
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic understanding of lithium-anode protection by organosulfide-based solid-electrolyte interphases and its implications†
Amruth
Bhargav
 ,
Hooman Yaghoobnejad
Asl
and
Arumugam
Manthiram
*
,
Hooman Yaghoobnejad
Asl
and
Arumugam
Manthiram
*
Materials Science and Engineering Program, Texas Materials Institute, The University of Texas at Austin, Austin, TX 78712, USA. E-mail: manth@austin.utexas.edu
First published on 20th April 2023
Abstract
Maximizing the energy density of Li-based batteries to power our future is predicated on the use of Li-metal as the anode. To improve the longevity of the Li anode, it is critical to regulate the electrolyte–Li metal interface through the solid-electrolyte interphase (SEI). Organopolysulfides have been shown to form favorable SEIs but the mechanism of Li protection is poorly understood. In this work, we first elucidate how the organic thiolate-rich SEI homogenizes Li flux and enables uniform Li deposition. We then utilize methyl, phenyl, and allyl-based polysulfides as model compounds to investigate the effect of the functional groups on the electrodeposition of Li. It is found that aliphatic, conjugated functional groups promote the formation of thiolates, can form polymeric, flexible SEI, provide oxidative stability, and effectively incorporate inorganic phases, thus forming a robust SEI. Diallyl polysulfide is used as a model compound to demonstrate that the improved cycling of the Li-metal anode can prolong the life of Li‖LiFePO4 coin cells as well as Li‖S and anode-free Ni‖Li2S pouch cells when cycled with realistic cell-assembly parameters. This work provides guidelines for the design of future organosulfur materials that could enable long-lasting Li-metal batteries.
1. Introduction
The transition to greener and cleaner energy sources to power our civilization is critical for a sustainable future. In this regard, rechargeable batteries will be key in enabling the widespread electrification of the transportation sector and the assimilation of renewable energy sources, such as wind and solar into the electric grid.1–3 To enable this transformation, batteries need to be energy-dense, low-cost, and sustainable. Cathode materials like LiFePO4 and sulfur fit this bill as they are made of earth-abundant elements.4,5 To maximize the energy density, it is preferable to pair these cathodes with a Li-metal anode as it has the lowest reduction potential (−3.04 V vs. standard hydrogen electrode) and a high capacity of 3861 mA h g−1. Unfortunately, the usage of Li-metal anodes presents two formidable challenges. First, as the deposition of Li-metal on a planar current collector, such as copper foil, is unconstrained, the theoretical volume expansion is infinite.6,7 The unconstrained, non-uniform deposition leads to the formation of dendrites, which can cause an internal short-circuit and present a safety hazard. Second, the high reactivity of Li-metal leads to the decomposition of electrolyte salts and solvents to form an interphase known as the solid-electrolyte interphase (SEI).8,9The SEI forming process actively consumes the Li inventory in the cell, leading to capacity fade.10 Li is also rendered electrochemically inaccessible if encased in the insulative SEI, leading to the formation of “dead” Li.11 The continuous volume change during battery operation leads to SEI fracture, which exposes fresh Li-metal surfaces, causing an incessant growth of the SEI that compounds the Li loss problem.12,13 Several inventive approaches have been developed to combat these issues. The non-uniform growth of Li can be regulated by guiding the Li-flux through alloying elements, such as silver,14 or through materials with uniform pore structures like covalent organic frameworks.15 This approach is rendered more effective when seeding and confining the lithium deposition within a 3-D matrix by using lithiophilic materials such as gold,16 silicon,17 and MoTe2 to name a few.18–21 The high reactivity of the Li-metal can be minimized by secluding the electrolyte from the Li-metal surface by grafting an engineered SEI through ex situ methods. Artificial SEIs composed of inorganic compounds like lithium sulfide,22 polymers like polyvinylidene fluoride,23 and polymer–sulfide hybrids are some examples.23 SEIs can also be engineered in situ by tuning the electrolyte solvation structure to preferentially decompose the salt and by using additives.24–26
A popular class of additive that has been explored for the in situ generation of a favorable SEI is organosulfides. The organopolysulfides are electrolyte soluble and hence they reduce on the Li-metal surface to a thiolate, which serves to protect the anode. Different organopolysulfides with varying functional groups have been reported, such as the triallylamine-based polymer,27 di(tri)sulfide-polyethylene glycol,28 3,5-bis(trifluoromethyl)thiophenol,29 and 1,3,5-benzenetrithiol30 to list a few. Other studies using organosulfur materials as the active material or as additives in lithium–sulfur (Li–S) batteries have also highlighted the positive influence of the reduced organosulfides on the Li-metal anode.31–35 These studies clearly show that the simultaneous presence of Li2S/Li2S2 plasticized with the thiolates forms a mechanically robust interface that regulates Li-ion flux and thus enables stable cycling of the anode. Unfortunately, as most studies focus on a single functional group, a deep understanding of the role of organosulfide-based SEI, especially the effect of different functional groups on the anode protection ability is still lacking.
In this work, we seek to bridge this gap in knowledge by first elucidating how organosulfides change the composition of the SEI layer and the morphology of Li deposition. Materials characterization and first-principles calculations help understand how the organosulfide-based SEI regulates Li flux. The effect of the functional groups on SEI composition and Li-deposition is studied to determine the molecular design rules to generate a favorable SEI. The impact of the stabilized Li-metal anode on LFP and sulfur cathode is presented along with the promising performance of anode-free, lean-electrolyte Li2S-based pouch cells.
2. Results and discussion
2.1 Mechanism of anode protection by organosulfide-rich SEI
In this study, we opt to use ether-based electrolytes as they are stable in the presence of polysulfides and present better compatibility with the Li-metal anode as compared to carbonate-based electrolytes.36,37 The additive-free baseline electrolyte is based on the commonly used composition used in Li–S batteries consisting of 1 M of lithium bis(trifluoromethanesulfonimide) (LiTFSI) dissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v ratio of 1,2-dimethoxyethane (DME) and 1,3-dioxolane (DOL), henceforth referred to as E1. It has been shown that lithium nitrate (LiNO3) and lithium polysulfides (LiPSs) synergistically stabilize lithium deposition.38,39 Therefore, 0.2 M each of LiNO3 and LiPSs in the form of Li2S8 was studied to determine how additives behave in the polysulfide-rich environment of Li–S cells. The inorganic LiPS was replaced with the organic analog, dimethyl polysulfide (DMPS) in the form of (CH3)2S8. DMPS was used as the model compound as the methyl group is the simplest functional group and thus would clearly showcase the benefits of using organosulfur compounds.
:1 v/v ratio of 1,2-dimethoxyethane (DME) and 1,3-dioxolane (DOL), henceforth referred to as E1. It has been shown that lithium nitrate (LiNO3) and lithium polysulfides (LiPSs) synergistically stabilize lithium deposition.38,39 Therefore, 0.2 M each of LiNO3 and LiPSs in the form of Li2S8 was studied to determine how additives behave in the polysulfide-rich environment of Li–S cells. The inorganic LiPS was replaced with the organic analog, dimethyl polysulfide (DMPS) in the form of (CH3)2S8. DMPS was used as the model compound as the methyl group is the simplest functional group and thus would clearly showcase the benefits of using organosulfur compounds.
