
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic and ionic effects of sulphur and other acidic adsorbates on the surface of an SOFC cathode material†
Matthäus
Siebenhofer
 *ab,
Andreas
Nenning
b,
George E.
Wilson
c,
John A.
Kilner
c,
Christoph
Rameshan
d,
Markus
Kubicek
b,
Jürgen
Fleig
b and
Peter
Blaha
e
*ab,
Andreas
Nenning
b,
George E.
Wilson
c,
John A.
Kilner
c,
Christoph
Rameshan
d,
Markus
Kubicek
b,
Jürgen
Fleig
b and
Peter
Blaha
e
aCentre for Electrochemistry and Surface Technology, CEST, Wr. Neustadt, Austria. E-mail: matthaeus.siebenhofer@tuwien.ac.at
bInstitute of Chemical Technologies and Analytics, TU Wien, Vienna, Austria
cDepartment of Materials, Imperial College, London, UK
dChair of Physical Chemistry, Montanuniversität Leoben, Leoben, Austria
eInstitute of Materials Chemistry, TU Wien, Vienna, Austria
First published on 15th March 2023
Abstract
The effects of sulphur adsorbates and other typical solid oxide fuel cell (SOFC) poisons on the electronic and ionic properties of an SrO-terminated (La,Sr)CoO3 (LSC) surface and on its oxygen exchange kinetics have been investigated experimentally with near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS), low energy ion scattering (LEIS) and impedance spectroscopy as well as computationally with density functional theory (DFT). The experiment shows that trace amounts of sulphur in measurement atmospheres form SO2−4 adsorbates and strongly deactivate a pristine LSC surface. They induce a work function increase, indicating a changing surface potential and a surface dipole. DFT calculations reveal that the main participants in these charge transfer processes are not sub-surface transition metals, but surface oxygen atoms. The study further shows that sulphate adsorbates strongly affect oxygen vacancy formation energies in the LSC (sub-)surface, thus affecting defect concentrations and oxygen transport properties. To generalize these results, the investigation was extended to other acidic oxides which are technologically relevant as SOFC cathode poisons, such as CO2 and CrO3. The results unveil a clear correlation of work function changes and redistributed charge with the Smith acidity of the adsorbed oxide and clarify fundamental mechanistic details of atomic surface modifications. The impact of acidic adsorbates on various aspects of the oxygen exchange reaction rate is discussed in detail.
Introduction
Solid oxide fuel and electrolysis cells (SOFCs/SOECs) are promising representatives of innovative and clean electrochemical energy-conversion technologies. They pose interesting opportunities for a variety of applications due to their high efficiency and their ability to operate on a range of different hydrocarbon fuels.1–4 While cell performance steadily increases and new materials are being developed to drive SOFCs towards lower operating temperatures,5–9 a key hindrance for a wide applicability lies in their inherent susceptibility to performance degradation, in particular with regard to the oxygen exchange reaction (OER) on the cathode side.10–12Apart from morphological degradation processes such as delamination or cracking, cathode degradation factors can be broadly divided into two categories: (1) materials change chemically under operating conditions, affecting the catalytic activity of the surface (e.g. Sr segregation in Sr-containing perovskites);13–15 (2) environmental impurities (e.g. S, CO2 or Cr) accumulate on the cathode and poison its surface, leading to second phase formation and to a performance decrease.16–19 Regarding the atomic mechanism of degradation processes, many suggestions have been brought forward, ranging from the blocking of active reaction sites by adsorbates20 or secondary phases17,21 to element depletion or accumulation in (sub-)surface regions of the material.22
In a recent study, the authors could show that trace amounts of sulphur, omnipresent in measurement gases in the high ppb range23 and also in air in the low ppb range,18,24,25 readily adsorb on the surface of La0.6Sr0.4CoO3−δ (LSC), a highly active but degradation prone SOFC cathode material.23 There, they form SO2−4 surface species and cause a sudden, strong drop of the OER rate. The study also revealed that this phenomenon is not unique to Sr-containing perovskites but also occurs on other potential SOFC cathode materials, such as Pr0.1Ce0.9O2−δ. Further investigations identified this SO2−4 formation as the starting point of long-term degradation processes in the form of second phase formation which are commonly associated with Sr segregation.26 In conjunction with recent advances identifying the acidity of oxidic surface modifications as indicative for their catalytic activity,27 these results strongly emphasize the importance of the outermost surface for the final performance of a material. However, while it is possible to qualitatively predict the surface activity of modified surfaces based on their acidity, the atomistic mechanism behind such modifications is still unclear.
The present study investigates in detail the effects of acidic adsorbates on the LSC surface and discusses their implications for the OER kinetics. Utilizing near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) and low energy ion scattering (LEIS), a detailed picture of the surface chemistry is drawn. Based on these experimental findings, ab initio calculations are employed to examine charge redistribution processes at the surface of LSC upon acidic adsorbate formation. Supported by density functional theory (DFT), the impact of acidic adsorbates on the electronic and ionic properties of LSC, as well as on the adsorption of molecular oxygen is discussed, illustrating various effects of surface modifications on the OER kinetics.
Methodology
Experimental methods
| Material | LSC dense | LSC porous | GDC |
|---|---|---|---|
| Substrate-target distance (cm) | 6.0 | 5.0 | 6.0 |
| Background O2 pressure (mbar) | 0.04 | 0.4 | 0.04 |
| Temperature (°C) | 600 | 450 | 600 |
| Frequency (Hz) | 2 | 5 | 1 |
Computational methods
DFT calculations of (La,Sr)CoO3−δ were performed using the full-potential augmented plane wave plus local orbitals method33 as implemented in the WIEN2k code.34,35 For all calculations, the PBE GGA (generalized gradient approximation) functional36 was used. For a suitable treatment of the correlated 3d-electrons of Co and Cr atoms, a Hubbard U correction was employed, using a potential Ueff = U − J of 3.35 eV for both atoms, considering previous investigations of similar materials.37–39 A volume optimization for LaCoO3 yielded a pseudocubic lattice parameter of 3.83 Å, being in good agreement with experiment.40,41 As a base model for all calculations we used a symmetric 2 × 2 × 3 LaO terminated (001) surface slab of LaCoO3 with the top La atoms replaced by Sr (see Fig. 1). Thereby, not only the stoichiometry is corrected towards realistic LSC cathodes with the La![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sr:Co ratio being 1:1:1.5 (1:0.7 : 1.7 for La0.6Sr0.4CoO3−δ), the model also considers the experimentally found SrO termination of LSC under solid oxide cell operating conditions.42,43 A symmetric slab was chosen over a two termination structure to avoid overall dipole formation which might interfere with electrostatic effects induced by adsorbates. Overall, the slab contains 8 Sr, 8 La, 12 Co and 40 O atoms. A vacuum region of 20 Å separates the slabs to minimize interactions between the surfaces.44,45
:Sr:Co ratio being 1:1:1.5 (1:0.7 : 1.7 for La0.6Sr0.4CoO3−δ), the model also considers the experimentally found SrO termination of LSC under solid oxide cell operating conditions.42,43 A symmetric slab was chosen over a two termination structure to avoid overall dipole formation which might interfere with electrostatic effects induced by adsorbates. Overall, the slab contains 8 Sr, 8 La, 12 Co and 40 O atoms. A vacuum region of 20 Å separates the slabs to minimize interactions between the surfaces.44,45
 | ||
| Fig. 1 (a) Unrelaxed slab model of LSC with a full SrO termination layer; (b) relaxed slab model of LSC with a full SrO termination layer (LSC-S0); (c) relaxed slab model of LSC with a full SrO termination layer and two sulphate adsorbates on each surface (“half-coverage”, LSC-S2). | ||
During calculations, different surface modifications were placed on the LSC slab, an exemplary structure with two sulphate adsorbates is shown in Fig. 1 c. To find the most stable configurations, a structure relaxation was performed for all structures, employing a Γ-centered 5 × 5 × 1 k-mesh. For a valid comparison of total energies, final calculations were performed with the same basis-set size corresponding to RminMTKmax = 6, with RminMT being the smallest atomic sphere radius (2.2, 2.2, 1.87, 1.15, 1.15, 1.38 and 1.38 bohr for La, Sr, Co, O, C, S and Cr atomic spheres respectively) and Kmax the largest reciprocal lattice vector. All relaxations were performed until residual forces did not exceed 3 mRy per bohr for three consecutive iterations. As energy separation between core and valence states, −6.0 Ry was used.
Depending on the environmental conditions, i.e. temperature and p(O2), the charge imbalance introduced by Sr-doping in LSC can be compensated electronically or by oxygen vacancies (VO). To investigate the interplay of acidic adsorbates and oxygen vacancies in the LSC surface and the LSC bulk, single oxygen atoms were removed from the structure and structure relaxation was performed. For a closer investigation of oxygen adsorption, O2 molecules were placed in and above the surface. Energetics of adsorption and oxygen vacancy formation have been evaluated by comparing the total energies of the final structures with the sum of the constituent structures, e.g. for O2 adsorption:  . The factors of 1/2 and 2 arises from the symmetric slab with two surfaces. DFT calculations of O2 and SO2 molecules have been performed with the same RminMTKmax and RMT values to ensure compatibility of the total energy values.
. The factors of 1/2 and 2 arises from the symmetric slab with two surfaces. DFT calculations of O2 and SO2 molecules have been performed with the same RminMTKmax and RMT values to ensure compatibility of the total energy values.
