
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltralow thermal conductivity in the mixed-anion solid solution Sn2SbS2−xSexI3†
Justin
Mark‡
 a,
Wenhao
Zhang‡
ab,
Kazuhiko
Maeda
cf,
Takafumi
Yamamoto
d,
Hiroshi
Kageyama
e and
Takao
Mori
*ab
a,
Wenhao
Zhang‡
ab,
Kazuhiko
Maeda
cf,
Takafumi
Yamamoto
d,
Hiroshi
Kageyama
e and
Takao
Mori
*ab
aInternational Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), Tsukuba, Ibaraki 305-0044, Japan. E-mail: MORI.Takao@nims.go.jp
bGraduate School of Pure and Applied Science, University of Tsukuba, Tsukuba, Ibaraki 305-8671, Japan
cDepartment of Chemistry, School of Science, Tokyo Institute of Technology, Tokyo, Japan
dLaboratory for Materials and Structures, Institute of Innovative Research, Tokyo Institute of Technology, Yokohama, Japan
eGraduate School of Engineering, Kyoto University, Kyoto, Japan
fLiving Systems Materialogy (LiSM) Research Group, International Research Frontiers Initiative (IRFI), Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama, Kanagawa 226-8502, Japan
First published on 28th April 2023
Abstract
The mixed-anion compounds Sn2SbS2−xSexI3 (x = 0, 0.2, 0.5) and Sn2BiS2I3 were synthesized and characterized. Rietveld refinement, elemental analysis, and diffuse reflectance measurements indicate successful substitution of Se for S and potential for bandgap tunability. Thermal conductivity measurements reveal ultralow thermal conductivities as low as 0.22 W m−1 K−1 at 573 K within this family of compounds. Such exceptionally low thermal conductivities are anomalous when compared to the previously reported heavier Pb2BiS2I3 (0.7 W m−1 K−1). Computational analysis in the form of electron localization function and Grüneisen parameter reveal Sn to be the most influential factor that controls the lattice thermal conductivity, due to its localized antibonding electrons and large contribution to structural anharmonicity.
1 Introduction
In the past, materials design has focused largely on cation substitutions, resulting in structure types often containing multiple cations. Materials design focusing on multiple anions and structure–property trends in these mixed-anion materials is far less established, despite mixed-anion materials providing a large platform for materials design.1–4 For example, substituting an octahedrally coordinated metal for another metal results in similar metal octahedra which will serve as the building block for a solid-state structure. However, with anion substitutions the options in coordination polyhedra are much more varied, such as cis-, trans-, fac-, or mer-octahedra.1,3 This allows for a variety of structural building blocks which can influence materials properties in unexpected ways. Not only are the coordination environments varied, but a degree of polar distortion can also be introduced to the metal polyhedral building blocks due to the differing electronegativities of the anions, adding another tunable variable to the material design process.1,3The diverse structures of mixed-anion materials remain relatively under explored compared to single anion compounds, however, they have shown to be promising for energy applications. The irregular coordination polyhedra of mixed-anion materials have shown to be reliable building blocks for novel noncentrosymmetric materials, a prerequisite for second harmonic generation, as seen in Ba2[GaF4(IO3)2](IO3), BaGeOSe2, and Sr3Ge2O4Se3.5–7 Other examples of intriguing mixed-anion applications include ionic conductors and thermoelectrics. Na2(CB9H10)(CB11H12) demonstrates significantly improved ionic conductivity compared to the single anion parent compounds, reaching conductivities 7× greater than the well-known superionic conductor Li10GeP2S12, while BiCuSeO is a promising thermoelectric material with ultralow thermal conductivity achieving zT values > 1.8–12
Despite the promising properties and diverse structural opportunities in mixed anion materials, most work is reported for oxygen containing materials (oxyhalides, oxychalcogenides, etc.).1,3 However, a recent computational work has shown non-oxide mixed anion materials can exhibit a much more diverse set of crystal structures. This is attributed to the high bond strength of metal-oxide bonds which limit variability compared to those of sulfides, tellurides, etc.13 Thus, we seek to expand on the structure–property relationships in non-oxide mixed anion materials for future materials design.
Given the complexity which can arise in mixed-anion crystal structures, they can serve as a promising platform for low thermal conductivity materials. Indeed, some of the studied mixed-anion materials such as Pb2BiS2I3, BiCuSeO, and Bi4O4SeCl2 have exhibited exceptionally low thermal conductivities of 0.7 W m−1 K−1, 0.5 W m−1 K−1, and 0.1 W m−1 K−1 respectively.9,14,15 Thermal conductivity can be divided into two components, electronic (κe) and lattice (κl) thermal conductivities. The reduction of thermal conductivity by extrinsic methods has been extensively studied as it is easier to manipulate κl by extrinsic methods, for example, by various nano–microstructuring16–18 and/or introducing various defects.19–22 However, intrinsic thermal conductivity is closely related to the crystal structure and bonding, making it much more difficult to pinpoint and modify. Commonly, heavy elements in a complex crystal structure can lead to complex phonon branching, small phonon velocities and high rates of phonon–phonon scattering, all of them beneficial for achieving lower lattice thermal conductivity.23 Other structural factors, such as a large interatomic space, weak bonding, partial occupancy, and bonding heterogeneity can provide sources of lattice anharmonicity, further reducing the thermal conductivity.24–27
With these factors in mind, we consider the large family of non-oxide mixed-anion Tt2PnCh2I3 (Tt = Sn, Pb; Pn = Sb, Bi; Ch = S, Se) compounds for which thermal conductivity is only reported for Pb2BiS2I3.14,28–30 In general they can be described as van der Waals layered compounds with I atoms loosely bonded as bridging atoms within the inter-layer space. Site symmetry is mostly low, indicating potentially large anharmonicity. Furthermore, the large polarizability of iodine can be expected to introduce additional bonding dynamics and reduce lattice thermal conductivity.31 These interesting structural features and the relative lack of experimental results give us the motivation to investigate into these compounds.
