
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Triquinoxalinediol as organic cathode material for rechargeable aqueous zinc-ion batteries†
Svit
Menart
 ab,
Klemen
Pirnat
*a,
David
Pahovnik
a and
Robert
Dominko
abc
ab,
Klemen
Pirnat
*a,
David
Pahovnik
a and
Robert
Dominko
abc
aNational Institute of Chemistry, Ljubljana, Slovenia. E-mail: klemen.pirnat@ki.si
bFaculty of Chemistry and Chemical Technology, University of Ljubljana, Ljubljana, Slovenia
cALISTORE-European Research Institute, Amiens, France
First published on 4th May 2023
Abstract
Aqueous zinc-ion batteries are one of the most promising candidates for large-scale energy storage applications. Herein, we report a rationally designed triquinoxalinediol (TQD) organic cathode material for aqueous zinc-ion batteries. Integration of hydroquinone units with hexaazatriphenylene (HAT) core enabled a high theoretical capacity of 669 mA h g−1 (based on a twelve-electron exchange reaction) and a practical reversible capacity of 503 mA h g−1 at 100 mA g−1. Both values are among the highest available in the literature. The charge storage mechanism was systematically studied using ex situ FT-IR and various electrochemical measurements revealing a co-insertion mechanism involving protons and zinc cations. The TQD suffers from significant capacity fading, which was attributed to the dissolution of active material in the electrolyte. That was supported by using in situ UV-vis spectroscopy. This work adds a promising material in a rapidly expanding mosaic of pyrazine/quinone organic cathode materials.
Introduction
Due to ever-increasing demands for the storage of surplus electrical energy generated by intermittent renewable energy sources, the development of new grid-scale storage technologies is of great importance. Currently, the electrochemical state-of-the-art storage is based on lithium-ion technology, which is limited by the availability of raw materials, high cost, and safety aspects connected with flammability of organic electrolytes.In recent years aqueous zinc-ion batteries have attracted a lot of attention as one of the most promising candidates for large-scale energy storage applications. The use of zinc metal offers advantages in terms of high specific capacity (820 mA h g−1), low toxicity, low cost, and high world resources, which are at least 20 times greater than Li. Its low redox potential (−0.76 V vs. SHE) and large overpotential for hydrogen evolution makes it one of the most suitable metal anodes for use with aqueous electrolytes typically showing much higher conductivities compared to nonaqueous electrolytes. Furthermore, aqueous zinc-ion battery components are stable in an ambient atmosphere, which simplifies production and decreases the cost per kW h of stored energy.1–3
The research of suitable zinc battery cathodes is predominantly focused on inorganic cathode materials such as manganese dioxides, Prussian blue analogs, and vanadium oxides. The mentioned cathode materials enable high capacities followed by fast capacity fading due to strong electrostatic interactions with a rigid lattice, large structural deformations, and material dissolution.4,5
Recently, several studies showed that organic cathode materials present a viable alternative to inorganic cathode materials. The flexibility of organic materials circumvents the slow solid-state diffusion limitations of inorganic cathode materials and enables efficient cycling even at higher rates. Organic cathode materials can accommodate different cations and have been proven to work in multivalent systems such as magnesium,6,7 aluminum,8,9 and zinc.10,11 Among many types of organic compounds tested in zinc batteries, such as stable organic radicals,12 conducting polymers,13 thiolates,14 triphenyl amines,15 dinitrobenzenes16 and phenothiazines,17 compounds based on carbonyls (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)11 and imines (CN)18 have attracted the most attention.
O)11 and imines (CN)18 have attracted the most attention.
Hexaazatrinaphthalene (HATN), a small organic cathode material,19 and its derivatives have already been explored as cathode materials in aqueous zinc-ion batteries.20 Herein, we present a novel HATN-based small organic cathode material derived by a synergistic combination of hexaazatriphenylene (HAT) core21 and hydroquinone motif enabling an increased theoretical capacity of 669 mA h g−1. The material triquinoxalinediol (TQD) exhibited the highest reported initial reversible capacity of 503.4 mA h g−1 at a current density of 100 mA g−1 (Table S1†). The charge storage mechanism was systematically studied using ex situ FT-IR and various electrochemical measurements revealing a coordination of the reduced species with protons or divalent zinc cations. Similar to other small organic cathode materials it suffers from significant capacity fading, which was attributed to the dissolution of active material in the electrolyte.