To quantitatively assess charge transfer upon adsorption of acidic molecules or O2, Bader's quantum theory of atoms in molecules was used.46 Here, a charge is assigned to every atom in the slab, whereas the atom is defined as the region bounded by a zero flux surface 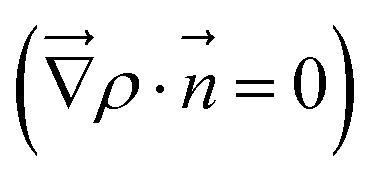 , and the electron density ρ is integrated over this region. To differentiate between geometric and electronic effects, the results are compared with a superposition of neutral atoms.
, and the electron density ρ is integrated over this region. To differentiate between geometric and electronic effects, the results are compared with a superposition of neutral atoms.
For investigations of the adsorption barrier for O2, an O2 molecule was placed far above a surface VO (O–Co distance ∼4–6 Å). From this starting point, a constraint minimization technique was employed, slowly reducing the distance between the lower O and the Co below by applying a pseudoforce on both atoms. Thereby, adsorption processes can be simulated and adsorption barriers can be estimated.
Results and discussion
Investigation of real LSC surfaces
 | ||
| Fig. 2 Evolution of the surface coverage with sulphate adsorbates and the corresponding change of the surface exchange resistance on a dense, (100) oriented LSC thin film with respect to temperature and time in 0.005 mbar O2. Sulphate groups were quantified from XPS measurements.23 | ||
The detailed effects of sulphate adsorbates on the surface chemistry of LSC are shown in Fig. 3. XPS measurements were first performed at 600 °C and 8 × 10−6 mbar O2, warranting very low impurity contents. Then, the samples were exposed to 1 mbar of O2, where an immediate saturation with sulphate adsorbates is obtained. The signals of La, Sr and Co remain nearly unchanged, indicating that the effect of sulphate adsorbates is not directly related to significant changes of Co oxidation states or the cation stoichiometry on the surface. The signals of O and S evolve as expected for sulphate formation with the sulphur signal increasing together with the appearance of an oxygen satellite feature at 532 eV, which has already been attributed to S-related compounds in the past.23,47 A ratio of S 2p and 532 eV O 1s atomic fractions of 0.23 agrees excellently with SO2−4 species.23 The slight shift of the S 2p species observed upon changing the oxygen partial pressure is likely correlated with a different surface potential induced by different oxygen vacancy concentrations and oxygen adsorbate coverages at different p(O2).
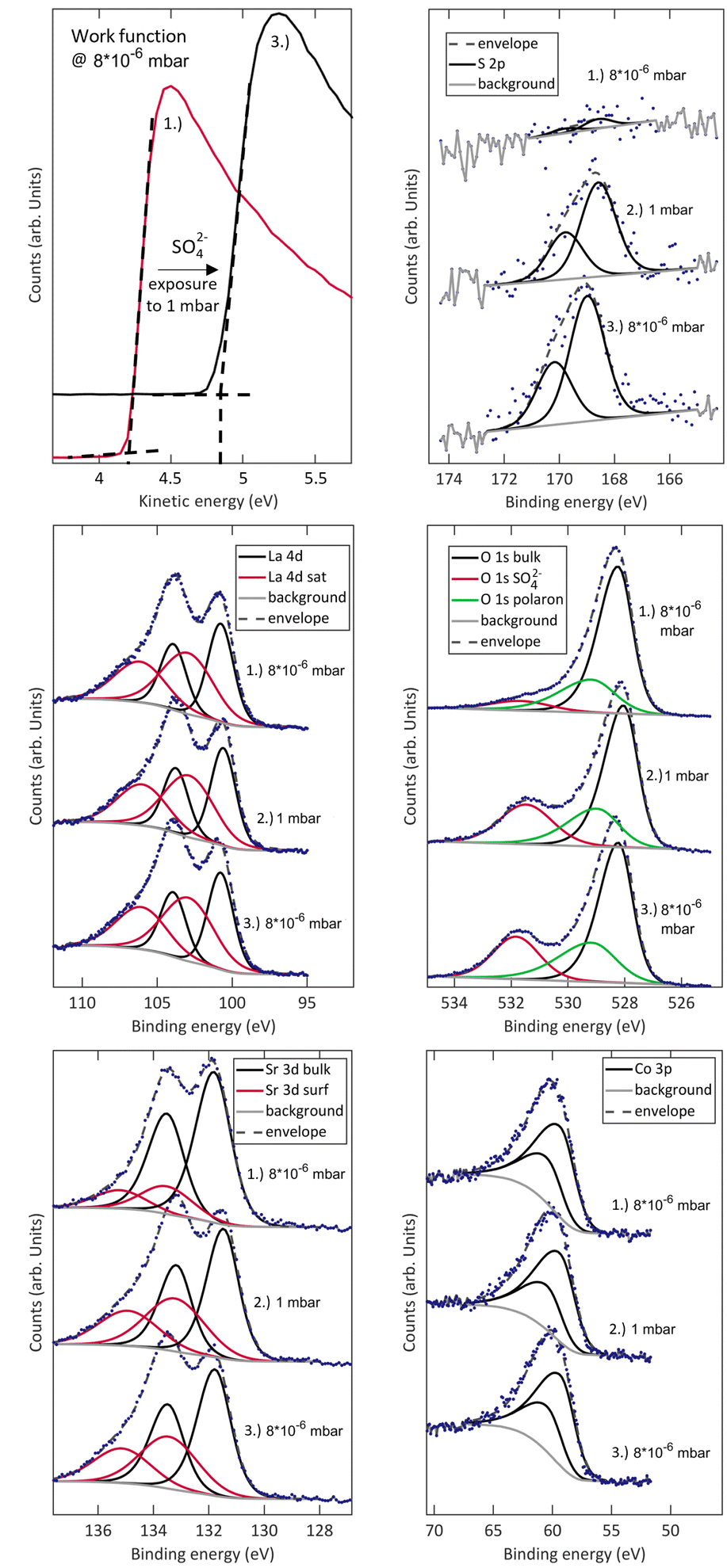 | ||
| Fig. 3 NAP-XPS measurements of S 2p, La 4d, O 1s, Sr 3d, Co 3p and the work function of a (001)-oriented dense LSC thin film at 8 × 10−6 mbar O2 and 600 °C before (1.) and after (3.) exposure to 1 mbar of O2 at 600 °C (2.). | ||
While this evolution of the surface chemistry has already been reported, a closer investigation of low kinetic energy regions reveals an additional change induced by SO2−4 adsorbate formation. The same measurement routine of measuring a clean sample at 8 × 10−6 mbar O2, allowing sulphates to accumulate at 1 mbar and measuring again at 8 × 10−6 mbar O2 unveils a significant change of the work function from 4.2 to 4.8 eV. This is a clear indication for the formation of a charged adsorbate layer and a space charge in the material. As a very rough estimate for the reach of such electrostatic effects, the Debye length can be calculated by48
 | (1) |
Prior to measurement, the samples were exposed to high vacuum (≈10−9 mbar) overnight, for an initial surface cleaning by desorption of loosely bound surface adsorbates. In addition, the samples were exposed to an in situ heating step to 400 °C (2 K per minute) to remove adventitious carbonates from the surface. The resulting LEIS spectra are shown in Fig. 4 and are denoted as LSC(-S) after thermal cleaning. For both samples, Sr clearly dominates the surface cation distribution, even more so for sulphate covered LSC. The measurements also yield the first direct proof of sulphur on the surface, albeit with a rather weak intensity. We strongly suspect that this is due to the SO2−4 environment which shields the sulphur from approaching ions. Additionally, NaCl traces appeared on the surface (much more pronounced on LSC-S2, where they are likely also the cause for the generally lower main cation intensities). We suspect that these traces stem either from contaminations on the heating stage or from handling during sample transfer. Na is also a common impurity found in oxide materials and very small amounts suffice to yield detectable contamination in LEIS measurements.52
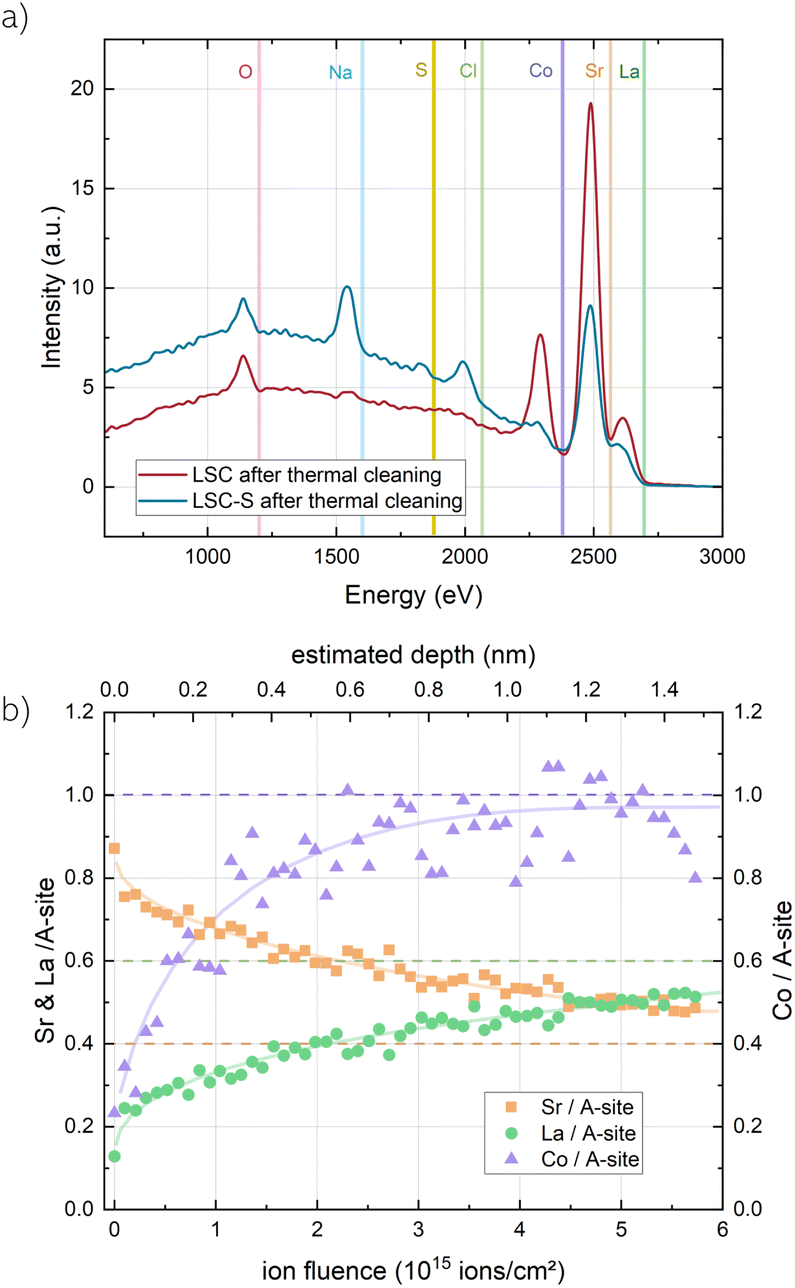 | ||
| Fig. 4 (a) LEIS spectra (3 keV 4He+ ions, 5 nA) of LSC directly out of the PLD chamber and sulphate-covered LSC-S (after exposure to 1 mbar O2 in the XPS chamber at 600 °C) after thermal cleaning by in situ heating to 400 °C. Vertical lines show the onset scattering energy of each surface atom. (b) depth-profiles of La, Sr and Co for the first 1.5 nm of LSC-S obtained by sputtering with a 500 eV 40Ar+ beam with a beam current of 100 nA. Signals were normalized to the bulk stoichiometry of a reference measurement in greater depth. | ||
In Fig. 4b, a depth profile of the cation ratios in the first 1.5 nm of a sulphate covered LSC thin film is shown (cation ratios were quantified by identifying the plateau reached after sufficient sputtering with the ideal bulk stoichiometry51). Similar to earlier investigations of LSC with LEIS,43 the results emphasize the Sr enrichment and show a strong Co depletion in the first layer, as well as a broader La depletion zone. It is worth mentioning that preferential sputtering will alter the ion yield,53 chiefly affecting lighter species, such as oxygen. Therefore, the measurements will slightly overestimate the La content in the LSC surface, however, this effect is difficult to correct for and we only suspect negligible deviations due to the low ion fluence. While these measurements illustrate that real LSC surfaces can be rather complicated, the results agree with NAP-XPS measurements and also with computational findings which are described below:
– LSC exhibits a Sr-rich termination layer and the surface is depleted of Co an La.
