
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning proton kinetics in BaCo0.4Fe0.4Zr0.2–XYXO3–δ triple ionic-electronic conductors via aliovalent substitution†
Jack H.
Duffy
 ab,
Harry W.
Abernathy
b and
Kyle S.
Brinkman
*ab
ab,
Harry W.
Abernathy
b and
Kyle S.
Brinkman
*ab
aDepartment of Materials Science and Engineering, Clemson University, 515 Calhoun Drive, Clemson, SC 29634, USA. E-mail: ksbrink@clemson.edu
bNational Energy Technology Laboratory, United States Department of Energy, 3610 Collins Ferry Road, Morgantown, WV 26507, USA
First published on 3rd April 2023
Abstract
The BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY0.1) triple ionic-electronic conductor (TIEC) has received thorough investigation as a potential cathode in protonic ceramic fuel cells (PCFCs) due to its excellent oxygen reduction reaction and concurrent conduction of electrons, oxygen ions, and protons. Proton conductivity and surface reactivity are paramount in PCFC cathodes to improve the active reaction area. However, few instances of direct proton kinetic measurements have been reported. In this work, a suite of BaCo0.4Fe0.4Zr0.2−XYXO3−δ (X = 0, 0.1, 0.2) materials is synthesized and evaluated through hydrogen permeation and electrical conductivity relaxation measurements to investigate the effect of aliovalent substitution of Y3+ for Zr4+ on bulk proton conductivity and surface kinetics. The permeation results suggest that aliovalent substitution significantly improves the proton conductivity upon a 10% B-site doping of Y, while further incorporation of Y slightly decreases conductivity from the 10% optimum. Through three separate conductivity relaxation measurements, oxidation, hydration, and isotopic switching, an improvement in the proton kinetics with Y-doping is observed in humidified oxidizing conditions, emulating conditions in intermediate-temperature electrochemical devices. These observations suggest that aliovalient doping plays an important role in the incorporation and mobility of protons in TIEC materials.
1. Introduction
Triple ionic-electronic conductors (TIECs) are an emerging class of materials for low to intermediate temperature electrochemical applications such as protonic ceramic fuel cells (PCFCs), catalysis, and membrane reactors.1–4 TIECs as currently defined in the literature are predominantly electron hole conductors, but also concurrently transport oxygen ions and protons under certain atmospheric conditions. The conduction of all three species makes TIECs excellent candidates as cathodes in PCFCs due to their high activity, good stability, and facile synthesis routes.5–8 Unlike in cathodes for oxide-ion-conducting solid oxide fuel cells (SOFCs), proton mobility is essential in cathodes for the operation of PCFCs; expanding the active area for water formation at the cathode improves overall reaction rate and reduces the potential for delamination at the electrolyte–cathode interface.9,10Numerous methods have been employed to study bulk proton mobility in materials with protonic, oxygen ionic, and electronic conductivity.11 Indirect methods such as proton uptake measurements via thermogravimetric analysis12–14 and proton exchange measurements via electrical conductivity relaxation (ECR)15–17 have been used to estimate proton mobility. In predominantly ionic-conducting materials, such as BaCeO3–BaZrO3-based solid solutions, total conductivity through electrochemical impedance spectroscopy (EIS) and electromotive force measurements (EMF) across varying atmospheres can directly probe the conductivities of protons, oxygen ions, and electrons.18–20 However, in TIECs, these methods prove difficult to deconvolute the ionic conductivities from the electronic conductivity, which typically accounts for nearly all the total conductivity.
Direct methods for defining proton conductivity in TIECs are available but have only been used to a limited extent. Hydrogen separation via gas permeation is well-studied in many perovskite ceramics and cermet materials.21 Utilizing a combination of gas permeation studies for protonic and oxygen-ionic carriers with DC four-point conductivity for electronic carriers has fully compared transport across varying atmospheric conditions in Sr2Sc0.1Nb0.1Co1.5Fe0.3O6−δ (SSNCF) and nanocomposite materials BaCe0.16Y0.04Fe0.8O3−δ (BCYF) and BaCo0.7Ce0.24Y0.06O3−δ (BCCY).22–24 Zhong, et al. recently utilized Hebb-Wagner polarization with blocking electrodes for estimation of proton conductivity of La2NiO4-based Ruddlesden–Popper oxides, though only at temperatures below 300 °C where protonic blocking electrodes could be realized.25
The BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY0.1) class of materials is widely studied as a model single-phase cubic perovskite and air-electrode for ceramic fuel cells and electrolysis applications. Beginning from BaCo0.4Fe0.4Zr0.2O3−δ (BCFZ), the substitution of Y3+ for Zr4+ was expected to impart greater protonic conductivity and improve oxygen kinetics.6,26,27 Our recent work probed this Y substitution and revealed the tradeoff between increasing surface kinetics and decreasing bulk oxygen ion conductivity with Y substitution.28 Numerous recent studies have probed protonic conductivity using gas permeation on BCFZY0.1 and its modified derivatives.29–33 However, systematic studies on the bulk protonic conductivity by varying aliovalent doping, like those performed in ionic conductors such as BaZr1−XYXO3−δ (BZY),34 are lacking in this system.
In this work, the substitution of Y3+ for Zr4+ in BaCo0.4Fe0.4Zr0.2−XYXO3−δ (BCFZYX, X = 0, 0.1, 0.2) is investigated for its effect on proton surface kinetics and bulk conductivity through electrical conductivity relaxation (ECR) and hydrogen permeation measurements. Maxima in protonic conductivity and surface kinetics are observed for the Zr and Y co-doped BCFZY0.1, while further Zr4+ or Y3+ substitution hinders the proton kinetics and bulk conductivity. Furthermore, the incorporation of water, and therefore protonic defects, decreases the activation energy of conductivity and surface exchange experiments. These results quantify and reveal the optimal concentration of aliovalent doping for proton mobility in the BCFZYX system while furthering understanding of high-performance TIEC materials.