The performance of these electrolytes was characterized by repeated plating and stripping of the Li-metal on a bare current collector, such as a Ni foil. 2 mA h cm−2 of Li was cycled corresponding to a thickness of roughly 9.7 μm at a current density of 2 mA cm−2. Coulombic efficiency (CE), which is the ratio of the charge extracted during the stripping process to the charge passed during the plating process, is used as a metric to assess the extent of the parasitic side reactions occurring at the Li-metal anode. The CE of the three electrolytes is plotted in Fig. 1a–c along with the scanning electron microscopy (SEM) images of the Li-metal surfaces after 25 stripping/plating cycles in Fig. 1d–f. The Li-metal stripping/plating with the E1 electrolyte is erratic with the CE, ranging from 99.8% to as low as 8%, which is corroborated by the rough surface of the lithium. The uncontrolled reaction of the electrolyte with the fresh Li surface generated in each cycle leads to fine, grainy Li deposits that are only a few microns wide. The high porosity and tortuosity of the deposits entrap the electrolyte and thus exacerbate the parasitic reactions. The addition of the commonly used LiNO3 additive greatly stabilizes lithium deposition as reflected in the improvement of CE to 94.8% (Fig. S1a†). Unfortunately, this improvement in CE is sub-optimal as the morphology of the deposited lithium remains grainy with the typical particle size below 5 μm (Fig. S1b†). However, the combination of LiNO3 and Li2S8 additives synergistically stabilizes the anode with an average CE of 96.15% with the anode suffering failure only after 130 cycles.40 This is also reflected in the morphology of the deposited Li showing long, elongated grains that are typically 5 to 10 μm in length. The reduced porosity also correlates well with the improved CE. The replacement of Li2S8 with DMPS leads to a sharp increase in CE with the average being about 97.64%. Consequently, the Li deposition is dramatically improved with the SEM, showing wide islands of Li stretching more than 20 μm in width. Cross-sectional SEM images in Fig. S2† of the lithium deposits also confirm the dramatic difference in the Li plating morphology between E1 electrolyte and the DMPS-containing electrolyte. It appears that the organosulfide-based SEI can guide a uniform deposition of Li, while maintaining a low active surface area, which prevents the reduction of electrolyte.
 | ||
| Fig. 1 Efficiency of Li-metal stripping/plating in the form of coulombic efficiency in (a) the baseline electrolyte, (b) with LiNO3 and Li2S8 additives, and (c) with LiNO3 and DMPS additives. (d–f) The corresponding SEM micrographs showing the morphology of the Li surface on Ni after 25 cycles of plating and stripping. | ||
To understand how the additives alter the composition of the SEI, the Li surface was analyzed with X-ray photoelectron spectroscopy (XPS) as it is an ideal surface-sensitive technique most commonly used to study SEI composition.8,41–43 Spectra were acquired after 25 plating/stripping cycles to get a representative idea of the various components and the data are presented in Fig. 2a and b. The S 2p spectra of the baseline E1 electrolyte show that the LiTFSI salt breaks down to form trace amounts of Li2S and lower-order LiPSs. The dominant components are oxidized sulfur species, such as Li2SO3 and Li2SO4 (indicated as Li2SOx) along with the peaks for the LiTFSI that is trapped in the crevices of the Li deposits, which persist even after washing the electrode. With the addition of just the LiNO3 additive, no appreciable increase is seen in the content of Li2S (Fig. S3†). However, a substantial increase in the amount of Li2SOx species is observed. As expected, the addition of LiNO3 and Li2S8 increases the content of Li2S, LiPS, and Li2SOx within the SEI. In the presence of DMPS, Li2S is present along with the thiolate, while Li2SOx is notably absent.
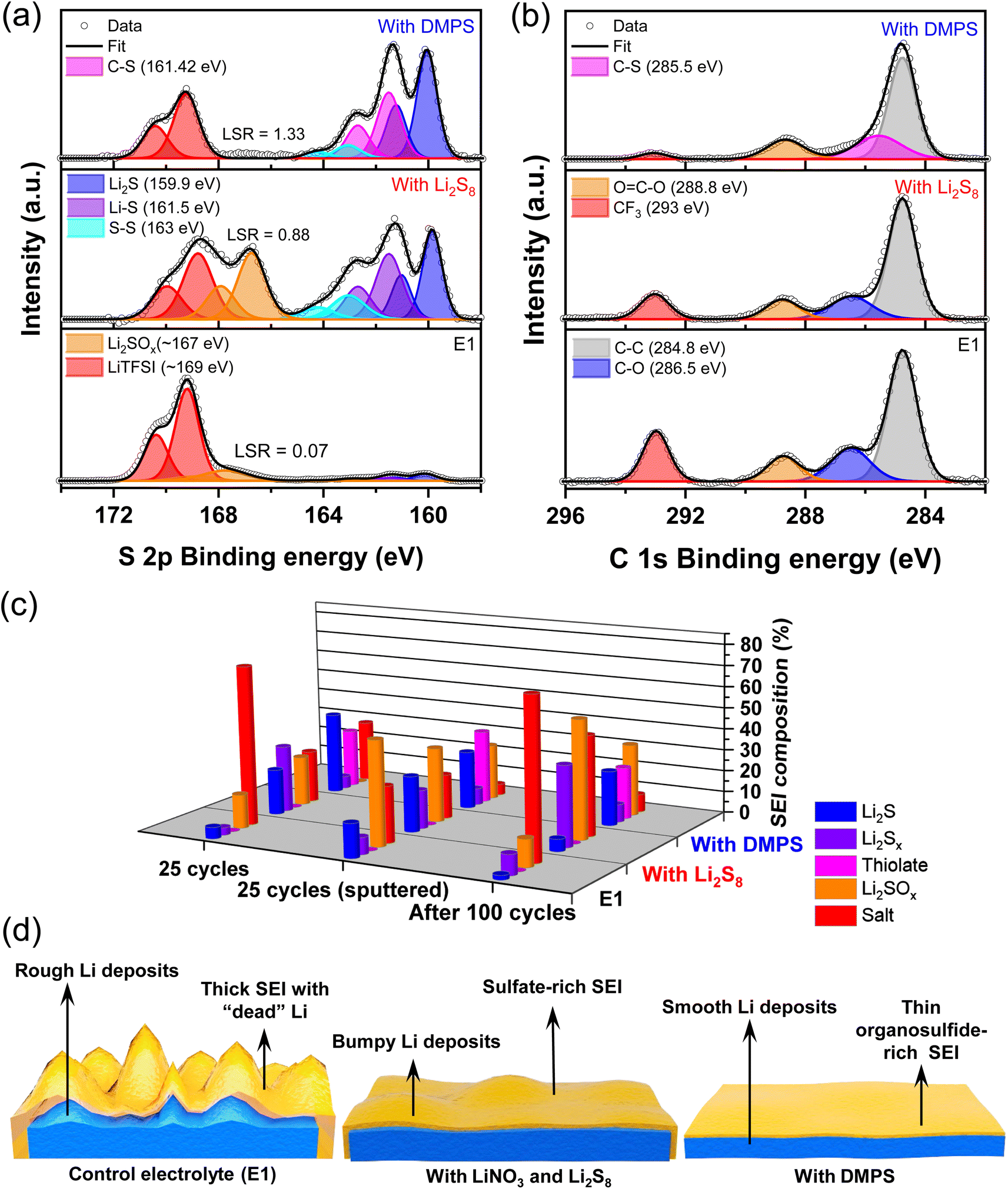 | ||
| Fig. 2 (a) Sulfur 2p and (b) carbon 1s XPS data of the SEI on the Li-metal deposited on Ni after 25 cycles in different electrolytes. LSR corresponds to Li2S to salt ratio. (c) Evolution in the composition of the sulfur species in the SEI across the electrolytes. (d) Schematic illustration of Li-metal deposition and SEI evolution with different electrolytes. | ||
In the baseline E1 and LiPS-containing electrolyte, the C 1s spectra indicates the presence of C–O bonds that could potentially stem from the breakdown of the ether group in the electrolyte solvents.10,27,38,44 Interestingly, this peak is absent in the presence of DMPS, suggesting that the solvent breakdown is suppressed. In order to correlate the CE values and the Li morphology shown in Fig. 1 with the SEI composition, we use a semi-quantitative metric called the Li2S to salt ratio or LSR, which is derived from the ratio of peak areas in the XPS data. The LSR captures the ratio of favorable SEI components, such as Li2S that is known to form a protective interface to that of unwanted components, such as Li2SOx and LiTFSI, which represent the Li lost in the SEI formation process and the SEI encasing the “dead” Li.10,11,45 The low LSR of 0.07 for the E1 electrolytes indicates the formation of thick SEI with electrochemically inaccessible Li, in agreement with the CE and SEM data. This is further supported by the observation of strong signals for metallic Li along with that of Li+ from SEI components in the Li 1s XPS data (Fig. S4†) only upon sputtering while being minimal on the surface of the anode. Li protection by Li2S increases the LSR to 0.88 for LiPS-bearing electrolyte. As evidenced by the high CE and dense Li deposits, the LSR for the DMPS-containing electrolyte is the highest indicating an effective suppression of the parasitic reactions. This is supported by the clear presence of metallic Li on the surface as well as in the bulk in Li 1s XPS.