– LSC-S is covered with acidic sulphate adsorbates, which are bound in an SO2−4 environment and are surrounded by oxygen atoms, likely accounting for the low sulphur signal.
Summarizing the surface analytical results of this study, NAP-XPS and LEIS investigations agree with the model that real LSC surfaces are largely SrO terminated and that sulphur readily adsorbs in an SO2−4 environment, likely associated with surface Sr. We find that SO2−4 formation leads to a significant shift of the LSC work function, indicating the formation of a surface dipole. In the following, we will employ DFT calculations to obtain a detailed understanding of the impact of SO2−4 adsorbates on the electronic and ionic structure of LSC.
Ab initio calculations of SO2−4 adsorbates on LSC
To understand the detailed effects of sulphate adsorbates on the electronic and ionic properties of LSC, ab initio calculations have been performed with WIEN2k.34,35 We investigated stable structures with different coverages of sulphate adsorbates and examined energetic and electrostatic effects. In particular, we evaluated adsorption energies of SO3 and O2 molecules on SrO-terminated LSC surfaces, the adsorption barrier for an approaching O2 molecule, as well as oxygen vacancy formation energies. Electrostatic effects were investigated by a Bader charge analysis,46 by evaluating work function changes upon SO3 adsorption and by analyzing the densities of states of specific atoms. The results of these calculations not only clarify the experimentally found surface dipole formation but also illustrate the manifold ways acidic adsorbates affect the oxygen exchange reaction.SO2−4 adsorbate structures and adsorption energies. The foundation for all calculations was a relaxed LSC surface slab with a full SrO termination (LSC-S0, see Fig. 1b). The structure exhibits a clear octahedral tilting, in agreement with earlier experimental studies on LSC.54 Moreover, a slight surface buckling is visible with the oxygen atoms positioned ≈0.2 Å above the Sr atoms. Starting from there, SO3 groups were placed directly above a surface oxygen and structure relaxation calculations were performed. The relaxed structure is shown in Fig. 1c. S–O bond distances and O–S–O bond angles in the sulphate adsorbate range between 1.46–1.48 Å and 112–117° for the three top oxygens and between 1.62–1.64 Å and 103–104° for the bottom oxygen in the LSC surface, respectively. Upon sulphate adsorption, the bottom oxygen gets slightly pulled out of the surface by ≈0.2 Å and the buckling increases correspondingly. As a consequence, the Co–O bond length to the subsurface also increases by the same 0.2 Å at sulphate covered sites. The octahedral tilting of the LSC structure gets stronger upon sulphate adsorption with the tilting angle increasing from 6° to 10° (final coordinates for different structures are given in S.1†). The core-level shift to higher binding energies observed in the O 1s species of bulk LSC and SO2−4 during XPS is reproduced by the calculations but slightly underestimated (2.5 eV vs. 3.5 eV experimentally). Calculated adsorption energies of SO3 groups for LSC with a 25% (LSC-S1) and a 50% sulphate coverage (LSC-S2), as well as for LSC-S2 with a surface vacancy are given in Table 2. As adsorption mechanism we suggest the adsorption of SO2 and the subsequent oxidation to SO3. Therefore, the reaction enthalpy for the oxidation reaction
 of −1.025 eV55 was subtracted from the energy difference:
of −1.025 eV55 was subtracted from the energy difference:  . The results show that SO3 groups are very strongly adsorbed on SrO-terminated LSC, with the adsorption strength decreasing with increasing coverage. For reduced surfaces with oxygen vacancies, an even stronger adsorption is found.
. The results show that SO3 groups are very strongly adsorbed on SrO-terminated LSC, with the adsorption strength decreasing with increasing coverage. For reduced surfaces with oxygen vacancies, an even stronger adsorption is found.
| LSC-S1 | LSC-S2 | LSC-S2 + VO | |
|---|---|---|---|
| Adsorption energy per SO3 (eV) | −3.31 | −2.46 | −2.96 |
The high calculated adsorption energies are also in line with experimental results which show that it is very hard to remove sulphate adsorbates from LSC surfaces.23 A decrease of sulphate coverage can be observed at 0.005 mbar O2 at a temperature of 700 °C, but sulphate groups adsorbed at 600 °C tend to remain on the surface. With regard to mitigation strategies, it is particularly interesting that samples directly after the PLD process do not exhibit sulphur signatures during XPS measurements. Instead, after transfer in ambient air, the surfaces are covered with substantial amounts of adventitious carbonates and we strongly suspect that these act as a protective layer against sulphates. This might indicate the possibility to modify SOFC cathode materials towards a sulphur resistant surface, potentially utilizing compounds which are only slightly acidic relative to the host lattice.
Vacancy formation energies. To assess the effects of SO2−4 adsorbates on the ionic properties of LSC surfaces, vacancy formation energies have been investigated. Oxygen vacancies (VO) have been created in the outermost surface (VO,surf, placed on a free surface O site for LSC-S2) and in the LaO layer (VO,bulk) and the total energies of the relaxed structures were compared. The resulting formation energies are given in Table 3.
| Surface (eV) | “Bulk” (eV) | |
|---|---|---|
| LSC-S0 | 1.30 | 1.12 |
| LSC-S2 | 0.19 | 2.15 |
Regarding the importance of VO for the oxygen exchange kinetics,56–58 this result requires further discussion. From an electrostatic point of view, it is reasonable that a negatively charged SO2−4 layer promotes the formation of positive VO,surf, as charge compensation is limited to short distances, evident by the small formation energy for surface vacancies on LSC-S2. However, the correlation of vacancy concentration and oxygen exchange kinetics has been topic of an ongoing discussion.59,60 Moreover, the much larger formation energy for a bulk vacancy might indicate a certain blocking of vacancy migration between the bulk and the surface of LSC. The absolute impact of surface modifications on the reaction rate will be discussed in a broader context below, including electrostatic as well as chemical contributions.