2 Experimental section
2.1 Synthesis
The starting materials, Sn shot (Wako, 99.9%), Sb powder (Sigma-Aldrich, 99.5%), S flakes (Sigma-Aldrich, 99.99%) Se shot (Alfa Aesar, 99.999%), Bi granules (5N Plus Inc., 99.999%), and I2 beads (Wako, 99.8%) were used as received. Stoichiometric amounts of the elements were loaded into silica ampoules and flame sealed under vacuum. The ampoules were placed in a muffle furnace and heated to 923 K over 12 h and annealed at this temperature for 36 h before allowing the furnace to naturally cool to room temperature. The resulting products are silvery looking ingots which were subsequently ground with a mortar and pestle before further use.2.2 Powder X-ray diffraction
Powder X-ray diffraction was performed using a Rigaku SmartLab3 with Cu-Kα radiation and operating at 40 kV and 40 mA. Sample powder was prepared using a well holder and compacted using a glass slide. Diffraction patterns were collected in the 2θ range of 5–80° with a step size of 0.01° and scan rate of 2° per minute. Rietveld refinement was performed using the GSAS-II software package.322.3 SEM-EDS
Elemental analysis of samples was performed using a FE-SEM SU8000 (Hitachi) equipped with a Quantax FQ5060 EDS detector (Bruker). Measurements were performed on polished pieces of sintered pellets mounted on carbon tape and sputtered for 30 s with a carbon coating. Bulk compositions were determined using an average of 3 spots.2.4 UV/vis spectroscopy
UV/vis diffuse reflectance spectra were collected using a V-770 UV-vis/NIR spectrophotometer equipped with an integrating sphere (JASCO) in the range of 500–2500 nm. Samples were finely ground into powder with a mortar and pestle and pressed against a glass slide for measurements. Data was then converted using the Kubelka–Munk function to determine the bandgap.2.5 Spark plasma sintering (SPS)
High density pellets of 10 mm diameter were sintered using a SPS-1080 (SPS SYNTEX Inc.). Finely ground powders were loaded into a graphite die between graphite foil and graphite plungers. The pellets were sintered by heating to 573 K over 5 minutes and annealing at this temperature for 5 minutes, all under an argon atmosphere and a uniaxial pressure of 76 MPa. After sintering, pellets were removed from the graphite die and polished to remove traces of graphite. The geometrical density of the resulting pellets was found to be ∼96% of their theoretical density.2.6 Thermal transport properties
For thermal diffusivity measurements samples were cut into disks of 10 mm diameter with a thickness of approximately 1.5 mm. The thermal diffusivity and specific heat were measured under a nitrogen atmosphere using a Laser Flash Analyzer (LFA) 467 Hyperflash (NETZSCH) and a Pyroceram standard in the temperature range of 303–573 K. The thermal conductivity (k) was then determined by the equation k = ρDCp where ρ, D, and Cp are the sample density, thermal diffusivity, and specific heat, respectively. Three shots were measured at each temperature and the average values are shown in this work. Low temperature thermal conductivity was measured from 6.5 to 300 K using the thermal transport option (TTO) of a Physical Property Measurement System (PPMS, Quantum Design). For the TTO measurement, we used the four-probe lead configuration with a bar shaped sample (2 mm × 2 mm × 8 mm, parallel to the sintering direction) and thermal conductivity was measured automatically in the continuous mode. We used an emissivity of 1.0 to correct the strong radiation heat loss occurring above 150 K.2.7 Quantum chemical calculations
The electronic structure of the compounds was calculated self-consistently using density functional theory (DFT) with plane waves, as implemented in the Quantum Espresso (QE) package.33 We used the generalized gradient approximation (PBE-GGA) pseudopotential in the projector augmented-wave (PAW) method.34,35 Considering the large interatomic distances presented in the layered structure, we employed a non-local correction for the van der Waals interaction (vdW-DF).36 A cutoff of 140 Ry and 1120 Ry was used for the kinetic energy and charge density, respectively, and k-grid sampling was done using a grid of 7 × 3 × 6 in the Brillouin zone of the primitive cell. Bader's charge analysis was performed using the Bader code.37 Electron localization function calculation and wave function projection was performed within QE. VESTA was used for visualization.For the phonon calculation, we relaxed the structure until the force on each atom is smaller than 1.5 × 10−5 Ry bohr−1. Interatomic force for the phonon calculation was extracted using the frozen phonon method as implemented in the phonopy packages.38 A displacement of 0.015 Å was used to generate the displacement pattern in a 4 × 1 × 3 supercell with 384 atoms. Reciprocal space was sampled using Γ point only when calculating the force.
3 Result and discussion
3.1 Crystal structure determination
Within the Tt2PnCh2I3 (Tt = Sn, Pb; Pn = Sb, Bi; Ch = S, Se) family, there are four different reported structural configurations.14,28,29 In the 80's Sn2SbS2I3 was reported to crystallize in the Cmcm space group, based on experiments performed at 293 K and 173 K (Fig. 1a).29 In 2007, Doussier et al. reported the crystal structure of Pb2SbS2I3 at 293 K to crystallize in the Cmcm space group similar to Sn2SbS2I3, but the Sb position for Sn2SbS2I3 is occupied by Pb in Pb2SbS2I3, while the Sn site is split into two partially occupied overlapping Pb and Sb sub-positions (Fig. 1c).28 Further experiments at 100 K found the structure to crystallize in the P21/c space group with distinct separate Pb and Sb sites (Fig. 1d).28 The room temperature (RT) and low temperature (LT) phases have similar structures; however, the LT phase eliminates the observed disorder in the RT structure. Using these structure models and bond valence analysis, Doussier et al. suggest that the reported Sn2SbS2I3 structure to be in error and crystallographic elemental assignments should be similar to Pb2SbS2I3.28,29 Furthermore, Islam et al. more recently reported a fourth structural arrangement for Pb2BiS2I3 and Sn2BiS2I3 at 293 K (Fig. 1b). This crystal structure is nearly identical to that of RT Pb2SbS2I3, but the original split crystallographic sites are resolved as single atom positions.14 Note that the P21/c space group is a subgroup of Cmcm. This group–subgroup relation can be described by a translationengleich of index 2 (t2) followed by a klassengleich of index 2 (k2), as seen in Fig. S6.†39,40 This relationship is represented by the following cell transformation: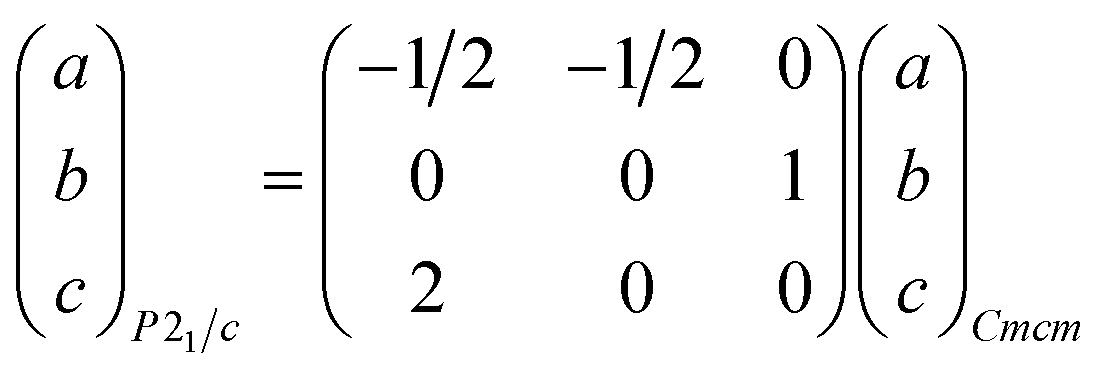