Results and discussion
Among many types of organic cathode materials, the integration of quinone and pyrazine units shows the best characteristics in terms of high capacity and stability.20,22–25 In this work, we aimed to improve the characteristics of recently reported small organic cathode material hexaazatrinaphthalenequinone (HATNQ), which delivered a high specific capacity of 482.5 mA h g−1.20 HATNQ possesses three quinone and three pyrazine redox-active centers, which enable a theoretical capacity of 515 mA h g−1 (Fig. 1). We envisaged that the removal of redox inactive benzene rings while keeping the redox centers intact could simultaneously increase the capacity and voltage of the material. Newly derived material triquinoxalinediol (TQD) enables an increased theoretical capacity of 669 mA h g−1 (Fig. 1). | ||
| Fig. 1 Design optimization strategy. | ||
TQD material was obtained through a multi-step synthesis (Fig. 2a and S1†) starting from a widely available 1,4-dimethoxybenzene precursor, which was first nitrated using nitric acid and afterward reduced to a diamine precursor using metallic tin. The synthesis of the HAT core was achieved through a condensation reaction between 3,6-dimethoxybenzene-1,2-diamine and hexaketocyclohexane octahydrate, a reaction analogous to the synthesis of HATNQ (Fig. S1†).20 In the last step of the synthesis, methyl ethers were deprotected using boron tribromide yielding the final hydroquinone TQD material. The structure of the TQD was confirmed with the use of various techniques. 1H NMR spectrum revealed two singlets with equal integral areas at 9.79 and 7.32 ppm, corresponding to –OH and aromatic protons, respectively. A small peak at 4.13 ppm is attributed to residual O–CH3 groups due to the incomplete deprotection of aromatic methyl ethers (Fig. 2b). The comparison between the integral area for –OH protons and –OCH3 revealed that only around 5% of the ethers remained protected. In the 13C NMR spectrum four distinguishable peaks at 146.4, 142.5, 133.6, and 113.6 match with four types of C atoms in TQD (Fig. 2c). Additionally, the synthesis of TQD is confirmed with MALDI-TOF mass spectrometry, which revealed two major sets of mass peaks corresponding to the [TQD + H]+ at 480.98 Da (calculated exact mass 481.09) and [TQD–CH3 + H]+ (TQD with one methyl-ether protecting group) at 494.99 Da (calculated exact mass 495.10 Da) (Fig. 2d). Smaller distributions belong to the same species ionized with Na+ instead of H+. The peaks corresponding to [M + 2H]+, [M + 3H]+, etc. species are observed due to the laser-induced photoreduction.26 Fourier transform infrared spectra (FT-IR) exhibited a broad peak between 3600 cm−1 and 3000 cm−1, which is attributed to O–H stretching and H2O vibrations. FT-IR spectrum exhibits a strong sharp peak at 1135 cm−1 attributed to C–O stretching vibration (Fig. 2e).
 | ||
| Fig. 2 (a) Synthetic route to TQD. (b) 1H NMR (DMSO-d6) spectrum of TQD. (c) 13C NMR (DMSO-d6) spectrum of TQD. (d) MALDI-TOF MS spectrum of TQD. (e) FT-IR spectra of TQD with an inset of zoomed-out region between 3600 and 600 cm−1. | ||
We obtained the electronic structures of TQD, oxidized TQD, and HATNQ with the use of the density functional theory (DFT) computational methods (Fig. 3). The pristine state of TQD represents a half-reduced state, which makes the comparison with HATNQ in its fully oxidized state irrelevant. To obtain a meaningful comparison, we calculated the electronic structure of hypothetical fully oxidized TQD. The comparison between the electronic structure of oxidized TQD and HATNQ shows a slightly lower energy gap (Eg) between HOMO and LUMO orbital (3.51 eV vs. 3.65 eV, respectively), suggesting that the intrinsic electronic conductivity of oxidized TQD is higher. The calculations showed that the LUMO energy of the oxidized TQD is lower (−3.88 eV) than that of HATNQ (3.48 eV) indicating greater electronic affinity corresponding to a higher reduction potential, which could also be explained by lower aromaticity of oxidized TQD in comparison with HATNQ. Similar observations have been shown in the literature, where LUMO differences between benzoquinone, naphthoquinone, and anthraquinone were correlated to aromaticity.27 The greater electronic affinity of oxidized TQD has been observed in galvanostatic measurements, where it delivered a higher initial discharge voltage than HATNQ (Fig. 4a).