O2 adsorption energies. O2 adsorption energies have been evaluated for an O2 molecule placed in a VO,surf. The results are shown in Table 4, revealing a substantial difference between LSC-S0 and LSC-S2. While for the sulphur free LSC-S0, the adsorption of an O2 molecule is energetically favourable, it costs energy to place an oxygen molecule between the sulphate groups while annihilating a surface vacancy. In addition, the bond distance of the O2 molecule is considerably increased on LSC-S0 (1.43 Å vs. 1.34 Å on LSC-S2), possibly indicating inhibited dissociation of the molecule on LSC-S2.
| LSC-S0 + VO | LSC-S2 + VO | |
|---|---|---|
| Adsorption energy per O2 (eV) | −1.40 | 0.48 |
O2 adsorption barriers. Next to electrostatic and energetic changes, sulphate adsorbates might also affect the adsorption barrier of an O2 molecule. While this study does not aim for a realistic modelling of O2 adsorption processes on LSC surfaces, DFT calculations allow for a qualitative assessment of changing adsorption barriers upon sulphate adsorption. For this purpose, an O2 molecule was placed far away from the surface and pulled towards its final position (see computational methods). The total energy was recorded as a function of the distance to the final position of the O2 molecule (see Fig. 5).
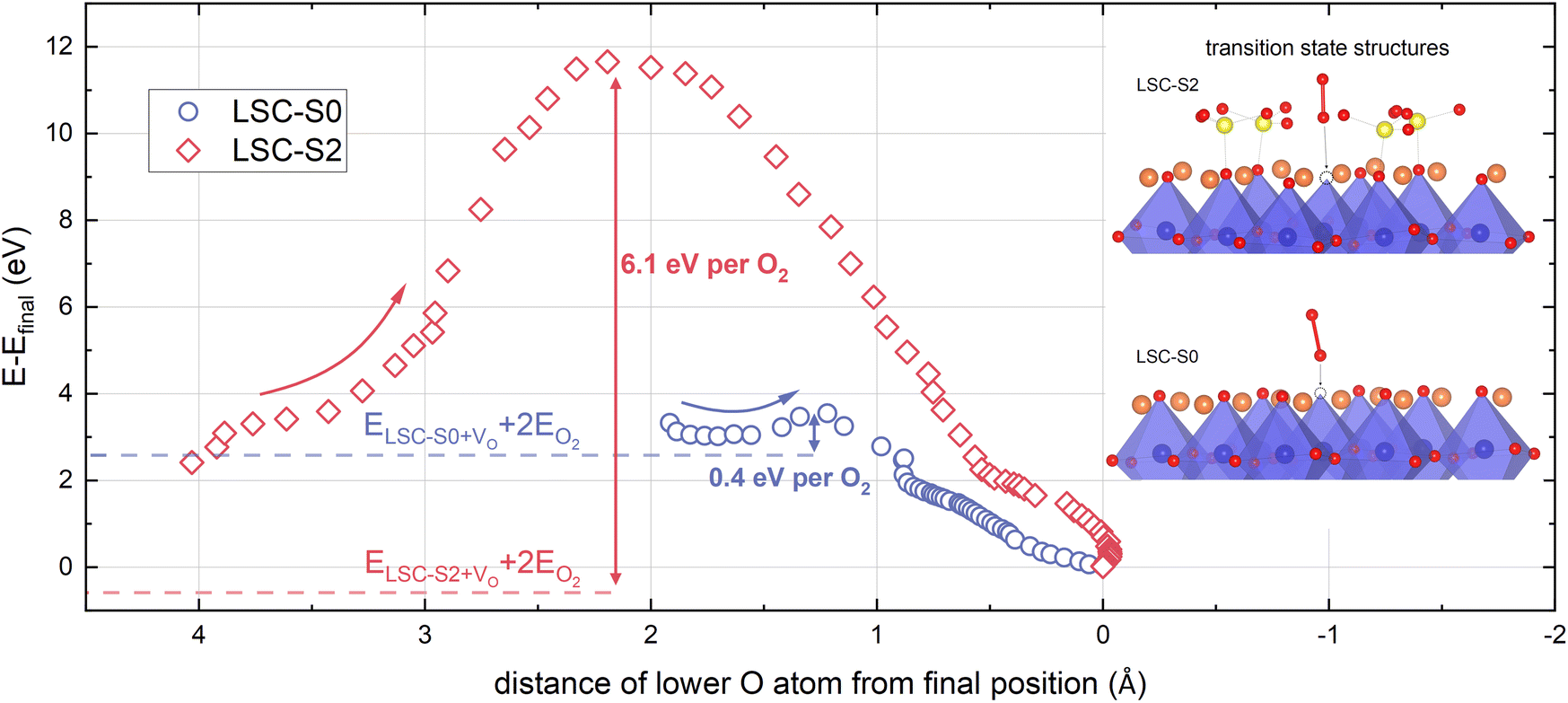 | ||
| Fig. 5 Evolution of the total energy during the approach of an O2 molecule to its final position in a surface vacancy on LSC-S0 and LSC-S2. The energy barriers are indicated in the graph and transition state structures are shown in the inset. The curves consist of ≈1300 (LSC-S0) and ≈1800 (LSC-S2) data points and the data has been averaged over 30 points. For barrier calculations, an RminMTKmax value of 5 was used for higher throughput, accounting for minor differences in adsorption energies. | ||
The method itself makes use of different simplifications. The O2 molecule does not possess initial momentum, all atoms are in static equilibrium positions and a pseudo-force is exerted on the lower O atom and the Co atom below (which might actually mimic oscillating of the atoms in a real adsorption situation) which is fully omitted after the barrier maximum is passed. Still, the analysis of the energy barrier illustrates that sulphate adsorbates change the energy landscape during O2 adsorption drastically. The energy barrier for this specific adsorption process changes from 0.4 eV to 6.1 eV per adsorbed O2 molecule. Also, the adsorption barrier occurs at a much larger distance from the LSC-S2 surface compared to LSC-S0.
O2 dissociation pathways. The dissociation of an O2 molecule in a surface vacancy on LSC-S0 and the subsequent lateral migration of the dissociated O atom were investigated computationally. Three different dissociation pathways were examined: dissociation via (i) a Sr bridge position between two nearest neighbour surface O atoms, (ii) a Sr bridge position in the second direction (these two directions differ by their orientation towards the tilted O-octahedra), (iii) a Sr top position, between two next nearest O atoms (relaxed structures are shown in S.2†). The dissociated O atom was constrained in x and y direction and structure relaxation was performed. The energy differences between the molecular O2 adsorbate and the dissociated structure give an indication about energetically favourable dissociation paths (Table 5).
| Sr bridge 1 | Sr bridge 2 | Sr top | |
|---|---|---|---|
| ΔE (eV) | 1.60 | 1.68 | 2.95 |
According to the calculations, the least likely dissociation path proceeds on top of a surface Sr atom. Instead, the dissociated atom is more likely to move between two surface Sr atoms. For the calculated structures of LSC-S2, these favourable dissociation pathways between two Sr atoms are blocked by SO2−4 adsorbates and thus only the energetically unfavorable dissociation path via the Sr top position remains, however, we also expect a higher dissociation/migration barrier due to the steric hindrance caused by SO2−4 adsorbates.
Work functions. As the work function is the prime experimental indication for the formation of a surface potential and a surface dipole, it has also been evaluated computationally for sulphate covered SrO-terminated LSC surfaces. The work function was calculated as the difference between the Fermi energy of the surface slab and the vacuum coulomb potential for the electron, evaluated in the middle of the vacuum between two neighbouring slabs. The results are shown in Table 6 and are in qualitative agreement with the experimental results. Starting with a relatively low work function, it increases steadily upon sulphate adsorption. Quantitatively, the experiment shows an increase of the work function from 4.2 to 4.8 eV for a SO2−4 coverage of ≈50%. However, a direct comparison of experimental and computational results is not straightforward, as XPS measurements were performed at elevated temperature at 8 × 10−6 mbar O2. Instead of a highly idealized surface with a perfectly (001) oriented SrO termination with no steps or kinks, we expect a surface with certain reconstructions, a substantial amount of surface vacancies, especially on sulphate covered LSC (which decrease the work function by supplying electrons, see also Table 6), and a non-negligible surface roughness. To exclude a dependency of the work function on the vacuum size, the work function of LSC-S0 was recalculated with a bigger vacuum (+20 bohr), yielding a value of 3.65 eV, demonstrating the validity of the analysis. In conclusion, theory confirms the experimental results that sulphate adsorption leads to a substantially increased work function. It is worth mentioning, that a similar phenomenon has already been discussed in the context of surface acidity and an extension to other acidic oxides will be discussed below .27
| LSC-S0 | LSC-S1 | LSC-S2 | LSC-S2 + VO | |
|---|---|---|---|---|
| WF (eV) | 3.69 | 6.18 | 7.65 | 6.78 |
Charge redistribution. As was shown experimentally and confirmed computationally, surface adsorption of sulphur results in the formation of SO2−4 surface species and an increase of the work function, indicating the formation of a surface dipole and charge transfer at the surface. To further investigate such charge transfer processes, the atomic charges in LSC-S0 and LSC-S2 have been compared using Baders theory of atoms in molecules.46 Selected results of this analysis are shown in Fig. 6a. Surprisingly, the charge of subsurface Co atoms is hardly affected by sulphate formation on the SrO-terminated LSC surface. Instead, a significant change of the charge of surface oxygens can be observed upon the addition of SO3 groups. According to the analysis, free surface oxygens transfer substantial amounts of charge towards oxygen in the sulphate group, leading to a charge depletion in the free outermost surface and a negative charge accumulation in the sulphate adsorbates. For LSC-S2, a charge of ∼0.2 e per free surface oxygen is transferred from the surface towards the adsorbates. With regard to the range of these redistribution effects, LSC-S1 was investigated as well, where only one of the 4 surface oxygens in the cell is covered with an SO3 group. Here, only the 2 nearest oxygen neighbours donate charge towards the adsorbate, while the diagonal oxygen is not affected. It is also noteworthy that, during all Bader charge investigations, the La charges remained completely unaffected.