 | ||
| Fig. 1 Reported crystal structures of (a) Sn2SbS2I3, (b) Pb2BiS2I3, and (c and d) Pb2SbS2I3. Crystal structure configuration in (d) is the theoretically determined structure configuration for Sn2SbS2I3. | ||
The difficulty in experimental crystal structure determination mainly arises from the fact that X-ray scattering factors of Sn and Sb (or Pb and Bi) are nearly identical, making their atomic positions ambiguous and difficult to be truly distinguished by X-ray diffraction. However, the crystal structure is of crucial importance for our later theoretical analysis. Therefore, we attempted to determine the stability of the different possible crystal structures for Sn2SbS2I3 by comparing the energy of different relaxed structural models from first principles calculation. The structural models were chosen so that they do not include mixed occupancies (Pb2SbS2I3 LT) or split sites (reducing the split sites to fixed position), which are difficult to treat in DFT calculations. The key point here is the determination of the positions of Sn and Sb. We considered three models: (1) the original Cmcm structure with the split Sb reduced from a half occupied 8f to a fully occupied 4c site, (2) a P21/c (lower) symmetry structure with staggered occupancy of the 8f Sb site and (3) the LT-P21/c configuration of Pb2SbS2I3, reported by Doussier et al. with a Sn2SbS2I3 composition.28 We denote them as Cmcm-4c, P21/c-(Cmcm), and LT-P21/c, respectively. The Cmcm-4c and P21/c-(Cmcm) are derived from the reported Cmcm structure of Sn2SbS2I3 but treat the split of the Sb 8f sites in different ways, while LT-P21/c represents the configuration in which half the Sn positions are swapped with the Sb position compared to the reported structure. Atomic positions and the lattice parameters for each structure model were optimized using variable-cell optimization routine implemented in Quantum Espresso while keeping the symmetry unchanged. The three crystal structures considered, structure parameters and their respective phonon dispersion are shown in Fig. S7, S8 and Table S2.† We found the Cmcm-4c structure is dynamically unstable with the appearance of imaginary phonon modes occurring at most parts of the Brillouin zone, while the two structures in P21/c are dynamically stable. DFT calculations of the ground state energies shows that LT-P21/c has the lowest energy, with P21/c-(Cmcm) and Cmcm-4c being 0.38 eV and 0.64 eV higher per formula unit, respectively. The calculation results agree with the bond valence analysis given by the previous report.28 With these results, we propose that Sn2SbS2I3 shares the same P21/c structure as Pb2SbS2I3 in its ground state (Fig. 1d) and use it for our later theoretical calculations.
3.2 Experimental synthesis
The solid-state synthesis of Sn2SbS2I3 was performed similarly to the procedure reported by Starosta et al.41 As the measured properties within this family of compounds are scarce, in order to build upon the structure–property relationships in these mixed-anion materials, we also attempted to synthesize the reported isostructural Sn2SbSe2I3 for comparison of anion effects.29 However, the synthesis was heavily contaminated with the competing Sn3SbSe2I5 phase, thus we resorted to partial Se substituted Sn2SbS2I3 for comparison and extrapolation of the Sn2SbSe2I3 phase.29 A series of samples of Sn2SbS2−xSexI3 (x = 0, 0.2, 0.5) were synthesized and are discussed herein. Note that attempts to synthesize the Sn2SbSSeI3 composition began to form significant amounts of undesired side phases.Powder X-ray diffraction indicates the three sample compositions are nearly identical and match well with the theoretical pattern of Sn2SbS2I3 (Fig. 2a). Upon closer comparison of the three experimental patterns, small peak shifts are observed for certain peaks in the direction of lower 2θ with increasing Se content. This indicates an expansion of the unit cells, consistent with the substitution of the larger Se in place of S. Furthermore, the magnitude of the peak shift is strongly correlated to the l index as observed in Fig. 2b, indicating the unit cell expansion is predominantly along the crystallographic c-axis. Rietveld refinement and determined unit cell parameters are shown in Fig. S1 and Table S1.† Compared to the reported single crystal unit cells of Sn2SbS2I3 and Sn2SbSe2I3, the determined unit cells for Sn2SbS2I3 and Se substituted samples are in good agreement.29 The Se substituted samples retain consistent a- and b-parameters, while a significant increase in c is observed, which is expected when comparing the pure S and Se analogs.29
 | ||
| Fig. 2 Powder X-ray diffraction patterns of (a) the as synthesized Sn2SbS2−xSexI3 (x = 0, 0.2, 0.5) samples and (b) zoomed in view of the blue highlighted section in (a) emphasizing observed peak shifts between samples. The calculated pattern for Sn2SbS2I3 is shown in red for reference.29 | ||
To further ensure phase composition, energy dispersive X-ray spectroscopy (EDS) was performed. EDS reveals that although the bulk samples match the nominal stoichiometries quite well, there are secondary phases present (Fig. S2†). This may explain the abnormally high wR values of the Rietveld refinements in which some peak intensities are significantly different, despite factoring in possible preferred orientation (Fig. S1†). However, no possible overlapping binary or ternary phases (SnS, Sn2S3, Sn2SI2, etc.) could explain the difference in intensities, even based on measured EDS compositions. It is possible that the overlapping phase(s) could be related unidentified ternary or quaternary structures in this complex system.