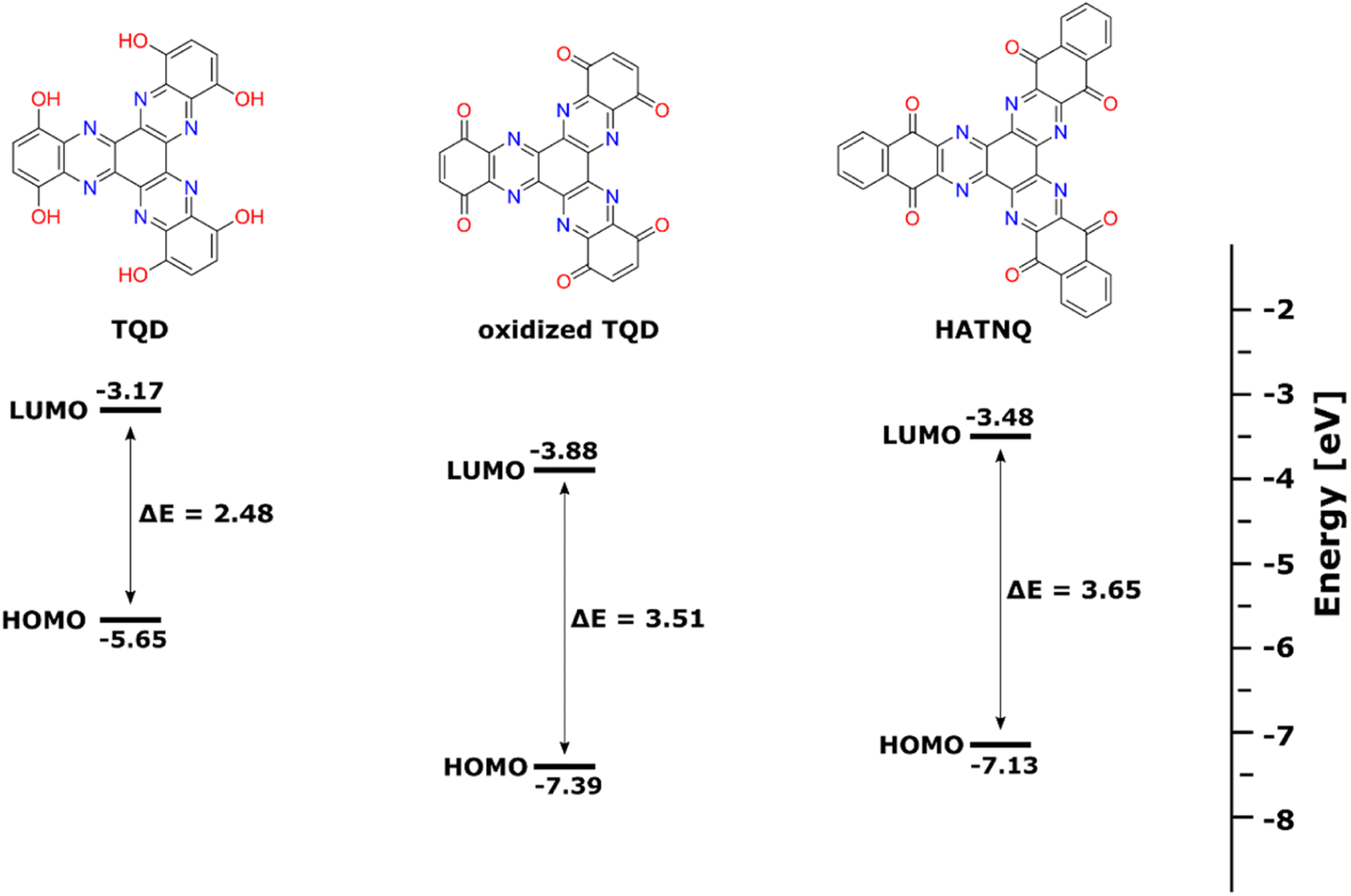 | ||
| Fig. 3 HOMO/LUMO energy levels of TQD, oxidized TQD, and HATNQ. | ||
 | ||
| Fig. 4 (a) Comparison of galvanostatic discharge/charge curves between TQD and HATNQ at 100 mA g−1, (b) cycling stability of TQD and HATNQ at 100 mA g−1, (c) discharge/charge curves of TQD at different cycles. | ||
Battery performance
The electrochemical performance of the synthesized TQD material was evaluated in a Swagelok Zn–organic battery cell using 4 M aqueous ZnSO4 solution as the electrolyte. The comparison between galvanostatic cycling of HATNQ and TQD at 100 mA g−1 shows that TQD slightly outperforms HATNQ in terms of capacity (503.4 mA h g−1vs. 471.2 mA h g−1, respectively) and average voltage (0.67 V vs. 0.64 V, respectively) (Fig. 4a). Both materials experience fast capacity fading, which is more pronounced in the case of TQD having capacity retention of 50.9% after 40 cycles (Fig. 4b). Charge/discharge curves of TQD show changes in the voltage plateaus in the first ten cycles, after which they exhibit constant shape with two less distinct sloping plateaus around 1.1 V and 0.4 V (Fig. 4c).The use of highly concentrated 4 M ZnSO4 electrolyte enabled slightly better cycling stability. In the comparison, the use of more diluted 1 M ZnSO4 caused faster capacity fading (Fig. 5a). A similar effect has already been observed in lithium organic batteries, where the use of concentrated electrolytes suppressed the dissolution of small organic cathode materials.28,29 The cycling stability of small organic cathode materials is influenced by the depth of discharge, which influences the amount of active material dissolution.22 To improve the cycling stability different cutoff voltages of 0.8 V, 0.6 V, 0.4 V, and 0.25 V were tested. After 150 cycles at 100 mA g−1 the capacity retention reached the values of 89.1%, 74.5%, 71.4%, and 36.2%, respectively (Fig. 5b and c). We have chosen a cutoff voltage of 0.4 V as the best compromise between capacity retention and specific capacity. The rate performance of TQD was performed in the cutoff voltage of 0.4 V, where the material reached capacities of 346 mA h g−1, 310 mA h g−1, 273 mA h g−1, 238 mA h g−1, 222 mA h g−1, 195 mA h g−1, 84 mA h g−1 at currents of 50 mA g−1, 100 mA g−1, 200 mA g−1, 500 mA g−1, 1 A g−1, 2 A g−1, and 5 A g−1 respectively (Fig. 5d and e). Many reports show long-term cycling stability at higher rates, which are of limited application for stationary grid storage, where moderate rates between 0.25C and 2C are needed.1 The use of higher rates can mask the instability of the material by utilizing a lower depth of discharge and shortened time period, which can lead to less dissolution.22 In comparison with the lower rate, the cycling stability of TQD at 1 A g−1 is expectedly better, reaching 70.7% capacity retention after 400 cycles (Fig. 5f).
 | ||