 | ||
| Fig. 6 (a) Bader charges of subsurface Co atoms and surface O atoms for different scenarios. (b) Bader charges of O atoms in an adsorbed O2 molecule for LSC-S0 and LSC-S2 surfaces. | ||
In addition, a Bader charge analysis was also performed for structures containing a surface vacancy and structures with an O2 molecule adsorbed in the surface vacancy. For vacancy containing structures, the additional two electrons introduced by the surface vacancy are distributed across both, O and Co atoms, indicating that polaronic charge carriers in LSC are not exclusively localized on Co and that O atoms play a substantial role in charge compensation in LSC. This is also in line with a recent tendency to assign electron holes in La-based perovskites a strong oxygen character and to accredit surface oxygen a high importance during charge transfer steps in catalytic redox reactions.61–67
This is further confirmed by the Bader charge analysis for an adsorbed O2 molecule on LSC-S0 and LSC-S2 (see Fig. 6b). The amount of charge which is transferred to the O2 molecule is substantially higher for the sulphate free surface than it is for the sulphate covered surface, underlining the importance of surface O for charge transfer during the oxygen reduction reaction. This result is also in line with previous investigations, describing the formation of superoxo species in oxygen vacancies on AO-terminated surfaces of different mixed conducting perovskites (ABO3) and Ruddlesden-Popper materials (A2BO4).68,69 Interestingly, the difference between the charge localized at the adsorbed oxygen molecule on LSC-S0 compared to LSC-S2 amounts to 0.93 e, possibly establishing a tie to mechanistic discussions of the oxygen exchange reaction where single electron transfer is often considered as an essential reaction step.57,70
The spatial distribution of charge is also visualized in difference density plots in Fig. 7, i.e. the difference of the self consistent electron density and the superposition of spherical neutral atoms densities. Here, apart from the orbital configuration and the strong covalent contribution to the Co–O bonds, one can observe a stronger buckling of the sulphate-covered surface as well as the strong bond between the SO3 group and the surface atom below, forming the SO2−4 adsorbate.
 | ||
| Fig. 7 Difference electron density maps of a vertical CoO2 plane for LSC-S0 (a) and LSC-S2 (b). Blue regions denote less electron density relative to the neutral lattice. | ||
Densities of state (DOS). For more information about the electrostatic effects induced by SO2−4 adsorbates, the partial density of states (PDOS) of different surface and subsurface atoms on LSC-S0 and LSC-S2 was investigated. In particular, the PDOS of O 2p and Co 3d electrons were examined (Fig. 8), as these are the prime candidates to be affected by sulphate adsorption. A first examination of the results confirms the picture suggested by the Bader charge analysis – while the Co DOS is only slightly affected when SO3 is adsorbed on the LSC surface, the surface O PDOS change substantially. In particular, a strong shift to lower energies for the O 2p state can be observed (it is also noteworthy that the O 2p bandwidth at the surface with 4 eV is significantly narrower than in the subsurface with 6 eV).
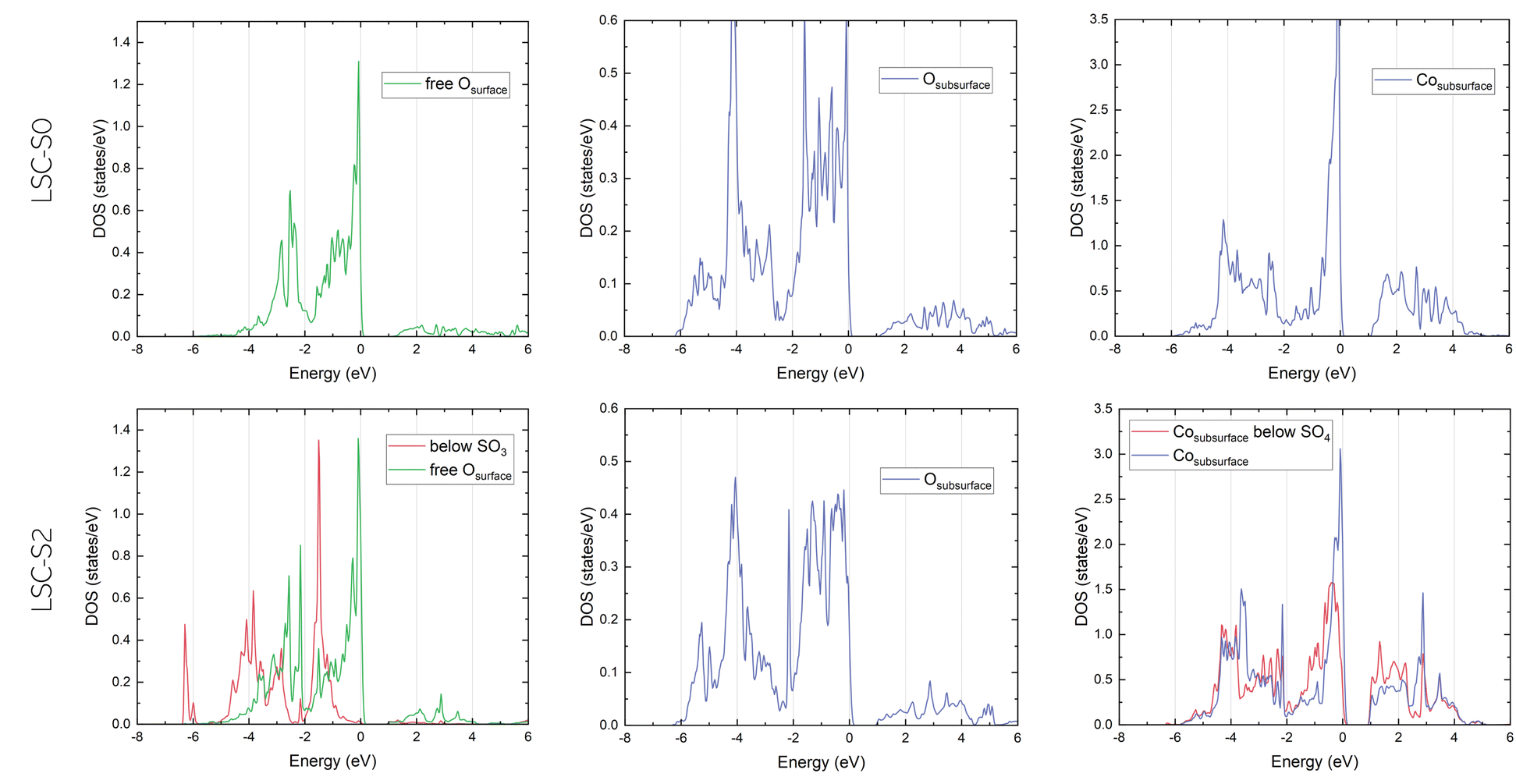 | ||
| Fig. 8 Densities of state for O-2p and Co-3d electrons for sulphate-free (top) and sulphate-covered (bottom) surfaces. | ||
This shift of the O 2p band is especially interesting in light of the ongoing discussion about the suitability of the O 2p band center as a descriptor for the oxygen reduction activity of a mixed conducting surface.71–74 For the present calculation, adsorption of an SO3 group moves the O 2p band center (identified as the centroid position of the O 2p band) from −1.44 eV on the sulphate free surface to −1.59 eV for a free surface O atom on a sulphate-covered surface and to −2.75 eV for an O below an SO3 group. Considering that reduction of molecular oxygen requires charge transfer to oxygen in a surface vacancy, it seems reasonable that this shift of the O 2p band of potentially charge transferring surface oxygens indeed contributes to a decrease of the reaction kinetics.
At the same time, these results also call for a critical examination of the idea of such a descriptor for the oxygen exchange activity. Due to the strong effect on the oxygen exchange kinetics observed upon sulphate adsorption (and surface modification in general27), it is questionable if the bulk O 2p band center is able to describe the oxygen activity of a material beyond a truly pristine state. Additionally, several studies have demonstrated the importance of surface terminations for the total oxygen exchange activity which is also not reflected by a bulk descriptor.44,51,75 Therefore, we suggest to further investigate the upper band edge of O atoms of a specific surface structure, which might more accurately describe the energetics of the oxygen reduction reaction.
Summarizing the so far obtained results, it becomes clear that sulphate adsorbates affect the surface properties of SrO-terminated LSC in manifold ways. The main underlying process upon sulphate adsorption is a charge transfer from surface oxygen towards the SO2−4 adsorbates. This introduces a surface dipole which is reflected in a substantial increase of the LSC workfunction, as was also observed experimentally. Additionally, sulphate adsorbates strongly affect the adsorption of O2 molecules, both via the adsorption barrier and via the final configuration of the adsorbate itself, affecting charge transfer and potentially also dissociation steps.