3.3 Change in band gap
We characterized the optical band gap using diffuse reflectance spectroscopy. Fig. 3 shows an indirect bandgap of 1.64(2) eV for Sn2SbS2I3, in good agreement with the 1.5 eV previously determined through photoconductivity measurements on a single crystal.41 As Se substitution increases, a reduction of the bandgap to 1.60(2) eV and 1.55(2) eV are observed for Sn2SbS1.8Se0.2I3 and Sn2SbS1.5Se0.5I3, respectively (Fig. 3). This is consistent with physical inspection of the samples as the ground parent compound is a dark maroon colour which progresses to a brown-maroon colour at 0.2 Se, and even further into a gray-maroon colour at 0.5 Se. This also provides further evidence of successful substitution of S by Se.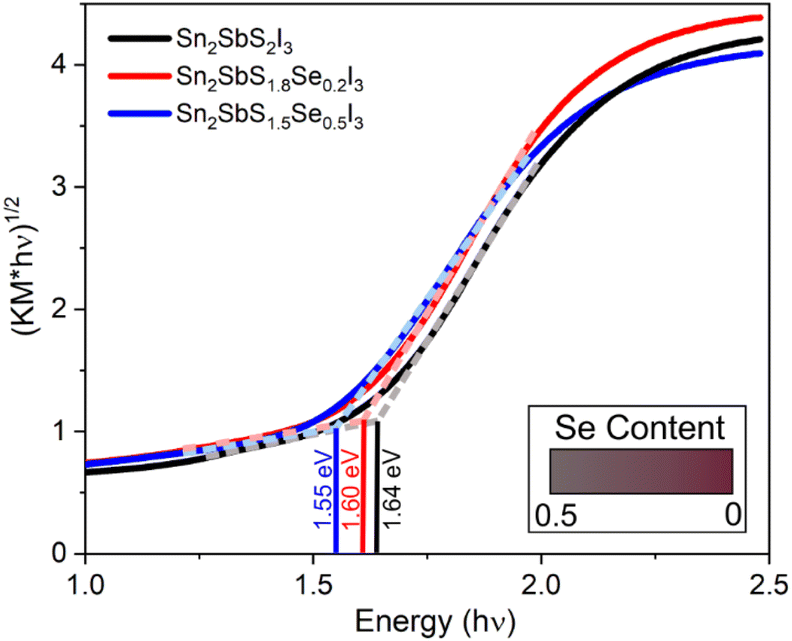 | ||
| Fig. 3 Indirect bandgap Tauc plots of solid-state UV-vis Kubelka–Munk diffuse reflectance spectra for Sn2SbS2−xSexI3 (x = 0, 0.2, 0.5). Colour gradient in the bottom right illustrates observed sample colours based on Se substitution amount. | ||
This trend is contradictory to that observed when substituting Sn for Pb (increase in bandgap), but consistent with the substitution of Sb for Bi (decrease in bandgap) observed for related Tt2PnS2I3 (Tt = Sn, Pb; Pn = Sb, Bi) compounds.14 From an electronegativity standpoint, substitution of S for Se should shift the valence band energy higher and decrease the bandgap due to the lower electronegativity of Se, secondly, comparing the DFT relaxed structure with Sn2SbS2I3 and Sn2SbSe2I3, it is found that the difference between the two in structure mainly come from the components S(Se) and its nearest neighbours. Apart from the longer interatomic distance between S(Se) and Sn or Sb, their relative position is also distorted slightly. The change in bandgap is therefore due to both a change in electronegativity, as well as local structure, and both affect the covalent part of bonding and thus the bandgap. Such an ability to fine tune the bandgap in this family of materials may be beneficial as Pb2SbS2I3 was recently shown to be a robust material for solar cell applications.42
3.4 Thermal conductivity
Given the reported thermal conductivity of Pb2BiS2I3 ranging between 0.7 and 1.2 W m−1 K−1 in the temperature range of 373–673 K, the related Sn2SbS2I3 is expected to exhibit similarly low thermal conductivity.14 Low thermal conductivities on the order of 0.5 W m−1 K−1 were also recently observed in the mixed anion compounds MnSbS2Cl and MnBiS2Cl which have similar elemental composition to the title compounds.43 However, given the lighter elemental composition, thermal conductivity of Sn2SbS2I3 is expected to be slightly higher to its Pb containing counterpart.Thermal diffusivity measurements reveal exceptionally low thermal conductivity as low as 0.28 W m−1 K−1 for Sn2SbS2I3 at 573 K (Fig. 4a). A further reduction of thermal conductivity is observed with the substitution of Se, achieving 0.22 W m−1 K−1 at 573 K for Sn2SbS1.5Se0.5I3 (Fig. 4a). This can be expected from the increase of structural disorder and phonon scattering from substitution and defects. The measured thermal conductivity shows a slight decrease with temperature for the parent sample but a very weak temperature dependence for the Se substituted samples. This suggests that phonon–phonon Umklapp scattering is so strong that the phonon mean free path reaches the amorphous limit, and the weakly temperature dependent coherent part of thermal conductivity plays an important role.44
 | ||
| Fig. 4 (a) High temperature thermal conductivity of Sn2SbS2−xSexI3 (x = 0, 0.2, 0.5) and Sn2BiS2I3 with respective pellet densities and (b) low temperature thermal conductivity for Sn2SbS2I3. | ||
To confirm such unexpectedly low thermal conductivity measurements, we conducted two further experiments. First, we synthesized another member of the Tt2PnCh2I3 (Tt = Sn, Pb; Pn = Sb, Bi; Ch = S, Se) family, namely Sn2BiS2I3 for direct experimental comparison. Using an identical synthesis procedure used for Sn2SbS2I3 similar purity product was confirmed through Rietveld refinement (Fig. S3†). Phase composition was confirmed through EDS and the optical bandgap matches well with previous reports (Fig. S4 and S5†).14 Measured thermal conductivity for Sn2BiS2I3 is shown in Fig. 4a and is of similar scale to Sn2SbS2I3, suggesting this low thermal conductivity is not specific to the Sn2SbS2−xSexI3 compositions, but can be observed in other members of the Sn containing family.