| Fig. 5 (a) Cycling stability of TQD in 1 M ZnSO4 and 4 M ZnSO4 at 100 mA g−1 in voltage window between 1.5 V and 0.25 V. (b) Charge/discharge curves of TQD in different cutoff voltages. (c) Cycling stability of TQD in different cutoff voltages at 100 mA g−1. (d and e) Rate performance of TQD in the cutoff voltages between 1.5 and 0.4 V. (f) Cycling stability of TQD at 1 A g−1 in the cutoff voltage of 0.4 V. *Measurements (b–f) were done using 4 M ZnSO4. | ||
Mechanism
The electrochemical reaction mechanism of TQD was investigated with the use of various techniques. Several recent works report on the co-storage mechanism of Zn2+ and H+ in small organic cathode materials.20,22–24Ex situ FT-IR spectra of the electrodes (60% TQD, 30% Printex XE2 carbon black, and 10% PTFE binder) in different charged states were measured in argon atmosphere after 5th full cycles using KBr pellets in transmittance mode. The broad peak at around 3430 cm−1, corresponding to the stretching vibration of O–H, was visible in both pristine (Fig. 6a, blue) and discharged states (Fig. 6a, red), while it disappeared in the charged state (Fig. 6a, black). Discharged state showed appearance of another peak at around 3240 cm−1 attributed to the N–H vibrations. Dynamic evolution of peaks associated with O–H and N–H vibrations indicates on H+ insertion mechanism. A peak at 1690 cm−1 attributed to CO stretching vibration emerged in a charged state and was not visible in the discharged state. The peak also overlaps with peaks in the pristine state, which could be explained by the partial oxidation of the material during electrode preparation and drying, since the pure TQD did not exhibit peaks in that area (Fig. 2e). A peak at around 1135 cm−1 was attributed to C–O vibrations. The C–O peak is less pronounced in the pristine state due to partial oxidation during electrode preparation and is noticeably increased in the discharged state. In the charged state, the peak completely disappeared. Attributing peaks in that area is less reliable due to overlapping peaks of PTFE binder, which exhibits two strong peaks at 1212 cm−1 and 1154 cm−1. Emergence and disappearance of peak attributed to C–O vibrations together with changing of O–H peaks serve as additional proof of activity of CO, which was also reported for HATNQ (Fig. 8).20
 | ||
| Fig. 6 (a) FT-IR spectra of TQD electrodes at different states of charge including pristine (blue), discharged to 0.25 V in 5th cycle (red) and charged to 1.5 V in 5th cycle (black). (b) Zoomed view of FT-IR spectra between 1800 and 600 cm−1. | ||
To further elucidate the possible H+ and Zn2+ co-insertion mechanism three-electrode cyclic voltammetry (CV) measurements with Ag/AgCl reference electrode and Pt counter electrode were performed. Four aqueous electrolytes were tested, including 4 M ZnSO4 with a measured pH value of 3.3, 4 M ZnSO4 + H2SO4 (pH = 1), H2SO4 (pH = 1), and H2SO4 (pH = 3.3) (Fig. 7a, b and S3†). CV measurement in 4 M ZnSO4 showed four couples of distinguishable cathodic and anodic peaks located at −0.46/−0.68 V, −0.06/−0.22 V, 0.10/0.03 