Effects of different acidic adsorbates
Apart from sulphur, several other oxides are known to degrade the oxygen exchange kinetics on SOFC cathode materials, such as CO220,76,77 or CrO3.78,79 A common property of all these oxides is their acidity relative to usual SOFC cathode material surfaces.27,80–82 While this fact has already been recognized previously,27,81 no conclusive mechanistic explanation for the correlation of surface acidity and oxygen exchange kinetics has been brought forward yet. While a detailed experimental and theoretical investigation of different combinations of surface oxides and host materials is far beyond the scope of this study, we suspect that the mechanism behind the here investigated sulphate induced degradation plays a major role in this discussion.For an initial assessment, the investigation is extended towards two other different, technologically relevant, acidic oxides, which may adsorb on the SrO-terminated LSC surface instead of SO3 groups. Computationally, SO3 groups have been replaced by CO2 and CrO3 groups, leading to carbonate (CO2−3) and chromate (CrO2−4) adsorbates. After a structure relaxation, the charge transfer has been assessed by performing a Bader charge analysis and calculating the difference between the charge of a free surface O atom and an adsorbate O atom. Again, Co atoms in the subsurface are not affected, confirming earlier investigations on SO3 groups. In addition, the work function has been evaluated for the CO2 and CrO3 covered LSC. The results are shown in Fig. 9. Very interestingly, we find that both the transferred charge and the work function correspond very well to the Smith acidity80 of the respective oxides, strongly indicating that the suggested phenomenon of charge transfer and surface dipole formation truly is the underlying process upon adsorption of acidic oxides.
 | ||
| Fig. 9 Effect of different acidic adsorbates on the work function and on the charge difference between surface oxygen atoms and adsorbate oxygen atoms on SrO-terminated LSC surfaces. | ||
In addition, the formation energies of oxygen vacancies on acidic surfaces have been evaluated, with the results shown in Table 7. Again, computational results confirm that the qualitative behaviour of sulphate covered LSC translates to other acidic adsorbates, i.e. the formation energy of surface oxygen vacancies is reduced substantially compared to the unperturbed SrO-termination. Quantitatively, the correlation with the Smith acidity is not as clear as for charge transfer and work function, in particular CrO2−4 adsorbates yield a low vacancy formation energy compared with SO2−4 adsorbates. The reason for this, however, requires further investigation of the LSC-CrO3 system.
| LSC-S0 | CO2−3 | CrO2−4 | SO2−4 | |
|---|---|---|---|---|
| VO,surf formation energy (eV) | 1.30 | 0.47 | 0.18 | 0.19 |
Impact of acidic adsorbates on the oxygen exchange reaction kinetics
To correlate theoretical predictions with experimental findings, it is necessary to discuss the impact of acidic adsorbates on the oxygen exchange kinetics in more detail. While significant advances have been achieved recently with regard to the clarification of the oxygen exchange mechanism,44,56,57,70,83 there is still no final consensus about the specific roles of the participating defects in the single reaction steps. It appears agreed upon that oxygen vacancies play an essential role in the incorporation of molecular oxygen, however, the discussion about whether charge transfer occurs before, during and/or after dissociation is still ongoing. Therefore, the here presented discussion tentatively assumes that charge transfer is coupled to the binding environments of the adsorbed oxygen molecule/atom and that the oxygen incorporation reaction follows the scheme (i) molecular adsorption (ii) dissociation (iii) incorporation.Based on the experimental and computational results we suggest that the following aspects need to be considered when discussing the oxygen exchange kinetics on LSC surfaces covered with acidic adsorbates:
– Acidic adsorbates affect the adsorption energy of O2 molecules as well as the adsorption barrier, thus strongly affecting upstream reaction steps of the incorporation direction. The same might hold for upstream reaction steps of the oxygen evolution direction, as the concentration of surface vacancies combined with a high acidic adsorbate coverage might strongly affect the number of surface oxygens available for O2 molecule formation. A more in-depth discussion of the connection between equilibrium concentrations and reaction energetics is presented in S.3.†
– If the dissociation of adsorbed O2 is the rate limiting step of the incorporation direction (different O2 dissociation pathways are discussed above), the kinetic barrier of this step will be affected by acidic adsorbates, indicated by the smaller bond length and reduced charge of the adsorbed O2 on LSC-S2 as well as different migration barriers or blocked dissociation pathways.
– Defect concentrations in and beneath the LSC surface are altered considerably. Oxygen vacancies clearly accumulate on surfaces with acidic adsorbates, however, subsurface vacancies are very unfavourable. This might strongly affect the oxygen transport to and from the surface. The situation for electronic charge carriers is even more complicated, as it depends on which particular species participates in charge transfer to the O2 molecule. While the oxygen exchange reaction is commonly written under the assumption that charge transfer equals a reduction or oxidation of a metal cation (e.g. Co), as mentioned earlier, recent studies indicate that electron holes have a strong oxygen character.62,67 While DFT calculations suggest that transition metals are not affected substantially by acidic adsorbates, for the latter case, with oxygen participating in the charge transfer, acidic adsorbates will have a strong effect on the availability of charge carriers for the reaction.
– Lastly, depending on the mechanism of the oxygen exchange reaction, also the surface dipole/potential itself, which is established by the charge transfer to the adsorbates will have an impact on the final reaction rate if charge is transferred across this electric field.
Conclusions
The electronic and ionic effects of acidic adsorbates on (La,Sr)CoO3−δ (LSC) surfaces have been investigated experimentally and computationally. Near ambient pressure XPS and low energy ion scattering have been employed to examine the surface chemistry of LSC surfaces upon exposure to sulphur containing trace impurities in the measurement atmosphere. The experiment found that sulphur is bound in an SO2−4 environment and that these adsorbates induce a significant shift of the work function by 0.6 eV. Impedance spectroscopy revealed that such sulphate formation processes lead to a strong and sudden degradation of the oxygen surface exchange kinetics in equilibrium. Ab initio calculations were then performed to gain insight into the detailed processes occurring on LSC surfaces upon sulphate adsorption. These calculations found strongly bound sulphate adsorbates which cause various changes in the surface chemistry of LSC: (i) charge is transferred from surface oxygen atoms to the sulphate groups; (ii) vacancy formation energies are significantly reduced in a sulphate covered surface and increased in the sub-surface; (iii) sulphate adsorbates inhibit the adsorption of molecular O2 into a surface vacancy by strongly increasing the adsorption barrier and by rendering an O2 adsorbate configuration energetically unfavourable. Exemplary calculations have been performed for two additional acidic adsorbates CO2−3 and CrO2−4 and reveal a quantitative agreement of electrostatic changes (amount of transferred charge and work function changes) with the Smith acidity scale, which has recently been proposed as a descriptor for the oxygen exchange kinetics on mixed conducting surfaces. Several processes have been identified which might affect the oxygen exchange kinetics of LSC surfaces in contact with acidic adsorbates, expanding our understanding of mixed conducting surfaces and the oxygen exchange reaction itself.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors like to acknowledge the financial support and open access funding provided by the Austrian Science Fund (FWF) project P31654-N37. M. S. was also partly supported by the Competence Center for Electrochemical Surface Technology (CEST) in the framework of the COMET scheme of the Austrian Research Promotion Agency (FFG, project 865864).References
- P. Boldrin and N. P. Brandon, Progress and outlook for solid oxide fuel cells for transportation applications, Nat. Catal., 2019, 2, 571–577 CrossRef CAS.
- R. M. Ormerod, Solid oxide fuel cells, Chem. Soc. Rev., 2003, 32, 17–28 RSC.
- M. Singh, D. Zappa and E. Comini, Solid oxide fuel cell: Decade of progress, future perspectives and challenges, Int. J. Hydrogen Energy, 2021, 46, 27643–27674 CrossRef CAS.
- E. D. Wachsman and K. T. Lee, Lowering the temperature of solid oxide fuel cells, Science, 2011, 334, 935–939 CrossRef CAS PubMed.
- J. Zhang, C. Lenser, N. H. Menzler and O. Guillon, Comparison of solid oxide fuel cell (SOFC) electrolyte materials for operation at 500 °C, Solid State Ionics, 2020, 344, 115138 CrossRef CAS.
- J. Patakangas, Y. Ma, Y. Jing and P. Lund, Review and analysis of characterization methods and ionic conductivities for low-temperature solid oxide fuel cells (LTSOFC), J. Power Sources, 2014, 263, 315–331 CrossRef CAS.
- A. J. Abd Aziz, N. A. Baharuddin, M. R. Somalu and A. Muchtar, Review of composite cathodes for intermediatetemperature solid oxide fuel cell applications, Ceram. Int., 2020, 46, 23314–23325 CrossRef CAS.
- M. Z. Ahmad, S. H. Ahmad, R. S. Chen, A. F. Ismail, R. Hazan and N. A. Baharuddin, Review on recent advancement in cathode material for lower and intermediate temperature solid oxide fuel cells application, Int. J. Hydrogen Energy, 2022, 47, 1103–1120 CrossRef CAS.
- A. Jun, J. Kim, J. Shin and G. Kim, Perovskite as a cathode material: a review of its role in solid-oxide fuel cell technology, ChemElectroChem, 2016, 3, 511–530 CrossRef CAS.
- S. P. Simner, M. D. Anderson, M. H. Engelhard and J. W. Stevenson, Degradation Mechanisms of La–Sr–Co–Fe–O3 SOFC Cathodes, Electrochem. Solid-State Lett., 2006, 9, A478 CrossRef CAS.
- Q. Fang, L. Blum and D. Stolten, Electrochemical performance and degradation analysis of an SOFC short stack following operation of more than 100,000 hours, J. Electrochem. Soc., 2019, 166, F1320 CrossRef CAS.
- I. Sreedhar, B. Agarwal, P. Goyal and A. Agarwal, An overview of degradation in solid oxide fuel cells-potential clean power sources, J. Solid State Electrochem., 2020, 24, 1239–1270 CrossRef CAS.
- B. Koo, K. Kim, J. K. Kim, H. Kwon, J. W. Han and W. Jung, Sr segregation in perovskite oxides: why it happens and how it exists, Joule, 2018, 2, 1476–1499 CrossRef CAS.