Another interesting aspect is the difference in Li2SOx levels. It is seen that the LiPS-containing electrolyte shows the highest intensity of Li2SOx, which precludes the breakdown of LiTFSI as the sole source of the SOx2− species. This points to the oxidation of the Li2S in the SEI to Li2SOx by LiNO3, as suggested by its increased Li2SOx presence in the electrolyte containing only the LiNO3 additive (Fig. S3†), which is also corroborated by other studies.10,46 Therefore, the incessant conversion of Li2S to Li2SOx is potentially undesirable as it would be a major source of Li inventory loss in the long term.10,44,46,47 Interestingly, this transformation is absent on the surface of the lithium in the presence of DMPS-containing electrolyte highlighting that the thiolate resists oxidation, thus maintaining adequate Li-ion conduction through the sulfide-rich SEI. The increase in the LSR also correlates well with the reduction in the contribution of the CF3 peak belonging to LiTFSI in the C 1s spectra. Furthermore, based on the F 1s XPS data in Fig. S5,† the thiolate containing SEI fosters the growth of Li-ion conducting inorganic phases, such as LiF.
Ar+ sputtering was employed to probe the compositional changes in the sulfur species with the SEI at a depth of roughly 400 nm as indicated in Fig. 2c. With the baseline electrolyte, the Li2S content increases from 5.4% to 16% and the Li2SOx content increases from 16% to nearly 50% compared to the surface. This confirms that the highly reducing nature of high-surface-area, grainy Li leads to extensive decomposition of the Li salt. With the addition of LiNO3 and Li2S8 additives, the composition stays consistent between the surface and the bulk. In the case of DMPS, an increase in the thiolate content is observed in the bulk. It is interesting to observe the appearance of Li2SOx within the bulk of lithium deposits with DMPS. This is, however, accompanied by a reduction in salt peaks, suggesting that the source of Li2SOx is Li2S oxidation rather than salt decomposition. The evolution of the SEI after 100 cycles was also probed as shown in Fig. 2c. Consistent with the improvement in CE, the salt peak decreases in the order E1 > LiPS > DMPS. The Li2S content in the SEI with the LiPS-containing electrolyte drops to ∼6%, while the Li2SOx content increases to 56%, which reaffirms that the oxidation of reduced sulfur species is the primary mechanism of Li loss. In contrast, with DMPS, the Li2S and thiolate content stays almost constant, while maintaining a high LSR, which proves the improved lithium protection capability of thiolates.
To better understand the difference between sulfide-rich and thiolate-rich SEIs in homogenizing the Li-ion flux at the Li-metal surface, we turn to first principles density functional theory (DFT) calculations. Li2S and CH3SLi species were placed atop a lithium slab and left to equilibrate. The distortion in the electric field is visualized in Fig. S6† and the potential difference between the slab with the species of interest and a pure lithium slab was calculated and is presented in Fig. 3. In the potential difference maps, the blue color indicates increased electron density, and the red color indicates the reduction of electron density. In the case of sulfide-rich SEI (Fig. 3a), strong electric field distortions exist through 4 layers of Li atoms. When looking at the top-down perspective, large, widespread distortions are observed. With this non-uniformity, several electron-rich areas can act as the preferred site for the reduction of Li+ to Li-metal during the plating process, thus increasing the propensity for dendrite formation. In contrast, in the case of thiolate-rich SEI (Fig. 3b), the distortions are minimal after 2 layers of Li atoms. Even the top-down perspective shows only minor potential differences. This arises as the covalent C–S bond withdraws most of the electron cloud away from sulfur, thus reducing the electrons withdrawn from the Li slab. The improved uniformity of the electric field minimizes the potential to form dendrites.
 | ||
| Fig. 3 Visualization of the potential difference along the depth and on the surface of a lithium slab with (a) a sulfide SEI and with (b) a thiolate SEI. | ||
The behavior of the three electrolytes is schematically summarized in Fig. 2d. The native SEI in the additive-free E1 electrolyte is composed of the partial breakdown of the LiTFSI salt. This SEI is not robust enough to arrest the continuous reaction of the electrolyte, leading to the formation of a thick SEI with entrapped Li. The addition of LiPS and LiNO3 leads to a sulfide-rich SEI, but the anode protection capability of this electrolyte deteriorates over cycling owing to the oxidation of Li2S to Li2SOx. When LiPS is substituted with DMPS, the thiolate-rich SEI homogenizes the electric field, resists oxidation, and promotes a uniform Li deposition.