V, and 0.29/0.22 V, which roughly coincide with d(Q − Q0)/dE peaks obtained with galvanostatic cycling in Zn–TQD battery (Fig. 7c). The comparison showed differences in the peak intensities, which could arise due to the differences in the used scan rate (galvanostatic mode used approximately 110 times slower scan rate than CV). Solution of H2SO4 (pH = 3.3), which simulates the concentration of H+ ions in 4 M ZnSO4 was used to test the possibility of H+ insertion into TQD without the interference of Zn2+ ions. The measurement revealed that TQD can react with H+ ions even at very low H+ concentrations (Fig. 7b). On the other hand, it showed approximately much lower electroactivity in comparison with 4 M ZnSO4 electrolyte, which indicates the need of Zn2+ co-insertion for complete utilization (Fig. 7a, green). The use of more concentrated H2SO4 (pH = 1) electrolyte provided peak currents comparable to the 4 M ZnSO4 electrolyte (Fig. 7a, blue). The measurement exhibited four sets of cathodic and anodic peaks, which after shifting according to the Nernst equation for the conditions in H2SO4 (pH = 3.3) roughly coincide with CV peaks in 4 M ZnSO4 electrolyte further suggesting the possibility of H+ insertion in 4 M ZnSO4 electrolyte (Fig. S3a†).
 | ||
| Fig. 7 (a) CV curves of TQD measured by a three-electrode system at 10 mV s−1 in 4 M ZnSO4 (black), 4 M ZnSO4 + H2SO4 (pH = 1) (red), H2SO4 pH = 1 (blue), and H2SO4 pH = 3.3 (green). (b) CV curve of TQD measured at 2 mV s−1 in H2SO4 pH = 3.3. (c) Comparison between d(Q − Q0)/dE curve obtained from the 5th galvanostatic cycle in Zn-battery using 4 M ZnSO4 electrolyte (black) and a CV curve measured by a three-electrode system at 10 mV s−1 and shifted according to Zn/Zn2+vs. Ag/AgCl = −0.957 V vs. Ag/AgCl. (d) Charge/discharge curves of TQD at 100 mA g−1 in different electrolytes including 4 M ZnSO4 (red, 3rd cycle), 2.2 M Zn(OTf)2 in 70% PEG (black, 5th cycle) and 1 M Zn(TFSI)2 in G2 (blue, 1st cycle). (e) Galvanostatic charge/discharge curves of TQD in 2.2 M Zn(OTf)2 in 70% PEG at 100 mA g−1. (f) Comparison of cycling stability of TQD in 2.2 M Zn(OTf)2 in 70% PEG and 4 M ZnSO4 at 100 mA g−1. | ||
Difference of CV curves in 4 M ZnSO4 (pH = 1) and H2SO4 (pH = 1) electrolytes with same H+ activity further indicate on Zn2+ co-insertion in electrolytes containing Zn2+ ions (Fig. 7a, red). To prove the possibility of Zn2+ insertion into TQD we employed Zn–TQD battery with water-free electrolyte 1 M zinc bis(trifluoromethylsulfonyl)imide Zn(TFSI)2 in diglyme (G2),22 which excluded the presence of H+ ions (Fig. 7d, blue curve). Although the system exhibited significantly lower capacity in comparison with 4 M ZnSO4 electrolyte (78.5 mA h g−1vs. 503.4 mA h g−1), it proved the possibility of Zn2+ insertion without H+ co-insertion. Because the choice of the electrolyte can have a profound effect on galvanostatic cycling additional “Zn2+ ion favoring” electrolyte 2.2 M zinc triflate (Zn(OTf)2) in 70% polyethylene glycol (PEG) was tested, where insertion of H+ ions is suppressed due to the strong interactions between PEG and H2O.30 In comparison with 1 M Zn(TFSI)2 in G2 the system exhibited a lot higher specific capacity of 324.8 mA h g−1, which was still significantly lower than the value obtained in 4 M ZnSO4. These results serve as additional proof of the synergistic role of the Zn2+ and H+ co-insertion mechanism. Although galvanostatic cycling of TQD in 2.2 M Zn(OTf)2 in 70% PEG in water exhibited lower specific capacity than in 4 M ZnSO4, it delivered a higher average voltage (0.83 V vs. 0.67 V, respectively) and higher capacity retention after 80 cycles at 100 mA g−1 (72.9% vs. 42.7%) (Fig. 7f). Utilization of water-suppressing electrolytes could therefore serve as another tool for the stabilization of cycling stability of small organic cathode materials, where a possible explanation could lie in a lower solubility of the discharged products in less polar PEG.
Based on the aforementioned results and results from the literature,20,22 we propose a redox mechanism where TQD is reversibly switching from a fully oxidized quinone state to a fully reduced state utilizing CN and CO redox centers with H+ and Zn2+ co-insertion (Fig. 8).
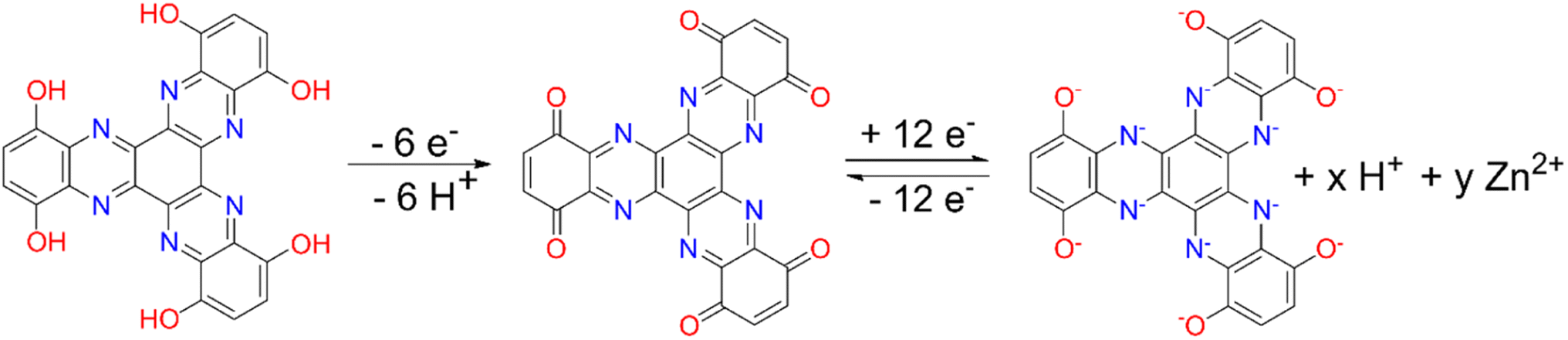 | ||
| Fig. 8 Proposed redox mechanism of TQD in aqueous Zn battery. | ||
Capacity fading
Small organic cathode materials are prone to significant capacity fading due to the dissolution of the active material in electrolyte.11,14 We observed the brown coloring of the separators during the Swagelok disassembly process (Fig. 9a). Additionally, we performed an in situ UV-vis experiment to link the capacity fading during the galvanostatic cycling experiment with the dissolution of the active material in the electrolyte. The experiment was conducted in a UV-vis cuvette with zinc strip as anode and TQD electrode slurry pressed on stainless steel mesh as cathode (Fig. 9c). Although we were unable to determine the structure of soluble species using NMR spectroscopy due to too low concentration of the solutes, the gradual increase of absorbance during cycling could serve as an indirect proof of dissolution of the active material in the electrolyte and a probable explanation for capacity fading (Fig. 9b). The evolution of absorbance at 280 nm in an in situ UV-vis experiment shows the correlation between capacity fading and an increase in absorbance (Fig. 9d). Ex situ UV-vis experiment of cathodes in charged (1.5 V) and discharged (0.25 V) states submerged into 3 M ZnSO4 for 2 days revealed dissolution of the active material in the discharged state with maximum absorbance at 280 nm and no apparent dissolution in a charged state (Fig. 9e), which could explain greater cycling stability in higher cut-off voltages. | ||