- W. Jung and H. L. Tuller, Investigation of surface Sr segregation in model thin film solid oxide fuel cell perovskite electrodes, Energy Environ. Sci., 2012, 5, 5370–5378 RSC.
- M. Kubicek, G. M. Rupp, S. Huber, A. Penn, A. K. Opitz, J. Bernardi, M. Stöger-Pollach, H. Hutter and J. Fleig, Cation diffusion in La0.6Sr0.4CoO3−δ below 800 °C and its relevance for Sr segregation, Phys. Chem. Chem. Phys., 2014, 16, 2715–2726 RSC.
- E. Bucher, C. Gspan, F. Hofer and W. Sitte, Post-test analysis of silicon poisoning and phase decomposition in the SOFC cathode material La0.58Sr0.4Co0.2Fe0.8O3−δ by transmission electron microscopy, Solid State Ionics, 2013, 230, 7–11 CrossRef CAS.
- E. Bucher, C. Gspan, F. Hofer and W. Sitte, Sulphur poisoning of the SOFC cathode material La0.6Sr0.4CoO3−δ, Solid State Ionics, 2013, 238, 15–23 CrossRef CAS.
- F. Wang, H. Kishimoto, T. Ishiyama, K. Develos-Bagarinao, K. Yamaji, T. Horita and H. Yokokawa, A review of sulfur poisoning of solid oxide fuel cell cathode materials for solid oxide fuel cells, J. Power Sources, 2020, 478, 228763 CrossRef CAS.
- C. Harrison, P. Slater and R. Steinberger-Wilckens, A review of Solid Oxide Fuel Cell cathode materials with respect to their resistance to the effects of chromium poisoning, Solid State Ionics, 2020, 354, 115410 CrossRef CAS.
- A. Yan, M. Cheng, Y. Dong, W. Yang, V. Maragou, S. Song and P. Tsiakaras, Investigation of a Ba0.5Sr0.5Co0.8Fe0.2O3−δ based cathode IT-SOFC: I. The effect of CO2 on the cell performance, Appl. Catal., B, 2006, 66, 64–71 CrossRef CAS.
- A. K. Opitz, C. Rameshan, M. Kubicek, G. M. Rupp, A. Nenning, T. Götsch, R. Blume, M. Hävecker, A. Knop-Gericke and G. Rupprechter, et al., The chemical evolution of the La0.6Sr0.4CoO3−δ surface under SOFC operating conditions and its implications for electrochemical oxygen exchange activity, Top. Catal., 2018, 61, 2129–2141 CrossRef CAS PubMed.
- R. Budiman, S. Liu, K. Bagarinao, T. Ishiyama, H. Kishimoto, K. Yamaji, T. Horita and H. Yokokawa, Determination of factors governing surface composition and degradation of La0.6Sr0.4Co0.2Fe0.8O3−δ electrode under sulfur-contained air, J. Electrochem. Soc., 2019, 166, F414 CrossRef CAS.
- C. Riedl, M. Siebenhofer, A. Nenning, A. Schmid, M. Weiss, C. Rameshan, A. Limbeck, M. Kubicek, A. K. Opitz and J. Fleig, In situ techniques reveal the true capabilities of SOFC cathode materials and their sudden degradation due to omnipresent sulfur trace impurities, J. Mater. Chem. A, 2022, 10, 14838–14848 RSC.
- M. Yoshikawa, T. Yamamoto, K. Yasumoto and Y. Mugikura, Degradation analysis of SOFC stack performance: investigation of cathode sulfur poisoning due to contamination in air, ECS Trans., 2017, 78, 2347 CrossRef CAS.
- A. J. Schuler, Z. Wuillemin, A. Hessler-Wyser and J. Van Herle, Sulfur as pollutant species on the cathode side of a SOFC system, ECS Trans., 2009, 25, 2845 CrossRef CAS.
- M. Siebenhofer, U. Haselmann, A. Nenning, G. Friedbacher, A. E. Bumberger, S. Wurster, W. Artner, H. Hutter, Z. Zhang and J. Fleig, et al., Surface Chemistry and Degradation Processes of Dense La0.6Sr0.4CoO3−δ Thin Film Electrodes, J. Electrochem. Soc., 2023, 170, 014501 CrossRef.
- C. Nicollet, C. Toparli, G. F. Harrington, T. Defferriere, B. Yildiz and H. L. Tuller, Acidity of surface-infiltrated binary oxides as a sensitive descriptor of oxygen exchange kinetics in mixed conducting oxides, Nat. Catal., 2020, 3, 913–920 CrossRef CAS.
- E. J. Crumlin, E. Mutoro, Z. Liu, M. E. Grass, M. D. Biegalski, Y.-L. Lee, D. Morgan, H. M. Christen, H. Bluhm and Y. Shao-Horn, Surface strontium enrichment on highly active perovskites for oxygen electrocatalysis in solid oxide fuel cells, Energy Environ. Sci., 2012, 5, 6081–6088 RSC.
- E. Mutoro, E. J. Crumlin, M. D. Biegalski, H. M. Christen and Y. Shao-Horn, Enhanced oxygen reduction activity on surface-decorated perovskite thin films for solid oxide fuel cells, Energy Environ. Sci., 2011, 4, 3689–3696 RSC.
- G. M. Rupp, A. Limbeck, M. Kubicek, A. Penn, M. Stöger-Pollach, G. Friedbacher and J. Fleig, Correlating surface cation composition and thin film microstructure with the electrochemical performance of lanthanum strontium cobaltite (LSC) electrodes, J. Mater. Chem. A, 2014, 2, 7099–7108 RSC.
- R. Rameshan, A. Nenning, J. Raschhofer, L. Lindenthal, T. Ruh, H. Summerer, A. K. Opitz, T. Martin Huber and C. Rameshan, Novel sample-stage for combined near ambient pressure x-ray photoelectron spectroscopy, catalytic characterization and electrochemical impedance spectroscopy, Crystals, 2020, 10, 947 CrossRef CAS.
- C. Ahamer, A. K. Opitz, G. Rupp and J. Fleig, Revisiting the temperature dependent ionic conductivity of yttria stabilized zirconia (YSZ), J. Electrochem. Soc., 2017, 164, F790 CrossRef CAS.
- D. J. Singh & L. P. Nordstrom, Pseudopotentials, and the LAPW Method, Springer Science & Business Media, 2006 Search PubMed.
- P. Blaha, K. Schwarz, G. K. Madsen, D. Kvasnicka, J. Luitz, et al.wien2k. An Augmented Plane Wave+local Orbitals Program for Calculating Crystal Properties, 2001 Search PubMed.
- P. Blaha, K. Schwarz, F. Tran, R. Laskowski, G. K. Madsen and L. D. Marks, WIEN2k: An APW+lo program for calculating the properties of solids, J. Chem. Phys., 2020, 152, 074101 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- L. Wang, T. Maxisch and G. Ceder, Oxidation energies of transition metal oxides within the GGA+U framework, Phys. Rev. B, 2006, 73, 195107 CrossRef.
- M. J. Gadre, Y.-L. Lee and D. Morgan, Cation interdiffusion model for enhanced oxygen kinetics at oxide heterostructure interfaces, Phys. Chem. Chem. Phys., 2012, 14, 2606–2616 RSC.
- Y.-L. Lee, J. Kleis, J. Rossmeisl and D. Morgan, Ab initio energetics of LaBO3 (001)(B=Mn,Fe,Co and Ni) for solid oxide fuel cell cathodes, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 224101 CrossRef.
- A. Wold and R. Ward, Perowskite-type oxides of cobalt, chromium and vanadium with some rare earth elements, J. Am. Chem. Soc., 1954, 76, 1029–1030 CrossRef CAS.
- S. Patil, H. Keer and D. Chakrabarty, Structural, electrical, and magnetic properties in the system BaxLa1−xCoO3, Phys. Status Solidi A, 1979, 52, 681–686 CrossRef CAS.
- M. A. Niania, A. K. Rossall, J. A. Van den Berg and J. A. Kilner, The effect of sub-surface strontium depletion on oxygen diffusion in La0.6Sr0.4Co0.2Fe0.8O3−δ, J. Mater. Chem. A, 2020, 8, 19414–19424 RSC.
- G. M. Rupp, H. Téllez, J. Druce, A. Limbeck, T. Ishihara, J. Kilner and J. Fleig, Surface chemistry of La0.6Sr0.4CoO3−δ thin films and its impact on the oxygen surface exchange resistance, J. Mater. Chem. A, 2015, 3, 22759–22769 RSC.
- Y. Cao, M. J. Gadre, A. T. Ngo, S. B. Adler and D. D. Morgan, Factors controlling surface oxygen exchange in oxides, Nat. Commun., 2019, 10, 1–15 CrossRef PubMed.
- J. W. Han and B. Yildiz, Mechanism for enhanced oxygen reduction kinetics at the (La,Sr)CoO3−δ/(La,Sr)2CoO3+δ hetero-interface, Energy Environ. Sci., 2012, 5, 8598–8607 RSC.
- R. F. Bader, Atoms in molecules, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- D. Zhang, M. L. Machala, D. Chen, Z. Guan, H. Li, S. Nemsak, E. J. Crumlin, H. Bluhm and W. C. Chueh, Hydroxylation and cation segregation in (La0.5Sr0.5)FeO3−δ electrodes, Chem. Mater., 2020, 32, 2926–2934 CrossRef CAS.
- K. W. Böer Introduction to Space Charge Effects in Semiconductors, Springer, 2010 Search PubMed.