2. Experimental
2.1 Synthesis of BCFZYX membranes
BCFZYX materials were synthesized using a wet chemistry sol–gel method.6 Stoichiometric amounts of precursor nitrates were dissolved in distilled water. Citric acid and EDTA were then added to the solution in a ratio of citric acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EDTA:metal of 1.5:1.5:1 as chelating agents. While stirring, ammonium hydroxide was added to the solution until the solution achieved a pH of 9. The solution was heated at 80 °C until a gel formed and was subsequently placed in an oven at 150 °C for 36 h. The resultant complex was fired at 1000 °C for 10 h to remove organic residues and calcine the perovskite phase.
:EDTA:metal of 1.5:1.5:1 as chelating agents. While stirring, ammonium hydroxide was added to the solution until the solution achieved a pH of 9. The solution was heated at 80 °C until a gel formed and was subsequently placed in an oven at 150 °C for 36 h. The resultant complex was fired at 1000 °C for 10 h to remove organic residues and calcine the perovskite phase.
Membranes for further experiments were fabricated by dry-pressing calcined powder. The BCFZYX powder was pressed at 160 MPa for two minutes into 15 mm pellets for permeation measurements and 20 mm pellets for ECR measurements. The pellets were then covered with calcined mother powder to reduce potential Ba-loss in sintering conditions. All pellets were sintered at 1275 °C for 8 h to form the dense membranes. All sintered samples had relative densities greater than 95%. The sintered pellets were then polished to 0.6 mm thickness for permeation and 1.1 mm thickness for ECR measurements, using progressively higher sanding paper grits up to 2000 grit to achieve a shiny surface and reduce surface effects during characterization.
2.2 Hydrogen permeation
Hydrogen permeation was measured using a homemade double-volume permeation cell. Each BCFZYX membrane was sealed to an alumina ceramic tube (O.D. = 0.5 in., I.D. = 0.375 in.) using ceramic bond paste (Ceramabond 552-VFG), covering the sides of the pellet to reduce potential side-wall permeation. The tube with the sealed pellet was then partially inserted into a larger tube (O.D. = 0.75 in., I.D. = 0.563 in.) and the interface between the two tubes was sealed with the same ceramic bond to create the fully sealed permeation cell. The 5% H2/95% N2 feed gas (fed through a room temperature water bubbler for some measurements) was fed through the large tube at 50 mL min−1. Ultra-high purity Argon (Ar, 99.999%) sweep gas was fed at 50 mL min−1 through the small tube. The sweep gas is then fed to an online gas chromatograph (GC, Inficon Micro GC) with a thermal conductivity detector. A schematic of the setup is displayed in Fig. 1. | ||
| Fig. 1 Schematic diagram for measuring hydrogen permeation flux for BCFZYX membranes. | ||
The GC was calibrated for hydrogen and nitrogen gases, using 500 ppm, 0.1%, 0.25%, 0.5% and 1% H2, and 0.75%, 1%, 2%, and 3% N2 gases. Calibration curves were formulated from these gas measurements, and the GC concentrations were calculated from the calibration curves. The hydrogen permeation flux was corrected by taking the total measured hydrogen flux and subtracting the calculated physical leakage from nitrogen concentration in the sweep gas. Measured leakage for all samples was less than 3% as measured by the GC.
A palladium layer was deposited on selected 0.9 mm thick BCFZYX membranes as a protective layer, emulating recent experiments.24,29,31 Extensive testing, including comparison of Pd-coated and bare membranes, suggested that dense, bare membranes were sufficiently stable during the measurement period to report their permeation and conductivity values. The detailed palladium deposition procedure is presented in the ESI.†
2.3 Structure and microstructure characterization
The crystalline structure of the materials was characterized by X-ray Diffraction (XRD, Hitachi SmartLab Powder Diffractometer) using Cu/kα radiation. The diffraction patterns were measured on crushed, pristine samples and crushed membranes previously exposed to 5% H2 in permeation conditions for 24 h and 50 h. The morphologies of the membranes were characterized pre- and post-exposure to 5% H2 using scanning electron microscopy (SEM, Hitachi SU5000). The composition of the membranes was also characterized pre- and post-exposure to 5% H2via energy dispersive X-ray spectroscopy (EDX).2.4 DC conductivity and conductivity relaxation
Electrical conductivity for all BCFZYX samples was measured via a DC four-point probe method using a digital multimeter (Keithley 2001 Series). Bar samples for the DC measurement were fabricated from 1.1 mm thick pellets, polished to create 16 × 5.25 × 1.1 mm bars. The measurement was wired using 0.25 mm diameter silver wire attached via silver conductive paste (99%). The DC conductivity measurement was carried out in 5% H2/95% N2 atmosphere at a flowrate of 50 mL min−1 to correspond with permeation measurement conditions. The ECR method was used to determine surface exchange coefficients, kchem, and diffusion coefficients, Dchem. To measure kO,chem and DO,chem, gas composition was rapidly changed in less than two seconds from pO2 of 0.1 to 0.21 at a flow rate of 150 mL min−1. To measure kOH,chem, 1000 ppm O2 base gas was switched between dry and wet (pH2O = 0.023) conditions. The kOH-OD,chem measurements were performed by switching 1000 ppm O2 base gas between bubbled H2O and bubbled D2O to probe the isotopic effect. In all experiments, the measured conductivity is normalized according to eqn (1) and fitted to a three-dimensional diffusion equation, eqn (2), derived from Fick's second law:34 | (1) |
 | (2) |
 | (3) |
Finally, αm, βn, γp are the non-zero roots of the following equations:
| αmtan(αm) = Lx, βntan(βn) = Ly, γptan(γp) = Lz | (4) |
Non-linear, least square fitting using the NETL GUI tool35 is used to solve the diffusion equation and fit kchem and Dchem according to the experimental relaxation curves.