2.2 Effect of functional groups on SEI
Having established the advantages of generating a thiolate-rich SEI in situ, it is critical to understand the effect of functional groups on the Li stabilization capability. For this purpose, we compare the methyl group with the phenyl and allyl groups. The phenyl group was chosen as is a common aromatic unit on which other functional groups can be substituted.29,30,48 The allyl group was chosen as it displays resonance like the phenyl group while being aliphatic like the methyl group, thus providing a good contrast of features. For the sake of simplicity, other functional groups and heteroatomic substituents were avoided. Furthermore, DFT simulation suggests that the electric field distortion between methyl and allyl groups is similar (Fig. S7†), so this choice of functional groups would highlight the differences in stability originating from just hydrocarbon substitution.0.2 M of DMPS, diphenyl polysulfide (DPPS, (C6H5)2S8), or diallyl polysulfide (DAPS, (C3H5)2S8) were added to E1 electrolyte containing 0.2 M LiNO3. The CE during repeated plating/stripping tests with different additives are indicated in Fig. 4a–c. DMPS can cycle with an average CE of 97.64% for 136 cycles before the cell failure due to dendrite formation. The introduction of DPPS improves the CE to 98.58% and consequently can cycle for 306 cycles before failure. DAPS shows the most improvement in CE with an average of 99.16% and the lifespan to 397 cycles. The improvement in CE is also subtly reflected in the Li morphology seen in the SEM images displayed in Fig. 4d–f. In dimethyl polysulfide-containing electrolyte, there are smaller islands (around 10–20 microns) of smooth Li deposits. In diphenyl polysulfide-containing electrolyte, these islands coalesce to larger patches of 20–30 microns in size. The Li surface also appears smoother. The Li surface is the smoothest in diallyl polysulfide-based electrolyte with the deposits extending up to 40–50 microns wide. Owing to the efficient Li protection by DAPS, the surface appears almost non-porous indicating that the SEI can sufficiently accommodate the volume change during cycling.
 | ||
| Fig. 4 Coulombic efficiency of Li‖Ni cells with an electrolyte containing (a) DMPS, (b) DPPS, and (c) DAPS additives. (d–f) The corresponding SEM micrographs show the morphology of the Li surface on Ni after 25 cycles of plating and stripping. | ||
To elucidate how the functional group alters the SEI composition, the XPS of the Li surface was analyzed. The S 2p spectra in Fig. 5a confirm that all three organopolysulfides reduce on the Li-metal surface to form a thiolate-laden SEI. Intriguingly, DMPS maintains a Li2S-rich SEI while both DPPS and DAPS form thiolate-rich SEI. The C–S bond in the C 1s spectra in Fig. 5b indicates that the amount of thiolate in the SEI follows the order DAPS > DPPS > DMPS. This suggests that the stabilization of the anion through the resonance effect strongly favors the reduction of the additive to thiolate as highlighted in Fig. 5c. The allyl group is highly sensitive to polysulfide radicals and can, therefore, undergo inverse vulcanization as previously reported in the literature.49–51 The addition of polysulfide radicals formed during battery cycling with the double bond of the allyl group as shown in Fig. 5c can lead to the formation of a cross-linked polymer on the anode. This could explain the anomalously high amount of neutral bridging sulfur (S–S) present in the S 2p spectra of DAPS. This could also explain the high ratio of C–S bonds seen in the C 1s spectra of DAPS.
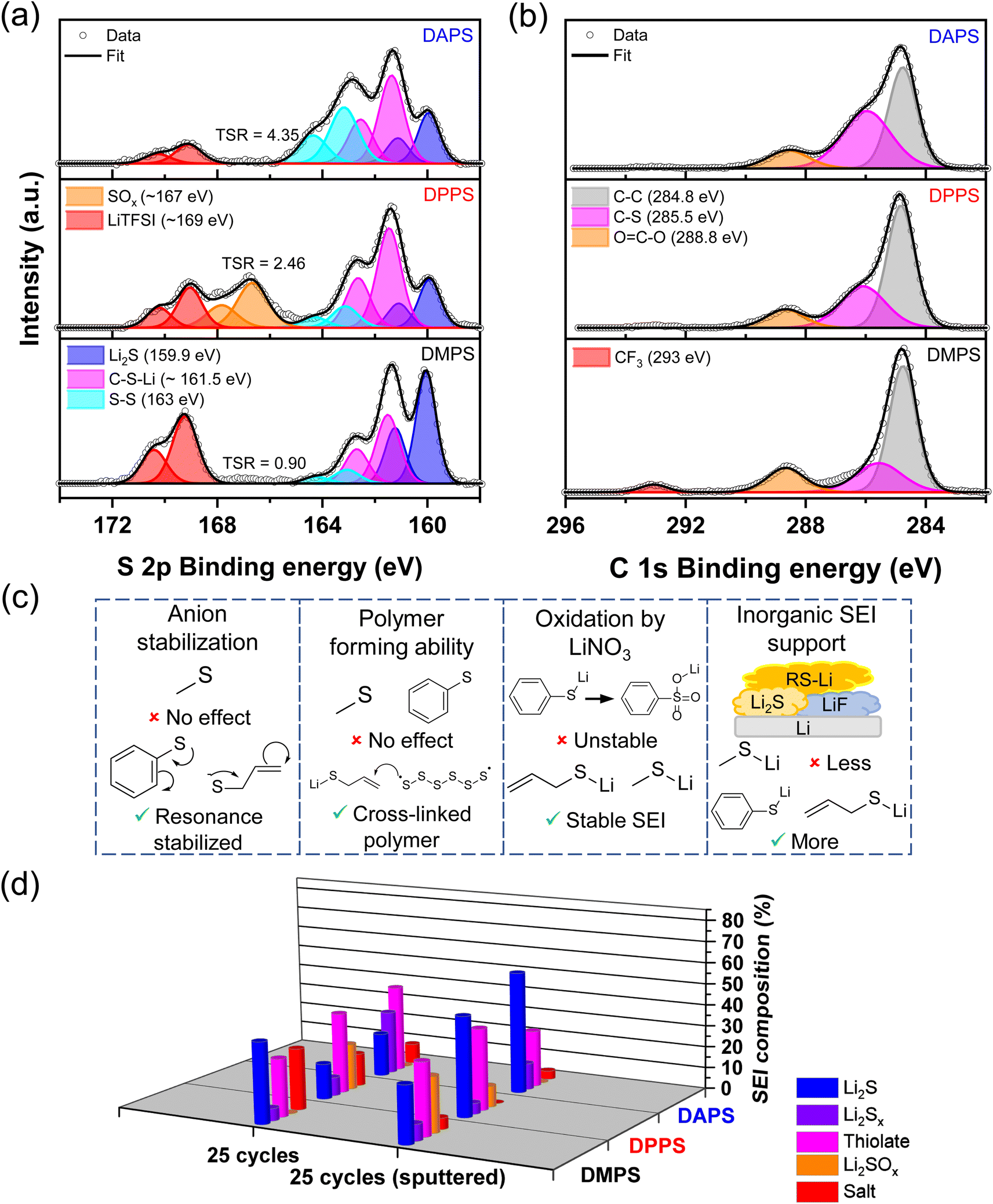 | ||
| Fig. 5 (a) Sulfur 2p and (b) carbon 1s XPS data of the SEI on the Li-metal deposited on Ni after 25 cycles in the presence of different additives. TSR corresponds to the thiolate to salt ratio. (c) Schematic illustrating how the various functional groups differ in the four critical properties that affect Li growth. (d) Differences in the composition of the sulfur species between the surface and the bulk. | ||
Another interesting observation is the presence of Li2SOx species in the case of DPPS and its notable absence in the aliphatic DMPS and DAPS. This suggests that the extended conjunction in the aromatic phenyl group can potentially stabilize the organosulfate anion. This stability could direct the conversion of organothiolates to organosulfates in the presence of LiNO3 as with the case of Li2S (Fig. 5c). While organosulfates appear to stabilize the lithium surface better than Li2SOx species based on the CE data, it would still lead to the unwarranted loss of Li and hence would not be preferred. For this reason, short range resonance bonds would be ideal. Inspection of the F 1s spectra in Fig. S8† shows that both DPPS and DAPS favor the formation of inorganic phases like LiF (Fig. 5c). The large size of the phenyl- and allyl thiolate anions might assist in the incorporation of compact anions like fluoride. It has been shown that LiF is a favorable SEI component as it could potentially boost the Li-ion conductivity and thus guide uniform Li deposition.52 Therefore, functional groups that encourage the formation of LiF are preferable.