- G. M. Rupp, A. Schmid, A. Nenning and J. Fleig, The superior properties of La0.6Ba0.4CoO3−δ thin film electrodes for oxygen exchange in comparison to La0.6Sr0.4CoO3−δ, J. Electrochem. Soc., 2016, 163, F564 CrossRef CAS.
- H. H. Brongersma, M. Draxler, M. De Ridder and P. Bauer, Surface composition analysis by low-energy ion scattering, Surf. Sci. Rep., 2007, 62, 63–109 CrossRef CAS.
- J. Druce, H. Tellez, M. Burriel, M. Sharp, L. Fawcett, S. Cook, D. McPhail, T. Ishihara, H. Brongersma and J. Kilner, Surface termination and subsurface restructuring of perovskite-based solid oxide electrode materials, Energy Environ. Sci., 2014, 7, 3593–3599 RSC.
- M. Burriel, H. Téllez, R. J. Chater, R. Castaing, P. Veber, M. Zaghrioui, T. Ishihara, J. A. Kilner and J.-M. Bassat, Influence of crystal orientation and annealing on the oxygen diffusion and surface exchange of La2NiO4+d, J. Phys. Chem. C, 2016, 120, 17927–17938 CrossRef CAS.
- M. Niania, M. Sharpe, R. Webb and J. Kilner, The surface of complex oxides; ion beam based analysis of energy materials, Nucl. Instrum. Methods Phys. Res. B, 2020, 480, 27–32 CrossRef CAS.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Water electrolysis on La1−xSrxCoO3−d perovskite electrocatalysts, Nat. Commun., 2016, 7, 1–11 Search PubMed.
- P. Linstorm, NIST chemistry webbook, NIST standard reference database number 69, J. phys. chem. ref. data, Monogr., 1998, 9, 1–1951 Search PubMed.
- D. Chen, Z. Guan, D. Zhang, L. Trotochaud, E. Crumlin, S. Nemsak, H. Bluhm, H. L. Tuller and W. C. Chueh, Constructing a pathway for mixed ion and electron transfer reactions for O2 incorporation in Pr0.1Ce0.9O2−x, Nat. Catal., 2020, 3, 116–124 CrossRef CAS.
- M. Siebenhofer, C. Riedl, A. Schmid, A. Limbeck, A. K. Opitz, J. Fleig and M. Kubicek, Investigating oxygen reduction pathways on pristine SOFC cathode surfaces by in situ PLD impedance spectroscopy, J. Mater. Chem. A, 2022, 10, 2305–2319 RSC.
- M. Mosleh, M. Søgaard and P. V. Hendriksen, Kinetics and mechanisms of oxygen surface exchange on La0.6Sr0.4FeO3−δ thin films, J. Electrochem. Soc., 2009, 156, B441 CrossRef CAS.
- N. Tsvetkov, Q. Lu, L. Sun, E. J. Crumlin and B. Yildiz, Improved chemical and electrochemical stability of perovskite oxides with less reducible cations at the surface, Nat. Mater., 2016, 15, 1010–1016 CrossRef CAS PubMed.
- L. Wang, R. Merkle, Y. A. Mastrikov, E. A. Kotomin and J. Maier, Oxygen exchange kinetics on solid oxide fuel cell cathode materials—general trends and their mechanistic interpretation, J. Mater. Res., 2012, 27, 2000–2008 CrossRef CAS.
- X. Wang, K. Huang, L. Yuan, S. Xi, W. Yan, Z. Geng, Y. Cong, Y. Sun, H. Tan and X. Wu, et al., Activation of surface oxygen sites in a cobalt-based perovskite model catalyst for CO oxidation, J. Phys. Chem. Lett., 2018, 9, 4146–4154 CrossRef CAS PubMed.
- J. Wang, J. Yang, A. K. Opitz, D. Kalaev, A. Nenning, E. J. Crumlin, J. T. Sadowski, I. Waluyo, A. Hunt and H. L. Tuller, et al., Strain-Dependent Surface Defect Equilibria of Mixed Ionic-Electronic Conducting Perovskites, Chem. Mater., 2022, 34, 5138–5150 CrossRef CAS.
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Perovskites in catalysis and electrocatalysis, Science, 2017, 358, 751–756 CrossRef CAS PubMed.
- D. N. Mueller, M. L. Machala, H. Bluhm and W. C. Chueh, Redox activity of surface oxygen anions in oxygendeficient perovskite oxides during electrochemical reactions, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- C. Yang and A. Grimaud, Factors controlling the redox activity of oxygen in perovskites: from theory to application for catalytic reactions, Catalysts, 2017, 7, 149 CrossRef.
- R. Zohourian, R. Merkle, G. Raimondi and J. Maier, Mixed-conducting perovskites as cathode materials for protonic ceramic fuel cells: understanding the trends in proton uptake, Adv. Funct. Mater., 2018, 28, 1801241 CrossRef.
- A. M. Ritzmann, A. B. Munnoz-Garcia, M. Pavone, J. A. Keith and E. A. Carter, Ab initio DFT+ U analysis of oxygen vacancy formation and migration in La1−xSrxFeO3−δ (x=0,0.25,0.50), Chem. Mater., 2013, 25, 3011–3019 CrossRef CAS.
- A. Staykov, H. Tellez, T. Akbay, J. Druce, T. Ishihara and J. Kilner, Oxygen activation and dissociation on transition metal free perovskite surfaces, Chem. Mater., 2015, 27, 8273–8281 CrossRef CAS.
- T. Akbay, A. Staykov, J. Druce, H. Tellez, T. Ishihara and J. A. Kilner, The interaction of molecular oxygen on LaO terminated surfaces of La2NiO4, J. Mater. Chem. A, 2016, 4, 13113–13124 RSC.
- R. Merkle and J. Maier, How is oxygen incorporated into oxides? A comprehensive kinetic study of a simple solid-state reaction with SrTiO3 as a model material, Angew. Chem., Int. Ed., 2008, 47, 3874–3894 CrossRef CAS PubMed.
- Y.-L. Lee, J. Kleis, J. Rossmeisl, Y. Shao-Horn and D. Morgan, Prediction of solid oxide fuel cell cathode activity with first-principles descriptors, Energy Environ. Sci., 2011, 4, 3966–3970 RSC.
- R. Jacobs, J. Booske and D. Morgan, Understanding and controlling the work function of perovskite oxides using density functional theory, Adv. Funct. Mater., 2016, 26, 5471–5482 CrossRef CAS.
- R. Jacobs, T. Mayeshiba, J. Booske and D. Morgan, Material discovery and design principles for stable, high activity perovskite cathodes for solid oxide fuel cells, Adv. Energy Mater., 2018, 8, 1702708 CrossRef.
- R. Jacobs, J. Hwang, Y. Shao-Horn and D. Morgan, Assessing correlations of perovskite catalytic performance with electronic structure descriptors, Chem. Mater., 2019, 31, 785–797 CrossRef CAS.
- Y. A. Mastrikov, R. Merkle, E. A. Kotomin, M. M. Kuklja and J. Maier, Surface termination effects on the oxygen reduction reaction rate at fuel cell cathodes, J. Mater. Chem. A, 2018, 6, 11929–11940 RSC.
- J. Hayd and E. Ivers-Tiffée, Detailed electrochemical study on nanoscaled La0.6Sr0.4CoO3−δ SOFC Thin-film cathodes in dry, humid and CO2-containing atmospheres, J. Electrochem. Soc., 2013, 160, F1197 CrossRef CAS.
- Z. Zhao, L. Liu, X. Zhang, W. Wu, B. Tu, D. Ou and M. Cheng, A comparison on effects of CO2 on La0.8Sr0.2MnO3+δ and La0.6Sr0.4CoO3−δ cathodes, J. Power Sources, 2013, 222, 542–553 CrossRef CAS.
- E. Bucher, M. Yang and W. Sitte, In situ investigations of the chromium-induced degradation of the oxygen surface exchange kinetics of IT-SOFC cathode materials La0.6Sr0.4CoO3−δ and La0.58Sr0.4Co0.2Fe0.8O3−δ, J. Electrochem. Soc., 2012, 159, B592 CrossRef CAS.
- N. Schrödl, E. Bucher, A. Egger, P. Kreiml, C. Teichert, T. Höschen and W. Sitte, Long-term stability of the ITSOFC cathode materials La0.6Sr0.4CoO3−δ and La02NiO4+δ against combined chromium and silicon poisoning, Solid State Ionics, 2015, 276, 62–71 CrossRef.
- D. W. Smith, An acidity scale for binary oxides, J. Chem. Educ., 1987, 64, 480 CrossRef CAS.
- C. Nicollet and H. L. Tuller, Perspective on the Relationship between the Acidity of Perovskite Oxides and Their Oxygen Surface Exchange Kinetics, Chem. Mater., 2022, 34, 991–997 CrossRef CAS.
- H. G. Seo, A. Staerz, D. S. Kim, D. Klotz, C. Nicollet, M. Xu, J. M. LeBeau & H. L. Tuller, Reactivation of Chromia Poisoned Oxygen Exchange Kinetics in Mixed Conducting Solid Oxide Fuel Cell Electrodes by Serial Infiltration of Lithia, Energy & Environmental Science, 2022 Search PubMed.
- W. Jung and H. L. Tuller, A New Model Describing Solid Oxide Fuel Cell Cathode Kinetics: Model Thin Film SrTi1−xFexO3−δ Mixed Conducting Oxides–a Case Study, Adv. Energy Mater., 2011, 1, 1184–1191 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta00978e |
| This journal is © The Royal Society of Chemistry 2023 |