3. Results & discussion
3.1 Hydrogen permeation performance and stability
All pristine BCFZYX compositions were characterized prior to hydrogen permeation measurements. From XRD, both BCFZY0.1 and BaCo0.4Fe0.4Y0.2O3−δ (BCFY) exhibit the cubic perovskite structure in the Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. BCFZ appears to exhibit the cubic perovskite structure, but also shows small shoulder peaks associated with an orthorhombic phase which has been indexed in the Pnma space group. There is no obvious atomic segregation in the BCFZ material system, and therefore BCFZ is observed as a mixture of both a dominant cubic phase and minor orthorhombic phase. The XRD spectra are shown in Fig. S1.† Consistent with our previous work, the addition of yttrium in the system increases the lattice parameter and the overall sinterability of the material, showing increased grain size with increasing yttrium content.28
m space group. BCFZ appears to exhibit the cubic perovskite structure, but also shows small shoulder peaks associated with an orthorhombic phase which has been indexed in the Pnma space group. There is no obvious atomic segregation in the BCFZ material system, and therefore BCFZ is observed as a mixture of both a dominant cubic phase and minor orthorhombic phase. The XRD spectra are shown in Fig. S1.† Consistent with our previous work, the addition of yttrium in the system increases the lattice parameter and the overall sinterability of the material, showing increased grain size with increasing yttrium content.28
Fig. 2 displays the hydrogen permeation flux as a function of membrane temperature for all BCFZYX compositions, normalized to 0.6 mm thickness. Error bars are calculated from measurement propagation error and the standard deviation of the final five representative measurement points. These error bars are generally large relative to the step-size between data points due to the low concentration of H2 relative to N2 in the feed stream, allowing the standard deviation of the N2 sweep composition to dominate the error calculation. It is observed that H2 flux increases from BCFZ to BCFZY0.1, followed by a small decrease in flux from BCFZY0.1 to BCFY. In addition, a significant increase in activation energy is observed from the Y-doped samples (EA ≈ 0.2 eV) to BCFZ (EA = 0.708 eV). This increase indicates that a greater energy barrier is required for H+ to move through BCFZ than the Y-doped compositions. A comparison of this work with other ceramic-based membranes is included in Table 1.
 | ||
| Fig. 2 Hydrogen permeation flux across BCFZYX membranes, normalized to 0.6 mm thickness. (a) Measurement across a dry 5% hydrogen|argon gradient for the BCFZYX compositions between 550 and 750 °C. (b) Measurement across a wet 5% hydrogen|argon gradient between 550 and 700 °C, with comparison to the dry gradient measurements. | ||
| Composition | T (°C) | Thickness (mm) | Feed gas | Sweep gas | J H2 (mL min−1·cm−2) | Reference |
|---|---|---|---|---|---|---|
| BCFZ | 600 | 0.62 | 5% H2/95% N2 | Ar | 0.015 | This work |
| BCFZY0.1 | 600 | 0.61 | 5% H2/95% N2 | Ar | 0.051 | This work |
| BCFY | 600 | 0.59 | 5% H2/95% N2 | Ar | 0.045 | This work |
| BCFZY0.1 | 650 | 0.65 | 10% H2/90% N2 | Ar | 0.05 | 30 |
| BaCe0.9Y0.1O3−δ − BCFZY/BCFZY | 650 | 0.65 | 10% H2/90% N2 | Ar | 0.1 | 30 |
| Pd|BCFZY0.1|Pd | 600 | 0.6 | 10% H2/90% N2 | Ar | 0.125 | 29 |
| Pd|SSNCF|Pd | 600 | 0.6 | 10% H2/90% N2 | Ar | 0.15 | 24 |
| Ba(Ce0.7Zr0.1Y0.1Yb0.1)0.95Ni0.05O3−δ | 600 | 0.6 | 10% H2/90% N2 | Ar | 0.09 | 36 |
| Composite SrCe0.9Y0.1O3–Ce0.8Gd0.2O2 | 800 | 1 | 20% H2/80% He | N2 | 0.04 | 37 |
| La5.5WO11.25−δ | 800 | 0.9 | 50% H2/50% He | Wet Ar (2.5% H2O) | 0.025 | 38 |
| 50% La5.5WO11.25−δ/50% La0.87Sr0.13CrO3−δ | 700 | 0.37 | Wet 50% H2/50% He | Wet Ar (2.5% H2O) | 0.15 | 39 |
| BaCe0.85Fe0.15O3−δ − BaFe0.85Ce0.15O3−δ | 850 | 1 | 50% H2/50% He | Ar | 0.42 | 40 |
Hydrogen permeation was also performed using the 5% H2/95% N2 feed gas bubbled through room temperature water for BCFZ and BCFZY0.1. The activation energy significantly decreases in both samples, from 0.71 to 0.48 eV for BCFZ and from 0.224 to 0.09 eV for BCFZY0.1. In conjunction with the decreased activation energy, the permeation flux slightly increased at temperatures below 600 °C. The introduction of water likely causes some incorporation and transport of oxygen in the membrane, which, at elevated temperatures, may compete with proton incorporation and transport due to increasing oxygen vacancy mobility, concentration, and surface kinetics.41,42 The introduction of water in the system increases the proton concentration via hydration, which is favored at lower temperatures.9,43,44 At these lower temperatures, the increase in oxygen content in the system therefore aids the transport of protons by increasing available hopping sites.43 Any co-permeation of oxygen is not observed because of the low sensitivity of oxygen and the inability to observe water in the GC measurement conditions.