We also used a PPMS as a second method to measure thermal conductivity as a comparison. The thermal conductivity of the Sn2SbS2I3 sample at low temperature is shown in Fig. 4b. Initially, an amorphous glass-like behaviour can be observed at low temperatures, however a T4 dependence becomes dominant at higher temperatures. This suggests significant radiation loss during heating of the sample, which significantly skews thermal conductivity data above 150 K.45 By applying a correction which accounts for additional radiative losses, the data collected above 150 K significantly changes and we observe a maximum value of thermal conductivity of about 0.54 W m−1 K−1 at around 100 K, which slowly decreases with further increase in temperature (see ESI† for detail of the postprocess correction method). After applying the correction, the measured thermal conductivity is approximately 0.49 W m−1 K−1, at 300 K. While still exceptionally low this value is considerably larger than that observed through the LFA method. This inconsistency may come from the still existing underestimation of radiation loss, which is difficult to estimate correctly above 150 K and they are further enlarged by the non-optimal sample geometry (see Discussion in the ESI† for estimation of error in thermal conductivity measurements). Despite the possible errors in this temperature range, the important information revealed from the low temperature data is the absence of a well-defined Umklapp peak and that the thermal conductivity at low temperature cannot be described by the commonly used Debye model of lattice thermal conductivity including boundary scattering and phonon scattering, indicating that the assumptions used in the Debye model, such as the linear phonon dispersion and dominant acoustic phonon scattering are not sufficient to describe the actual case.46,47 Nonetheless, the rounded thermal conductivity maximum and the rather weak temperature dependence suggests the early onset of strong phonon scattering from multiple sources.
Electrical property measurements of the samples proved unsuccessful using ZEM 2 and simple measurements with a multimeter were unable to produce readings, suggesting high resistivity. This is consistent with measurements done by Islam et al. on Sn2BiS2I3 and Pb2SbS2I3, which had resistivities above 1 MΩ.14 The high resistivity indicates that the electronic contribution to the thermal conductivity is negligible and that the total thermal conductivity and lattice thermal conductivity are nearly identical.
3.5 First principles phonon calculation
Phonon dispersion was calculated for Sn2SbS2I3 in the LT-P21/c phase (structure as in Fig. 1d) using force constants from the displacement method. Typical features of phonon dispersion for heavy, multi-element compounds are present, namely, a low-lying acoustic phonon branch below 1 THz, and a low optical phonon frequency with most of the phonon modes concentrated near the 2 THz region, mainly from the I vibration, as can be seen from the phonon density of state in Fig. 5b. Higher frequency optical phonons mainly come from the motion of S. These are separate from the lower phonon bands and are unimportant for thermal transport.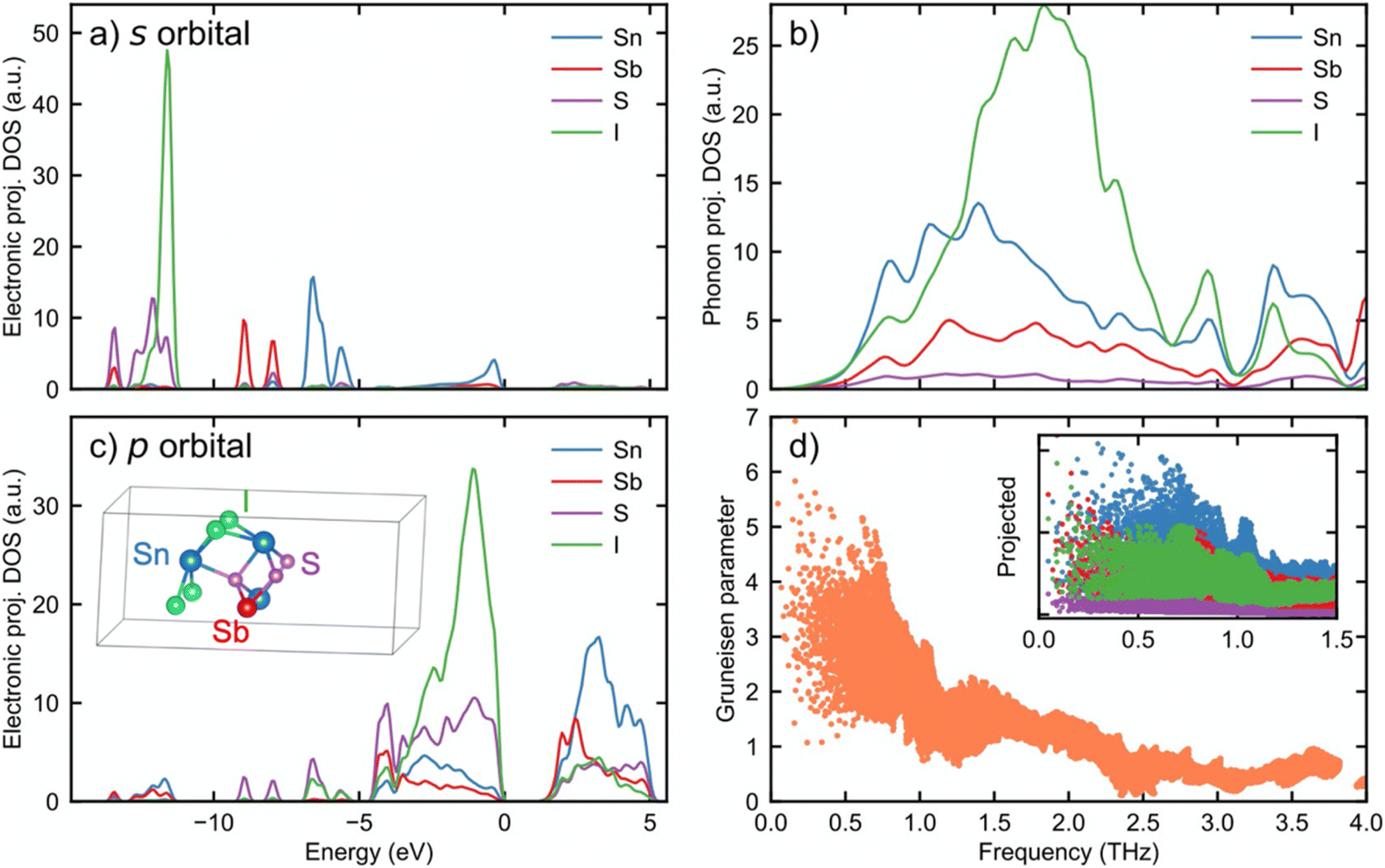 | ||
| Fig. 5 (a) Projected s components of the electronic density of state for Sn2SbS2I3 and (c) projected p components of the electronic density of state. (b) Projected phonon density of state onto different elements in the low frequency part and (d) Grüneisen parameter of each phonon mode sampled in the Brillouin zone. The inset in (d) projects Grüneisen parameter onto each atom with colours corresponding to (c). | ||
Although defects, mixed and partial occupations that could be expected in experiment samples should play an important role in the ultralow thermal conductivity of current series of compounds, we do not attempt to cover their quantitative effects. Rather, we focus on the chemical reasoning through the anharmonicity of the structure, using Grüneisen parameter as an indicator. Grüneisen parameter is defined as the change in phonon frequency corresponding to cell expansion and is closely related to phonon anharmonicity.48,49 The Grüneisen parameter of Sn2SbS2I3 in the low frequency region is plotted in Fig. 5d. It is surprising to find the mode average Grüneisen parameter of all low frequency phonon (<1 THz) to be 2.44, with the highest value reaching over 5 for acoustic modes. This is on the same level with the calculated Grüneisen parameter of the strongly anharmonic SnSe (4.1, 2.1, 2.3 for a-, b-, and c-axis, respectively) and much larger in comparison with conventional thermoelectric compounds like PbTe (1.45).50
