
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Covalent organic framework derived synthesis of Ru embedded in carbon nitride for hydrogen and oxygen evolution reactions†
Tianyu
Gao‡
a,
Kilaparthi Sravan
Kumar‡
b,
Zhen
Yan
c,
Maya
Marinova
d,
Martine
Trentesaux
a,
Mohammed A.
Amin
 e,
Sabine
Szunerits
b,
Yong
Zhou
a,
Vlad
Martin-Diaconescu
f,
Sébastien
Paul
*a,
Rabah
Boukherroub
*b and
Vitaly
Ordomsky
*a
e,
Sabine
Szunerits
b,
Yong
Zhou
a,
Vlad
Martin-Diaconescu
f,
Sébastien
Paul
*a,
Rabah
Boukherroub
*b and
Vitaly
Ordomsky
*a
aUniv. Lille, CNRS, Centrale Lille, ENSCL, Univ. Artois, UMR 8181 – UCCS – Unité de Catalyse et Chimie du Solide, F-59000 Lille, France. E-mail: sebastien.paul@centralelille.fr; vitaly.ordomsky@univ-lille.fr
bUniv. Lille, CNRS, Univ. Polytechnique Hauts-de-France, UMR 8520 – IEMN, F-59000 Lille, France. E-mail: rabah.boukherroub@univ-lille.fr
cE2P2L, UMI 3464 CNRS-Solvay, 3966 Jin Du Rd, 201108 Shanghai, China
dUniv. Lille, CNRS, INRAE, Centrale Lille, Univ. Artois, FR 2638 IMEC Institut Michel-Eugène Chevreul, F-59000 Lille, France
eDepartment of Chemistry, College of Science, Taif University, P.O. Box 11099, Taif 21944, Saudi Arabia
fALBA Synchrotron – CELLS, Carrer de la Llum 2-26, 08290 Cerdanyola del Vallès, Barcelona, Spain
First published on 7th August 2023
Abstract
The hydrogen and oxygen evolution reactions (HER and OER) are two steps in electrochemical water splitting for conversion of electric power to chemical energy. The main challenge remains the development of efficient, stable and cheap electrocatalysts able to perform both reactions under alkaline conditions. Single atom Ru stabilized by nitrogen-doped carbon and RuO2 are currently the materials of choice for the HER and OER, respectively. Here, we propose a strategy for the preparation of Ru embedded in a carbon nitride matrix for efficient HER and OER in KOH solution. It is based on the preparation of a covalent organic framework 2D CIN-1 structure with coordinated RuII, producing Ru oxide nanoparticles with low valence Ru sites arranged in the form of nanowires between layers of graphitic carbon nitride after pyrolysis. The material demonstrates smaller overpotentials for the HER and OER in comparison with benchmark Pt and RuO2 catalysts, and high catalytic stability.
1. Introduction
Hydrogen (H2) is considered as a promising and preferred alternative to fossil fuels due to its high energy density of 120 kJ g−1 and eco-friendly emission. Electrocatalytic water splitting, an effective and environmentally friendly technology for H2 production, has attracted great attention over the last few years1–4 and is based on the cathodic hydrogen evolution reaction (HER) and anodic oxygen evolution reaction (OER).5 Noble metal-based catalysts, especially Pt-based catalysts,6 are at the forefront in boosting the sluggish OER and HER kinetics and remain the benchmark materials for both reactions due to their favorable kinetics for the HER and OER.7 However, the high cost of Pt metal limits its consideration for commercial and large scale applications for water splitting.Over the last few decades, a large number of alternative electrocatalysts have been proposed, including non-noble metal and metal-free catalysts.8–12 Ruthenium is one of the best Pt analogs as it possesses a similar metal hydride (M–H) bond strength of 65 kcal mol−1,13,14 and is more available and five times cheaper than Pt, which makes it popular in catalysis.15,16 Various scientific reports revealed that the HER performance of Ru is comparable to or even better than that of Pt, in both neutral and alkaline media. Recently, Baek et al. synthesized Ru nanoparticles (∼2 nm) uniformly dispersed on graphene nanoplatelets (Ru@GnP), and underlined the outstanding HER performance in both acidic and alkaline electrolytes with small overpotentials of 13 mV in 0.5 M H2SO4 and 22 mV in 1 M KOH at a current density of 10 mA cm−2.17 Feng et al. reported the electrochemical performance of ruthenium/N-doped carbon (Ru/CN), prepared by electrochemical polymerization of aniline on graphite foam (GF) and dipping the obtained aniline/GF in Ru salt, followed by subsequent pyrolysis of the Ru3+/aniline complex at 900 °C. The resulting material has shown excellent electrocatalytic HER activity with an overpotential of 21 mV (10 mA cm−2) in 1 M KOH.18 Qiu et al. synthesized Ru-based electrocatalysts with abundant Ru active sites using bimetallic MOFs (CuRu-MOF) through pyrolysis and etching of Cu. The as-prepared ultrafine Ru nanoparticles anchored onto hierarchically porous carbon (Ru–HPC) exhibited outstanding HER activity with a low Tafel slope value of 33.9 mV dec−1, even lower than that of commercial 20% Pt/C (41 mV dec−1).19
Although recent publications proposed the use of transition metals for the OER,20 the rutile phase of ruthenium oxide (RuO2) still serves as a benchmark material for the OER in both acidic and alkaline media.21,22 The OER activities have been studied for different particle sizes, different crystal structures, and different degrees of hydration and electronic states.23–25 RuO2 is limited by its low anodic stability under acidic conditions, especially at high overpotentials as well as by its reduced energy density (≈400 W h kg−1, theoretically), and related low mass activity.25
Considering that Ru could be used for both the HER and OER, it would be highly desirable to develop a Ru-based electrocatalyst for efficient water splitting. Ru@RuO2 core–shell nanorods, synthesized by oxidation of Ru nanorods, indeed demonstrated high efficiency for both the OER and HER reaching overpotentials of 320 and 137 mV, respectively, at a current density of 10 mA cm−2.26 A possible strategy to improve the electrocatalytic performance would be through the design of catalysts containing highly dispersed Ru stabilized by nitrogen-doped carbon to provide both Ru and RuO2 at the same time. Ru–N bonding provides several important advantages in comparison with other materials related to the favorable electronic structure of ruthenium, the synergistic effects between Ru and N-doped carbon in reducing water dissociation energy, improved adsorption properties of intermediate reactants and high stability of the Ru species avoiding metal sintering.27,28
Recently, solid-state pyrolysis of porous organic networks such as MOFs was widely applied for the facile preparation of metal nanostructures incorporated in porous carbon materials.29–33 Covalent organic frameworks (COFs) are a new type of microporous materials which are constructed by the covalent linkage of organic building blocks.34,35 Because of ordered pores, carbon-rich layers and diverse heteroatoms, COFs are promising precursors for the synthesis of carbon-based materials. Surprisingly, they have not been yet used for the preparation of carbon nitrides.