For TIEC systems, the hydrogen permeation flux is estimated using the following:11,45
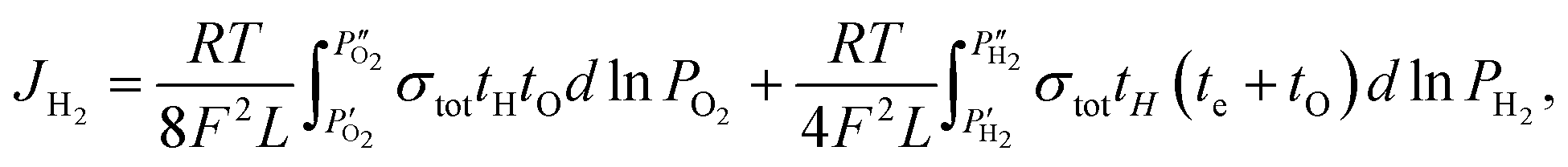 | (5) |
 | (6) |
Assuming that tO = 0 under the measurement conditions, and that te ≫ tH which is confirmed via measurement and shown in Fig. 3a, the equation reduces further to:24
 | (7) |
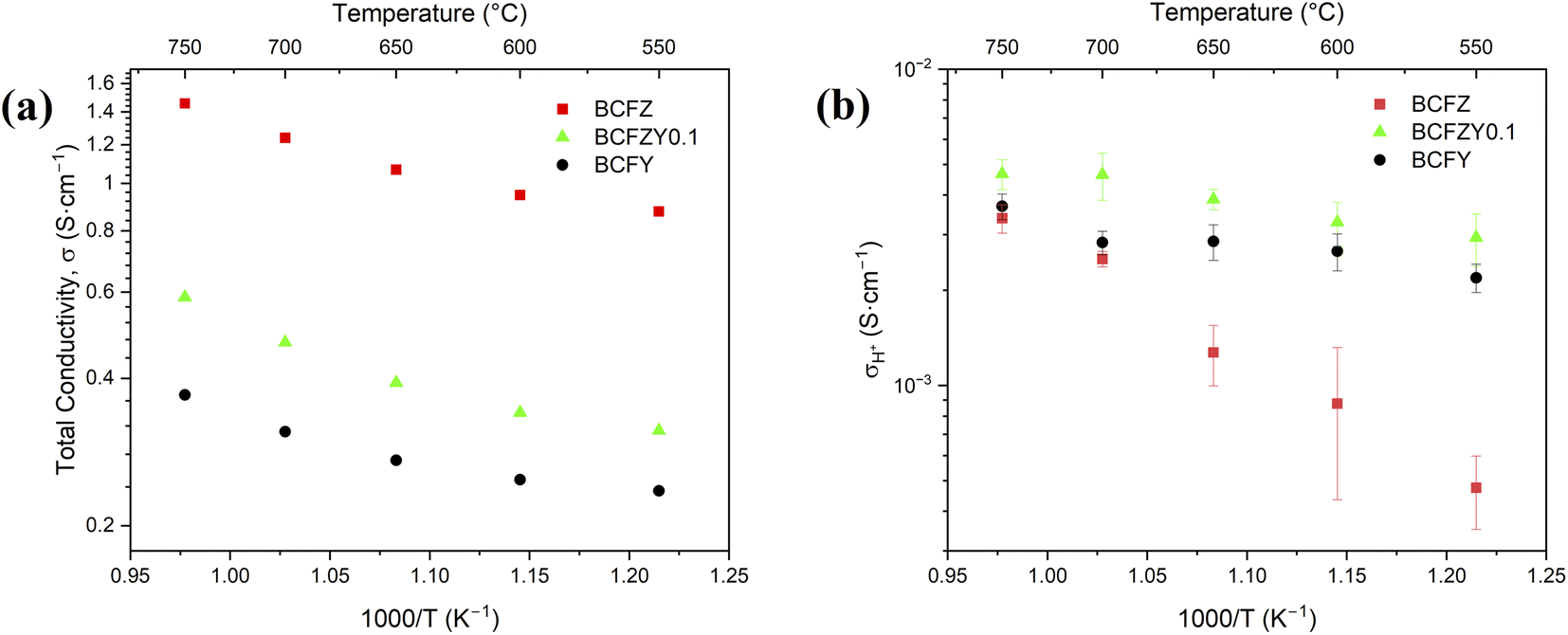 | ||
| Fig. 3 (a) Total conductivity of BCFZYX measured in 5% H2/95% N2 atmospheric conditions. (b) The protonic conductivity in the given reducing atmospheric gradient, estimated using the hydrogen permeation flux and eqn (7), normalized to 0.6 mm thickness. | ||
Using the GC to estimate the sweep-side hydrogen gas concentration, eqn (7) was then utilized to estimate the proton conductivity, σH, of each material, which is displayed in Fig. 3b. In agreement with permeation measurements, BCFZY0.1 exhibits the highest proton conductivity, estimated at 3.2 × 10−3 S cm−1 at 600 °C, which is four times greater than the estimated proton conductivity for BCFZ at 8.5 × 10−4 S cm−1 at 600 °C. The estimated conductivity of BCFZYX is lower than predominantly ionic conductors in similar atmospheric conditions, such as BaZr0.8Y0.2O3−δ46,47 and BaCe0.7Zr0.1Y0.1Sm0.1O3−δ.48 However, it should be noted that the ionic conductors' conductivity is measured via EIS, which has a much smaller chemical gradient and different measurement conditions and therefore is not a wholly direct comparison. BCFZYX does, however, exhibit protonic conductivity on the same order of magnitude of other TIECs such as BaCo0.4Fe0.4Zn0.1Y0.1O3−δ in similar measurement conditions using gas permeation.32
It was hypothesized that increasing Y concentration would increase oxygen vacancies in the material and increase the basicity of the oxygen site, thus increasing the proton uptake (carrier concentration) and mobility.1,13 The increase in oxygen vacancies is estimated in the equation written in Kröger–Vink notation:
 | (8) |
The increase in conductivity through increases in carrier concentration and mobility appears to hold true for the BCFZ and BCFZY0.1 concentrations. However, Y-substitution up to BCFY further increases oxygen vacancy concentration and may introduce potential trapping sites of protons in the system, analogous to electrolyte systems such as BZY.46,49–51 The trapping effect would decrease proton mobility, which decreases overall proton conductivity even when coupled with the increasing carrier concentration. Greater Y concentration also expands the lattice parameter through the increased size of Y3+ against Zr4+, increasing the protonic hopping distance between oxygen sites.52 These results suggest that an optimum concentration exists when applying aliovalent doping to change bulk protonic conductivity in TIEC materials.