A microscopic picture of the anharmonicity is revealed by comparing the projected Grüneisen parameter of each atom, obtained by multiplying the mode Grüneisen parameter with the atomic component of phonon eigen-vectors, in the inset of Fig. 5d. From the projected Grüneisen parameter, it is found that Sn-involving phonon modes show high Grüneisen parameters. Fig. 5b also shows that Sn contributes to the largest phonon density of states in the lower frequency optical phonon region (0.5–1 THz). The phonon in these branches should scatter acoustic phonons effectively.47,51,52 Therefore, we can conclude that Sn atoms are the most important source of the high anharmonicity we found in the structure and lead to a low thermal conductivity. This result also explains the low thermal conductivity of the Sn containing Sn2BiS2I3 measured in the current work.
We attempt to establish the chemical reason for the high anharmonicity of the Sn atom from electronic structure calculation. Using the Bader's analysis method, we obtained the Bader's charge of each atoms Sn+1, Sb+1.85, I−0.5 and S−1.25.53–55 Incomplete charge transfer for S and I, compared with their formal charge of −2 and −1 indicates covalent character, i.e., the orbital components from Sn and Sb atoms are also important in the valence region. Indeed, the projected electronic density of state of Sn2SbS2I3 in Fig. 5a and b shows the hybridization of p orbitals in the energy window from −5 to 0 eV. We would like to point out that some density of state from s orbital of Sn can be seen just below the valence band maximum. The s component can be identified to be anti-bonding since the main bonding region of Sn s orbital is below −5 eV in energy as shown in projected density of states. This antibonding state would involve p orbital of Sn and S along their bonding direction due to the square pyramid like coordination of the Sn and stabilized due to the large interatomic space in the opposite side of S atom.56
The above anti-bonding Sn s component is exhibited in the electron localization function plotted in Fig. 6 with surface of ELF = 0.6.53,57 The crescent moon shaped region near the Sn atom with high electron localization shows the antibonding states containing Sn s orbital in real space. Using integrated local density of state (ILDOS) from −2 to 0 eV, it can be shown that this electron localized region corresponds to the anti-bonding s orbital component discussed above (Fig. S9†). It is known that such “lone pair like” localized antibonding electronic states are important in suppressing lattice thermal conductivity as they introduce restoring force strongly deviated from harmonic approximation, thus leading to the large anharmonicity observed for Sn and the entire crystal structure of Sn2SbS2I3.58–61 In comparison, Pb2BiS2I3 does not show strong localized antibonding states both in real space, as shown in Fig. 6 and in electronic density of states (Fig. S9†), highlighting the important role of the Sn antibonding s states in suppressing the lattice thermal conductivity.
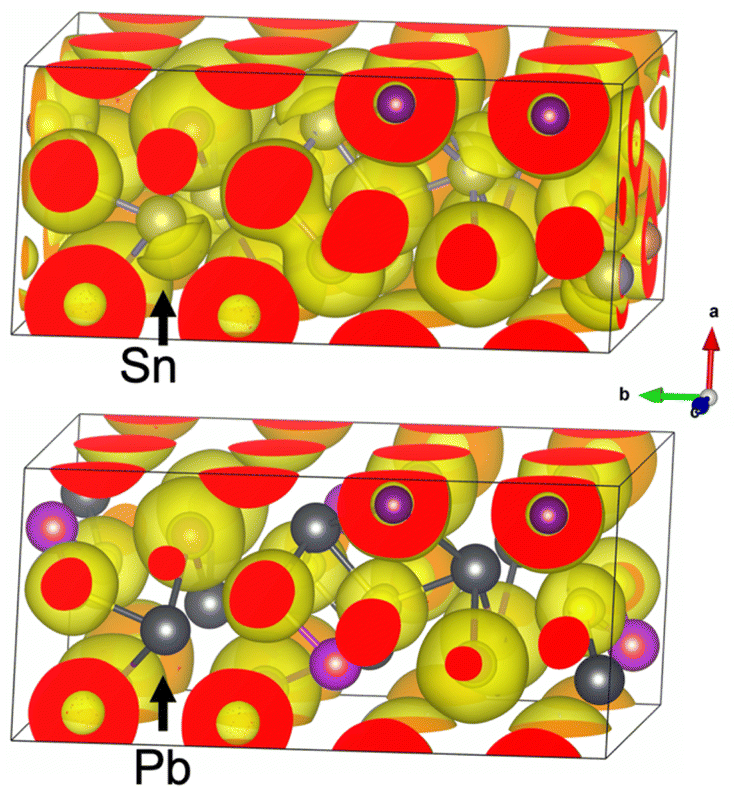 | ||
| Fig. 6 Electron localization function (η = 0.6 surface) of the calculated LT-P21/c structure of Sn2SbS2I3, compared with that of Pb2BiS2I3 counterpart. Sn and Pb atoms are pointed out using black arrows. | ||
4 Conclusions
The series Sn2SbS2−xSexI3 (x = 0, 0.2, 0.5) was successfully synthesized and characterized by UV-vis diffuse reflectance spectroscopy and thermal diffusivity measurements, revealing a tunable band gap ranging from 1.55(2) to 1.64(2) eV and ultralow thermal conductivity in the range of 0.25 W m−1 K−1 from 303–573 K. Computational efforts elucidate the most stable crystal structure configuration of Sn2SbS2I3 to be in a P21/c configuration rather than the previously reported Cmcm. Further calculations using this model suggest that the ultralow thermal conductivity is attributed to the Sn atoms, which contribute largely to structural anharmonicity with localized antibonding s electrons. Such low thermal conductivity results for Sn2SbS2I3 are counter-intuitive when compared to Pb2BiS2I3 (0.7 W m−1 K−1) suggesting that different electronic states can exist in the same family of compounds due to change in chemical composition and exploring such multicomponent systems can provide opportunity for rich chemistry and unique structure properties relationships.Author contributions
Justin Mark: conceptualization, investigation (lead), data curation, formal analysis, visualization and draft preparation. Wenhao Zhang: data curation, investigation (calculation), formal analysis, visualization and draft preparation. Kazuhiko Maeda: review & editing. Takafumi Yamamoto: review & editing. Hiroshi Kageyama: review & editing. Takao Mori: conceptualization, funding acquisition, supervision, review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI JP16H06441, JP19H00833, JP22H05147, JP22H05148 and JST Mirai Program Grant Number JPMJMI19A1. We thank Prof. Jean-François Halet (CNRS-Saint-Gobain-NIMS) for the discussion on the chemical bonding.References
- J. K. Harada, N. Charles, K. R. Poeppelmeier and J. M. Rondinelli, Adv. Mater., 2019, 31, 1805295 CrossRef PubMed.
- J. He, Z. Yao, V. I. Hegde, S. S. Naghavi, J. Shen, K. M. Bushick and C. Wolverton, Chem. Mater., 2020, 32, 8229–8242 CrossRef CAS.
- H. Kageyama, K. Hayashi, K. Maeda, J. P. Attfield, Z. Hiroi, J. M. Rondinelli and K. R. Poeppelmeier, Nat. Commun., 2018, 9, 772 CrossRef PubMed.
- K. Maeda, F. Takeiri, G. Kobayashi, S. Matsuishi, H. Ogino, S. Ida, T. Mori, Y. Uchimoto, S. Tanabe, T. Hasegawa, N. Imanaka and H. Kageyama, Bull. Chem. Soc. Jpn., 2022, 95, 26–37 CrossRef CAS.
- J. Chen, C.-L. Hu, F.-F. Mao, J.-H. Feng and J.-G. Mao, Angew. Chem., Int. Ed., 2019, 58, 2098–2102 CrossRef CAS PubMed.
- B.-W. Liu, X.-M. Jiang, G.-E. Wang, H.-Y. Zeng, M.-J. Zhang, S.-F. Li, W.-H. Guo and G.-C. Guo, Chem. Mater., 2015, 27, 8189–8192 CrossRef CAS.
- W. Xing, P. Fang, N. Wang, Z. Li, Z. Lin, J. Yao, W. Yin and B. Kang, Inorg. Chem., 2020, 59, 16716–16724 CrossRef CAS PubMed.
- W. S. Tang, K. Yoshida, A. V. Soloninin, R. V. Skoryunov, O. A. Babanova, A. V. Skripov, M. Dimitrievska, V. Stavila, S. Orimo and T. J. Udovic, ACS Energy Lett., 2016, 1, 659–664 CrossRef CAS.
- F. Li, J.-F. Li, L.-D. Zhao, K. Xiang, Y. Liu, B.-P. Zhang, Y.-H. Lin, C.-W. Nan and H.-M. Zhu, Energy Environ. Sci., 2012, 5, 7188 RSC.
- J. Li, J. Sui, Y. Pei, C. Barreteau, D. Berardan, N. Dragoe, W. Cai, J. He and L.-D. Zhao, Energy Environ. Sci., 2012, 5, 8543 RSC.
- Y. Liu, L.-D. Zhao, Y. Liu, J. Lan, W. Xu, F. Li, B.-P. Zhang, D. Berardan, N. Dragoe, Y.-H. Lin, C.-W. Nan, J.-F. Li and H. Zhu, J. Am. Chem. Soc., 2011, 133, 20112–20115 CrossRef CAS PubMed.
- L.-D. Zhao, J. He, D. Berardan, Y. Lin, J.-F. Li, C.-W. Nan and N. Dragoe, Energy Environ. Sci., 2014, 7, 2900–2924 RSC.
- M. Amsler, L. Ward, V. I. Hegde, M. G. Goesten, X. Yi and C. Wolverton, Phys. Rev. Mater., 2019, 3, 035404 CrossRef CAS.
- S. M. Islam, C. D. Malliakas, D. Sarma, D. C. Maloney, C. C. Stoumpos, O. Y. Kontsevoi, A. J. Freeman and M. G. Kanatzidis, Chem. Mater., 2016, 28, 7332–7343 CrossRef CAS.
- Q. D. Gibson, T. Zhao, L. M. Daniels, H. C. Walker, R. Daou, S. Hébert, M. Zanella, M. S. Dyer, J. B. Claridge, B. Slater, M. W. Gaultois, F. Corà, J. Alaria and M. J. Rosseinsky, Science, 2021, 373, 1017–1022 CrossRef CAS PubMed.
- K. Biswas, J. He, I. D. Blum, C.-I. Wu, T. P. Hogan, D. N. Seidman, V. P. Dravid and M. G. Kanatzidis, Nature, 2012, 489, 414–418 CrossRef CAS PubMed.
- J. Mao, Z. Liu, J. Zhou, H. Zhu, Q. Zhang, G. Chen and Z. Ren, Adv. Phys., 2018, 67, 69–147 CrossRef.
- Z. Liu, W. Gao, H. Oshima, K. Nagase, C.-H. Lee and T. Mori, Nat. Commun., 2022, 13, 1120 CrossRef CAS PubMed.
- X. Zhou, G. Wang, L. Zhang, H. Chi, X. Su, J. Sakamoto and C. Uher, J. Mater. Chem., 2012, 22, 2958–2964 RSC.
- Z. Liu, J. Mao, T.-H. Liu, G. Chen and Z. Ren, MRS Bull., 2018, 43, 181–186 CrossRef.
- Y. Jiang, J. Dong, H.-L. Zhuang, J. Yu, B. Su, H. Li, J. Pei, F.-H. Sun, M. Zhou, H. Hu, J.-W. Li, Z. Han, B.-P. Zhang, T. Mori and J.-F. Li, Nat. Commun., 2022, 13, 6087 CrossRef CAS PubMed.
- Z. Liu, N. Sato, W. Gao, K. Yubuta, N. Kawamoto, M. Mitome, K. Kurashima, Y. Owada, K. Nagase, C.-H. Lee, J. Yi, K. Tsuchiya and T. Mori, Joule, 2021, 5, 1196–1208 CrossRef CAS.
- J. M. Ziman, Electrons and Phonons: The Theory of Transport Phenomena in Solids, Clarendon Press, Oxford University Press, Oxford: New York, 2001 Search PubMed.
- Z. Liu and T. Mori, in System-Materials Nanoarchitectonics, ed. Y. Wakayama and K. Ariga, Springer, Japan, Tokyo, 2022, pp. 199–231 Search PubMed.
- Z. Liu, W. Zhang, W. Gao and T. Mori, Energy Environ. Sci., 2021, 14, 3579–3587 RSC.
- E. S. Toberer, A. Zevalkink and G. J. Snyder, J. Mater. Chem., 2011, 21, 15843 RSC.
- G. A. Slack, Semiconductors and Semimetals, Academic Press, New York, 1979, vol. 34 Search PubMed.
- C. Doussier, Y. Moëlo, P. Léone, A. Meerschaut and M. Evain, Solid State Sci., 2007, 9, 792–803 CrossRef CAS.
- A. Ibanez, J.-C. Jumas, J. Olivier-Fourcade and E. Philippot, J. Solid State Chem., 1984, 55, 83–91 CrossRef CAS.
- J. Olivier-Fourcade, J. C. Jumas, M. Maurin and E. Philippot, Z. Anorg. Allg. Chem., 1980, 468, 91–98 CrossRef CAS.
- K. Ogawa, H. Suzuki, C. Zhong, R. Sakamoto, O. Tomita, A. Saeki, H. Kageyama and R. Abe, J. Am. Chem. Soc., 2021, 143, 8446–8453 CrossRef CAS PubMed.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- M. I. Aroyo, D. Orobengoa, G. de la Flor, E. S. Tasci, J. M. Perez-Mato and H. Wondratschek, Acta Crystallogr., Sect. A: Found. Adv., 2014, 70, 126–137 CrossRef CAS PubMed.
- E. Kroumova, J. M. Perez-Mato and M. I. Aroyo, J. Appl. Crystallogr., 1998, 31, 646 CrossRef.
- V. I. Starosta, J. Kroutil and L. Beneš, Cryst. Res. Technol., 1990, 25, 1439–1442 CrossRef CAS.
- R. Nie, B. Kim, S.-T. Hong and S. I. Seok, ACS Energy Lett., 2018, 3, 2376–2382 CrossRef CAS.
- N. Sato, N. Kuroda, S. Nakamura, Y. Katsura, I. Kanazawa, K. Kimura and T. Mori, J. Mater. Chem. A, 2021, 9, 22660–22669 RSC.
- M. Simoncelli, N. Marzari and F. Mauri, Nat. Phys., 2019, 15, 809–813 Search PubMed.
- A. L. Pope, B. Zawilski and T. M. Tritt, Cryogenics, 2001, 41, 725–731 CrossRef CAS.
- J. Yang, G. P. Meisner, D. T. Morelli and C. Uher, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 63, 014410 CrossRef.
- S. Misra, C. Barreteau, J.-C. Crivello, V. M. Giordano, J.-P. Castellan, Y. Sidis, P. Levinský, J. Hejtmánek, B. Malaman, A. Dauscher, B. Lenoir, C. Candolfi and S. Pailhès, Phys. Rev. Res., 2020, 2, 043371 CrossRef CAS.
- N. W. Ashcroft and N. D. Mermin, Solid State Physics, Holt, Rinehart and Winston, New York, 1976 Search PubMed.
- C. H. Lee and C. K. Gan, Phys. Rev. B, 2017, 96, 035105 CrossRef.
- L.-D. Zhao, S.-H. Lo, Y. Zhang, H. Sun, G. Tan, C. Uher, C. Wolverton, V. P. Dravid and M. G. Kanatzidis, Nature, 2014, 508, 373–377 CrossRef CAS PubMed.
- W. Li, J. Carrete, G. K. H. Madsen and N. Mingo, Phys. Rev. B, 2016, 93, 205203 CrossRef.
- M. M. Koza, M. R. Johnson, R. Viennois, H. Mutka, L. Girard and D. Ravot, Nat. Mater., 2008, 7, 805–810 CrossRef CAS PubMed.
- G. Frenking, The Chemical Bond, Wiley-VCH, Weinheim, 2014 Search PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- P. S. V. Kumar, V. Raghavendra and V. Subramanian, J. Chem. Sci., 2016, 128, 1527–1536 CrossRef CAS.
- T. A. Albright, J. K. Burdett and M.-H. Whangbo, Orbital Interactions in Chemistry, Wiley, Hoboken, New Jersey, 2nd edn, 2013 Search PubMed.
- A. Savin, R. Nesper, S. Wengert and T. F. Fässler, Angew. Chem., Int. Ed. Engl., 1997, 36, 1808–1832 CrossRef CAS.
- O. Cherniushok, R. Cardoso-Gil, T. Parashchuk, R. Knura, Y. Grin and K. T. Wojciechowski, Chem. Mater., 2022, 34, 6389–6401 CrossRef CAS PubMed.
- M. D. Nielsen, V. Ozolins and J. P. Heremans, Energy Environ. Sci., 2013, 6, 570–578 RSC.
- E. J. Skoug and D. T. Morelli, Phys. Rev. Lett., 2011, 107, 235901 CrossRef PubMed.
- W. Zhang, N. Sato, K. Tobita, K. Kimura and T. Mori, Chem. Mater., 2020, 32, 5335–5342 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta00609c |
| ‡ J. M. and W. Z. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |