taken by a SPECS EA10P hemispherical analyzer with a monochromatic Al Kα source. The X-ray Absorption Spectroscopy (XAS) data were obtained at the Mn and Zn K-edges at the CLAESS beamline of the ALBA synchrotron light source.41 The XAS spectra have been acquired in transmission mode by means of ionization chambers. The Si (111) double crystal monochromator was employed and the higher harmonic were rejected by appropriately choosing the incident angle and coating of the collimating and focusing mirror. The beam size was around 1 × 1 mm2 with a total incoming flux around 1013 photons per s. The radiation damage was suppressed by an approximately 90% attenuation of the incoming beam using Al filters. In situ measurements were performed using modified coin cells with a 6 mm diameter Kapton windows and a 0.5 M ZnSO4 with 0.05 M MnSO4 aqueous electrolyte.Electrochemical performance
To prepare cathodes, MnO2 based materials, Super P and PVDF were mixed at a weight ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 in 1-methyl-2-pyrrolidone. Then the slurry was coated on a carbon paper and was dried in the oven (60 °C) for 24 h. The total loading on the carbon paper is ca. 1.0 mg cm−2. The capacity is calculated by the mass of MnO2 based materials. The Zn ion battery electrochemical performance of cathode materials were studied in Swagelok cells. The cells were assembled with Zn foil as the anode, glass fibre filter as the separator, 2 M ZnSO4 with 0.2 M MnSO4 aqueous electrolyte, and MnO2 based materials as the cathode. The cyclic voltammetry measurements (CV) and electrochemical impedance spectroscopy (EIS, frequency range from 100 kHz to 100 mHz and an AC amplitude of 5 mV) measurements were performed by a Bio-logic VMP3 multichannel potentiostat. Galvanostatic discharge/charge tests were conducted by a battery cycling equipment (LANHE M340A).
:2:1 in 1-methyl-2-pyrrolidone. Then the slurry was coated on a carbon paper and was dried in the oven (60 °C) for 24 h. The total loading on the carbon paper is ca. 1.0 mg cm−2. The capacity is calculated by the mass of MnO2 based materials. The Zn ion battery electrochemical performance of cathode materials were studied in Swagelok cells. The cells were assembled with Zn foil as the anode, glass fibre filter as the separator, 2 M ZnSO4 with 0.2 M MnSO4 aqueous electrolyte, and MnO2 based materials as the cathode. The cyclic voltammetry measurements (CV) and electrochemical impedance spectroscopy (EIS, frequency range from 100 kHz to 100 mHz and an AC amplitude of 5 mV) measurements were performed by a Bio-logic VMP3 multichannel potentiostat. Galvanostatic discharge/charge tests were conducted by a battery cycling equipment (LANHE M340A).
Results and discussion
N-doped carbon was synthesized from the carbonization of urea-soaked BC and carbon-w was prepared from the pyrolysis of pure BC without urea. The morphologies and structure of prepared samples were compared by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). N-doped carbon displays nanosheet structure and high porosity (Fig. 1a and d), and is labeled as NCS. In comparison, carbon-w shows more compact structure (Fig. S1a and c†), implying that urea works as a porogen for carbon. After NCS and carbon-w reacted with NaMnO4, their morphologies barely changed (Fig. 1a–c, S1a and b†). TEM images further reveal some nanorods on NCS (Fig. 1d–f) and the corresponding selected area electron diffraction (SAED) depicts rings corresponding to interlayer distances of 0.24 and 0.21 nm (Fig. S2a†), corresponding to (006) and (−112) planes of MnO2.42,43 The energy-dispersive spectroscopy (EDS) elemental line profiles of NCS/MnO2 confirm the existence of the elements of Mn and O for the nanorod (Fig. S2b†). At larger scale, EDS mapping images show that the elements C, N, Mn and O distribute uniformly in NCS/MnO2 (Fig. 1g). The morphology of commercial MnO2 instead is dominated by large irregular particles of several tens of micrometers along with smaller particles of less than one micrometer (Fig. S1a–f†). | ||
| Fig. 1 Electron microscopy of prepared materials: SEM images of (a) NCS; (b) NCS/MnO2. TEM images of (c) NCS; (d and e) NCS/MnO2. (f) Scanning TEM of NCS/MnO2. (g) Element mapping images of NCS/MnO2. | ||
X-ray diffraction (XRD) measurement was carried out to investigate the phase structures of NCS, carbon-w, C/MnO2 and NCS/MnO2. As shown Fig. 2a, carbon-w and NCS show a broad peak at around 26°, referring to the plane (002) of graphite carbon.44,45 Both C/MnO2 and NCS/MnO2 exhibit a weak and broad peak at 36°, which can be indexed to the plane (006) of MnO2.46 X-ray absorption spectroscopy (XAS) reveals that the energy position of Mn peak for NCS/MnO2 is close to reference MnO2, further confirming the formation of MnO2 on NCS (Fig. S3a†). Commercial MnO2 was also investigated by XAS and XRD (Fig. S3†). Raman spectrum was used to quantify the graphitization degrees of carbon-w and NCS. There are in both samples two peaks located at 1350 cm−1 (D band) and 1600 cm−1 (G band), representing disordered and graphitized carbon (Fig. 2b).47,48 The intensity ratio G to D band (ID/IG) is used to evaluate the degree of graphitization. The value of ID/IG for NCS is 1.12, which is lower than carbon-w (1.21). It implies that a higher graphitization can be obtained in the presence of urea, which is expected to infer better electron conduction.49 After reacting with NaMnO4, a new peak appearing at 640 cm−1 can be associated with the Mn-O stretching vibration of MnO6 groups,50 demonstrating the formation of MnO2 both on carbon-w and NCS. Compared with carbon-w and NCS, the ratios of ID/IG for NCS/MnO2 (1.18) and C/MnO2 (1.27) increase. This indicates that the introduction of MnO2 causes more defects into carbon-w and NCS, leading to more defective graphitic structures and lower degree of graphitization.51,52