Following extended exposure to reducing conditions, BCFZYX compositions exhibited significant changes from its pristine structure. Fig. 4a displays a cross-sectional SEM micrograph of BCFY after 50 h of exposure to reducing conditions during permeation experiments. It is evident from the EDX analysis in Fig. 4b that Co segregates from the bulk of the material and forms small Co-rich nanoparticles across the grain. This Co segregation is also evident in the other compositions, though the occurrence of nanoparticle formation appears to increase with the introduction of yttrium from a visual analysis with EDX. The nanoparticles also appear to remain at the grain boundary interfaces in BCFZY0.1 as shown in Fig. S4.† From a purely thermodynamic perspective, Co has the least stable oxide form of the constituent oxides of the solid solution, and therefore would be expected to be the first element to exsolve.53,54 Despite the formation of a Co-containing phase, secondary phase formation is not obvious through XRD analysis, shown for BCFY in Fig. 4c, suggesting that the amount of secondary phase is nominal compared to the dominant perovskite phase. The main evidence of secondary phase formation comes from the peak shift to higher angles after 24 h of H2 exposure, indicating the exsolution of Co from the lattice, followed by shift to lower angles following reduction of multivalent ions.
 | ||
| Fig. 4 (a) BSE-EDX image of BCFY cross-section after 50 h of exposure to 5% H2/95% N2|BCFY|Ar gradient, (b) EDX mapping of 50 h-exposed BCFY for Co, showing cobalt-rich phase segregation. (c) XRD of pristine BCFY against 24 h- and 50 h-exposed BCFY, showing no significant secondary-phase peaks. | ||
In addition, exposure to reducing gas does not cause destruction of the perovskite structure. Increasing reduction times appears to cause peak broadening in the diffraction pattern, which is especially evident in BCFZ and BCFZY0.1. This peak broadening may indicate reduced crystallinity in the perovskite structure. BCFZ appears to show more pronounced peak-splitting after 24 h exposure as well, which is associated with decreased symmetry to the orthorhombic Pnma space group. The peak broadening may also be associated with a reduction in symmetry to the orthorhombic Pnma phase based on additional reflections present in the XRD spectrum after 50 h exposure. BCFY maintains a much more crystalline structure in comparison after extended reduction with no obvious structural symmetry changes, which is notable due to its greater incidence of Co-segregated nanoparticle formation. In all samples, long-term reduction causes a lattice expansion with a shift toward lower angles, likely due to the reduction of multivalent metal ions Co and Fe.29,30
In tandem with the XRD and EDX studies, the permeation flux was measured against exposure time. Although temperature changes were taken in between measurement times, the flux at the same temperature over the course of 50 h remained relatively constant, and within the range of error of each sampling period. The peak broadening and peak splitting observed in XRD after long-term exposures does not coincide with a significant change in the permeation flux, suggesting that despite a potential reduction in crystallinity and symmetry in the material, the permeation measurements accurately describe the bulk material properties. These correlations help to confirm the validity of the measurements over the 50 h period despite the exsolution of a Co-rich phase and apparent changes in symmetry. These results are further detailed in ESI with Fig. S2–S8.†
Hydrogen permeation was also measured for BCFZYX coated with thin layers of palladium as a protective barrier layer.24,29,31 While these membranes were significantly thicker than the uncoated membranes, the estimated conductivity between the coated and uncoated membranes was within the range of error of the measurement for BCFZY0.1 and BCFY. For BCFZ, the estimated conductivity was significantly higher in Pd-coated samples than uncoated samples. This difference suggests that surface exchange may be enhanced in the coated BCFZ membranes leading to an apparent higher proton conductivity in contrast to BCFZY0.1 and BCFY membranes, as Pd would catalyze the surface dissociation and oxidation of hydrogen.55,56 Pd-coated membranes showed no notable differences in phase stability from the uncoated membranes. These results are further elucidated in Fig. S9–S11.†
3.2 Protonic kinetics through surface exchange experiments
To access the effect of protons and hydroxyl groups on surface exchange in BCFZYX, electronic conductivity of each composition was measured under rapid gas switching. At elevated temperatures in humid conditions, TIEC materials exchange protons in two hypothesized mechanisms:13,57 | (9) |
 | (10) |
Eqn (9) represents the hydration reaction, which utilizes oxygen vacancies for proton uptake, typically in reducing conditions, though some evidence suggests that it may occur in oxidizing conditions as well.57Eqn (10) represents the hydrogenation reaction, which utilizes lattice oxygen and holes for proton uptake in oxidizing conditions. In oxidizing conditions, oxygen exchange via the oxygen reduction reaction (ORR) also occurs according to the following:
 | (11) |
To probe the protonic surface kinetics, a conductivity relaxation experiment was performed by switching atmospheric conditions under constant 1000 ppm O2 through a room-temperature-bubbler from H2O to D2O. Low oxygen partial pressure increases the proton concentration through the increase in oxygen vacancies, allowing simulation of eqn (9) by exchanging all protons with deuterons.58 The incorporation of deuterons decreases the total conductivity of the material due to the increased mass of the deuteron, as shown in the inset of Fig. 5a for BCFZ. BCFZY0.1 exhibited the fastest relaxation upon switching, and, when fit, exhibited the highest kOH-OD,chem of 2.52 × 10−4 cm s−1 at 600 °C, as shown in Fig. 5b. This surface exchange is markedly similar to proton-conducting-electrolyte material BaCe0.7Zr0.1Y0.1Yb0.1O3−δ (kOH-OD = 2.06 × 10−4 s−1 at 600 °C) measured via in situ Raman spectroscopy.59 The surface exchange from OH and OD groups closely follows the trend exhibited in the hydrogen permeation experiments, with maximum protonic surface kinetics in the co-doped BCFZY0.1 sample, decreasing with further Y- or Zr-doping. This result further suggests that Y-doping beyond the BCFZY0.1 composition decreases the mobility of protons in the system despite the increase in carrier concentration.