| Fig. 9 (a) Photo of separators after cycling in 3 M ZnSO4. (b) In situ UV-vis spectrum during galvanostatic cycling of TQD at 100 mA g−1 in 3 M ZnSO4. (c) In situ UV-vis cell configuration. (d) Evolution of absorbance at 280 nm and discharge capacity during in situ UV-vis experiment. (e) Ex situ UV-vis spectrum of electrodes submerged in 3 M ZnSO4 in charged (red) and discharged (black) state. | ||
Conclusions
In summary, we have rationally designed a novel high-performance small organic cathode material in which we successfully integrated hydroquinone and hexaazatriphenylene motifs. Newly synthesized TQD delivered a high initial reversible capacity of 503.4 mA h g−1 at 100 mA g−1 (Ctheo = 669 mA h g−1). Investigation of the redox mechanism revealed a synergistic effect of Zn2+/H+ co-insertion for full capacity utilization. We believe that in the future the materials synthesis could be further optimized yielding completely deprotected material with even higher reversible capacity. With the use of UV-vis in situ spectroscopy, we show that material dissolution has a detrimental effect on cycling stability. We observed stabilizing effect of water-suppressing electrolyte 2.2 M Zn(OTf)2 in 70% PEG, which could in the future be coupled with other dissolution-suppressing strategies, such as the utilization of Nafion membranes.11,14 All in all our study adds another new material to the rapidly expanding mosaic of high-performance quinone/pyrazine organic cathode materials.Author contributions
S. Menart conceived the research and wrote original draft. K. Pirnat supervised the research and is corresponding author. D. Pahovnik conducted MALDI-TOF and it's interpretation. R. Dominko is head of the lab, project administration, funding acquisition. All authors contributed to the writing, revising and editing of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Slovenian Research Agency (ARRS) young researcher scheme and research core funding [P2-0423], [P2-0145] and projects [N2-0214], [N2-0165], and Ministry of Education, Science and Sport (MIZS) for funding M.Era-net project InsBioration (call 2021).References
- C. Li, S. Jin, L. A. Archer and L. F. Nazar, Joule, 2022, 6, 1733–1738 CrossRef.
- B. Tang, L. Shan, S. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 RSC.
- L. Hu, P. Xiao, L. Xue, H. Li and T. Zhai, EnergyChem, 2021, 3, 100052 CrossRef CAS.
- D. Selvakumaran, A. Pan, S. Liang and G. Cao, J. Mater. Chem. A, 2019, 7, 18209–18236 RSC.
- C. Li, X. Zhang, W. He, G. Xu and R. Sun, J. Power Sources, 2020, 449, 227596 CrossRef CAS.
- A. Vizintin, J. Bitenc, A. K. Lautar, J. Grdadolnik, A. R. Vitanova and K. Pirnat, ChemSusChem, 2020, 13, 2328–2336 CrossRef CAS PubMed.
- T. Bančič, J. Bitenc, K. Pirnat, A. K. Lautar, J. Grdadolnik, A. R. Vitanova and R. Dominko, J. Power Sources, 2018, 395, 25–30 CrossRef.
- J. Bitenc, N. Lindahl, A. Vizintin, M. E. Abdelhamid, R. Dominko and P. Johansson, Energy Storage Mater., 2020, 24, 379–383 CrossRef.
- X. Peng, Y. Xie, A. Baktash, J. Tang, T. Lin, X. Huang, Y. Hu, Z. Jia, D. J. Searles, Y. Yamauchi, L. Wang and B. Luo, Angew. Chem., Int. Ed., 2022, 61, e202203646 CAS.
- S. Zheng, Q. Wang, Y. Hou, L. Li and Z. Tao, J. Energy Chem., 2021, 63, 87–112 CrossRef CAS.
- Q. Zhao, W. Huang, Z. Luo, L. Liu, Y. Lu, Y. Li, L. Li, J. Hu, H. Ma and J. Chen, Sci. Adv., 2018, 4, eaao1776 Search PubMed.
- E. Schröter, L. Elbinger, M. Mignon, C. Friebe, J. C. Brendel, M. D. Hager and U. S. Schubert, J. Power Sources, 2023, 556, 232293 CrossRef.
- H.-Y. Shi, Y.-J. Ye, K. Liu, Y. Song and X. Sun, Angew. Chem., 2018, 130, 16597–16601 CrossRef.
- M. R. Tuttle, C. Walter, E. Brackman, C. E. Moore, M. Espe, C. Rasik, P. Adams and S. Zhang, Chem. Sci., 2021, 12, 15253–15262 RSC.
- H. Glatz, E. Lizundia, F. Pacifico and D. Kundu, ACS Appl. Energy Mater., 2019, 2, 1288–1294 CrossRef CAS.
- Z. Song, L. Miao, H. Duan, L. Ruhlmann, Y. Lv, D. Zhu, L. Li, L. Gan and M. Liu, Angew. Chem., 2022, 134, e202208821 Search PubMed.
- H. Cui, T. Wang, Z. Huang, G. Liang, Z. Chen, A. Chen, D. Wang, Q. Yang, H. Hong, J. Fan and C. Zhi, Angew. Chem., 2022, 134, e202203453 Search PubMed.
- J. Li, L. Huang, H. Lv, J. Wang, G. Wang, L. Chen, Y. Liu, W. Guo, F. Yu and T. Gu, ACS Appl. Mater. Interfaces, 2022, 14, 38844–38853 CrossRef CAS PubMed.
- Z. Tie, L. Liu, S. Deng, D. Zhao and Z. Niu, Angew. Chem., Int. Ed., 2020, 59, 4920–4924 CrossRef CAS PubMed.
- Y. Chen, J. Li, Q. Zhu, K. Fan, Y. Cao, G. Zhang, C. Zhang, Y. Gao, J. Zou, T. Zhai and C. Wang, Angew. Chem., Int. Ed., 2022, e202116289 CAS.
- J. L. Segura, R. Juárez, M. Ramos and C. Seoane, Chem. Soc. Rev., 2015, 44, 6850–6885 RSC.
- Y. Gao, G. Li, F. Wang, J. Chu, P. Yu, B. Wang, H. Zhan and Z. Song, Energy Storage Mater., 2021, 40, 31–40 CrossRef.
- W. Wang, V. S. Kale, Z. Cao, Y. Lei, S. Kandambeth, G. Zou, Y. Zhu, E. Abouhamad, O. Shekhah, L. Cavallo, M. Eddaoudi and H. N. Alshareef, Adv. Mater., 2021, 33, 2103617 CrossRef CAS PubMed.
- Y. Wang, X. Wang, J. Tang and W. Tang, J. Mater. Chem. A, 2022, 10, 13868–13875 RSC.
- L. Lin, Z. Lin, J. Zhu, K. Wang, W. Wu, T. Qiu and X. Sun, Energy Environ. Sci., 2023, 16, 89–96 RSC.
- M. P. Napolitano, P. C. Kuo, J. V. Johnson, J. Arslanoglu and R. A. Yost, Int. J. Mass Spectrom., 2017, 421, 14–24 CrossRef CAS.
- K. C. Kim, T. Liu, S. W. Lee and S. S. Jang, J. Am. Chem. Soc., 2016, 138, 2374–2382 CrossRef CAS PubMed.
- T. Cai, Y. Han, Q. Lan, F. Wang, J. Chu, H. Zhan and Z. Song, Energy Storage Mater., 2020, 31, 318–327 CrossRef.
- Z. Liu, T. Xu, X. Meng, Y. Zhao, Q. Lan, Y. Liu, Z. Song, J. Qin and H. Zhan, Batteries Supercaps, 2022, e202200436 Search PubMed.
- C. Li, R. Kingsbury, L. Zhou, A. Shyamsunder, K. A. Persson and L. F. Nazar, ACS Energy Lett., 2022, 7, 533–540 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta01400b |
| This journal is © The Royal Society of Chemistry 2023 |