A metric akin to LSR, termed as thiolate to salt ratio (TSR), was used to correlate the CE and morphology to the amount of thiolate present on the surface of the SEI. As indicated in Fig. 5a, the TSR follows the order DAPS > DPPS > DMPS, which correlates well with the improvements in CE. This trend is also reflected in the amount of CF3 belonging to LiTFSI present in the C 1s spectra. The anodes were also subjected to Ar+ sputtering to understand the differences between the surfaces and the bulk of Li deposits with the various functional groups (Fig. 5d). Interestingly, the Li2S content in the bulk follows the same trend as TSR, suggesting that as the stability of the thiolate anion improves, the thiolate prefers to be on the surface, while facilitating a Li2S-rich bulk leading to a bilayer SEI. Another key difference is that the stability of the thiolate anion reduces the propensity to form sulfate-rich species within the SEI.
The CE, SEM, and XPS data help us formulate design rules for the functional groups that can be used to develop the most favorable thiolate-rich SEI based on the key properties indicated in Fig. 5c. Ideally, the functional group should be conjugated and aliphatic as it favors the formation of the thiolate. Such a thiolate would be resistant to oxidation and help incorporate other inorganic phases. Critically, the presence of vinylic or allylic groups would facilitate the formation of cross-linked thiolate polymer in the SEI. This organic layer could potentially interpenetrate with the inorganic phases like Li2S and LiF, thus boosting conductivity while simultaneously imparting flexibility to the SEI, which would help accommodate the volume change occurring during cycling without fracturing the SEI. Such a robust, flexible SEI would exclude the electrolyte from contacting the freshly deposited Li, while maintaining a uniform ion flux to help form smooth, dendrite-free Li morphology. The aforementioned rules also explain and can help delineate the various benefits observed across different functional groups employed in previous reports of organosulfide-based SEIs.27–30
2.3 Implications of organosulfide-rich SEI on Li-metal batteries
It appears that DAPS possesses most of the desirable traits to produce a stable thiolate-based SEI. Symmetric cells with DAPS containing electrolyte show a high CE of 96.84% even when cycled at a high current density of 10 mA cm−2 (Fig. S9†). Improving the stability of the Li-metal anode enables its paring with existing Li-ion cathode materials, such as LiFePO4 (LFP) to improve the cell energy density. Additionally, stable Li-metal anodes would also unlock longer life in next-generation cathodes, such as sulfur. To determine the suitability of the DAPS additive for practical applications, it was compared against the E1 electrolyte containing only 0.2 M LiNO3 additive referred to as the control electrolyte. First, DAPS was tested with an LFP cathode having a loading of 6.5 mg cm−2, cycled with an electrolyte loading of 6 μL mg−1 and at a 1C rate. The long-term stability is shown in Fig. 6a along with the representative voltage profiles in Fig. 6b. With the control electrolyte, LFP displays a higher peak capacity of 151 mA g−1, but can only retain 45.4% of this capacity after 400 cycles. In contrast, the DAPS gradually reaches its peak capacity of 142 mA g−1 over the course of 150 cycles potentially due to the lowered conductivity of the electrolyte in the presence of DAPS, but can retain 71.1% of the capacity after 400 cycles. The improved CE in the cells with DAPS indicates that the improvement in life stems from the improved Li stripping/plating despite the presence of excess Li. The improved cycling of the anode is also reflected in the reduced cell overpotential in the voltage profiles.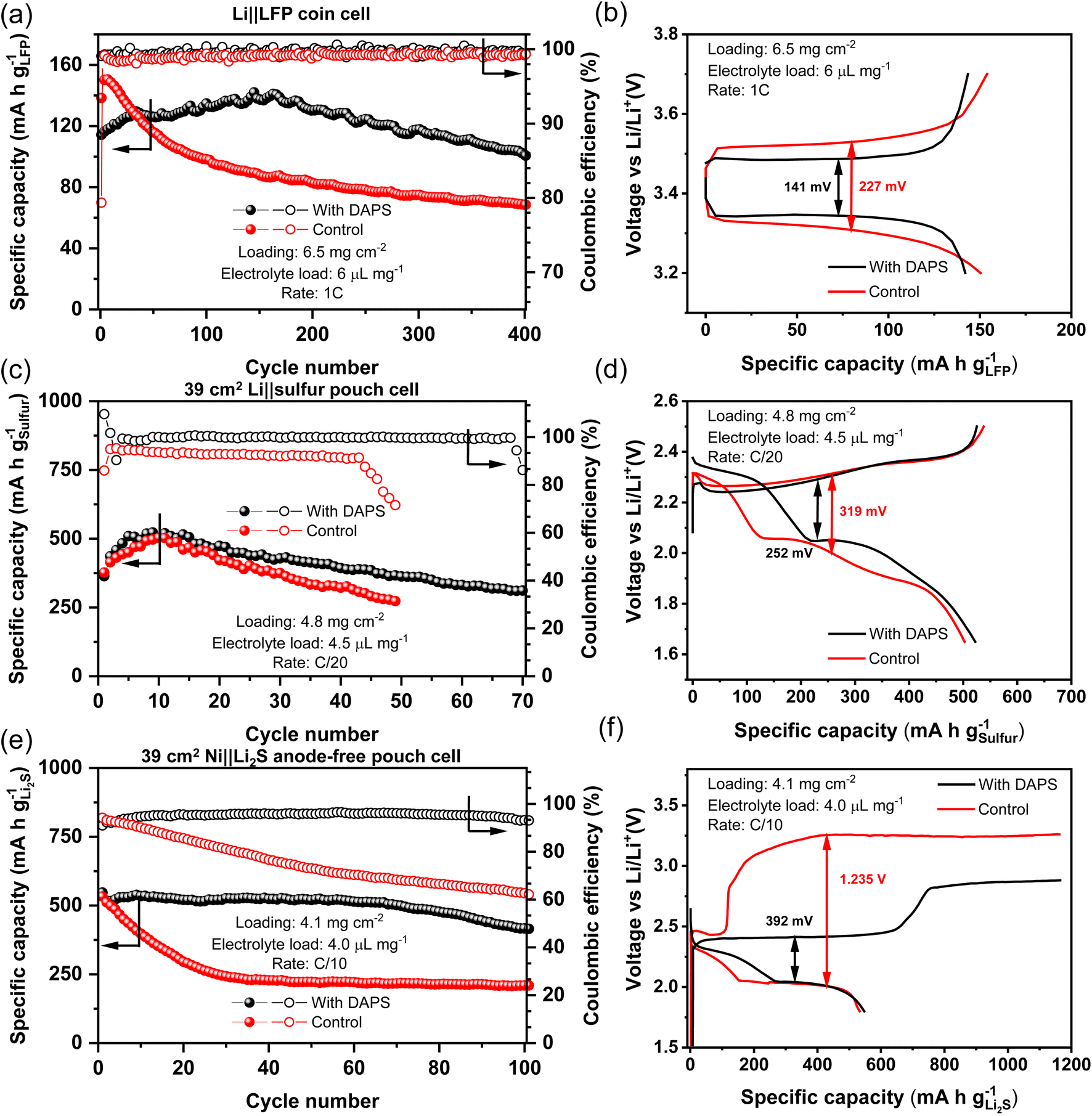 | ||
| Fig. 6 Long term cycling stability of (a) Li‖LFP coin cells, (c) Li‖S pouch cells, and (e) anode-free Ni‖Li2S pouch cells. Here, the control electrolyte E1 with 0.2 M LiNO3 additive was tested against the E1 electrolyte containing 0.2 M DAPS with 0.2 M LiNO3 additive. (b), (d), and (f) show the representative voltage profiles, respectively, from (a), (c), and (e). | ||
As DAPS already consists of polysulfides, the Li–S system is suited to test the efficacy of this additive. Single-stack pouch cells were fabricated with cathodes containing a sulfur loading of 4.8 mg cm−2, a lean electrolyte amount of 4.5 μL mg−1 paired with a 200 μm thick Li-metal anode, corresponding to a N/P ratio of 5.2. The cycle life of the cells when cycled at a C/20 rate is reported in Fig. 6c. As the cells are cycled with a high sulfur loading and lean electrolyte condition, wherein the sulfur redox is typically sluggish, they show a low but nearly identical initial capacity. Further, the cells also show an activation period of 10 cycles wherein the access to sulfur slowly increases with improved access to the electrolyte over repeated cathode expansion–contraction cycles. However, the cell with the control electrolyte fails at cycle 49. The low