 | ||
| Fig. 5 (a) Conductivity relaxation switching from room-temperature bubbled H2O to D2O under 1000 ppm O2 atmosphere, with inset showing decreasing total conductivity of BCFZ by switching from proton to heavier deuteron, and (b) fitting of kOH-OD for BCFZYX from 500–700 °C. | ||
To better emulate conditions in protonic ceramic fuel cells, another conductivity relaxation experiment was conducted to probe the effect of water incorporation on oxygen surface exchange. In dry, oxidizing conditions, rapid switching of oxygen partial pressure changes the total conductivity according to eqn (11). Increasing Y concentration generally increases kO,chem and decreases DO,chem, consistent with our previous study.28 Exceptions occur in the low-temperature regime, where BCFZY0.1 exhibits the highest kO,chem and DO,chem in part due to its low activation energy compared to BCFZ and BCFY. The reduced ORR kinetics for BCFZ compared to Y-doped compositions are consistent with previous studies on functional devices.6 The full ORR kinetics in dry conditions are included in Fig. S13.†
When probing the same samples under constant humidified conditions of pH2O = 0.023, evidence of the competition between hydration and ORR is observed. In BCFZY0.1 and BCFY, the addition of water vapor decreased kO,chem across all temperatures, while DO,chem remains approximately the same, consistent with a previous study of BCFZY0.1,60 and suggesting decreased ORR in humidified conditions, also observed in other materials.61–63 In contrast, for BCFZ at temperatures ≤600 °C, the relaxation time decreased and kO,chem increased in humidified conditions, suggesting that water vapor improved the ORR at lower temperatures. Fig. 6a shows these changes in kO,chem for BCFZ from 500–700 °C. Comparisons for BCFZY0.1 and BCFY are included in Fig. S15.†Fig. 6b displays the difference in kO, chem in dry and wet conditions across the compositional range at 600 °C. In wet conditions, kO,chem follows a similar trend to the bulk protonic conductivity and the H2O-D2O exchange and suggests that the co-doped BCFZY0.1 exhibits the best oxygen exchange in PCFC conditions. The greater incorporation of protonic carriers likely lowers the available oxygen reduction sites as more Y is substituted in the system. The activation energy of kO,chem also decreases with the introduction of humidity, as presented in Fig. 5c, with BCFZY0.1 exhibiting the lowest activation energy in both dry and humidified environments. This low activation energy suggests that BCFZY0.1 will have the best surface kinetics at lower temperatures for electrochemical functional devices.
 | ||
| Fig. 6 (a) Plot of kchem and Dchem under switching from 10% to 21% O2 for BCFZ with comparison of each under dry and wet conditions, (b) comparison of kchem under dry and wet conditions for all BCFZYX compositions, (c) activation energy of kchem from 500–700 °C for BCFZYX under dry and wet conditions. | ||
In a separate experiment, the exchange of hydroxyl groups is also observed via an ECR measurement. Oxygen concentration is held constant at 1000 ppm and the atmosphere is switched from dry to 2.3% H2O. In this experiment, the conductivity follows a non-monotonic, two-fold relaxation, as shown in the inset in Fig. 7a, similar to recently observed measurements.57,64,65 Upon switching from dry to humidified atmosphere, the conductivity of these samples increases significantly, followed by a slow decrease back to equilibrium. This phenomenon is likely ascribed to eqn (1) and (2) in some combination. The fast proton surface kinetics are expected to account for the initial conductivity spike,66 while the slower proton-lattice oxygen incorporation accounts for the slow kinetic shift back to equilibrium.67 From the slow kinetic shift, the relaxation curves are fitted across the temperature range, as shown for the three compositions in Fig. 7a. Following curve fitting, it was observed that the substitution of Y increases the kOH,chem across the temperature range, with a maximum difference in surface exchange at 600 °C, as shown in Fig. 7b. This increase continues the trend of improved proton kinetics with the introduction of Y in the BCFZYX system.
 | ||
| Fig. 7 (a) Fitted curves from conductivity relaxation switching from 0% to 2.3% H2O under 1000 ppm O2 atmosphere, with inset displaying BCFY conductivity spike upon switching from the fast proton uptake kinetics, followed by the relaxation resulting in decrease in total conductivity. (b) Fitting of kOH,chem for BCFZYX from 500–700 °C. | ||
4. Conclusions
By varying the ratio of Zr:Y, compositions of BCFZYX were probed for direct comparison of bulk proton conductivity and proton surface kinetics. The substitution of Y3+ for Zr4+ improved the bulk proton conductivity, likely due to improved carrier concentration and proton uptake capability. However, full Y3+ substitution in BCFY reduced the proton conductivity, potentially due to trapping effects from the high vacancy concentration. In reducing conditions, BCFZYX exhibited Co segregation to the grain boundaries, which increased in magnitude with the increase in Y3+ substitution. However, the main perovskite structure was maintained across the compositional suite. Three distinct conductivity relaxation experiments in oxidizing conditions, via isotope exchange, hydration, and oxidation under humidified conditions, revealed improved proton and water surface kinetics with Y3+ substitution. These results underscore the importance of aliovalent doping in a model, single perovskite system, and help to further understand the superior performance of BCFZY0.1 in intermediate-temperature functional devices.
Author contributions
Jack H. Duffy: investigation, methodology, conceptualization, writing – original draft, review & editing. Harry W. Abernathy: funding acquisition, supervision, writing – review & editing. Kyle S. Brinkman: conceptualization, funding acquisition, project administration, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by the Department of Energy, National Energy Technology Laboratory an agency of the United States Government, through an appointment administered by the Oak Ridge Institute for Science and Education. Neither the United States Government nor any agency thereof, nor any of its employees, nor the support contractor, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof. The authors also gratefully acknowledge Dr Jon Ihlefeld and Ian Brummel at the University of Virginia for performing the Pd deposition. KSB was supported in part by an appointment to the NETL Research Participation Program, sponsored by the U.S. Department of Energy and administered by the Oak Ridge Institute for Science and Education.Notes and references
- M. Papac, V. Stevanović, A. Zakutayev and R. O'Hayre, Nat. Mater., 2021, 20, 301–313 CrossRef CAS PubMed.
- J. Irvine, J. L. M. Rupp, G. Liu, X. Xu, S. Haile, X. Qian, A. Snyder, R. Freer, D. Ekren, S. Skinner, O. Celikbilek, S. Chen, S. Tao, T. H. Shin, R. O’Hayre, J. Huang, C. Duan, M. Papac, S. Li, V. Celorrio, A. Russell, B. Hayden, H. Nolan, X. Huang, G. Wang, I. Metcalfe, D. Neagu and S. G. Martín, J. Phys.: Energy, 2021, 3, 031502 CAS.
- X. Li, L. He, X. Zhong, J. Zhang, S. Luo, W. Yi, L. Zhang, M. Hu, J. Tang, X. Zhou, X. Zhao and B. Xu, Scanning, 2018, 2018, 1341608 Search PubMed.
- J. Sunarso, S. S. Hashim, N. Zhu and W. Zhou, Prog. Energy Combust. Sci., 2017, 61, 57–77 CrossRef.
- C. Duan, R. J. Kee, H. Zhu, C. Karakaya, Y. Chen, S. Ricote, A. Jarry, E. J. Crumlin, D. Hook, R. Braun, N. P. Sullivan and R. O'Hayre, Nature, 2018, 557, 217–222 CrossRef CAS PubMed.
- C. Duan, J. Tong, M. Shang, S. Nikodemski, M. Sanders, S. Ricote, A. Almansoori and R. O’Hayre, Science, 2015, 349, 1321–1326 CrossRef CAS PubMed.
- S. Choi, C. J. Kucharczyk, Y. Liang, X. Zhang, I. Takeuchi, H.-I. Ji and S. M. Haile, Nat. Energy, 2018, 3, 202–210 CrossRef CAS.
- K. Park, H. Bae, H. K. Kim, I. G. Choi, M. Jo, G. M. Park, M. Asif, A. Bhardwaj, K. S. Lee, Y. C. Kim, S. J. Song, E. D. Wachsman and J. Y. Park, Adv. Energy Mater., 2022, 13(2), 2202999 CrossRef.
- R. Zohourian, R. Merkle, G. Raimondi and J. Maier, Adv. Funct. Mater., 2018, 28, 1801241 CrossRef.
- K. Y. Park, Y. D. Kim, J. I. Lee, M. Saqib, J. S. Shin, Y. Seo, J. H. Kim, H. T. Lim and J. Y. Park, ACS Appl. Mater. Interfaces, 2019, 11, 457–468 CrossRef CAS PubMed.
- Y. Huang, R. Qiu, W. Lian, L. Lei, T. Liu, J. Zhang, Y. Wang, J. Liu, J. Huang and F. Chen, J. Power Sources, 2022, 528, 231201 CrossRef CAS.
- D. Poetzsch, R. Merkle and J. Maier, Phys. Chem. Chem. Phys., 2014, 16, 16446–16453 RSC.
- D. Poetzsch, R. Merkle and J. Maier, Faraday Discuss., 2015, 182, 129–143 RSC.
- R. Zohourian, R. Merkle and J. Maier, Solid State Ionics, 2017, 299, 64–69 CrossRef CAS.
- Y. Xu, F. Hu, Y. Guo, J. Zhang, Y. Huang, W. Zhou, J. Sun, B. He and L. Zhao, Sep. Purif. Technol., 2022, 297, 121482 CrossRef CAS.
- P. Wang, D. Xu, J. Cheng and T. Hong, Ionics, 2021, 27, 1185–1192 CrossRef CAS.
- Y. Chen, T. Hong, P. Wang, K. Brinkman, J. Tong and J. Cheng, J. Power Sources, 2019, 440, 227122 CrossRef CAS.
- X. Qi and Y. S. Lin, Solid State Ionics, 2000, 130, 149–156 CrossRef CAS.
- T. Yajima, H. Suzuki, T. Yogo and H. Iwahara, Solid State Ionics, 1992, 51, 101–107 CrossRef CAS.
- Z. Zhao, M. Zou, H. Huang, H. Wofford and J. Tong, Ceram. Int., 2021, 47, 32856–32866 CrossRef CAS.
- S. S. Hashim, M. R. Somalu, K. S. Loh, S. Liu, W. Zhou and J. Sunarso, Int. J. Hydrogen Energy, 2018, 43, 15281–15305 CrossRef CAS.
- Y. Song, Y. Chen, W. Wang, C. Zhou, Y. Zhong, G. Yang, W. Zhou, M. Liu and Z. Shao, Joule, 2019, 3, 2842–2853 CrossRef CAS.
- D. Zou, Y. Yi, Y. Song, D. Guan, M. Xu, R. Ran, W. Wang, W. Zhou and Z. Shao, J. Mater. Chem. A, 2022, 10, 5381–5390 RSC.
- C. Zhou, J. Sunarso, Y. Song, J. Dai, J. Zhang, B. Gu, W. Zhou and Z. Shao, J. Mater. Chem. A, 2019, 7, 13265–13274 RSC.
- P. Zhong, K. Toyoura, L. Jiang, L. Chen, S. A. Ismail, N. Hatada, T. Norby and D. Han, Adv. Energy Mater., 2022, 12, 2200392 CrossRef CAS.
- C. Duan, J. Huang, N. Sullivan and R. O'Hayre, Appl. Phys. Rev., 2020, 7, 011314 CAS.
- J. Tong, W. Yang, B. Zhu and R. Cai, J. Membr. Sci., 2002, 203, 175–189 CrossRef CAS.
- J. H. Duffy, Y. Meng, H. W. Abernathy and K. S. Brinkman, Membranes, 2021, 11, 766 CrossRef CAS PubMed.
- C. Zhou, J. Sunarso, J. Dai, R. Ran, Y. Song, F. He, W. Zhou and Z. Shao, J. Membr. Sci., 2020, 596, 117709 CrossRef CAS.
- D. Zhang, X. Zhang, X. Zhou, Y. Song, Y. Jiang and B. Lin, Ceram. Int., 2022, 48, 9946–9954 CrossRef CAS.
- M. Liang, F. He, C. Zhou, Y. Chen, R. Ran, G. Yang, W. Zhou and Z. Shao, Chem. Eng. J., 2020, 420, 127717 CrossRef.
- X. Wang, W. Li, C. Zhou, M. Xu, Z. Hu, C.-W. Pao, W. Zhou and Z. Shao, ACS Appl. Mater. Interfaces, 2023, 15, 1339–1347 CrossRef CAS PubMed.
- M. Liang, Y. Song, D. Liu, L. Xu, M. Xu, G. Yang, W. Wang, W. Zhou, R. Ran and Z. Shao, Appl. Catal., B, 2022, 318, 121868 CrossRef CAS.
- H. J. M. Bouwmeester, M. W. den Otter and B. A. Boukamp, J. Solid State Electrochem., 2004, 8, 599–605 CrossRef CAS.
- B. T. Na, T. Yang, J. Liu, S. Lee, H. Abernathy, T. Kalapos and G. Hackett, Solid State Ionics, 2021, 361, 115561 CrossRef CAS.
- M. Yang, F. He, C. Zhou, F. Dong, G. Yang, W. Zhou and Z. Shao, J. Membr. Sci., 2021, 620, 118980 CrossRef CAS.
- B. Meng, H. Wang, H. Cheng, X. Wang, X. Meng, J. Sunarso, X. Tan and S. Liu, Sep. Purif. Technol., 2019, 213, 515–523 CrossRef CAS.
- S. Escolástico, C. Solís, T. Scherb, G. Schumacher and J. M. Serra, J. Membr. Sci., 2013, 444, 276–284 CrossRef.
- S. Escolástico, C. Solís, C. Kjølseth and J. M. Serra, Energy Environ. Sci., 2014, 7, 3736–3746 RSC.
- S. Cheng, Y. Wang, L. Zhuang, J. Xue, Y. Wei, A. Feldhoff, J. Caro and H. Wang, Angew. Chem., Int. Ed., 2016, 55, 10895–10898 CrossRef CAS PubMed.
- J. Guan, S. E. Dorris, U. Balachandran and M. Liu, Solid State Ionics, 1997, 100, 45–52 CrossRef CAS.
- D. K. Lim, H. N. Im, S. J. Song and H. I. Yoo, Sci. Rep., 2017, 7, 1–9 CrossRef PubMed.
- K. D. Kreuer, Annu. Rev. Mater. Res., 2003, 33, 333–359 CrossRef CAS.
- R. Merkle, M. F. Hoedl, G. Raimondi, R. Zohourian and J. Maier, Annu. Rev. Mater. Res., 2021, 51, 461–493 CrossRef CAS.
- T. Norby and Y. Larring, Solid State Ionics, 2000, 136–137, 139–148 CrossRef CAS.
- D. Han and T. Uda, J. Mater. Chem. A, 2018, 6, 18571–18582 RSC.
- D. Han, Y. Noda, T. Onishi, N. Hatada, M. Majima and T. Uda, Int. J. Hydrogen Energy, 2016, 41, 14897–14908 CrossRef CAS.
- Y. Meng, J. Gao, H. Huang, M. Zou, J. Duffy, J. Tong and K. S. Brinkman, J. Power Sources, 2019, 439, 227093 CrossRef CAS.
- Y. Yamazaki, F. Blanc, Y. Okuyama, L. Buannic, J. C. Lucio-Vega, C. P. Grey and S. M. Haile, Nat. Mater., 2013, 12, 647–651 CrossRef CAS PubMed.
- C. Y. Regalado Vera, H. Ding, D. Peterson, W. T. Gibbons, M. Zhou and D. Ding, J. Phys.: Energy, 2021, 3, 032019 Search PubMed.
- J. Ding, J. Balachandran, X. Sang, W. Guo, J. S. Anchell, G. M. Veith, C. A. Bridges, Y. Cheng, C. M. Rouleau, J. D. Poplawsky, N. Bassiri-Gharb, R. R. Unocic and P. Ganesh, Chem. Mater., 2018, 30, 4919–4925 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- T. B. Reed, Free Energy of Formation of Binary Compounds: an Atlas of Charts for High-Temperature Chemical Calculations, 1971 Search PubMed.
- D. R. Stull and H. Prophet, JANAF Thermochemical Tables, NSRDS-NBS 37, U.S. Department of Commerce, National Bureau of Standards, 2nd edn, 1971 Search PubMed.
- S. N. Paglieri and J. D. Way, Sep. Purif. Methods, 2002, 31, 1–169 CrossRef CAS.
- M. R. Rahimpour, F. Samimi, A. Babapoor, T. Tohidian and S. Mohebi, Chem. Eng. Process., 2017, 121, 24–49 CrossRef CAS.
- T. Hong, W. Lu, K. Ren and T. Liu, Ionics, 2020, 26, 5293–5297 CrossRef CAS.
- D. Poetzsch, R. Merkle and J. Maier, J. Electrochem. Soc., 2015, 162, F939 CrossRef CAS.
- J. Gao, Y. Meng, J. H. Duffy and K. S. Brinkman, Adv. Energy Sustainability Res., 2021, 2, 2100098 CrossRef CAS.
- Y. Meng, J. Duffy, B. T. Na, J. Gao, T. Yang, J. Tong, S. Lee and K. S. Brinkman, Solid State Ionics, 2021, 368, 115639 CrossRef CAS.
- C. Zhou, D. Liu, M. Fei, X. Wang, R. Ran, M. Xu, W. Wang, W. Zhou, R. O. Hayre and Z. Shao, J. Power Sources, 2023, 556, 232403 CrossRef CAS.
- Y. L. Huang, C. Pellegrinelli and E. D. Wachsman, J. Electrochem. Soc., 2016, 163, F171–F182 CrossRef CAS.
- C. Zhou, X. Shen, D. Liu, J. Cui, Y. Yi, M. Fei, J. Zhou, L. Zhang, R. Ran, M. Xu, W. Zhou and Z. Shao, J. Power Sources, 2022, 530, 231321 CrossRef CAS.
- A. Falkenstein, R. A. de Souza, W. A. Meulenberg and M. Martin, Phys. Chem. Chem. Phys., 2020, 22, 25032–25041 RSC.
- S. Zhu, M. Drazkowski, B. A. Boukamp and H. J. M. Bouwmeester, Solid State Ionics, 2022, 384, 115994 CrossRef CAS.
- A. Nenning, E. Navickas, H. Hutter and J. Fleig, J. Phys. Chem. Lett., 2016, 7, 2826–2831 CrossRef CAS PubMed.
- D. Poetzsch, R. Merkle and J. Maier, Adv. Funct. Mater., 2015, 25, 1542–1557 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD comparison of pristine BCFZYX samples, total conductivity vs. time in reducing conditions, hydrogen flux vs. time for 50 h measurement, SEM/EDX of BCFZ and BCFZY0.1 after hydrogen permeation, XRD comparisons of pristine and reduced BCFZ and BCFZY0.1 samples, palladium deposition technique, SEM/EDX characterization of Pd-coated membranes, estimated conductivity of Pd-coated BCFZYX membranes from hydrogen permeation measurements, fitted ECR parameters kchem and Dchem for dry, oxidizing conditions, activation energy comparison of Dchem, complete comparison of dry and wet condition oxidation ECR for BCFZY0.1 and BCFY compositions. See DOI: https://doi.org/10.1039/d3ta00654a |
| This journal is © The Royal Society of Chemistry 2023 |