 | ||
| Fig. 2 Structural and surface characterization: (a) XRD patterns. (b) Raman spectra. (c) Pore size distribution. XPS spectra (d) C 1s, (e) N 1s, (f) Mn 2p3/2. | ||
The porous structure of NCS was inspected by N2 adsorption–desorption measurement (Fig. S4a and Table S1†). Although the surface area of NCS (431 m2 g−1) is lower than that of carbon-w (1009 m2 g−1), its pore volume is much larger (0.86 vs. 0.19 cm3 g−1). The pore size distribution curves show that the pore structure of NCS is predominately composed by mesopores and macropores (Fig. S4b†), leading to a high surface external area, while the pore structure of carbon-w mainly consists of micropores.39 The high porosity of NCS can be attributed to the released ammonia and carbon dioxide gas from urea during cellulose carbonization.53,54 After MnO2 deposition, the surface area and pore volume of both carbons decrease (Table S1†). This is probably because some parts of NCS and carbon-w were consumed and the introduction of MnO2 blocked pores to some extent, resulting in decreased surface areas and pore volumes, although after MnO2 depositions trends are similar to those found with carbon substrates. Despite NCS/MnO2 does not have a larger surface area (229 m2 g−1) than C/MnO2 (401 m2 g−1), NCS/MnO2 exhibits a larger pore volume (0.42 cm3 g−1) than C/MnO2 (0.12 cm3 g−1). In addition, the porous structure of NCS/MnO2 mainly consists of mesopores and macropores, while the pore structures of C/MnO2 and commercial MnO2 are dominated by micropores (Fig. 2c). These results demonstrate that NCS/MnO2 possesses higher porosity than C/MnO2 and commercial MnO2. Electrodes of high porosity is more favorable for fast ion transfer, which can shorten the diffusion path.55,56 This has effect also on the content of MnO2 in NCS/MnO2, revealed by thermogravimetric analysis (TGA), which is about 42% (Fig. S5†), much higher than in C/MnO2, about 11%. The high porosity of NCS/MnO2 favors the diffusion of NaMnO4 into the bulk and the MnO2 deposition can take place on the surface and inside of NCS at the same time.57,58 In contrast, the diffusion path of NaMnO4 into carbon-w with low porosity is difficult and the reaction between carbon-w and NaMnO4 mainly occurs on the surface of carbon-w. As a consequence, the larger pore volume of NCS is better filled by MnO2, resulting in much higher mass content of MnO2 in NCS/MnO2 than in C/MnO2.
To detect the surface chemical composition and chemical state of these samples, X-ray photoelectron spectroscopy (XPS) was performed. N 1s peak is observed in the full spectrum of NCS (Fig. S6†), indicating that N was successfully doped into carbon. The nitrogen content of NCS is 4.8 at%. After NCS reacted with NaMnO4, C 1s, N 1s, Mn 2p and O 1s peaks are obvious in the full spectrum of NCS/MnO2 (Fig. S6†). The C 1s spectrum of NCS/MnO2 can be fitted into 4 peaks at 284.6, 285.2, 285.9, 288.3 and 292.5 eV (Fig. 2d), corresponding to C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–N, C–O, CO and O–CO.59,60 The N 1s spectrum of NCS/MnO2 can be divided into three peaks (Fig. 2e), attributed to pyridinic-N (398.6 eV), pyrrolic-N (399.7 eV) and graphitic-N (400.9 eV).61 It has been shown that pyridinic-N and pyrrolic-N can enhance the electronic conductivity of carbon and hence promote the battery performance.62,63 The binding energy of Mn 2p of C/MnO2 and NCS/MnO2 are similar to commercial MnO2 (Fig. 2f and S7†), confirming the presence of Mn oxides in the prepared composites. Mn 2p peaks were deconvoluted into two multiplet components attributed to Mn3+ and Mn4+ according to the method proposed by Ilton et al.64 The area of these peaks indicates significant Mn3+ fractions at the surface of these samples (Table S2†), particularly in the case of C/MnO2. This could be attributed to the reductive environment provided by the carbon excess that supports the Mn oxides.
C, C–N, C–O, CO and O–CO.59,60 The N 1s spectrum of NCS/MnO2 can be divided into three peaks (Fig. 2e), attributed to pyridinic-N (398.6 eV), pyrrolic-N (399.7 eV) and graphitic-N (400.9 eV).61 It has been shown that pyridinic-N and pyrrolic-N can enhance the electronic conductivity of carbon and hence promote the battery performance.62,63 The binding energy of Mn 2p of C/MnO2 and NCS/MnO2 are similar to commercial MnO2 (Fig. 2f and S7†), confirming the presence of Mn oxides in the prepared composites. Mn 2p peaks were deconvoluted into two multiplet components attributed to Mn3+ and Mn4+ according to the method proposed by Ilton et al.64 The area of these peaks indicates significant Mn3+ fractions at the surface of these samples (Table S2†), particularly in the case of C/MnO2. This could be attributed to the reductive environment provided by the carbon excess that supports the Mn oxides.
To evaluate the electrochemical performance of MnO2 electrodes, full batteries with Zn foil anodes were assembled in Swagelok cells. As shown in Fig. 3a, cyclic voltammetry curve (CV) of all three cathodes tested present two pairs of reduction/oxidation peaks, suggesting that they have similar redox reactions. But the currents of these peaks for NCS/MnO2 are larger than C/MnO2 and commercial MnO2, demonstrating that NCS/MnO2 possesses higher electrochemical activity and fast reaction kinetic.65 The rate capabilities of cathodes were measured at different current densities (Fig. 3b). NCS/MnO2 provides 226 mA h g−1 at 0.1 A g−1 (Fig. 3c). Even at high current densities of 1 A g−1, NCS/MnO2 still can deliver 210 mA h g−1. C/MnO2 and commercial MnO2 were also measured as a comparison. C/MnO2 and commercial MnO2 demonstrate much lower capacities than NCS/MnO2 at various current densities (Fig. 3b and S8†). In particular, the capacities of commercial MnO2 (from 187 mA h g−1 at 0.1 A g−1 to 127 mA h g−1 at 1 A g−1) and C/MnO2 (from 77 mA h g−1 at 0.1 A g−1 to 53 mA h g−1 at 1 A g−1) decrease greatly with the increment of the current density. The capacity at 1 A g−1 for commercial MnO2 and C/MnO2 are respectively 68% and 69% of that at 0.1 A g−1, considerably lower than for NCS/MnO2 (90%). The lower capacities compared to NCS/MnO2 can be explained with the lower MnO2 content of C/MnO2 (as determined by TGA), and with the large number of coarse particles in the commercial MnO2 sample. These results show that NCS/MnO2 possesses an excellent rate performance.
 | ||
| Fig. 3 Electrochemical behavior in ZIB cathodes: (a) CV curves at a scan rate of 0.2 mV s−1. (b) Rate performance. (c) Galvanostatic discharge/charge profiles of NCS/MnO2 at different current densities. (d) Cycling performance at 0.2 A g−1. (e) Cycling performance at 2 A g−1. | ||
To further evaluate the cycling stability, the electrodes were cycled at 0.2 A g−1 (Fig. 3d). The discharge capacities gradually increase during the cycling, which can be associated with an electrochemical activation process also reported by other authors on ZIB systems.66 The capacity of NCS/MnO2 increased to a larger extent than C/MnO2 and commercial MnO2, probably because of the high porosity of NCS/MnO2, which can facilitate the deposition of MnO2 during charge.67 Among these electrodes, NCS/MnO2 shows a higher capacity (358 mA h g−1) than commercial MnO2 (177 mA h g−1) and C/MnO2 (74 mA h g−1) after 60 cycles. The NCS/MnO2 capacity is even larger above 100 cycles (over 400 mA h g−1, see Fig. S9†). The capacity of MnO2-free NCS and carbon-w were also investigated (Fig. S10†). The capacities of NCS (60 mA h g−1) and carbon-w (48 mA h g−1) are similar at 0.2 A g−1 after 60 cycles. To evaluate the role of Mn2+ in the electrolyte, the Swagelok cell with NCS/MnO2 as cathode was assembled without using MnSO4 additive under the same conditions (Fig. S11†). The capacity of NCS/MnO2 declines rapidly with cycle number. It is obvious that the addition of MnSO4 can enhance the electrochemical reversibility. This confirms that the MnSO4 additive can generate a proper equilibrium between Mn2+ dissolution and the re-oxidation of the Mn2+, which improves the stability of the cathode.68 In addition, the test without MnO2 in the electrode demonstrates that capacity can even be provided by Mn2+ in the electrolyte, which precipitates as oxide during charge. However, the larger difference with the NCS sample shows the important contribution of MnO2 already present inside the pores of the electrodes, which only the NCS texture allows to great extent.
The cycling stability of all electrodes were also investigated at a high current density of 2 A g−1. There is a noticeable decay in capacity for NCS/MnO2 (Fig. 3e) and the reason for attenuation was investigated. As shown in Fig. S12,† the 2nd discharge capacity (232 mA h g−1) is lower than the 1st charge capacity (252 mA h g−1). It demonstrates that less MnO2 is dissolved on the 2nd discharge than is deposited on the 1st charge.69 So there is some residual MnO2 on the electrode after the 2nd discharge. This phenomenon continues during the early cycles. In other words, the dissolution of MnO2 does not catch up with the deposition of MnO2 during the early stage of cycling. So there is more residual MnO2 on the electrode with cycling, decreasing the conductivity of the electrode.70 As a result, the capacity fades. As seen in Fig. 3e, the capacity of for NCS/MnO2 still can be maintained at 114 mA h g−1 after 1800 cycles. For a comparison, low capacities are attained for C/MnO2 (22 mA h g−1) and commercial MnO2 (37 mA h g−1). The excellent performance of NCS/MnO2 is also superior to most of reported Mn-based cathodes (Table S3†).
To further manifest the advantages of BC as carbon source, filter paper (FP) and printer paper (PP) were also immersed in urea solution following the same protocol as BC. The FTIR spectra of dried urea/filter paper and dried urea/printer paper are similar as their pristine ones and just display a few weak peaks coming from urea (Fig. S13†), implying that urea is also absorbed on these papers, but the amount is very small. Conversely, the FTIR spectra of dried urea/BC is completely consistent with urea. The mass of urea absorbed by filter paper and printer paper are respectively 0.004 and 0.005 g cm−2 (Table S4†), which is negligible in comparison with the BC case (0.15 g cm−2). The mass ratio between absorbed urea and BC is 25, much higher than urea/FP (0.4) and urea/PP (0.6). These results clearly indicate the large BC water-absorbing capacity compared to regular microfibrous paper, allowing to incorporate large solute amounts that are retained and well distributed after drying. Given the small amount of added urea, there is no significant morphologic difference between MnO2 derived from papers immersed or not immersed in urea, although a small capacity enhancement is still observed in treated samples (Fig. S14 and S15†).
Electrochemical impedance spectroscopy was conducted to further analyze the reaction kinetics of NCS/MnO2, C/MnO2 and commercial MnO2 (Fig. S16†). The Nyquist plots is composed of one semicircle in the high frequency region and one straight line in the low frequency region. The semicircle is associated with charge-transfer resistance and the line is related to ion diffusion process.71 NCS/MnO2 shows a smaller semicircle than other C/MnO2 and commercial MnO2, revealing a smaller charge transfer resistance for NCS/MnO2. The diffusion coefficient of these electrodes can be reflected by the Warburg coefficient σ, which is inversely proportional to the diffusion coefficient.72 The σ values of these electrodes can be obtained by fitting the linear relation between Z′ and ω−1/2. The σ values of NCS/MnO2, C/MnO2 and commercial MnO2 are respectively 26, 50, and 77 Ω s−1. The σ value of C/MnO2 and commercial MnO2 are nearly 2–3 times higher than NCS/MnO2, showing the fast ion diffusion for NCS/MnO2. The fast charge transfer and the ion diffusion of NCS/MnO2 can be attributed to the optimal architecture of NCS. N-doping can enhance the electronic conductivity of carbon73 and the high porosity can offer numerous channels for transporting ions.74
To better comprehend the electrochemical reaction mechanism of NCS/MnO2, CV tests were conducted at different scan rates ranging from 0.2 to 1.0 mV s−1 (Fig. S17a†). The peaks slightly broaden without significant change of the CV. According to previous literature,75 the charge storage kinetics can be expressed by the following equation: i = a·νb (i: peak current; v: scan rate; a, b: variable values). The b value can be calculated by the slopes of the fitting curves of logi versus logv (Fig. S17b†). The b values of 1.0 and 0.5 respectively correspond to the capacitive-like behavior and the diffusion-controlled process.76,77 The b value of these peaks are respectively 0.74, 0.79, 0.63 and 0.7 (Fig. S17c†), suggesting that the electrochemical reactions kinetics of NCS/MnO2 contains both capacitive and diffusion-controlled processes. As the scan rate increases from 0.2 to 1.0 mV s−1, the fraction of capacitive contribution gradually increases from 46 to 58% (Fig. S18†). This small variation with a 5-fold increase in scan rate confirms the good rate capability of our system.
Considering the capacity measured on the basis of the MnO2 content as determined by TGA (Fig. S5†), we estimate up to 558 mA h (gMnO2)−1 at 0.2 A g−1 after 1st discharge. This value is considerably superior to the capacity expected for 1-electron reduction of MnO2 (308 mA h (gMnO2)−1), and approaching the quantitative 2-electron reduction of MnO2 to Mn2+ according to eqn (1):
| MnO2 + 4H+ + 2e ⇌ Mn2+ + 2H2O | (1) |
However, such simple dissolution/precipitation mechanism is contradicted by the test reported above on the MnO2-free carbons. The very small capacity they exhibited demonstrate that only a small amount of MnO2 forms on charge from Mn2+ in solution, while samples with a substantial initial MnO2 loading have much larger capacity. According to recent literature,78 a key role is played by formation and dissolution zinc sulfate hydroxide hydrate (Zn4(OH)6SO4·5H2O, ZSH) in parallel to MnO2 dissolution and precipitation.
To confirm if this mechanism also applies to NCS/MnO2, its structural evolution was followed during the process of discharge and charge. Ex situ SEM, TEM, XRD, XPS and XAS analyses were conducted during the initial 2 cycles of galvanostatic discharge/charge at 0.2 A g−1 (Fig. S19†). After the first full discharge, compared with the XRD pattern of pristine NCS/MnO2 electrode, there are new peaks located at 32.6° and 58.5° (Fig. 4a and S20†), which are consistent with formation of ZSH.79 SEM images show that large amounts of flakes emerge on the electrode of NCS/MnO2 (Fig. 4e and S21b†) and these flakes can be assigned to ZSH.80 TEM shows that the MnO2 nanorods initially present in the NCS/MnO2 cathode have vanished after the 1st full discharge (compare Fig. 4g and h). Consistently, only traces of Mn are still present on the surface, as indicated by XPS Mn 2p (Fig. 4b), demonstrating that MnO2 actually dissolves during the discharge.79 The generation of ZSH has been attributed to the dissolution reaction of MnO2 into Mn2+, which locally increases the pH, triggering the ZSH precipitation.81,82 Upon the first full charge, these large ZSH flakes almost disappear (Fig. 4f and S21c†) and many new nanosheets show up. The energy of Mn XPS and XAS peaks almost return to that of the pristine state (Fig. 4b and c), further suggesting that re-deposition of MnO2 occurs. To address the roles of ZSH and Mn2+ on the charge process, two NCS/MnO2 cathodes (electrolyte: 2 M ZnSO4 and 0.2 M MnSO4) at the 1st full discharge were extracted from the cell. They were respectively removed of Mn2+ by washing, and of ZSH by acetic acid treatment (10 vol%). The Mn2+-free cathode was assembled with 2 M ZnSO4 electrolyte. The ZSH-free cathode (Fig. S22†) was assembled with the 2 M ZnSO4 and 0.2 M MnSO4 electrolyte. Then both cathodes were fully charged. The charge capacities of both electrodes are small and there are not obvious plateaus (Fig. S23†), suggesting that the occurrence of plateaus is indeed related to the simultaneous presence of both ZSH and Mn2+. Images and XRD collected at the end of the second full discharge and charge suggest that the overall reactions are similar to those occurring during the 1st cycle (Fig. S24 and 25†).
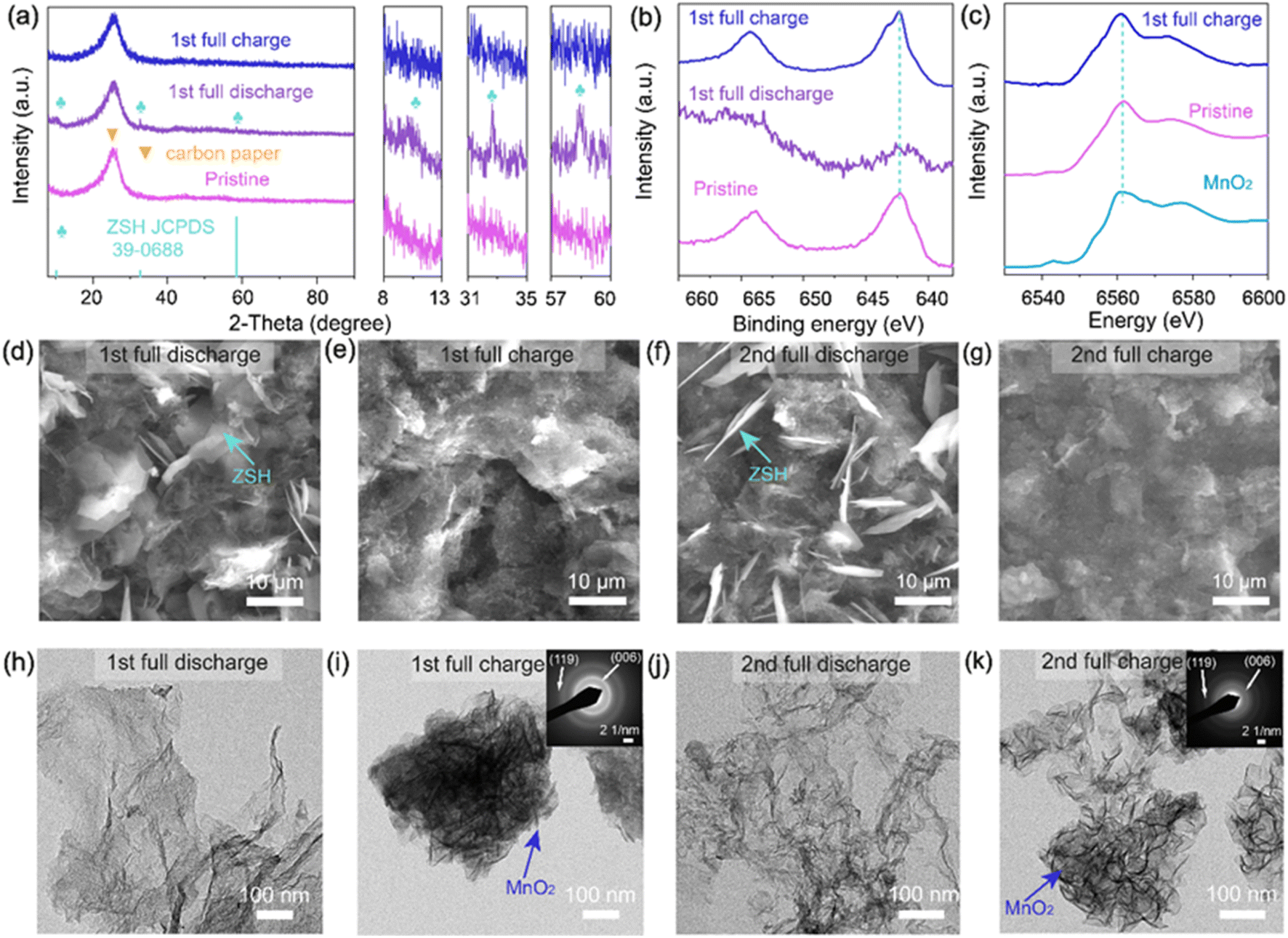 | ||
| Fig. 4 Structural evolution of NCS/MnO2 electrode during discharge/charge process: (a) XRD patterns; (b) XPS spectra Mn 2p; (c) XAS; (d–g) SEM images (h–k) TEM images. | ||
The dissolution-precipitation mechanism is definitely confirmed by in situ Mn and Zn K-edge XAS spectra of NCS/MnO2 electrode, reported in Fig. 5. Normalized Mn K-edge XANES spectra, exhibit a clear transfer from the component at 6561 eV that can be attributed to the Mn(IV) in the pristine MnO2 to the one at 6553 eV after discharge, indicating the formation at its expense of reduced Mn2+ species. After charge the Mn environment is again very close to that found in the pristine electrode demonstrating the reversible Mn redox activity during the charge–discharge process. Unlike the Mn K-edge spectra, the Zn K-edge XANES spectra exhibit only small changes in the shape of the edge due to changes in the Zn local environment but they do not show any shift on the edge energy at all states of charge confirming that Zn ions are not redox active. Fig. 5c and d show the corresponding Fourier transform of the k2 weighted Mn and Zn K-edge extended X-ray absorption fine structure (EXAFS) spectra, respectively. The strong peak located around 1.5 Å is attributed to the closest oxygen shell (Mn–O and Zn–O bond), while the peak around 2.5 Å is assigned to the metal–metal shell, absent in the case of solvated cations. At the fully discharged state the intensity of the Mn–Mn peak in suppressed, while the Mn-O distance slightly shifts to higher distances. This result confirms the formation of Mn2+ and reversible dissolution/precipitation of MnO2 by discharging. Conversely, the Zn EXAFS is consistent with the reversible formation of a solid ZSH phase at the end of discharge without any redox activity. In light of this possible mechanism, we relate the long cycle life of the NCS/MnO2 to its large area and high conductivity favoring optimal distribution of interfacial currents and dispersion of the precipitates.
 | ||
| Fig. 5 In situ measurements of NCS/MnO2 electrode: (a and b) Mn and Zn K-edge XANES spectra at selected states of charge and the Fourier transforms of their corresponding k2-weighted EXAFS oscillations (c and d). | ||
Conclusions
In summary, we have reported the unexpected formation porous N-doped carbon nanosheet from carbonization of urea-impregnated BC. The optimal texture and electric properties of NCS are demonstrated in aqueous ZIBs. NCS is employed as a support for the growth of highly dispersed nanostructured MnO2. The obtained NCS/MnO2 processed as cathode provides a superior performance (114 mA h g−1 at 2 A g−1 after 1800 cycles) compared to commercial MnO2 and many cathodes reported in literature. With advantages of highly porous and conductive carbon networks, NCS/MnO2 exhibits remarkable capacity, excellent rate capability and long cycle life. Besides, the charge storage mechanism and the corresponding phase transformation of NCS/MnO2 were evaluated by multiple characterization methods, supporting a MnO2 2-electron full dissolution-deposition mechanism that appears highly reversible thanks to the unique properties of this bacterial cellulose derived composite. Considering the simple, green and scalable fabrication, NCS/MnO2 can be a promising cathode candidate for high-performance ZIBs.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
This research was funded by the Spanish Agency for Research (AEI) with ERDF cofunding, through the “Severo Ochoa” Programme for Centers of Excellence in R&D (FUNFUTURE, CEX2019-000917-S), the projects RTI2018-096273-B-I00, PID2021-124681OB-I00, and TED2021-132707B-I00. This research has been partially developed within the CSIC Interdisciplinary Thematic Platform (PTI+) Transición Energética Sostenible+ (PTI-TRANSENER+) as part of the CSIC program for the Spanish Recovery, Transformation and Resilience Plan funded by the Recovery and Resilience Facility of the European Union, established by the Regulation (EU) 2020/2094. W. W. and C. L. are grateful for the support from the China Scholarship Council (CSC No.: 201808340076 and 202106370079). This work has been performed within the framework of the doctoral program in materials science of UAB (W. W. and C. L.). XAS experiments were performed at CLAESS beamline of ALBA Synchrotron with the collaboration of ALBA staff.References
- N. Liu, X. Wu, Y. Yin, A. Chen, C. Zhao, Z. Guo, L. Fan and N. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 28199–28205 CrossRef CAS.
- H. Jia, Z. Wang, B. Tawiah, Y. Wang, C.-Y. Chan, B. Fei and F. Pan, Nano Energy, 2020, 70, 104523 CrossRef CAS.
- P. Ruan, X. Chen, L. Qin, Y. Tang, B. Lu, Z. Zeng, S. Liang and J. Zhou, Adv. Mater., 2023 DOI:10.1002/adma.202300577.
- Z. Yi, G. Chen, F. Hou, L. Wang and J. Liang, Adv. Energy Mater., 2020, 11, 2003065 CrossRef.
- H. Zhang, X. Liu, H. Li, I. Hasa and S. Passerini, Angew. Chem., Int. Ed., 2021, 60, 598–616 CrossRef CAS PubMed.
- Y. Lv, Y. Xiao, L. Ma, C. Zhi and S. Chen, Adv. Mater., 2022, 34, 2106409 CrossRef CAS.
- N. Zhang, X. Chen, M. Yu, Z. Niu, F. Cheng and J. Chen, Chem. Soc. Rev., 2020, 49, 4203–4219 RSC.
- J. Huang, Y. Li, R. Xie, J. Li, Z. Tian, G. Chai, Y. Zhang, F. Lai, G. He, C. Liu, T. Liu and D. J. L. Brett, J. Energy Chem., 2021, 58, 147–155 CrossRef CAS.
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 CrossRef CAS.
- L. Godeffroy, I. Aguilar, J. Médard, D. Larcher, J. M. Tarascon and F. Kanoufi, Adv. Energy Mater., 2022, 12, 2200722 CrossRef CAS.
- X. Chen, P. Ruan, X. Wu, S. Liang and J. Zhou, Acta Phys. -Chim. Sin., 2021, 38, 2111003 Search PubMed.
- Y. Liu, X. Chi, Q. Han, Y. Du, J. Huang, Y. Liu and J. Yang, J. Power Sources, 2019, 443, 227244 CrossRef CAS.
- Y. Zhao, Y. Zhu and X. Zhang, InfoMat, 2019, 2, 237–260 CrossRef.
- J. Huang, Z. Wang, M. Hou, X. Dong, Y. Liu, Y. Wang and Y. Xia, Nat. Commun., 2018, 9, 2906 CrossRef PubMed.
- C. Wang, Y. Zeng, X. Xiao, S. Wu, G. Zhong, K. Xu, Z. Wei, W. Su and X. Lu, J. Energy Chem., 2020, 43, 182–187 CrossRef.
- X. Shen, X. Wang, Y. Zhou, Y. Shi, L. Zhao, H. Jin, J. Di and Q. Li, Adv. Funct. Mater., 2021, 31, 2101579 CrossRef CAS.
- Y. Fu, Q. Wei, G. Zhang, X. Wang, J. Zhang, Y. Hu, D. Wang, L. Zuin, T. Zhou, Y. Wu and S. Sun, Adv. Energy Mater., 2018, 8, 1801445 CrossRef.
- C. Hu, Y. Lin, J. W. Connell, H. M. Cheng, Y. Gogotsi, M. M. Titirici and L. Dai, Adv. Mater., 2019, 31, 1806128 CrossRef PubMed.
- H. Yu, G. Liu, M. Wang, R. Ren, G. Shim, J. Y. Kim, M. X. Tran, D. Byun and J. K. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 5820–5830 CrossRef CAS.
- D. Hursan, A. A. Samu, L. Janovak, K. Artyushkova, T. Asset, P. Atanassov and C. Janaky, Joule, 2019, 3, 1719–1733 CrossRef CAS PubMed.
- Z. Sheng, P. Qi, Y. Lu, G. Liu, M. Chen, X. Gan, Y. Qin, K. Hao and Y. Tang, ACS Appl. Mater. Interfaces, 2021, 13, 34495–34506 CrossRef CAS PubMed.
- A. Samanta, B. K. Barman, S. Mallick and C. R. Raj, ACS Appl. Energy Mater., 2020, 3, 10108–10118 CrossRef CAS.
- B. He, Z. Zhou, P. Man, Q. Zhang, C. Li, L. Xie, X. Wang, Q. Li and Y. Yao, J. Mater. Chem. A, 2019, 7, 12979–12986 RSC.
- S. Wang, C. M. McGuirk, A. d'Aquino, J. A. Mason and C. A. Mirkin, Adv. Mater., 2018, 30, 1800202 CrossRef PubMed.
- Z. Wang, J. Huang, J. Mao, Q. Guo, Z. Chen and Y. Lai, J. Mater. Chem. A, 2020, 8, 2934–2961 RSC.
- S. M. J. Rogge, M. Waroquier and V. Van Speybroeck, Acc. Chem. Res., 2018, 51, 138–148 CrossRef CAS PubMed.
- S. Roig-Sanchez, E. Jungstedt, I. Anton-Sales, D. C. Malaspina, J. Faraudo, L. A. Berglund, A. Laromaine and A. Roig, Nanoscale Horiz., 2019, 4, 634–641 RSC.
- A. Alonso-Díaz, J. Floriach-Clark, J. Fuentes, M. Capellades, N. S. Coll and A. Laromaine, ACS Biomater. Sci. Eng., 2019, 5, 413–419 CrossRef.
- M. Gao, J. Li, Z. Bao, M. Hu, R. Nian, D. Feng, D. An, X. Li, M. Xian and H. Zhang, Nat. Commun., 2019, 10, 437 CrossRef CAS PubMed.
- J. Caro-Astorga, K. T. Walker, N. Herrera, K. Y. Lee and T. Ellis, Nat. Commun., 2021, 12, 5027 CrossRef CAS PubMed.
- E. Tsouko, S. Maina, D. Ladakis, I. K. Kookos and A. Koutinas, Renewable Energy, 2020, 160, 944–954 CrossRef CAS.
- C. Chen, W. Ding, H. Zhang, L. Zhang, Y. Huang, M. Fan, J. Yang and D. Sun, Carbohydr. Polym., 2022, 278, 118995 CrossRef CAS PubMed.
- J. Wang, J. Tavakoli and Y. Tang, Carbohydr. Polym., 2019, 219, 63–76 CrossRef CAS PubMed.
- L. Ma, Z. Bi, Y. Xue, W. Zhang, Q. Huang, L. Zhang and Y. Huang, J. Mater. Chem. A, 2020, 8, 5812–5842 RSC.
- J. Xu, J. Rong, F. Qiu, Y. Zhu, K. Mao, Y. Fang, D. Yang and T. Zhang, J. Colloid Interface Sci., 2019, 555, 294–303 CrossRef CAS PubMed.
- Y. Zhang, J. Tan, F. Wen, Z. Zhou, M. Zhu, S. Yin and H. Wang, Int. J. Hydrogen Energy, 2018, 43, 6167–6176 CrossRef CAS.
- A. Dobashi, J. Maruyama, Y. Shen, M. Nandi and H. Uyama, Carbohydr. Polym., 2018, 200, 381–390 CrossRef CAS PubMed.
- Y. Gao, D. He, L. Wu, Z. Wang, Y. Yao, Z.-H. Huang, H. Yang and M.-X. Wang, Chem. Eng. J., 2021, 420, 127411 CrossRef CAS.
- W. Wang, S. Khabazian, S. Roig-Sanchez, A. Laromaine, A. Roig and D. Tonti, Renewable Energy, 2021, 177, 209–215 CrossRef.
- W. Wang, S. Khabazian, M. Casas-Papiol, S. Roig-Sanchez, A. Laromaine, A. Roig and D. Tonti, Int. J. Hydrogen Energy, 2022, 47, 29753–29761 CrossRef CAS.
- L. Simonelli, C. Marini, W. Olszewski, M. Ávila Pérez, N. Ramanan, G. Guilera, V. Cuartero, K. Klementiev and N. L. Saini, Cogent Phys., 2016, 3, 1231987 Search PubMed.
- G. Wang, Y. Sun, D. Li, W. Wei, X. Feng and K. Mullen, Small, 2016, 12, 3914–3919 CrossRef CAS PubMed.
- K. Zhang, H. Zhao, Z. Zhang, J. Chen, X. Mu, X. Pan, Z. Zhang, J. Zhou, J. Li and E. Xie, Carbon, 2015, 95, 746–755 CrossRef CAS.
- Y. Zhang, Z. Wang, K. Hu, J. Ren, N. Yu, X. Liu, G. Wu and N. Liu, Energy Storage Mater., 2021, 34, 311–319 CrossRef.
- P. P. Ghimire, M. Gao and M. Jaroniec, Carbon, 2019, 153, 206–216 CrossRef CAS.
- J. Li, Y. Chen, J. Guo, F. Wang, H. Liu and Y. Li, Adv. Funct. Mater., 2020, 30, 2004115 CrossRef CAS.
- H. Wang, F. X. Yin, N. Liu, R. H. Kou, X. B. He, C. J. Sun, B. H. Chen, D. J. Liu and H. Q. Yin, Adv. Funct. Mater., 2019, 29, 1901531 CrossRef.
- A. Singhania and A. N. Bhaskarwar, Renewable Energy, 2018, 127, 509–513 CrossRef CAS.
- S. Li, Z. Lei, G. Yu, Q. Xu, W. Xu, R. Wu and M. K. Banks, ACS Sustainable Chem. Eng., 2020, 8, 12768–12774 CrossRef CAS.
- M. Yuan, F. Luo, Y. Rao, J. Yu, Z. Wang, H. Li and X. Chen, Carbon, 2021, 183, 128–137 CrossRef CAS.
- K. Wang, Y. Chen, R. Tian, H. Li, Y. Zhou, H. Duan and H. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 11333–11342 CrossRef CAS.
- R. Wang, M. He, Y. Zhou, S. Nie, Y. Wang, W. Liu, Q. He, W. Wu, X. Bu and X. Yang, Carbon, 2020, 156, 378–388 CrossRef CAS.
- Z. Weng, K. Zhang, Y. Qi, T. Zhang, M. Xia, F. Hu, S. Zhang, C. Liu, J. Wang and X. Jian, Carbon, 2020, 159, 495–503 CrossRef CAS.
- A. Eftekhari and Z. Fan, Mater. Chem. Front., 2017, 1, 1001–1027 RSC.
- A. R. Selvaraj, A. Muthusamy, C. Inho, H.-J. Kim, K. Senthil and K. Prabakar, Carbon, 2021, 174, 463–474 CrossRef CAS.
- L. Jin, X. Guo, R. Gong, J. Zheng, Z. Xiang, C. Zhang and J. P. Zheng, Energy Storage Mater., 2019, 23, 409–417 CrossRef.
- T. Le, Y. Yang, L. Yu, Z. H. Huang and F. Kang, Sci. Rep., 2016, 6, 37368 CrossRef CAS PubMed.
- A. E. Fischer, K. A. Pettigrew, D. R. Rolison, R. M. Stroud and J. W. Long, Nano Lett., 2007, 7, 281–286 CrossRef CAS PubMed.
- G. Li, J. Sun, W. Hou, S. Jiang, Y. Huang and J. Geng, Nat. Commun., 2016, 7, 10601 CrossRef CAS PubMed.
- J. Zhang, W. Zhu, Y. Pei, Y. Liu, Y. Qin, X. Zhang, Q. Wang, Y. Yin and M. D. Guiver, ChemSusChem, 2019, 12, 4165–4169 CrossRef CAS PubMed.
- J. Wang, Z. Yang, Y. Li, X. Fan, F. Zhang, G. Zhang, W. Peng and S. Wang, Carbon, 2020, 163, 43–55 CrossRef CAS.
- F. Zheng, Y. Yang and Q. Chen, Nat. Commun., 2014, 5, 5261 CrossRef CAS PubMed.
- H. Huang, R. Xu, Y. Feng, S. Zeng, Y. Jiang, H. Wang, W. Luo and Y. Yu, Adv. Mater., 2020, 32, 1904320 CrossRef CAS.
- E. S. Ilton, J. E. Post, P. J. Heaney, F. T. Ling and S. N. Kerisit, Appl. Surf. Sci., 2016, 366, 475–485 CrossRef CAS.
- Y. Zhao, P. Zhang, J. Liang, X. Xia, L. Ren, L. Song, W. Liu and X. Sun, Energy Storage Mater., 2022, 47, 424–433 CrossRef.
- F. Tang, J. Gao, Q. Ruan, X. Wu, X. Wu, T. Zhang, Z. Liu, Y. Xiang, Z. He and X. Wu, Electrochim. Acta, 2020, 353, 136570 CrossRef CAS.
- H. Moon, K. H. Ha, Y. Park, J. Lee, M. S. Kwon, J. Lim, M. H. Lee, D. H. Kim, J. H. Choi, J. H. Choi and K. T. Lee, Adv. Sci., 2021, 8, 2003714 CrossRef CAS PubMed.
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS.
- D. Wu, L. M. Housel, S. J. Kim, N. Sadique, C. D. Quilty, L. Wu, R. Tappero, S. L. Nicholas, S. Ehrlich, Y. Zhu, A. C. Marschilok, E. S. Takeuchi, D. C. Bock and K. J. Takeuchi, Energy Environ. Sci., 2020, 13, 4322–4333 RSC.
- Y. Zhou, X. Cheng, B. Tynan, Z. Sha, F. Huang, M. S. Islam, J. Zhang, A. N. Rider, L. Dai, D. Chu, D.-W. Wang, Z. Han and C.-H. Wang, Carbon, 2021, 184, 504–513 CrossRef CAS.
- R. Liang, J. Fu, Y.-P. Deng, Y. Pei, M. Zhang, A. Yu and Z. Chen, Energy Storage Mater., 2021, 36, 478–484 CrossRef.
- B. Sambandam, S. Kim, D. T. Pham, V. Mathew, J. Lee, S. Lee, V. Soundharrajan, S. Kim, M. H. Alfaruqi, J.-Y. Hwang and J. Kim, Energy Storage Mater., 2021, 35, 47–61 CrossRef.
- H. He, D. Huang, Y. Tang, Q. Wang, X. Ji, H. Wang and Z. Guo, Nano Energy, 2019, 57, 728–736 CrossRef CAS.
- M. Yang, Y. Wang, Z. Sun, H. Mi, S. Sun, D. Ma and P. Zhang, J. Energy Chem., 2022, 67, 645–654 CrossRef CAS.
- J. Wang, J. G. Wang, X. Qin, Y. Wang, Z. You, H. Liu and M. Shao, ACS Appl. Mater. Interfaces, 2020, 12, 34949–34958 CrossRef CAS PubMed.
- D. Chao, C. R. Zhu, M. Song, P. Liang, X. Zhang, N. H. Tiep, H. Zhao, J. Wang, R. Wang, H. Zhang and H. J. Fan, Adv. Mater., 2018, 30, 1803181 CrossRef PubMed.
- G. Fang, C. Zhu, M. Chen, J. Zhou, B. Tang, X. Cao, X. Zheng, A. Pan and S. Liang, Adv. Funct. Mater., 2019, 29, 1808375 CrossRef.
- C. Li, X. Chi, J. Huang, J. Wu and Y. Liu, ACS Appl. Energy Mater., 2022, 5, 1478–1486 CrossRef CAS.
- X. Zeng, J. Liu, J. Mao, J. Hao, Z. Wang, S. Zhou, C. D. Ling and Z. Guo, Adv. Energy Mater., 2020, 10, 1904163 CrossRef CAS.
- J. Zhou, M. Xie, F. Wu, Y. Mei, Y. Hao, L. Li and R. Chen, Adv. Mater., 2022, 34, 2106897 CrossRef.
- X. Xie, H. Fu, Y. Fang, B. Lu, J. Zhou and S. Liang, Adv. Energy Mater., 2022, 12, 2102393 CrossRef CAS.
- X. Guo, J. Zhou, C. Bai, X. Li, G. Fang and S. Liang, Mater. Today Energy, 2020, 16, 100396 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta01487h |
| This journal is © The Royal Society of Chemistry 2023 |