overall CE of ∼92.5% and its precipitous drop after 43 cycles are emblematic of poor SEI at the anode leading to its eventual failure.53 On the other hand, DAPS-based cells can last for up to 70 cycles, while cycling with a CE of ∼99.8%, highlighting the superior Li stripping/plating. Inspection of the voltage profile during peak performance in Fig. 6d reveals that DAPS is also able to mediate sulfur redox leading to lowered overpotentials and improved energy efficiency. This behavior is also reflected in lower loading coin cells cycled at a high rate of C/2 (Fig. S10†). The additive-free coin cell shows a capacity fade of 42% after 200 cycles, while the anode stabilized cell in the presence of DAPS fades by only 14% after 200 cycles. Electrochemical impedance spectroscopy of the pristine cells (Fig. S11†) show lower charge-transfer resistance (RCT) in the DAPS-containing electrolyte compared to the blank electrolyte, reconfirming that DAPS facilitates sulfur redox. After 40 cycles, the RCT increases owing to the build-up of the SEI at the anode (Fig. S12†). Despite this, the RCT in the DAPS electrolyte is 22 Ω, while that of the blank electrolyte is 73 Ω, indicating that facile ion transport through the SEI is continually maintained in the case of DAPS. While this serves to demonstrate improved cyclability owing to a more stable Li-metal anode, the capacity of the pouch cells may be improved in the future by incorporating agents that improve sulfur redox, such as electrocatalysts.
The encouraging results with Li‖S cells prompted us to study the performance in anode-free Ni‖Li2S cells. As the anode-free systems start with the stoichiometric amount of Li, the cycle life mirrors the effectiveness of Li cycling. Pouch cells with a Li2S loading of 4.1 mg cm−2 and a lean electrolyte amount of 4.0 μL mg−1, paired with a bare Ni foil as the current collector, were fabricated and cycled at a C/10 rate. As with sulfur cathodes, both cells show identical material utilization in the first cycle (Fig. 6d). However, in the control cell, the capacity drops by 50% in just 24 cycles and can retain only 39% of its initial capacity by 100 cycles. In sharp contrast, the DAPS-bearing cell can retain 76% of its initial capacity after 100 cycles. The monotonic drop in CE from 94.2% to 62.2% in a span of 100 cycles showcases the ineffectiveness of sulfide-rich SEI in protecting the Li-metal. Meanwhile, the CE with DAPS is stable throughout with an average of ∼96%, showcasing the robustness of the thiolate-rich SEI. In addition to anode protection, the dramatic lowering of the first charge voltage by nearly 850 mV reaffirms the secondary benefit of accelerating sulfur redox. This makes organopolysulfide additives the ideal candidate for Li protection in the Li–S system.
3. Conclusion
The successful deployment of Li-metal anode hinges on the ability to stabilize the metal–electrolyte interface through the SEI. By delineating the source of the different SEI components, this study establishes that organosulfide-rich SEI improves the stability of the Li-metal anode. Computational studies confirm that the reduced electric field distortions on the Li-metal surface in the presence of a thiolate-rich SEI facilitate a uniform, dense growth of Li. By using methyl, phenyl, and allyl-based polysulfides, the profound effect that a simple hydrocarbon substitution has on the Li-anode is uncovered. It is shown that an ideal organosulfide consists of conjugated, aliphatic chains. DAPS as a model additive highlights the significant improvement in performance that can be achieved through the engineering of the organic functional group. While the functional groups presented here are simple, further studies based on the design rules uncovered in this study could reveal the hidden potential of heteroatomic functional groups. The richness of organic chemistry also presents the opportunity to fine-tune the properties of such interphases so as to enable long-life Li-metal batteries.4. Experimental section
Materials
Sulfur (S8, 99.5+%, Acros Organics), lithium sulfide (Li2S, 99.9%, Alfa Aesar), dimethyl trisulfide (DMTS, ≥98.0%, Sigma Aldrich), diphenyl disulfide (DPDS, 99%, Sigma Aldrich), diallyl trisulfide (DATS, ≥98%, Sigma Aldrich), lithium bis(trifluoromethanesulfonimide) (LiTFSI, LiN(CF3SO2)2, 99%, Acros Organics), lithium nitrate (LiNO3, 99+%, Acros Organics), 1,2-dimethoxyethane (DME, 99+%, Acros Organics), 1,3-dioxolane (DOL, 99.5%, Acros Organics), lithium foil (99.9%, 200 μm thickness, Goodfellow), Super P (TIMCAL), aligned carbon nanotubes (CNT, >95%, OD = 10–20 nm, L = 30–100 μm), (Nanostructured & Amorphous Materials, Inc.), Ketjenblack (KB, EC-600JD, AkzoNobel), polyethylene oxide (PEO, average MW ∼ 4000000, Sigma Aldrich) and polyvinylpyrrolidone (PVP, average MW ∼ 1300000, Acros Organics), polyvinylidene fluoride (PVDF, Kynar HSV 1800, Arkema), lithium iron phosphate (LFP, MTI Corp.), 1-methyl-2-pyrrolidinone (NMP, 99.5%, Sigma Aldrich), nickel foil (MF-NiFoil-25u, MTI Corp.), and carbon coated Al-foil (MTI Corp.) were used as received.
Electrolyte preparation
The baseline (E1) electrolyte was prepared by dissolving 1 M LiTFSI in a 1:1 v/v ratio of DME and DOL. The LiPS-containing electrolyte was prepared by dissolving appropriate quantities of Li2S, elemental S, and LiNO3 through overnight stirring in the E1 electrolyte to yield 0.2 M Li2S8 and 0.2 M LiNO3. The DMPS electrolyte was prepared by dissolving DMTS, Li2S, elemental S, and LiNO3 in the E1 electrolyte to yield 0.2 M DMPS and 0.2 M LiNO3. DPPS and DAPS electrolyte were similarly prepared by substituting DMTS with DPDS and DATS, respectively, while adjusting the stoichiometry of Li2S and elemental S. The nominal order of the polysulfides is as indicated in the figures.
Cathode fabrication
A slurry of LFP, Super P, CNT, and PVDF was made in a 85:5:5:5 wt ratio in NMP. The slurry was blade cast onto an Al foil with a loading of about 6.5 mg cm−2. The electrodes were dried at 120 °C overnight under a vacuum before being punched into 12 mm dia discs to be used in cells.
Sulfur and KB were ground in 9:1 wt ratio before being transferred to a Teflon-lined autoclave. The autoclave was heated to 160 °C and kept for 10 h to melt-diffuse the sulfur into the pores of KB. The S/KB powder was ground finely before the slurry preparation. A slurry consisting of S/KB composite, Super P, CNT, PEO, and PVP was mixed in 75:10:5:8:2 wt ratio in water. The slurry was blade-cast onto Al foil and dried at 60 °C overnight under vacuum before being punched into pouch cell electrodes. The sulfur content was 67.5% and the loading was 4.8 ± 0.2 mg cm−2.
A slurry consisting of Li2S, Super P, CNT, PEO, and PVP in 70:10:10:8:2 wt ratio was made in an acetonitrile, DME, DOL mixture, and blade-cast onto an Al foil. The electrodes were dried at 60 °C overnight before being punched for pouch cell. The loading was 4.1 ± 0.3 mg cm−2.
Cell fabrication
Coin cells were fabricated in the CR-2032 format in an Ar-filled glovebox. Li‖Ni symmetric cells were fabricated with a 0.5 in dia disc of Ni foil as the current collector, Celgard 2500 separator, 30 μL of the appropriate electrolyte, and a 7/16 inch dia Li chip attached to a Ni-foam spacer as the anode. The Ni foil was oversized to avoid alignment issues.LFP coin cells were fabricated similarly with the cathode discs, an appropriate amount of electrolyte, and a 14 mm dia Li chip as the anode.
Single-stack soft-packaging pouch cells having a dimension of 8.1 cm × 4.8 cm (∼39 cm2) were fabricated inside a glovebox. The cathodes were carefully wrapped in a Celgard 2500 separator, aligned with the anodes, and sealed at the tabs inside the soft packaging. An appropriate amount of electrolyte was added before being fully sealed. The sulfur cathodes were paired with a 200 μm thick Li anode mounted on a Ni foam support for welding tabs. The Li2S cathodes were paired with bare Ni foils.
At least 5 cells of each type were fabricated to ensure reproducibility. The representative data shown in the figure show the average for that cell along with the standard deviation for the experiment.
Electrochemical measurements
Li‖Ni symmetric cells were tested with current densities and areal capacities calculated based on the geometric area of the Li-chip as it was the limiting electrode dimension. LFP cells were cycled between 3.7 and 3.2 V with a current density corresponding to 1C = 170 mA g−1. Sulfur cells were tested in the 2.5–1.65 V range with a C rate corresponding to 1C = 1672 mA g−1. Li2S cells were first charged with a voltage cut-off of 3.6 V with a capacity cut-off equal to the theoretical capacity of the cell. In subsequent steps, the cells were tested in the 2.8–1.8 V range with a C rate corresponding to 1C = 1165 mA g−1. Electrochemical impedance spectroscopy (EIS) was performed on a BioLogic VSP potentiostat in the frequency range of 1 MHz to 100 mHz.Materials characterization
Li‖Ni symmetric cells were cycled for 25 cycles followed by a final Li-plating step before being disassembled for characterization. The electrodes were thoroughly rinsed with blank DME solvent before being mounted on the air-sensitive sample holder for the instruments. All characterizations were performed in triplicate to ensure the reproducibility.SEM was performed on a FEI Quanta 650 microscope with an acceleration voltage of 10 kV. XPS was performed with a Kratos AXIS Ultra DLD spectrometer with monochromatic Al Kα radiation having an energy of 1468.5 eV at 12 kV and 10 mA. Survey scans were performed on multiple spots on the sample to ensure accuracy of the spectra. Region scans were performed with 20 eV pass energy, a step size of 0.1 eV, and a dwell time of 2000 ms per step without utilizing the charge-neutralizer to avoid alterations of peak shape or position. Ar+ sputtering was performed to obtain the SEI composition of the bulk. Sputtering depth was estimated using pristine Li chips as a calibrant. Deconvolution of the acquired spectra was performed using the CASA XPS software. The adventitious carbon peak at 284.8 eV was used as an internal calibrant. SEI composition was normalized to the total sulfur species to account for and to minimize sample-to-sample variation.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Assistant Secretary for Energy Efficiency and Renewable Energy, Office of Vehicle Technologies of the U.S. Department of Energy through the Advanced Battery Materials Research (BMR) Program (Battery500 Consortium) award number DE-EE0007762.References
- A. Manthiram, Nat. Commun., 2020, 11, 1550 CrossRef CAS PubMed.
- J. B. Goodenough, Energy Environ. Sci., 2014, 7, 14–18 RSC.
- W. Li, E. M. Erickson and A. Manthiram, Nat. Energy, 2020, 5, 26–34 CrossRef CAS.
- S. Chung and A. Manthiram, Adv. Mater., 2019, 31, 1901125 CrossRef PubMed.
- S. S. Sharma and A. Manthiram, Energy Environ. Sci., 2020, 13, 4087–4097 RSC.
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X. Q. Yang and J. G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- X. B. Cheng, R. Zhang, C. Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- X.-B. B. Cheng, R. Zhang, C.-Z. Z. Zhao, F. Wei, J.-G. Zhang, Q. Zhang, X.-B. B. Cheng, R. Zhang, C.-Z. Z. Zhao, F. Wei, Q. Zhang and J.-G. Zhang, Adv. Sci., 2016, 3, 1500213 CrossRef PubMed.
- W. Liu, P. Liu and D. Mitlin, Adv. Energy Mater., 2020, 10, 2070177 CrossRef CAS.
- S. Nanda and A. Manthiram, Energy Environ. Sci., 2020, 13, 2501–2514 RSC.
- C. Fang, J. Li, M. Zhang, Y. Zhang, F. Yang, J. Z. Lee, M. H. Lee, J. Alvarado, M. A. Schroeder, Y. Yang, B. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 CrossRef CAS PubMed.
- K. N. Wood, M. Noked and N. P. Dasgupta, ACS Energy Lett., 2017, 2, 664–672 CrossRef CAS.
- X. Z. Fan, M. Liu, R. Zhang, Y. Zhang, S. Wang, H. Nan, Y. Han and L. Kong, Chin. Chem. Lett., 2022, 33, 4421–4427 CrossRef CAS.
- Y. G. Lee, S. Fujiki, C. Jung, N. Suzuki, N. Yashiro, R. Omoda, D. S. Ko, T. Shiratsuchi, T. Sugimoto, S. Ryu, J. H. Ku, T. Watanabe, Y. Park, Y. Aihara, D. Im and I. T. Han, Nat. Energy, 2020, 5, 299–308 CrossRef CAS.
- J. He, A. Bhargav and A. Manthiram, Angew. Chem., Int. Ed., 2022, 61, e202116586 CAS.
- K. Yan, Z. Lu, H. W. Lee, F. Xiong, P. C. Hsu, Y. Li, J. Zhao, S. Chu and Y. Cui, Nat. Energy, 2016, 1, 1–8 Search PubMed.
- Z. Liang, D. Lin, J. Zhao, Z. Lu, Y. Liu, C. Liu, Y. Lu, H. Wang, K. Yan, X. Tao and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2862–2867 CrossRef CAS PubMed.
- J. He, A. Bhargav and A. Manthiram, Adv. Energy Mater., 2022, 12, 2103204 CrossRef CAS.
- Y. Liu, Y. Zhai, Y. Xia, W. Li and D. Zhao, Small Struct., 2021, 2, 2000118 CrossRef CAS.
- E. Cha, J. H. Yun, R. Ponraj and D. K. Kim, Mater. Chem. Front., 2021, 5, 6294–6314 RSC.
- C. Yan, X.-Q. Zhang, J.-Q. Huang, Q. Liu and Q. Zhang, Trends Chem., 2019, 1, 693–704 CrossRef CAS.
- H. Chen, A. Pei, D. Lin, J. Xie, A. Yang, J. Xu, K. Lin, J. Wang, H. Wang, F. Shi, D. Boyle and Y. Cui, Adv. Energy Mater., 2019, 9, 1900858 CrossRef.
- S. Gao, F. Sun, N. Liu, H. Yang and P. F. Cao, Mater. Today, 2020, 40, 140–159 CrossRef CAS.
- L. Su, E. Jo and A. Manthiram, ACS Energy Lett., 2022, 2165–2172 CrossRef CAS.
- X. Cao, H. Jia, W. Xu and J.-G. Zhang, J. Electrochem. Soc., 2021, 168, 010522 CrossRef CAS.
- S. Nanda, A. Bhargav, Z. Jiang, X. Zhao, Y. Liu and A. Manthiram, Energy Environ. Sci., 2021, 14, 5423–5432 RSC.
- G. Li, Y. Gao, X. He, Q. Huang, S. Chen, S. H. Kim and D. Wang, Nat. Commun., 2017, 8, 1–10 CrossRef PubMed.
- J. Xie, Y. Song, B. Li, H. Peng, J. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2020, 59, 22150–22155 CrossRef CAS PubMed.
- J. Wei, X. Zhang, L. Hou, P. Shi, B. Li, Y. Xiao, C. Yan, H. Yuan and J. Huang, Adv. Mater., 2020, 32, 2003012 CrossRef CAS PubMed.
- W. Guo, W. Zhang, Y. Si, D. Wang, Y. Fu and A. Manthiram, Nat. Commun., 2021, 12, 1–13 CrossRef PubMed.
- A. Bhargav, Y. Ma, K. Shashikala, Y. Cui, Y. Losovyj and Y. Fu, J. Mater. Chem. A, 2017, 5, 25005–25013 RSC.
- A. Bhargav, C. H. Chang, Y. Fu and A. Manthiram, ACS Appl. Mater. Interfaces, 2019, 11, 6136–6142 CrossRef CAS PubMed.
- A. Bhargav and A. Manthiram, Adv. Energy Mater., 2020, 10, 2001658 CrossRef CAS.
- W. Guo, D. Y. Wang, Q. Chen and Y. Fu, Adv. Sci., 2022, 9, 2103989 CrossRef CAS PubMed.
- Z. Shadike, S. Tan, Q. C. Wang, R. Lin, E. Hu, D. Qu and X. Q. Yang, Mater. Horiz., 2021, 8, 471–500 RSC.
- T. Yim, M. S. Park, J. S. Yu, K. Kim, K. Y. Im, J. H. Kim, G. Jeong, Y. N. Jo, S. G. Woo, K. S. Kang, I. Lee and Y. J. Kim, Electrochim. Acta, 2013, 107, 454–460 CrossRef CAS.
- J. He, A. Bhargav, W. Shin and A. Manthiram, J. Am. Chem. Soc., 2021, 143, 20241–20248 CrossRef CAS PubMed.
- W. Li, H. Yao, K. Yan, G. Zheng, Z. Liang, Y. M. Chiang and Y. Cui, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- S. Nanda, A. Gupta and A. Manthiram, Adv. Energy Mater., 2018, 8, 1801556 CrossRef.
- J. Tan, M. Ye and J. Shen, Mater. Horiz., 2022, 9, 2325–2334 RSC.
- G. Zhang, Z. W. Zhang, H. J. Peng, J. Q. Huang and Q. Zhang, Small Methods, 2017, 1, 1700134 CrossRef.
- V. Shutthanandan, M. Nandasiri, J. Zheng, M. H. Engelhard, W. Xu, S. Thevuthasan and V. Murugesan, J. Electron Spectrosc. Relat. Phenom., 2019, 231, 2–10 CrossRef CAS.
- S. T. Oyakhire, H. Gong, Y. Cui, Z. Bao and S. F. Bent, ACS Energy Lett., 2022, 7, 2540–2546 CrossRef CAS.
- A. Jozwiuk, B. B. Berkes, T. Weiß, H. Sommer, J. Janek and T. Brezesinski, Energy Environ. Sci., 2016, 9, 2603–2608 RSC.
- S. Nanda and A. Manthiram, Adv. Energy Mater., 2021, 11, 2003293 CrossRef CAS.
- Y. Ye, M. K. Song, Y. Xu, K. Nie, Y. sheng Liu, J. Feng, X. Sun, E. J. Cairns, Y. Zhang and J. Guo, Energy Storage Mater., 2019, 16, 498–504 CrossRef.
- S. Nanda, A. Bhargav and A. Manthiram, Joule, 2020, 4, 1–15 CrossRef.
- W. Guo, Z. Wawrzyniakowski, M. Cerda, A. Bhargav, M. Pluth, Y. Ma and Y. Fu, Chem.–Eur. J., 2017, 23, 16941–16947 CrossRef CAS PubMed.
- I. Gomez, O. Leonet, J. A. Blazquez and D. Mecerreyes, ChemSusChem, 2016, 9, 3419–3425 CrossRef CAS PubMed.
- J. Zhou, T. Qian, N. Xu, M. Wang, X. Ni, X. Liu, X. Shen and C. Yan, Adv. Mater., 2017, 29, 1701294 CrossRef PubMed.
- Y. Zhang, R. S. Glass, K. Char and J. Pyun, Polym. Chem., 2019, 10, 4078–4105 RSC.
- J. Tan, J. Matz, P. Dong, J. Shen and M. Ye, Adv. Energy Mater., 2021, 11, 2100046 CrossRef CAS.
- X.-B. Cheng, C. Yan, J.-Q. Huang, P. Li, L. Zhu, L. Zhao, Y. Zhang, W. Zhu, S.-T. Yang and Q. Zhang, Energy Storage Mater., 2017, 6, 18–25 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional XPS, visualizations of electric fields from theoretical calculations, and EIS data. See DOI: https://doi.org/10.1039/d3ta00417a |
| This journal is © The Royal Society of Chemistry 2023 |