In this work, we synthesized a 2D imine-based COF with an incorporated RuII–2-diphenylphosphinobenzaldehyde complex. The pyrolysis of this material at 500 °C under a N2 atmosphere produced highly dispersed RuO2 nanoparticles containing low valence Ru sites between the layers of carbon nitride. The material provided a high activity for the HER and OER in 1 M KOH.
2. Experimental
2.1. Materials and reagents
RuCl3·H2O, 2-(diphenylphosphino)benzaldehyde (DBP), melamine, 1,3,5-triformylbenzene, 1,4-diaminobenzene, 1,4-dioxane and acetic acid were purchased from Sigma-Aldrich. Piperazinedicarbaldehyde, N,N-dimethylformamide (DMF), dry ethanol, acetone, dichloromethane (CH2Cl2) and tetrahydrofuran (THF) were procured from Alfa Aesar. Dimethyl sulfoxide (DMSO) was obtained from Acros Organics.2.2. Synthesis of materials
Ru@CIN-1 and pure CIN-1 were calcined under a N2 atmosphere at 500 °C for 2 h to yield Ru@g-CNx and g-CNx, respectively.
2.3. Characterization
The analysis of Ru and P was performed with the use of an energy dispersive micro X-ray fluorescence spectrometer M4 TORNADO (Bruker). CHNO analysis was performed using a FlashSmart automated analyzer. N, C and H were detected as N2, CO2 and H2O, respectively. The resulting gases were separated on a packed column and detected by using a TCD detector. Oxygen was analyzed separately by pyrolysis with CO analysis.Fourier transform infrared spectroscopy (FTIR) experiments were carried out with a Thermo Fisher Scientific Nicolet 6700 FTIR instrument (32 scans at a resolution of 4 cm−1) equipped with a mercury cadmium telluride (MCT) detector.
The Raman spectra were recorded on a XploRA Raman confocal microscope from Horiba Jobin Yvon. A 532 nm diode laser was used to excite the samples. The scattered light was guided through a 150 μm pinhole, dispersed and collected using a Peltier-cooled CCD. The laser power was reduced using density filters in order to avoid sample alteration.
The X-ray diffraction patterns (XRD) were recorded on a PANalytical Empyrean X-ray diffractometer in the Bragg–Brentano configuration with a 0.02° step size and 1 s step time. Cu Kα radiation (40 kV and 30 mA) was used as the X-ray source. The interplanar distances were calculated using the Bragg equation:  where λ is the wavelength of X-ray radiation (λ = 1.5406 Å) and θ is the diffraction angle of the peak.
where λ is the wavelength of X-ray radiation (λ = 1.5406 Å) and θ is the diffraction angle of the peak.
The Brunauer–Emmett–Teller (BET) surface area, pore volume, pore diameter and N2 sorption–desorption isotherms were measured on a Micromeritics Tristar Model 3020 Surface Area and Porosimetry analyzer. The samples (100 mg) were degassed under vacuum at 250 °C for 2 h prior to N2 physisorption.
Thermogravimetric analysis (TGA) measurements were performed using an SDT 2960 analyzer from TA Instruments under air flow (50 mL min−1) with a temperature ramping rate of 10 °C min−1.
Scanning transmission electron microscopy (STEM) analysis was performed on a Titan Themis 300 S/TEM equipped with a probe aberration corrector and a monochromator, allowing a special resolution of 70 pm and an energy resolution of 150 meV. The microscope is equipped with a Super-X windowless 4 quadrant SDD (silicon drift detector) detection system for the STEM-energy-dispersive X-ray (EDX) mapping and several annual dark field detectors. The experiment was performed with a spot size of about 500 pm, a semi-convergence angle of 21 mrad, and a probe current of approximately 100 pA. For the high-angle annular dark-field (HAADF) images, the collection angles were between 50 and 200 mrad. The STEM-EDX mapping was acquired with a dwell time of 15 μm per px with continuous scanning over several frames during a total time of 10–15 min per acquisition. The microscope is also equipped with a CETA camera with a 4k × 4k CMOS sensor for the TEM images and diffraction acquisition.
Ru K-edge X-ray absorption spectra (XAS) of samples diluted in cellulose were acquired at room temperature in the form of pellets. XAS spectra were collected in transmission mode for references Ru black, RuCl3 and RuO2 and fluorescence mode for Ru@ClN-1 and Ru@g-CNx employing a Si311 double crystal monochromator and a 6 channel multi-element SDD detector available at the CLAESS beamline of the ALBA Synchrotron. Several XAS repeats were collected to ensure reproducibility and statistics. The averaged spectra were treated by using the Athena software package.37 The energy scale was calibrated by setting the first inflection point of the Ru metal spectra taken as 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 117 eV. EXAFS spectra were extracted using the AUTOBK algorithm employing an Rbkg of 1.1 in the 0 to 15.3 Å−1 region of k-space. The FEFF6 code38,39 was used for scattering path generation, and multi (k1, k2, k3)-weighted fits of the data were carried out in r-space and a k-range of 3–14 Å−1. The S02 value was set to 0.9, and a global E0 was employed with the initial E0 value set to the first inflection point of the rising edge. Scattering paths were fit in terms of Δreff and σ2, which represent the deviation from the expected interatomic distances and the structural disorder, respectively. To assess the goodness of the fits, both the Rfactor (% R) and the reduced χ2 (χv2) were minimized, ensuring that the data were not over-fit. An increase in the number of variables is generally expected to improve the Rfactor; however χv2 may go through a minimum and then increase, which is an indication that the model is over-fitting the data.40 The best fit models were determined using a grid search with fixed values for path coordination numbers by employing Larch, the Python implementation of Artemis.41
117 eV. EXAFS spectra were extracted using the AUTOBK algorithm employing an Rbkg of 1.1 in the 0 to 15.3 Å−1 region of k-space. The FEFF6 code38,39 was used for scattering path generation, and multi (k1, k2, k3)-weighted fits of the data were carried out in r-space and a k-range of 3–14 Å−1. The S02 value was set to 0.9, and a global E0 was employed with the initial E0 value set to the first inflection point of the rising edge. Scattering paths were fit in terms of Δreff and σ2, which represent the deviation from the expected interatomic distances and the structural disorder, respectively. To assess the goodness of the fits, both the Rfactor (% R) and the reduced χ2 (χv2) were minimized, ensuring that the data were not over-fit. An increase in the number of variables is generally expected to improve the Rfactor; however χv2 may go through a minimum and then increase, which is an indication that the model is over-fitting the data.40 The best fit models were determined using a grid search with fixed values for path coordination numbers by employing Larch, the Python implementation of Artemis.41
The X-ray photoelectron spectroscopy (XPS) experiments were carried out using a Kratos Axis Ultra DLD spectrometer, equipped with a monochromatic Al Kα X-ray source (1486.6 eV) operating at 225 W (15 kV, 15 mA). The instrument base pressure was 5 × 10−10 torr and a charge neutralizing system was used for all acquisitions. Analysis was performed at a pass energy of 40 eV and a step size of 0.1 eV. Binding energies (BE) were referenced to the C peak at 285.5 eV.
2.4. Electrochemical methods
The electrocatalytic measurements were performed using a ModuLab-MTS electrochemical test station (Solartron, France) with a conventional three-electrode configuration consisting of a 3 mm diameter glassy carbon (GC) electrode coated with Ru@g-CNx, a saturated calomel electrode (SCE), Hg|Hg2Cl2, and Pt coil as the working, reference and counter electrodes, respectively. Before starting the experiments, an aqueous electrolyte solution (1 M KOH) was degassed using nitrogen for at least 30 min. Furthermore, linear sweep voltammetry (LSV) was performed at a scan rate of 5 mV s−1 to record the polarization curves.In order to determine the electrocatalytically active surface area (ECSA), double-layer capacitance (Cdl) was calculated by performing cyclic voltammetry (CV) at different scan rates in the non-faradaic potential range (0.11 to 0.24 V vs. RHE) of the material. The Cdl (μF cm−2) value is determined from the slope of the current density (mA cm−2) as a function of the scan rate (mV s−1) plot. Electrochemical impedance spectroscopy (EIS) was performed at an AC amplitude of 10 mV in a frequency range from 1 kHz to 0.1 Hz at −0.03 V vs. RHE. The long-term stability experiments were conducted using the chronopotentiometric technique at a constant current density of −10 mA cm−2. The reference electrode (SHE) was calibrated to the reversible hydrogen electrode (RHE) according to the Nernst equation:
| E(RHE) = E(Hg/Hg2Cl2) + (0.242 + 0.059 × pH) |
The pH of the electrolyte solution (1 M KOH) was experimentally determined using a Mettler Toledo pH meter and found to be 13.6 (average of three sets). Hence, we have used this value for calibration during all electrocatalytic experiments. All the electrochemical analyses were conducted without iR correction.
In order to prepare the catalyst-modified GC working electrode, 3 mg of the desired catalyst was dispersed in 1 mL of water by ultrasonication for 30 min and then 10 μL of the catalyst suspension was drop-casted onto the GC electrode and dried at 60 °C overnight. Hence, the mass loading on the electrode surface was 0.042 mg cm−2. Furthermore, all the measurements were repeated at least three times to confirm the reproducibility of the material. The turnover frequency (TOF, s−1) measurements were carried out using the formula  where I is the current (A), x is the number of electrons transferred during the HER (2) and OER (4), F is the Faraday constant (96485C) and n is the number of active sites calculated by using cyclic voltammograms.42
where I is the current (A), x is the number of electrons transferred during the HER (2) and OER (4), F is the Faraday constant (96485C) and n is the number of active sites calculated by using cyclic voltammograms.42
3. Results and discussion
3.1. COF derived synthesis of Ru@g-CNx
A covalent organic framework CIN-1 was synthesized by condensation of piperazinedicarbaldehyde with melamine (Fig. 1). The CHNO analysis revealed the absence of oxygen in CIN-1 with a composition close to the theoretical values (C, 50.5%; N, 44.2%; H, 5.3%) for condensation of an aldehyde with melamine (Table 1). The incorporation of 0.4 wt% of Ru was achieved by the addition of 2-(diphenylphosphino)benzaldehyde as a ligand coordinating with RuCl3 during the crystallization of CIN-1, keeping the composition similar to that of CIN-1. | ||
| Fig. 1 Schematic illustration of the synthesis of a Ru carbon nitride catalyst. | ||
| Materials | Composition (wt%) | S BET (m2 g−1) | Pore volume (cm3 g−1) | |||||
|---|---|---|---|---|---|---|---|---|
| C | O | N | H | P | Ru | |||
| CIN-1 | 47.0 | — | 48.7 | 4.3 | — | — | 419.5 | 0.26 |
| Ru@CIN-1 | 47.0 | 0.1 | 47.5 | 4.3 | 0.1 | 0.4 | 400.2 | 0.20 |
| g-CNx | 53.0 | 10.7 | 33.9 | 2.4 | — | — | 61.8 | 0.21 |
| Ru@g-CNx | 39.7 | 10.9 | 33.9 | 2.5 | 1.2 | 0.9 | 23.9 | 0.18 |
The pyrolysis of the prepared COF materials by treatment of CIN-1 and Ru@CIN-1 at 500 °C under N2 flow was used to prepare graphitic materials. The chemical analysis of g-CNx and Ru@g-CNx revealed a decrease in the hydrogen content and an increase in the contribution of carbon in comparison with that of nitrogen (Table 1), which could be assigned to the partial deamination of the material. The increase in the oxygen content could be ascribed to partial oxidation of the material during contact with air after pyrolysis. The content of Ru in Ru@g-CNx increased to 0.9 wt% during pyrolysis due to a decrease in the contribution of C, H and N in the material.
X-ray diffraction (XRD) analysis of pure CIN-1 (Fig. 2a) showed only one broad peak at 20° corresponding to the (001) reflection related to interplanar stacking.43 As reported in the literature, microporous polymers based on Schiff base chemistry are usually amorphous.44 It is interesting to note that Ru@CIN-1 additionally displayed a sharp peak at 11.9° ascribed to the (100) inplanar structural packing of CIN-1. The increased crystallinity could be assigned to the incorporation of the Ru–DBP complex into the CIN-1 structure which increases the size of the network layer.45 The XRD patterns of g-CNx and Ru@g-CNx, after pyrolysis (Fig. 2a), exhibited a distinct shift of the diffraction peak to 26.3° in comparison with those of parent materials; this peak is typical of the (002) diffraction plane for interlayer stacking in graphitic carbon nitrides with a distance between layers of about 0.34 nm, according to the Bragg equation.46
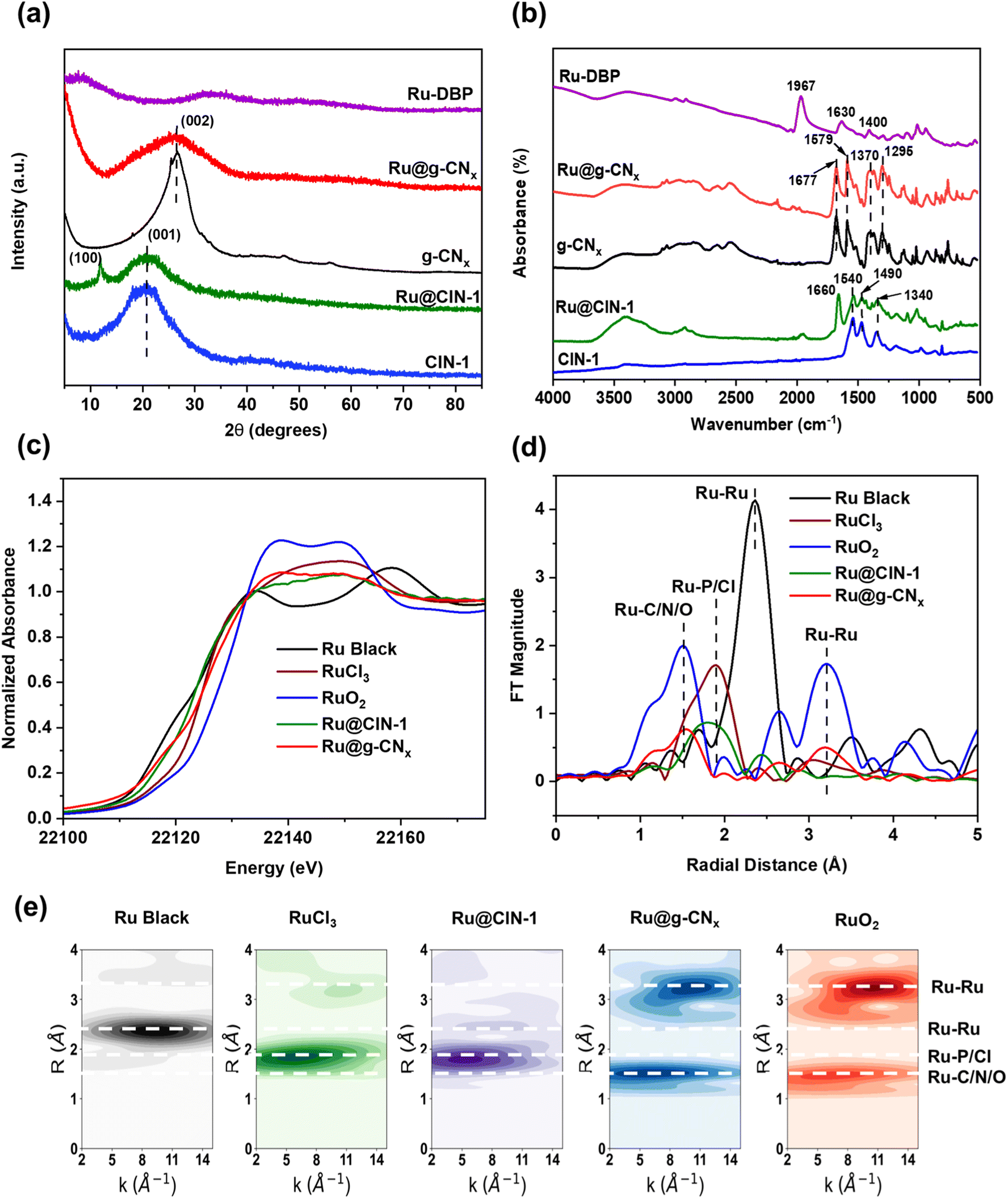 | ||
| Fig. 2 (a) XRD patterns, (b) FTIR spectra, (c) Ru K-edge XANES spectra, (d) Fourier transformed χ(k)-functions of the EXAFS spectra and (e) Cauchy WT-EXAFS of Ru–DBP, Ru@CIN-1, CIN-1, Ru, RuO2, g-CNx and Ru@g-CNx. | ||
As shown in Fig. 2b, the FTIR spectra of pure CIN-1 and Ru@CIN-1 depicted distinct peaks of triazine ring vibrations at 1540, 1490 and 1340 cm−1.36,47 The FTIR spectrum of the Ru–DBP complex comprised peaks at 1967 cm−1 assigned to coordinated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration, and 1630 and 1400 cm−1 due to carbon–carbon stretching vibrations of the aromatic rings.48 The disappearance of the CO group vibrations and the appearance of benzene aromatic ring vibrations during the crystallization of Ru@CIN-1 indicated the successful incorporation of the Ru–DBP complex into the CIN-1 structure. FTIR spectra of g-CNx and Ru@g-CNx (Fig. 2b) revealed the appearance of bands at 1677 and 1579 cm−1, and a group of peaks in the range of 1250–1500 cm−1 ascribed respectively to CN and aromatic C–N stretching vibrations.49,50
O stretching vibration, and 1630 and 1400 cm−1 due to carbon–carbon stretching vibrations of the aromatic rings.48 The disappearance of the CO group vibrations and the appearance of benzene aromatic ring vibrations during the crystallization of Ru@CIN-1 indicated the successful incorporation of the Ru–DBP complex into the CIN-1 structure. FTIR spectra of g-CNx and Ru@g-CNx (Fig. 2b) revealed the appearance of bands at 1677 and 1579 cm−1, and a group of peaks in the range of 1250–1500 cm−1 ascribed respectively to CN and aromatic C–N stretching vibrations.49,50
Raman spectra of pure CIN-1 and Ru@CIN-1 (Fig. S1, ESI†) confirmed the FTIR results by the presence of characteristic features of the triazine ring (975 cm−1), piperazine ring (1361 & 1429 cm−1) and imine (CN) stretching vibrations (1562 cm−1).47 This differs from the Raman plots of g-CNx and Ru@g-CNx which were composed of the characteristic D- and G-bands at around 1357 and 1580 cm−1 related to the lattice defects caused by graphite edges with sp3 bonds of the carbon and graphitic phase containing sp2 bonds of carbon, respectively.32,51,5213C CP/MAS NMR spectra (Fig. S2, ESI†) also demonstrated the conversion of the peaks corresponding to aliphatic carbon atoms in piperazine at 53.5 ppm and carbon in CN groups at 166 ppm to a broad asymmetric peak at δ = 150 ppm characteristic of sp2 aromatic carbons of a graphene sheet after pyrolysis.53,54
Transmission electron microscopy (TEM) images revealed a granular morphology of pure CIN-1 with a diameter of particles of about 20 nm in comparison with a layered-sheet structure of imine network layers of 50–200 nm for Ru@CIN-1 (Fig. S3, ESI†). EDX mapping images of the Ru@CIN-1 material confirmed the uniform distribution of Ru and P in the CIN-1 structure (Fig. S4, ESI†). TEM analysis demonstrated that pyrolysis resulted in the arrangement of Ru nanoparticles in the form of nanowires between the layers of graphitic carbon nitride for Ru@g-CNx in comparison with the generation of large carbon nitride particles for g-CNx material (Fig. 3, S5 and S6, ESI†). These Ru nanowires extend to the size of 500 nm and consist of Ru nanoparticles localized between carbon nitride layers, according to the proposed structure of the initial material (Fig. 1 and S5, ESI†). Elemental mapping demonstrated that Ru, N, P and C are present together in the material and pyrolysis does not affect the distribution of elements.
 | ||
| Fig. 3 STEM-HAADF of Ru@g-CNx with EDX mapping of C, N, P and Ru elements. | ||
The CIN-1 and Ru@CIN-1 materials exhibited a type II N2 adsorption isotherm with a sharp uptake below 0.05 P/P0, indicating the presence of micropores between the layers and a second uptake at a higher relative pressure originating from the textural pores generated by COF nanoparticles (Fig. S7, ESI†). The Brunauer–Emmett–Teller (BET) surface area values of CIN-1 and Ru@CIN-1 are 419 and 400 m2 g−1, respectively, which match well with the literature55 (Table 1). Pyrolysis induced a significant decrease in N2 adsorption in comparison with the parent materials reaching 62 and 23 m2 g−1 for g-CNx and Ru@g-CNx, respectively, which are usual values for carbon nitride materials56,57 (Fig. S7, ESI†).
The electronic state of Ru was studied by X-ray absorption spectroscopy (XAS) analysis (Fig. 2c–e, S8 and Table S1, ESI†). The Ru@CIN-1 X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra featured a similar profile to that of RuCl3 although the Ru centers are effectively more reduced in Ru@CIN-1 compared to RuCl3. The pyrolysis produced Ru@g-CNx possessing a spectrum with a shape reminiscent of that of RuO2, with significantly lower intensity and a broader white line. Similarly, Fourier transform (FT) EXAFS of the Ru@g-CNx sample showed patterns consistent with the presence of RuO2, with lower intensity, equivalent to ∼30% RuO2 character. Furthermore, the intensity ratios of the first coordination shell attributable to Ru–C/N/O at ∼1.97 Å in the sample relative to the longer scattering shells in the 3 to 6 Å range of 1.2:1 relative to the 1:1 found in the RuO2 reference inferred the presence of single Ru centers most probably stabilized by N of carbon nitride (Fig. 2 and Table S1, ESI†). The interaction of Ru with electronegative N in carbon nitride has been ascribed earlier to high dispersion of metals,58 high stability under working conditions59 and capture of electrons with optimized adsorption/desorption behaviour in the HER and OER.28,60,61 Together with the low coordination number, and rising edge energies indicating an average Ru oxidation of +0.5 for Ru@g-CNx, the data are consistent with low valence Ru centers,62 having a low coordination number together with a RuO2 lattice having O vacancies. XPS Ru 3d analysis confirms the presence of Ru2+ in Ru@CIN-1 and Ru–DBP with peaks at 281.4 and 280.9 eV, respectively (Fig. S9, ESI†). Ru@g-CNx has two doublets with BE Ru 3d5/2 280.7 and 282.4 eV, which can be assigned to RuO2 and Ru(OH)3, respectively.63
To conclude, the characterization results demonstrated Ru incorporation in the presence of a triphenylphosphine ligand in the structure of CIN-1 material. Pyrolysis of COF-based materials led to the generation of graphitic carbon nitride materials with RuO2 nanowires containing Ru in a low valence state.
3.2. Electrochemical properties
The performance of the Ru@g-CNx coated on a GC electrode for the HER in potassium hydroxide (1 M KOH) solution was investigated by performing LSV polarizations at a scan rate of 5 mV s−1. The overpotential versus the reversible hydrogen electrode (RHE) at 10 mA cm−2, the anticipated current density for a 12.3% efficient solar water-splitting device, was used to assess the HER performance. Fig. 4a reveals that Ru@g-CNx had superior electrocatalytic activity by requiring only 53.2 mV for driving a current density of 10 mA cm−2, which is far better than those of RuO2, g-CNx, Ru@CIN-1 and CIN-1. This interface also displayed better catalytic activity than the benchmarked Pt electrode exhibiting an overpotential of 63.7 mV at 10 mA cm−2, comparable to the performance of the most efficient materials reported in the literature64 (Table S2, ESI†). | ||
| Fig. 4 (a) HER polarization curves of Pt, Ru@g-CNx, commercial RuO2, Ru@CIN-1, Ru–DBP and g-CNx acquired at a scan rate of 5 mV s−1 in 1 M KOH solution. (b) OER polarization curves of commercial RuO2, Ru@g-CNx, Ru@CIN-1 and g-CNx. (c) Tafel plots and slope values of RuO2, Pt, Ru@g-CNx, Ru@CIN-1 and g-CNx derived from cathodic polarization LSV plots of (a). (d) Tafel plots and slope values of Ru@g-CNx, RuO2, Ru@CIN-1 and g-CNx derived from anodic polarization LSV plots of (b). | ||
The Tafel slope analysis gave us profound insight into the kinetic analysis of the HER derived from the LSV plots usually with lower Tafel slopes attributed to faster kinetics. Fig. 4c illustrates the Tafel slope of the Ru@g-CNx electrode, which is close to 30 mV dec−1, indicating a faster HER rate and Tafel–Volmer mechanism (1, 3) with electrochemical H2 desorption as the rate-determining step in the HER process. The Tafel slope values of Ru@CIN-1 (279.6 mV dec−1) and RuO2 (78.4 mV dec−1) were also determined from the LSV curves, which are significantly higher than those of Ru@g-CNx, suggesting that the HER proceeds via a primary discharge step.
| RuO + H2O + e− → RuOHads + OH− (Volmer) | (1) |
| RuOHads + H2O + e− → RuO + H2 + OH− (Heyrovsky) | (2) |
| RuOHads + RuOHads → 2-RuO + H2 (Tafel) | (3) |
Furthermore, the oxygen evolution performance of Ru@g-CNx was assessed in 1.0 M KOH aqueous solution in a three-electrode system. It could be seen from the LSV curves of Ru@g-CNx, RuO2, Ru@CIN-1 and g-CNx that oxygen evolution takes place at a much lower overpotential of 280 mV for Ru@g-CNx at a benchmark current density of 10 mA cm−2, as shown in Fig. 4. The Tafel slope value of Ru@g-CNx is around 49.5 mV dec−1, lower than that of commercial RuO2 (74.3 mV dec−1), Ru@CIN-1 (381.8 mV dec−1) and g-CNx (235.6 mV dec−1), which states that the kinetics of Ru@g-CNx for the water oxidation reaction follows the Volmer–Heyrovsky reaction pathway (Fig. 4). The Ru@g-CNx catalyst features a performance comparable to that of the most efficient materials for the OER65 (Table S3, ESI†).
The result was further supported by the Nyquist plots, showing that Ru@g-CNx has enhanced electron transfer ability compared to Ru@CIN-1 and g-CNx, as shown in Fig. 5. The three samples exhibited comparable solution resistance (Rs) values, as shown in EIS Nyquist graphs, but different charge transfer resistance (Rct) values. The sequence of HER activity is well supported by the fact that Ru@g-CNx has the smallest Rct of 45.8 Ω and Ru@CIN-1 and g-CNx displayed the largest Rct values. The smallest charge transfer implies fast electron transfer to the interface during the HER or OER, which, in turn, guarantees better catalytic activity. LSV measurements of the catalysts were subsequently carried out in the presence of blocking ligands like thiocyanate ions (SCN−) to identify the HER-active centers of Ru@g-CNx. According to Fig. S10, ESI,† the Ru@g-CNx electrocatalyst's LSV curve revealed a sharp cathodic shift when SCN− is present, indicating that Ru particles serve as the active sites.
 | ||
| Fig. 5 (a) Nyquist plots of various materials recorded in the range of 1 kHz to 0.1 Hz at a constant potential of 30 mV vs. RHE at 10 mV amplitude. (b) Chronopotentiometric plots of Ru@g-CNx at a current density of 10 mA cm−2 and −10 mA cm−2 for the OER and HER, respectively in 1 M KOH. (c) LSV plot of overall water splitting in 1 M KOH using Ru@g-CNx as both the anode and cathode. (d) Chronoamperometric measurements of overall water splitting in 1 M KOH aqueous solution at a constant current density of 10 mA cm−2 using Ru@g-CNx as both the anode and cathode. | ||
For the practical application of any catalyst, stability is one of the key factors being judged. The long-term stability of the Ru@g-CNx catalyst was probed by using potential–time plots in an alkaline environment. The chronopotentiometric plots of the Ru@g-CNx catalyst obtained in 1.0 M KOH aqueous solution at current densities of −10 mA cm−2 and 10 mA cm−2 for the HER and OER, respectively, are depicted in Fig. 5. After constant current density for 45 h, a limited decline (about 3%) of the catalyst performance for the OER was registered, while no obvious decrease in performance was observed during the HER stability test. The identical LSV polarization curves of Ru@g-CNx before and after 10000 CV cycles for both the HER and OER also confirmed the high stability of the catalyst (Fig. S11, ESI†). This robustness indicates that the catalyst is by far one of the best catalysts reported for water splitting. The analysis of hydrogen and oxygen production showed that Ru@g-CNx achieved a faradaic efficiency (FE) of almost 100% for actual water splitting, i.e. (99.78 and 99.73% for the HER and OER, respectively) (Fig. S12, ESI†). The microscopic analysis of the catalyst after the reaction revealed the presence of Ru in the form of nanowires in the carbon nitride matrix (Fig. S13, ESI†).
It is interesting to note that Ru according to XPS analysis is still in the form of a mixture of RuO2 and Ru(OH)3 after the OER; however, it is partially reduced to metallic Ru after the HER (Fig. S9, ESI†). The change in the oxidation state to adapt for the target reaction explains high efficiency in both the HER and OER. A variety of 2D or 1D metal nanostructured hydroxides/oxides of transition metals have been used as well for both the HER and OER.66,67
In order to understand the enhancement of electrocatalytic activity during the HER and OER for Ru@g-CNx compared to Ru@CIN-1 and g-CNx, the electrochemical active surface areas (ECSAs) were determined from the electrical double-layer capacitance (Cdl). Cyclic voltammograms were measured in a non-faradaic potential region between 0.1 and 0.25 V vs. RHE at different scan rates (Fig. S14, ESI†). Theoretically, it is acknowledged that the ECSA is proportional to its Cdl value for electrocatalysts with a similar composition.
The Cdl values of Ru@g-CNx (9.1 mF cm−2), g-CNx (0.25 mF cm−2), and CIN-1 (0.75 mF cm−2) were calculated from the slope of the straight line of the current density vs. scan rate curves in Fig. S14, ESI.† Hence, the ECSA experiments revealed that the Ru@g-CNx catalyst had the highest number of active sites caused by the lattice defects. This explains the catalytic behavior of the catalyst. The ECSA-normalized LSV curves for both the HER and OER were plotted to understand the intrinsic catalytic activity of the samples. In Fig. S15, ESI† we see that in both the HER and OER, Ru@g-CNx has higher catalytic activity than the precursor compounds. We have further employed per-site turnover frequency (TOF) analysis to compare the practical performance of the catalysts. The TOF curves, which vary with potential, revealed a higher intrinsic activity per site in the Ru@g-CNx catalyst when compared to both the Ru@CIN-1 and g-CNx catalysts (Fig. S15, ESI†).
Due to the robust catalytic activity of Ru@g-CNx, we were motivated to perform the overall water splitting in a 2-electrode system at 10 mA cm−2 for 26 h, as shown in Fig. 5. The LSV plot of the system recorded an overpotential of just 286 mV vs. RHE at a scan rate of 5 mV s−1. The chronopotentiometric plot showed that the catalyst had a good performance at a constant supply of 10 mA cm−2 in 1 M KOH electrolyte solution with a limited degradation of 1.35% after 26 h. The electrolyzer only required a cell voltage of 1.51 V to generate a current density of 10 mA cm−2, which is quite outstanding and comparable to that of the newly reported state-of-the-art materials for total water splitting (Table S4, ESI†).
4. Conclusion
In summary, we demonstrated the synthesis of a novel functional Ru electrocatalyst embedded in graphite carbon nitride prepared by pyrolysis of a covalent organic framework incorporated Ru complex. This resulted in the generation of Ru oxide nanowires with a low valence state of Ru between the layers of graphitic carbon nitride after pyrolysis. The resulting material exhibited a very low overpotential of just 53.2 mV to achieve a current density of 10 mA cm−2 and a Tafel slope of 33.2 mV dec−1 for the HER in an alkaline medium (1 M KOH). In addition, the catalyst performed very well during the OER with an overpotential of only 280 mV at 10 mA cm−2 and a low Tafel slope of 49.5 mV dec−1, exceeding the performance of commercially available RuO2. The improved catalytic activity was attributed to the generation of a large number of active sites, as witnessed by electrochemical active surface area and charge transfer resistance results from EIS. Furthermore, chronopotentiometry measurements concluded that the catalyst is highly stable in a corrosion-free environment for at least 45 h producing both hydrogen and oxygen. Furthermore, the robustness of the developed electrocatalyst was demonstrated in a full water splitting configuration with a slight degradation of 1.35% for at least 26 h for delivering a current density of 10 mA cm−2.Data availability
All data generated or analysed during this study are included in this published article (and its ESI file†).Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
The authors thank Solvay for the financial support of this work. The Chevreul Institute is thanked for its help in the development of this work through the ARCHICM project supported by the “Ministère de l'Enseignement Supérieur de la Recherche et de l'Innovation”, the region “Hauts-de-France”, the ERDF program of the European Union and the “Métropole Européenne de Lille” and the CPER “Wavetech”. The authors would like to thank Mr Florent Blanchard and Mr Ahmed Addad for their help in carrying out the work on the X-ray diffraction and electron microscopy facilities of the Advanced Characterization Platform of the Chevreul Institute. The authors also thank Mr Olivier Gardoll and Mrs Joelle Thuriot for their help in TGA and XRF. Tianyu Gao thanks the China Scholarship Council (CSC No. 201807030012) for the financial support. K. Sravan also acknowledges financial support from the Hauts-de-France region and CEFIPRA. The authors are also thankful to the Taif University Researchers Supporting Project number (TURSP-2020/03), Taif University, Taif, Saudi Arabia. XAS experiments were performed at the BL22-CLAESS beamline at ALBA Synchrotron with the collaboration of ALBA staff as part of projects 2022035778 and 2022035785.References
- H. Ishaq, I. Dincer and C. Crawford, Int. J. Hydrogen Energy, 2022, 47, 26238–26264 CrossRef CAS.
- P. J. Megía, A. J. Vizcaíno, J. A. Calles and A. Carrero, Energy Fuels, 2021, 35, 16403–16415 CrossRef.
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- W. Zhou, J. Jia, J. Lu, L. Yang, D. Hou, G. Li and S. Chen, Nano Energy, 2016, 28, 29–43 CrossRef CAS.
- M. F. Neira D'Angelo, V. Ordomsky, J. van der Schaaf, J. C. Schouten and T. A. Nijhuis, Catal. Sci. Technol., 2013, 3, 2834–2842 RSC.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- S. H. Ahn, S. J. Hwang, S. J. Yoo, I. Choi, H.-J. Kim, J. H. Jang, S. W. Nam, T.-H. Lim, T. Lim, S.-K. Kim and J. J. Kim, J. Mater. Chem., 2012, 22, 15153–15159 RSC.
- X. Wen, X. Yang, M. Li, L. Bai and J. Guan, Electrochim. Acta, 2018, 296, 830–841 CrossRef.
- A. Phuruangrat, D. J. Ham, S. J. Hong, S. Thongtem and J. S. Lee, J. Mater. Chem., 2010, 20, 1683–1690 RSC.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- W.-J. Jiang, S. Niu, T. Tang, Q.-H. Zhang, X.-Z. Liu, Y. Zhang, Y.-Y. Chen, J.-H. Li, L. Gu, L.-J. Wan and J.-S. Hu, Angew. Chem., Int. Ed., 2017, 56, 6572–6577 CrossRef CAS PubMed.
- S. Cherevko, S. Geiger, O. Kasian, N. Kulyk, J.-P. Grote, A. Savan, B. R. Shrestha, S. Merzlikin, B. Breitbach, A. Ludwig and K. J. J. Mayrhofer, Catal. Today, 2016, 262, 170–180 CrossRef CAS.
- B. Sarkar, D. Das and K. K. Nanda, J. Mater. Chem. A, 2021, 9, 13958–13966 RSC.
- Q. Wang, S. Santos, C. A. Urbina-Blanco, W. Y. Hernández, M. Impéror-Clerc, E. I. Vovk, M. Marinova, O. Ersen, W. Baaziz, O. V. Safonova, A. Y. Khodakov, M. Saeys and V. V. Ordomsky, Appl. Catal., B, 2021, 290, 120036 CrossRef CAS.
- D. Wu, Q. Wang, O. V. Safonova, D. V. Peron, W. Zhou, Z. Yan, M. Marinova, A. Y. Khodakov and V. V. Ordomsky, Angew. Chem., Int. Ed., 2021, 60, 12513–12523 CrossRef CAS PubMed.
- F. Li, G.-F. Han, H.-J. Noh, I. Ahmad, I.-Y. Jeon and J.-B. Baek, Adv. Mater., 2018, 30, 1803676 CrossRef PubMed.
- J. Zhang, P. Liu, G. Wang, P. P. Zhang, X. D. Zhuang, M. W. Chen, I. M. Weidinger and X. L. Feng, J. Mater. Chem. A, 2017, 5, 25314–25318 RSC.
- T. Qiu, Z. Liang, W. Guo, S. Gao, C. Qu, H. Tabassum, H. Zhang, B. Zhu, R. Zou and Y. Shao-Horn, Nano Energy, 2019, 58, 1–10 CrossRef CAS.
- Y. Wang, L. Zhao, J. Ma and J. Zhang, Energy Environ. Sci., 2022, 15, 3830–3841 RSC.
- H. Sun and W. Jung, J. Mater. Chem. A, 2021, 9, 15506–15521 RSC.
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- E. Tsuji, A. Imanishi, K.-i. Fukui and Y. Nakato, Electrochim. Acta, 2011, 56, 2009–2016 CrossRef CAS.
- K. Biswas and C. N. Rao, J. Nanosci. Nanotechnol., 2007, 7, 1969–1974 CrossRef CAS PubMed.
- Y. Qin, T. Yu, S. Deng, X.-Y. Zhou, D. Lin, Q. Zhang, Z. Jin, D. Zhang, Y.-B. He, H.-J. Qiu, L. He, F. Kang, K. Li and T.-Y. Zhang, Nat. Commun., 2022, 13, 3784 CrossRef CAS PubMed.
- R. Jiang, D. T. Tran, J. Li and D. Chu, Energy Environ. Mater., 2019, 2, 201–208 CrossRef CAS.
- J. Wang, X. Guan, H. Li, S. Zeng, R. Li, Q. Yao, H. Chen, Y. Zheng and K. Qu, Nano Energy, 2022, 100, 107467 CrossRef CAS.
- X. Gao, J. Chen, X. Sun, B. Wu, B. Li, Z. Ning, J. Li and N. Wang, ACS Appl. Nano Mater., 2020, 3, 12269–12277 CrossRef CAS.
- G. Kim, J. Yang, N. Nakashima and T. Shiraki, Chem.–Eur. J., 2017, 23, 17504–17510 CrossRef CAS PubMed.
- L. Chen, L. Zhang, Z. Chen, H. Liu, R. Luque and Y. Li, Chem. Sci., 2016, 7, 6015–6020 RSC.
- Q. Xu, Y. Tang, L. Zhai, Q. Chen and D. Jiang, Chem. Commun., 2017, 53, 11690–11693 RSC.
- X. Hu, Y. Long, M. Fan, M. Yuan, H. Zhao, J. Ma and Z. Dong, Appl. Catal., B, 2019, 244, 25–35 CrossRef CAS.
- Q. Xu, Y. Tang, X. Zhang, Y. Oshima, Q. Chen and D. Jiang, Adv. Mater., 2018, 30, e1706330 CrossRef PubMed.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- P. J. Waller, F. Gandara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS PubMed.
- T. Gao, Z. Yan, V. Ordomsky and S. Paul, ChemCatChem, 2022, 14, e202101450 CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 96–100 CrossRef CAS PubMed.
- J. J. Rehr and R. C. Albers, Rev. Mod. Phys., 2000, 72, 621–654 CrossRef CAS.
- P. R. Bevington and D. K. Robinson, Data Reduction and Error Analysis for the Physical Sciences, McGraw-Hill, New York, 2nd edn, 1992 Search PubMed.
- M. Newville, J. Phys.: Conf. Ser., 2013, 430, 1–7 CrossRef.
- Y.-R. Liu, X. Shang, W.-K. Gao, B. Dong, J.-Q. Chi, X. Li, K.-L. Yan, Y.-M. Chai, Y.-Q. Liu and C.-G. Liu, Appl. Surf. Sci., 2017, 412, 138–145 CrossRef CAS.
- S.-Y. Li, W.-H. Li, X.-L. Wu, Y. Tian, J. Yue and G. Zhu, Chem. Sci., 2019, 10, 7695–7701 RSC.
- M. G. Schwab, B. Fassbender, H. W. Spiess, A. Thomas, X. Feng and K. Müllen, J. Am. Chem. Soc., 2009, 131, 7216–7217 CrossRef CAS PubMed.
- S. Roy, T. Chatterjee, B. Banerjee, N. Salam, A. Bhaumik and S. M. Islam, RSC Adv., 2014, 4, 46075–46083 RSC.
- J. Liu, Y. Zhang, L. Zhang, F. Xie, A. Vasileff and S.-Z. Qiao, Adv. Mater., 2019, 31, 1901261 CrossRef PubMed.
- J. Xue, Y. Bai and H. Liu, Anal. Bioanal. Chem., 2019, 411, 3721–3729 CrossRef CAS PubMed.
- J. G. Małecki and A. Maroń, Transition Met. Chem., 2012, 37, 727–734 CrossRef.
- N. Bandara, Y. Esparza and J. Wu, Sci. Rep., 2017, 7, 11538 CrossRef PubMed.
- M. P. Kumar, T. Kesavan, G. Kalita, P. Ragupathy, T. N. Narayanan and D. K. Pattanayak, RSC Adv., 2014, 4, 38689–38697 RSC.
- Y. Zhang, Y. Ma, Y.-Y. Chen, L. Zhao, L.-B. Huang, H. Luo, W.-J. Jiang, X. Zhang, S. Niu, D. Gao, J. Bi, G. Fan and J.-S. Hu, ACS Appl. Mater. Interfaces, 2017, 9, 36857–36864 CrossRef CAS PubMed.
- X. Cui, Y. Long, X. Zhou, G. Yu, J. Yang, M. Yuan, J. Ma and Z. Dong, Green Chem., 2018, 20, 1121–1130 RSC.
- H. Darmstadt, C. Roy, S. Kaliaguine, G. Xu, M. Auger, A. Tuel and V. Ramaswamy, Carbon, 2000, 38, 1279–1287 CrossRef CAS.
- A. M. Panich, A. I. Shames and N. A. Sergeev, Appl. Magn. Reson., 2013, 44, 107–116 CrossRef CAS.
- M. K. Bhunia, S. K. Das, P. Pachfule, R. Banerjee and A. Bhaumik, Dalton Trans., 2012, 41, 1304–1311 RSC.
- E. Alwin, M. Zieliński, A. Suchora, I. Gulaczyk, Z. Piskuła and M. Pietrowski, J. Mater. Sci., 2022, 57, 15705–15721 CrossRef CAS.
- H. Wang, X. Yuan, Y. Wu, G. Zeng, X. Chen, L. Leng and H. Li, Appl. Catal., B, 2015, 174–175, 445–454 CrossRef CAS.
- Z. Yu, Y. Li, A. Torres-Pinto, A. P. LaGrow, V. M. Diaconescu, L. Simonelli, M. J. Sampaio, O. Bondarchuk, I. Amorim, A. Araujo, A. M. T. Silva, C. G. Silva, J. L. Faria and L. Liu, Appl. Catal., B, 2022, 310, 121318 CrossRef CAS.
- X. Zhang, Z. Xia, H. Li, S. Yu, S. Wang and G. Sun, J. Colloid Interface Sci., 2023, 640, 170–178 CrossRef CAS PubMed.
- B. Jiang, Z. Wang, L. Cheng, Z. Chen, Y. Dong, X. Wang, T. Wang, X. Mao, Y. Gao, Z. Xu, K. Yin and K. Wu, J. Alloys Compd., 2023, 939, 168717 CrossRef CAS.
- M. Gao, Z. Wang, S. Sun, D. Jiang, W. Wei and M. Chen, J. Alloys Compd., 2021, 864, 158174 CrossRef CAS.
- F. J. Escobar-Bedia, M. Lopez-Haro, J. J. Calvino, V. Martin-Diaconescu, L. Simonelli, V. Perez-Dieste, M. J. Sabater, P. Concepción and A. Corma, ACS Catal., 2022, 12, 4182–4193 CrossRef CAS.
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- J. Ding, Q. Shao, Y. Feng and X. Huang, Nano Energy, 2018, 47, 1–7 CrossRef CAS.
- C.-Z. Yuan, Y.-F. Jiang, Z.-W. Zhao, S.-J. Zhao, X. Zhou, T.-Y. Cheang and A.-W. Xu, ACS Sustainable Chem. Eng., 2018, 6, 11529–11535 CrossRef CAS.
- Y. Wang, J. Ma, J. Wang, S. Chen, H. Wang and J. Zhang, Adv. Energy Mater., 2019, 9, 1802939 CrossRef.
- Y. Wang, S. Chen and J. Zhang, Adv. Funct. Mater., 2019, 29, 1904955 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta01362f |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |