
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect detection of molecular hydrogen upon p- and n-doping of organic semiconductors with complex oxidants or reductants†
Francesca
Pallini
 a,
Sara
Mattiello
a,
Norberto
Manfredi
a,
Sara
Mecca
a,
Alexey
Fedorov
b,
Mauro
Sassi
a,
Khaled
Al Kurdi
c,
Yi-Fan
Ding
d,
Chen-Kai
Pan
d,
Jian
Pei
d,
Stephen
Barlow
ce,
Seth R.
Marder
cef,
Thuc-Quyen
Nguyen
g and
Luca
Beverina
*a
a,
Sara
Mattiello
a,
Norberto
Manfredi
a,
Sara
Mecca
a,
Alexey
Fedorov
b,
Mauro
Sassi
a,
Khaled
Al Kurdi
c,
Yi-Fan
Ding
d,
Chen-Kai
Pan
d,
Jian
Pei
d,
Stephen
Barlow
ce,
Seth R.
Marder
cef,
Thuc-Quyen
Nguyen
g and
Luca
Beverina
*a
aDepartment of Materials Science, Università di Milano-Bicocca, via Cozzi 55, 20125 Milan, Italy. E-mail: luca.beverina@unimib.it
bCNR-IFN, L-NESS, via Anzani 42, I-22100 Como, Italy
cSchool of Chemistry and Biochemistry, Center for Organic Photonics and Electronics, Georgia Institute of Technology, Atlanta, GA 30332-0400, USA
dBeijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Polymer Chemistry and Physics of Ministry of Education, Center of Soft Matter Science and Engineering, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China
eRenewable and Sustainable Energy Institute, University of Colorado Boulder, Boulder, CO 80303, USA
fDepartment of Chemical and Biological Engineering, Department of Chemistry, Materials Science and Engineering Program, University of Colorado Boulder, Boulder, CO 80303, USA
gCenter for Polymers and Organic Solids, Department of Chemistry & Biochemistry, University of California, Santa Barbara, CA 93106-9510, USA
First published on 8th March 2023
Abstract
Molecular doping can increase the conductivity of organic semiconductors and plays an increasingly important role in emerging and established plastic electronics applications. 4-(1,3-Dimethyl-2,3-dihydro-1H-benzimidazol-2-yl)-N,N-dimethylaniline (N-DMBI-H) and tris(pentafluorophenyl)borane (BCF) are established n- and p-dopants, respectively, but neither functions as a simple one-electron redox agent. Molecular hydrogen has been suggested to be a byproduct in several proposed mechanisms for doping using both N-DMBI-H and BCF. In this paper we show for the first time the direct detection of molecular hydrogen in the uncatalysed doping of a variety of polymeric and molecular semiconductors using these dopants. Our results provide insight into the doping mechanism, providing information complementary to that obtained from more commonly applied methods such as optical, electron spin resonance, and electrical measurements.
Introduction
Plastic electronics offers low-power and low-cost solutions for display, lighting,1 and solar-power generation applications.2 Thanks to the flexibility and stretchability of organic compounds, applications in wearable electronics for both sensing and thermoelectric power generation are also possible.3 Moreover, most organic semiconductors are almost or completely non-toxic, thus also enabling biosensing and even the realisation of edible devices.4,5 Despite these desirable features, the charge-transport properties of state-of-the-art materials often fall short of the specification requirements for practical devices. Doping can improve the conductivity of organic semiconductors by orders-of-magnitude.6 The most straightforward doping scheme consists of a direct electron transfer (ET) reaction from the dopant to the semiconductor in the case of n-type doping or from the semiconductor to the dopant in the case of p-type doping. p-Type doping via ET is generally more easily carried due to the relatively high lying highest occupied molecular orbital (HOMO) levels, i.e., low ionisation energies (IEs), of many device-relevant conjugated polymers, such as poly-3-hexylthiophene (P3HT). For example, electron-poor molecules like the fluorinated tetracyanoquinodimethane derivative F4TCNQ can be handled in air and provide efficient ET p-type doping both in solution and in the solid state,7,8 although it is insufficiently oxidising to dope high-IE polymers. Conversely, the n-type doping of conjugated polymers is much more challenging, even when applied to electron-poor substrates with comparatively high reduction potentials and electron affinities (EAs).9 Suitable dopants should feature IEs lower than 3.8 eV, the energy more or less corresponding to the EA of established semiconductors with electron-transporting applications, such as PC60BM or poly{[N,N′-bis(2-octyldodecyl)-naphthalene-1,4,5,8-bis(dicarboximide)-2,6-diyl]-alt-5,5′-(2,2′-bithiophene)} (P(NDI2OD-T2)).10–14 Such low-IE dopants cannot generally be handled in air and require specific precautions.The development of dopants in which simple ET is precluded and the doping reaction requires bond cleavage and/or formation has had a significant impact over such scenarios; the barriers associated with these reactions can lead to kinetic stability sufficiently high to enable handling (although not necessarily storage)15 in air, sometimes even permitting processing in air with subsequent thermal or photochemical activation of the doping process. 4-(1,3-Dimethyl-2,3-dihydro-1H-benzimidazol-2-yl)-N,N-dimethylaniline, N-DMBI-H, is by far the most widely studied n-dopant of this type,13,16–18 while BCF can effectively p-dope donor–acceptor polymers such as poly[2,6-(4,4-bis-(2-ethylhexyl)-4H-cyclopenta[2,1-b;3,4-b′]dithiophene)-alt-4,7(2,1,3-benzothiadiazole)] (PCPDTBT),19,20 as well as relatively high IE materials such as indenopyrazine polymers.21 The mechanism by which stabilised n- and p-dopants react and the details of the various steps leading to the generation of the semiconductor radical ions (polarons) have been the subjects of much speculation and computation. While in some cases the mechanism is now clearly understood, a general and comprehensive rationalization is still lacking.22–26
As mentioned above, N-DMBI-H is a widely used stabilised n-dopant to dope materials such as PC60BM (EA ∼ 3.9 eV)27 and P(NDI2OD-T2) (EA ∼ 3.9–3.8 eV, after heating)28 forming N-DMBI+ despite an IE of ca. 4.4 eV,29 which leads to air stability, but also precludes a simple ET doping mechanism. Several alternative, less straightforward, mechanisms have been proposed for the doping reaction and computationally investigated.16,18,29–32 There is comparatively little experimental evidence supporting these studies, but, at least in some cases, kinetic data indicate that the initial step is a hydride transfer to form AH−, which, as suggested by calculations, may undergo an electron transfer with a neutral semiconductor, A, to form the desired radical anion A˙− and AH˙.30,33 Furthermore, in some cases, calculations suggest that the subsequent reaction of two neutral open-shell AH˙ species could potentially liberate H2 and regenerate neutral semiconductor A.30 Some other proposed mechanisms involve cleavage of the N-DMBI–H bond to form the highly reducing N-DMBI˙ and H˙, the latter again presumably forming H2.16,18,30,34
H2 generation has also been suggested in the p-doping of polymers with Lewis acids such as BCF, in analogy to previous work on the oxidation of metallocenes in non-aqueous solvents by BCF·H2O, where the proton acts as a stronger oxidant than when solvated by water.35 Specifically the first step, supported by comparison with other Brønsted acids, such as CF3CO2H, is assumed to be a protonation of neutral polymer P to form PH+.19,36 The proposed subsequent steps mirror those proposed for hydride-n-doped semiconductors described above, i.e., ET from P to PH+, thus forming P˙+ ad PH˙,19 with two PH˙ species subsequently reacting to form H2 and regenerate the starting polymer. Thus, both uncatalysed n- and p-doping using N-DMBI-H and BCF respectively have been proposed to involve H2 formation; however, in neither case is there any direct experimental evidence of H2 formation, although H2has been detected when the doping of a perylene diimide by N-DMBI-H is catalysed by gold nanoparticles.32
Here we describe for the first time the direct detection of H2 evolution in n- and p-doping experiments carried out on a variety of small molecules and polymers using N-DMBI-H34 and BCF precursors. We confirm previous observations that the doping process is strongly dependent on the semiconductor in question and the conditions. In the specific case of the solid-state doping of P(NDI2OD-T2) with N-DMBI-H we also further investigated reaction pathways using a deuterated dopant, N-DMBI-D.29 Our findings support previously suggested mechanisms of doping for a wide variety of different electron-poor and electron-rich semiconductors. In addition, they also offer circumstantial evidence that doping in the solid occurs at the interfaces between segregated dopants and the host material. This study opens the way for the use of H2 detection as a direct method to study the doping process independently from electrical measurements.
Results and discussion
We carried out the study using two different setups. The first one (H2-GC setup) is aimed at the detection of H2 as a function of sample composition and doping conditions. The setup is based on a gas chromatographic column composed of molecular sieves and a thermal conductivity detector (TCD) that produces a voltage signal in response to a change in thermal conductivity of the carrier gas (argon) due to elution of a gaseous analyte. Specifically, we introduced the host material and the dopant precursor under an Ar atmosphere in a GC-MS vial, sealed with a PTFE/silicone septum equipped with a crimp cap. We then heated the vial and sampled the atmosphere via a gas-tight volumetric syringe at fixed times for analysis using the GC instrument (calibrated for H2, N2 and O2 detection). We used the H2-GC setup for measurements involving all polymers and small molecules shown in Fig. 1, when in mixture with the appropriate dopant selected amongst N-DMBI-H, (N-DMBI)2 (a dopant related to N-DMBI-H that also reacts to form N-DMBI+ but which is not expected to form H2),34 and BCF.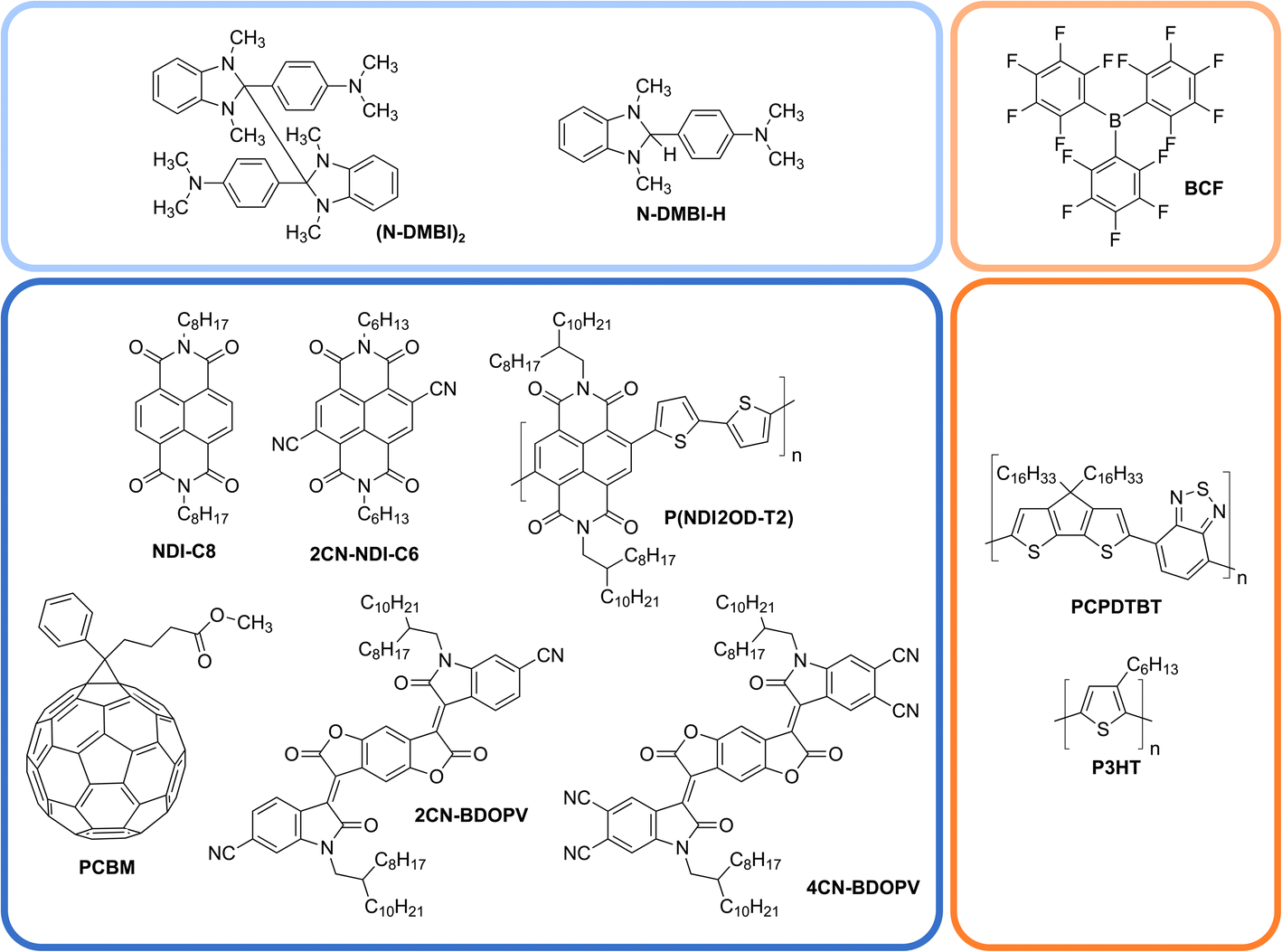 | ||
| Fig. 1 n-Dopable (bottom left) and p-dopable (bottom right) materials employed in this study and the corresponding n-dopants N-DMBI-H and (N-DMBI)2 and the p-dopant BCF. | ||
The second setup (D2-MS) is aimed at gaining insight into the specific mechanistic pathway leading to H2 evolution. This homemade setup is composed of a vacuum chamber equipped with a mass detector capable of discriminating gaseous species with low molecular weight and a remotely controlled lamp heater. It enables the simultaneous measurement of H2, D–H, and D2 levels in the chamber while heating the sample. This set up is only applicable for nonvolatile samples and we used it exclusively to characterize the solid-state doping of P(NDI2OD-T2), a process known to be efficient in the solid state when the sample is thermally annealed. For the purpose of the study, we synthesised the deuterated dopant N-DMBI-D.29
H2-GC setup for N-DMBI-H doping experiments
We selected the N-DMBI-H:P(NDI2OD-T2) blend as the reference system due to the vast literature dedicated to its electrical characterisation and the need for thermal activation.37 We sealed a mixture of powder sample of N-DMBI-H![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P(NDI2OD-T2) (in 2:1 molar ratio) in a GC-MS vial and subjected the sample to a 2 h thermal treatment at 150 °C in an oil bath. Such conditions ensure thorough mixing of the two powders (the melting point of N-DMBI-H is 110 °C)15 and correspond to the thermal treatment leading to the maximum increase in the conductivity of N-DMBI-H:P(NDI2OD-T2) blend thin films.15,37 We also performed control experiments using samples of pure N-DMBI-H and P(NDI2OD-T2) under the very same conditions. The direct detection of H2 from samples of N-DMBI-H/P(NDI2OD-T2) blends in the thin film form is not possible with our setup due to the very small amount of gas evolved. Fig. 2 shows the GC traces we obtained for the three samples.
:P(NDI2OD-T2) (in 2:1 molar ratio) in a GC-MS vial and subjected the sample to a 2 h thermal treatment at 150 °C in an oil bath. Such conditions ensure thorough mixing of the two powders (the melting point of N-DMBI-H is 110 °C)15 and correspond to the thermal treatment leading to the maximum increase in the conductivity of N-DMBI-H:P(NDI2OD-T2) blend thin films.15,37 We also performed control experiments using samples of pure N-DMBI-H and P(NDI2OD-T2) under the very same conditions. The direct detection of H2 from samples of N-DMBI-H/P(NDI2OD-T2) blends in the thin film form is not possible with our setup due to the very small amount of gas evolved. Fig. 2 shows the GC traces we obtained for the three samples.
 | ||
| Fig. 2 GC traces corresponding to the atmosphere of sealed vials containing: (A) N-DMBI-H:P(NDI2OD-T2) 2:1 blend; (B) pure N-DMBI-H; (C) pure P(NDI2OD-T2). Light blue arrows indicate peaks corresponding to H2, O2 and N2 in (A). H2 retention time is highlighted with a light blue dashed line in (B) and (C). | ||
The traces of the pure P(NDI2OD-T2) and N-DMBI-H samples only show two peaks at elution times of 0.88 and 1.57 min, corresponding to oxygen and nitrogen, respectively. Such air contamination most likely takes place in the syringe needle while transferring the sampled reaction atmosphere from the sealed vial to the GC instrument and thus after the doping experiment is over. There is no prior contamination of the reaction mixture with oxygen as the whole thermal treatment is performed in a sealed vial under an inert atmosphere.
The GC trace of the P(NDI2OD-T2):N-DMBI-H sample is dominated by an intense peak at 0.48 min attributed to the generation of H2. The integration of the calibrated trace enables an estimate of over 6000 ppm of H2 in the vial atmosphere, corresponding to a total production of 0.54 μmol of H2. The blend consists of 19 mg of polymer (19 μmol of repeat units) and 11 mg of N-DMBI-H (41 μmol). According to the mechanism shown in Fig. 3, the maximum theoretical amount of H2 produced if every repeating unit of the polymer were to react is 9.6 μmol.
 | ||
| Fig. 3 Possible doping mechanisms of n-dopable semiconductors with N-DMBI-H. After the first transfer step and subsequent reaction with a neutral acceptor (A), a radical AH˙ species forms, which might be capable of evolving hydrogen. | ||
The estimate is based on the observation that N-DMBI-H alone does not produce any hydrogen; thus, assuming that H2 is formed only through the reaction of two AH˙ species or the reaction of AH˙ and N-DMBI-H, the amount of hydrogen produced should be eighteen times greater than what was measured. There are two possible explanations for the discrepancy. First, there may be other parallel reaction pathways not involving hydrogen formation in addition to the two reactions above. We will discuss in more detail the fate of the AH˙ species while discussing the D2-MS experiment. Second, literature reports estimate the maximum doping level of P(NDI2OD-T2) to be around 20%, based on EPR38 and electrical measurements.39 In this case, the maximum expected amount of H2 evolved would be 1.9 μmol, a value much closer to the one we measured.
To further confirm the origin of the H2 evolution, we repeated the doping experiment under identical conditions, while using (N-DMBI)2 instead of N-DMBI-H as the dopant. As shown in Fig. 4, in this case we did not observe any H2 evolution. This is in complete agreement with the previous observation that (N-DMBI)2 behaves as a clean 2-electron reducing agent not involving hydrogen atom/ion transfer and/or abstraction reactions.40
 | ||
| Fig. 4 GC trace corresponding to the atmosphere of a sealed vial containing blends of (N-DMBI)2 and P(NDI2OD-T2). Light blue arrows point at retention times of H2, O2 and N2 species. | ||
We cross-checked the reliability of our data by performing the doping experiment on PC60BM/N-DMBI-H blends. The literature reports convincing evidence that in this case the reaction in solution proceeds via hydride transfer to PC60BM followed by ET from the reduced fullerene derivative (PC60BM-H−) to another PC60BM molecule, with formation of PC60BM˙− and PCBM-H˙. The further evolution of fullerene-H˙ to form multiply hydrogenated derivatives, rather than reacting with another fullerene-H˙ to give molecular hydrogen, has already been reported in the case of both C60 and PCBM.33,41 Consistent with these findings, Fig. 5, panel (i) shows that the thermal treatment of a PC60BM/N-DMBI-H solid blend does not lead to the formation of hydrogen in detectable amounts.
 | ||
| Fig. 5 GC traces corresponding to the atmosphere of sealed vials containing bends of N-DMBI-H with different acceptors: (i) PC60BM; (ii) 2CN-NDI-C6; (iii) NDI-C8; (iv) 4CN-BDOPV; (v) 2CN-BDOPV. | ||
Aside from PC60BM and P(NDI2OD-T2), we also considered other readily n-doped electron-transporting molecular semiconductors: the two extended isoindigo derivatives 2CN-BDOPV and 4CN-BDOPV and the naphthalenediimide derivatives NDI-C8 and 2CN-NDI-C6 (Fig. 1). A detailed investigation of the doping mechanism of such derivatives has already been reported.30 The molecular acceptor 2CN-BDOPV is known to be an efficient hydride acceptor but at least under some reaction conditions, notably excess dopant, [2CN-BDOPV-H]− is stable in solution to further reaction and can be directly observed by NMR.27 Under our experimental conditions, both 2CN-BDOPV and its close analogue 4CN-BDOPV produced only traces amounts of H2 (Fig. 4B, panels (v) and (iv), respectively), consistent with these previous findings. Based on such a result, it is possible that both 2CN-BDOPV-H˙ and 4CN-BDOPV-H˙ further evolve to multiply hydrogenated species, just like PC60BM does, or that the further reaction of [xCN-BDOPV-H]− with the corresponding undoped species is sufficiently slow that the hydride addition dominates the process and no neutral acceptor to be doped remains. To further characterise the mechanism of this class of acceptors, we performed a second experiment on 2CN-BDOPV using substoichiometric amounts of N-DMBI-H (see ESI, Fig. S2†). GC traces related to this experiment show no detection of H2, corroborating the hypothesis that either the doping process of this derivative does not happen prevalently via H2 formation in the solid state or, if the reaction between [2CN-BDOPV-H]− and the corresponding undoped 2CN-BDOPV happens, it is very slow under such experimental conditions.
Some of us have previously investigated the doping mechanism of 2CN-NDI-C6 and N-DMBI-H in solution.30 UV-vis-NIR data show a slow transformation from A to A˙−, but in contrast to the case of 2CN-BDOPV, there is no evidence of any detectable intermediates such as AH−. However, an AH− or AH˙ intermediate is implicated by a primary kinetic isotope effect and by incorporation of deuterium into the product when N-DMBI-D is used; this suggests that the rate at which any such intermediate reacts further is likely much faster than its rate of formation. In the present solid-state study we did observe H2 formation, but in an amount (108 ppm, 9.7 nmol) almost two orders-of-magnitude lower than that expected for stoichiometric reaction with the dopant. It is possible that the hydride-transfer mechanism remains dominant in the solid state but that in this case the neutral radical hydrogenated species does not exclusively evolve via multiple hydride transfer like in the case of PC60BM and CN-BDOPV but also with formation of molecular hydrogen.
Finally, the behavior of the naphthalenediimide NDI-C8 is intermediate between P(NDI2OD-T2) and all other acceptors. The blend with N-DMBI-H gave H2 evolution (1620 ppm, 146 nmol) on the same order of magnitude of the one we observed with P(NDI2OD-T2). In the overall, the dataset agrees with the previously observed trends, thus confirming that hydrogen measurements could usefully complement other characterisation tools such as electrical measurements and EPR data.30 In summary, the hydrogen detection experiments are consistent with previous suggestions that, in terms of the dominant doping mechanism and reaction kinetics, the semiconductor hydride affinity is more relevant than its electron affinity; i.e., the LUMO levels of the different targets are not the determining factor.
D2-MS setup for DMBI-D doping experiments
To gain insight into the fate of the hydrogen atom in N-DMBI-H doping of P(NDI2OD-T2), we synthesised the deuterated dopant N-DMBI-D and repeated the doping experiment using P(NDI2OD-T2) while working in a vacuum chamber equipped with a low mass detector capable of discriminating H2, H–D, and D2. We focused the attention on N-DMBI-D:P(NDI2OD-T2) because this polymer is routinely doped with N-DMBI-H in the literature and our H2 detection experiments demonstrated that this reaction involves the formation of the largest amounts of H2.Fig. 6 shows the evolution in time of the partial pressure difference for the H2 (black, dotted line), H–D (blue, dashed line) and D2 (light blue line) species with respect to the baseline, while heating the sample at a constant rate of 10 °C min−1 from 50 °C to 160 °C, and holding it at the latter temperature. The gas evolution starts around 90 °C, peaks at 120 °C and is complete at 160 °C. The traces show that all the possible species are formed, with a predominance of H–D over D2. It is also apparent that the gas evolution occurs in two steps: one (major) at 120 °C and the other at 135 °C. The relative intensity of the second peak is comparable for H2, weaker for H–D and barely noticeable for D2.
 | ||
| Fig. 6 Selective detection of D2 (light blue line), H–D (blue, dashed line), and H2 (black, dotted line) in a DMBI-D:P(NDI2OD-T2) blend as a function of time while heating at the constant rate of 10 °C min−1. | ||
The two distinct processes could be related to interfacial reaction between segregated domains, followed by thermally activated diffusion leading to further hydrogen evolution within the polymer phase.15 This hypothesis would also explain why of all the gas evolution traces, the one related to D2 evolution shows only a barely visible shoulder at 135 °C. The D2 formation reaction is much more likely to happen close to the dopant/acceptor interface where the concentration of D is higher thus increasing the probability that either of the possible hydrogen-generating steps depicted in Fig. 3 results in D2 formation (2AD˙ → 2A + D2 and AD˙ + N-DMBI-D → A˙− + N-DMBI+ + D2). Conversely, we speculate that the aforementioned thermally promoted D/H diffusion leading to further doping involves either H˙ or D˙ transfer between closely spaced different A sites as depicted in Fig. 7. Such a mechanism would imply the concentration of D would steeply decay according to a power low of the number of steps required for diffusion from D rich regions (located close to the N-DMBI-D/A phase boundary) to H rich ones (bulk of A phase). In this framework, the observation of a still detectable amount of D (H–D or D2) from such a process still pinpoints it in proximity of the phase boundary.
 | ||
| Fig. 7 Graphical representation of reaction and diffusion mechanisms leading to D2, H–D and H2 formation after the first doping step of A with N-DMBI-D. Pentagons represent N-DMBI derivatives, and circles represent A derivatives. Light blue color indicates the presence of H atoms only, an orange color indicates the presence of a D atom on the species, and a purple contour indicates the presence of a radical. | ||
In terms of discriminating between the different doping mechanisms, the observation that all species are formed requires at least some deuterium transfer to the acceptor. Once the AD˙ species are formed, the elimination of both H–D (mostly) and H2 (minor) involving hydrogenated neighboring sites becomes possible and is indeed observed. We note that if in the specific AD˙ species formed, the D is bound to a carbon that also bears a H atom (such as a naphthalene CH position), then a kinetic isotope effect will in fact favor the loss of the H over D in the subsequent reaction of 2AD˙ moieties or of AD˙ with N-DMBI-H. Our findings offer circumstantial evidence that the solid-state doping is an interfacial phenomenon involving the interphases between segregated dopants and the host material.
H2-GC setup for BCF doping experiments
The p-doping mechanism that has been proposed for polymers such as PCPDTBT using Lewis acids such as BCF has some similarities with the doping cascade involving hydride transfer in n-doping some electron-transporting polymers. The process is activated by the BCF·H2O complex, acting as a Brønsted acid in the protonation of the target polymer. The consequent local lowering of the LUMO of the polymer in solution makes it possible for a non-protonated polymer segment present in solution to act as an ET donor, with the formation of a radical cationic chain (the p-doped polymer) and a neutral radical one. As in the case of the n-type AH˙ species we discussed in the n-doping context, the fate of the PCPDTBT-H˙ chain can either be hydrogen evolution (by reaction of two PCPDTBT-H˙ chains) or multiple hydrogenations. Fig. 8A shows a schematic of the process.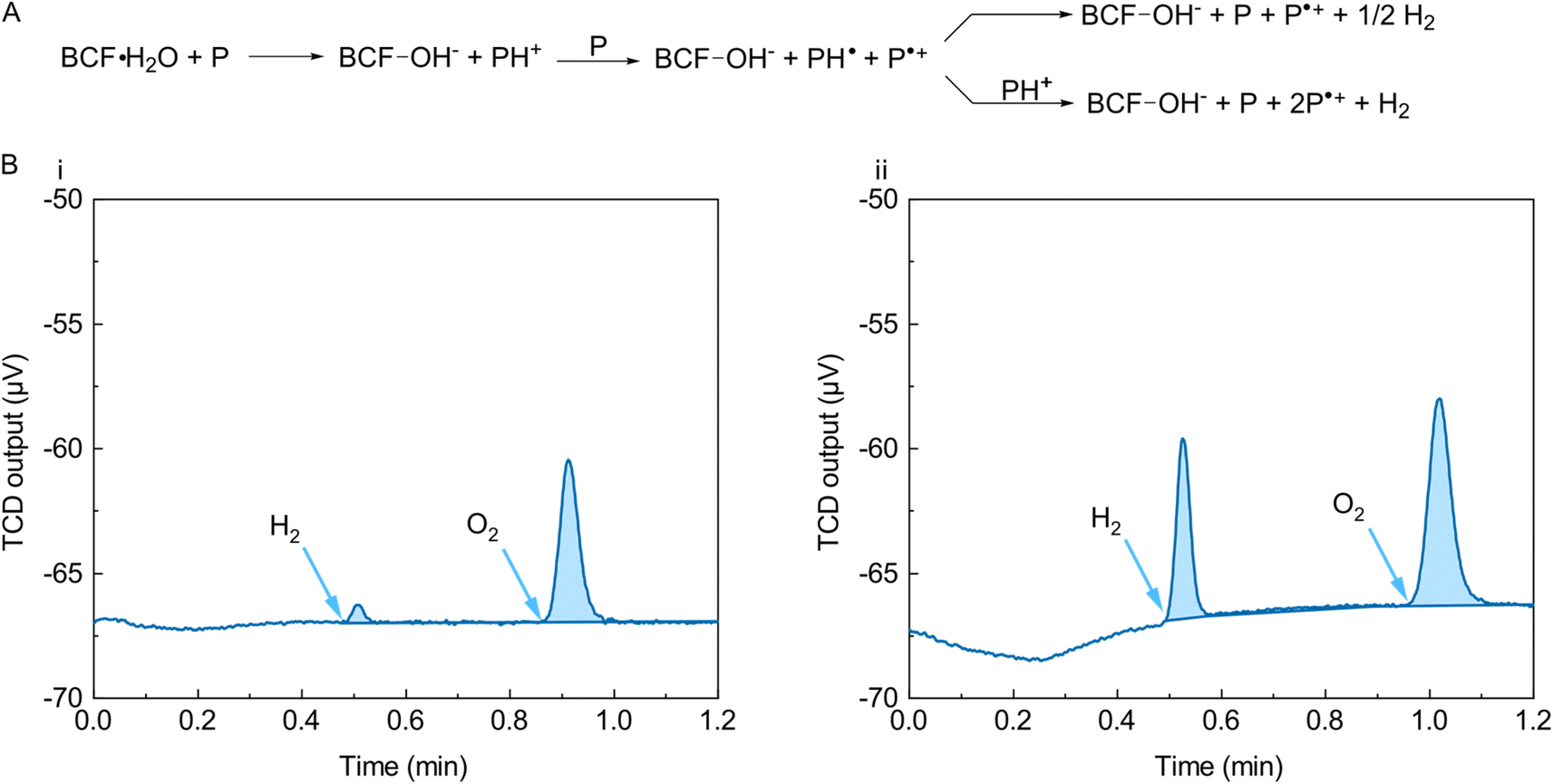 | ||
| Fig. 8 (A) Doping mechanism of a p-dopable polymer semiconductor (P) with BCF. After the first protonation step and subsequent reaction with a neutral polymer, a radical PH˙ species forms, which might be capable of evolving hydrogen. Note: DFT calculations suggest that the overall reaction is rendered more energetically favorable when an additional BCF or BCF and water molecules are involved leading to the formation of [BCF(OH)BCF]- or [BCF(OH)(OH2)BCF]- counterions;36 however, this is not shown here for simplicitity. (B) Bottom: H2 evolution experiments for the reaction of PCPDTBT with BCF in anhydrous (i) and H2O-saturated (ii) chlorobenzene solution in the H2-GC setup. | ||
We used the H2-GC setup on BCF:PCPDTBT solutions in chlorobenzene to measure if hydrogen was a significant side product of the doping cascade and if the presence of water had an impact on the doping efficiency. We decided to work in solution rather than with a solid blend of polymer and BCF in order to closely mimic the conditions some of us previously employed to study the process by NMR, EPR and UV-Vis.19,36 We did try to perform the experiment directly on the dry blend of polymer and BCF, but we observed neither a change in the colour of the sample, nor hydrogen evolution. One might speculate that this doping mechanisms requires the presence of a solvent due to the need for an equilibrium protonation of the polymer chain as the rate limiting step.36
Fig. 8B shows that we observed sizable H2 evolution only when the chlorobenzene we used for the reaction was saturated with water. This is in agreement with the electrical and UV-Vis characterization data previously published.19,36 The observation of H2 formation helps support the proposed p-doping mechanism of conjugated polymers by BCF. We obtained a similar result when performing an identical experiment on P3HT, suggesting a possible generality of the phenomenon.
Fig. 9 shows the time dependence of H2 generation for P3HT vs. PCPDTBT samples measured at the same polymer concentration with a 1:1 and 1:2 repeating unit:BCF ratio. The process requires over 48 h to be complete, in agreement with spectroscopical data previously reported. The H2 generation shows a similar time dependence for the two polymers and is slightly more efficient for the P3HT samples. There is also a clear dependency on the BCF stoichiometry, also in agreement with previous observations. It should be noted, however, that while the trends shown in Fig. 8 are reliable, they do not represent a quantitative measurement. In fact, we had to perform multiple injections on the same samples, each time a 250 μL volume, i.e., ¼ of the gas volume in the GC vial. The consequent loss in internal pressure as well as the inevitable leakages connected with each sampling leads to systematic error in the measurements. We are currently working on a different setup enabling the direct measurement of the H2 evolved without resorting to sampling with syringes.
 | ||
| Fig. 9 H2 detection as a function of time in samples containing 9 × 10−3 M PCPDTBT (light blue), and 9 × 10−3 M P3HT (blue) with 1 (squares) and 2 (circles) molar equivalents of BCF dopant. | ||
Conclusions
We have shown that H2 is formed during examples of both n- and p-doping reactions of organic semiconductors, specifically both when using N-DMBI-H as an n-dopant for a variety of electron-transporting polymers and small molecules and BCF as a p-dopant for hole-transporting conjugated polymers. These findings are consistent with previously hypothesised doping mechanisms but confirm the formation of H2 as a side product for the first time. Moreover, we show that additional insights may be gained from the use of an isotopically labeled dopant in conjunction with mass-selective detection, which provides evidence that, in the case of N-DMBI-D and P(NDI2OD-T2), deuteride or deuterium is transferred to the polymer. This study opens the way for the use of H2 detection as a direct method for investigating doping mechanisms and, in favorable cases, to study the doping process, including the formation of byproducts, independently from electrical measurements; it is anticipated to be particularly powerful when used in conjunction with spectroscopic detection of semiconductor radical-ion formation and electrical measurements.Author contributions
Francesca Pallini: investigation and methodology. Performed all doping experiments.Sara Mattiello: writing – review and editing.
Norberto Manfredi: methodology. Set up for hydrogen detection.
Sara Mecca: investigation. Synthesis of dopants.
Alexey Fedorov: methodology. Set up for the hydrogen vs. deuterium detection.
Mauro Sassi: conceptualization. Discussion of the possible routes leading to the formation of hydrogen.
Khaled Al Kurdi: investigation. Synthesis of some molecular n-dopable materials.
Yi-Fan Ding: Investigation. Synthesis of some molecular n-dopable materials.
Chen-Kai Pan: investigation. Synthesis of some molecular n-dopable materials.
Jian Pei: supervision of the synthesis of some molecular n-dopable materials.
Stephen Barlow: conceptualization and writing – review & editing.
Seth R. Marder: writing – review & editing.
Thuc-Quyen Nguyen: conceptualization and writing – review & editing.
Luca Beverina: conceptualization and writing of the original draft.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Italian Ministry of University (MUR, Grant Dipartimenti di Eccellenza-2017 “Materials for Energy” and Grant PRIN-2017 BOOSTER 2017YXX8AZ) and the National Science Foundation (through DMR-1807797/2216857 and through the DMREF program DMR-1729737). T.-Q. N. acknowledges the support from the US Department of Energy under Award no. DE-SC0017659.References
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, All-Organic Thermally Activated Delayed Fluorescence Materials for Organic Light-Emitting Diodes, Nat. Rev. Mater., 2018, 3(4), 18020, DOI:10.1038/natrevmats.2018.20.
- P. Bi, S. Zhang, Z. Chen, Y. Xu, Y. Cui, T. Zhang, J. Ren, J. Qin, L. Hong, X. Hao and J. Hou, Reduced Non-Radiative Charge Recombination Enables Organic Photovoltaic Cell Approaching 19% Efficiency, Joule, 2021, 4, 1988–2003, DOI:10.1016/j.joule.2021.06.020.
- U. Ail, M. J. Jafari, H. Wang, T. Ederth, M. Berggren and X. Crispin, Thermoelectric Properties of Polymeric Mixed Conductors, Adv. Funct. Mater., 2016, 26(34), 6288–6296, DOI:10.1002/adfm.201601106.
- A. S. Sharova, F. Melloni, G. Lanzani, C. J. Bettinger and M. Caironi, Edible Electronics: The Vision and the Challenge, Adv. Mater. Technol., 2021, 6(2), 2000757, DOI:10.1002/admt.202000757.
- G. E. Bonacchini, C. Bossio, F. Greco, V. Mattoli, Y.-H. Kim, G. Lanzani and M. Caironi, Tattoo-Paper Transfer as a Versatile Platform for All-Printed Organic Edible Electronics, Adv. Mater., 2018, 30(14), 1706091, DOI:10.1002/adma.201706091.
- H. Hase and I. Salzmann, 11 - Doping in Organic Semiconductors, in Handbook of Organic Materials for Electronic and Photonic Devices, ed. O. Ostroverkhova, Woodhead Publishing Series in Electronic and Optical Materials, Woodhead Publishing, 2nd edn, 2019, pp. 349–383, DOI:10.1016/B978-0-08-102284-9.00011-5.
- P. Pingel and D. Neher, Comprehensive Picture of p-Type Doping of P3HT with the Molecular Acceptor F4TCNQ, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87(11), 115209, DOI:10.1103/PhysRevB.87.115209.
- J. Li, G. Zhang, D. M. Holm, I. E. Jacobs, B. Yin, P. Stroeve, M. Mascal and A. J. Moulé, Introducing Solubility Control for Improved Organic P-Type Dopants, Chem. Mater., 2015, 27(16), 5765–5774, DOI:10.1021/acs.chemmater.5b02340.
- Y. Lu, J.-Y. Wang and J. Pei, Achieving Efficient N-Doping of Conjugated Polymers by Molecular Dopants, Acc. Chem. Res., 2021, 54(13), 2871–2883, DOI:10.1021/acs.accounts.1c00223.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, N-Type Organic Semiconductors in Organic Electronics, Adv. Mater., 2010, 22(34), 3876–3892, DOI:10.1002/adma.200903628.
- J. Liu, L. Qiu, R. Alessandri, X. Qiu, G. Portale, J. Dong, W. Talsma, G. Ye, A. A. Sengrian, P. C. T. Souza, M. A. Loi, R. C. Chiechi, S. J. Marrink, J. C. Hummelen and L. J. A. Koster, Enhancing Molecular N-Type Doping of Donor-Acceptor Copolymers by Tailoring Side Chains, Adv. Mater., 2018, 30(7), 1704630, DOI:10.1002/adma.201704630.
- A. Werner, F. Li, K. Harada, M. Pfeiffer, T. Fritz, K. Leo and S. Machill, N-Type Doping of Organic Thin Films Using Cationic Dyes, Adv. Funct. Mater., 2004, 14(3), 255–260, DOI:10.1002/adfm.200305053.
- V. A. Kolesov, C. Fuentes-Hernandez, W.-F. Chou, N. Aizawa, F. A. Larrain, M. Wang, A. Perrotta, S. Choi, S. Graham, G. C. Bazan, T.-Q. Nguyen, S. R. Marder and B. Kippelen, Solution-Based Electrical Doping of Semiconducting Polymer Films over a Limited Depth, Nat. Mater., 2017, 16(4), 474–480, DOI:10.1038/nmat4818.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dötz, M. Kastler and A. Facchetti, A High-Mobility Electron-Transporting Polymer for Printed Transistors, Nature, 2009, 457(7230), 679–686, DOI:10.1038/nature07727.
- F. Pallini, S. Mattiello, M. Cassinelli, P. Rossi, S. Mecca, W. L. Tan, M. Sassi, G. Lanzani, C. R. McNeill, M. Caironi and L. Beverina, Unexpected Enhancement of Molecular N-Doping Efficiency in Polymer Thin Films by a Degradation Product, ACS Appl. Energy Mater., 2022, 5(2), 2421–2429, DOI:10.1021/acsaem.1c03893.
- Y. Zeng, W. Zheng, Y. Guo, G. Han and Y. Yi, Doping Mechanisms of N-DMBI-H for Organic Thermoelectrics: Hydrogen Removal vs. Hydride Transfer, J. Mater. Chem. A, 2020, 8(17), 8323–8328, 10.1039/D0TA01087A.
- B. Saglio, M. Mura, M. Massetti, F. Scuratti, D. Beretta, X. Jiao, C. R. McNeill, M. Sommer, A. Famulari, G. Lanzani, M. Caironi and C. Bertarelli, N-Alkyl Substituted 1H-Benzimidazoles as Improved n-Type Dopants for a Naphthalene-Diimide Based Copolymer, J. Mater. Chem. A, 2018, 6(31), 15294–15302, 10.1039/C8TA04901G.
- B. D. Naab, S. Zhang, K. Vandewal, A. Salleo, S. Barlow, S. R. Marder and Z. Bao, Effective Solution- and Vacuum-Processed n-Doping by Dimers of Benzimidazoline Radicals, Adv. Mater., 2014, 26(25), 4268–4272, DOI:10.1002/adma.201400668.
- B. Yurash, D. X. Cao, V. V. Brus, D. Leifert, M. Wang, A. Dixon, M. Seifrid, A. E. Mansour, D. Lungwitz, T. Liu, P. J. Santiago, K. R. Graham, N. Koch, G. C. Bazan and T.-Q. Nguyen, Towards Understanding the Doping Mechanism of Organic Semiconductors by Lewis Acids, Nat. Mater., 2019, 18(12), 1327–1334, DOI:10.1038/s41563-019-0479-0.
- R. Schier, A. M. Valencia and C. Cocchi, Microscopic Insight into the Electronic Structure of BCF-Doped Oligothiophenes from Ab Initio Many-Body Theory, J. Phys. Chem. C, 2020, 124(26), 14363–14370, DOI:10.1021/acs.jpcc.0c03124.
- Y. Han, G. Barnes, Y.-H. Lin, J. Martin, M. Al-Hashimi, S. Y. AlQaradawi, T. D. Anthopoulos and M. Heeney, Doping of Large Ionization Potential Indenopyrazine Polymers via Lewis Acid Complexation with Tris(Pentafluorophenyl)Borane: A Simple Method for Improving the Performance of Organic Thin-Film Transistors, Chem. Mater., 2016, 28(21), 8016–8024, DOI:10.1021/acs.chemmater.6b03761.
- J.-M. Kim, S.-J. Yoo, C.-K. Moon, B. Sim, J.-H. Lee, H. Lim, J. W. Kim and J.-J. Kim, N-Type Molecular Doping in Organic Semiconductors: Formation and Dissociation Efficiencies of a Charge Transfer Complex, J. Phys. Chem. C, 2016, 120(17), 9475–9481, DOI:10.1021/acs.jpcc.6b01175.
- Y.-F. Ding, C.-Y. Yang, C.-X. Huang, Y. Lu, Z.-F. Yao, C.-K. Pan, J.-Y. Wang and J. Pei, Thermally Activated N-Doping of Organic Semiconductors Achieved by N-Heterocyclic Carbene Based Dopant, Angew. Chem., 2021, 133(11), 5880–5884, DOI:10.1002/ange.202011537.
- O. Bardagot, C. Aumaître, A. Monmagnon, J. Pécaut, P.-A. Bayle and R. Demadrille, Revisiting Doping Mechanisms of N-Type Organic Materials with N-DMBI for Thermoelectric Applications: Photo-Activation, Thermal Activation, and Air Stability, Appl. Phys. Lett., 2021, 118(20), 203904, DOI:10.1063/5.0047637.
- Y. Lu, Z.-D. Yu, Y. Liu, Y.-F. Ding, C.-Y. Yang, Z.-F. Yao, Z.-Y. Wang, H.-Y. You, X.-F. Cheng, B. Tang, J.-Y. Wang and J. Pei, The Critical Role of Dopant Cations in Electrical Conductivity and Thermoelectric Performance of N-Doped Polymers, J. Am. Chem. Soc., 2020, 142(36), 15340–15348, DOI:10.1021/jacs.0c05699.
- S. Guo, S. K. Mohapatra, A. Romanov, T. V. Timofeeva, K. I. Hardcastle, K. Yesudas, C. Risko, J.-L. Brédas, S. R. Marder and S. Barlow, N-Doping of Organic Electronic Materials Using Air-Stable Organometallics: A Mechanistic Study of Reduction by Dimeric Sandwich Compounds, Chem.–Eur. J., 2012, 18(46), 14760–14772, DOI:10.1002/chem.201202591.
- Z.-L. Guan, J. B. Kim, H. Wang, C. Jaye, D. A. Fischer, Y.-L. Loo and A. Kahn, Direct Determination of the Electronic Structure of the Poly(3-Hexylthiophene):Phenyl-[6,6]-C61 Butyric Acid Methyl Ester Blend, Org. Electron., 2010, 11(11), 1779–1785, DOI:10.1016/j.orgel.2010.07.023.
- S. Guo, S. B. Kim, S. K. Mohapatra, Y. Qi, T. Sajoto, A. Kahn, S. R. Marder and S. Barlow, N-Doping of Organic Electronic Materials Using Air-Stable Organometallics, Adv. Mater., 2012, 24(5), 699–703, DOI:10.1002/adma.201103238.
- B. D. Naab, S. Guo, S. Olthof, E. G. B. Evans, P. Wei, G. L. Millhauser, A. Kahn, S. Barlow, S. R. Marder and Z. Bao, Mechanistic Study on the Solution-Phase n-Doping of 1,3-Dimethyl-2-Aryl-2,3-Dihydro-1H-Benzoimidazole Derivatives, J. Am. Chem. Soc., 2013, 135(40), 15018–15025, DOI:10.1021/ja403906d.
- S. Jhulki, H.-I. Un, Y.-F. Ding, C. Risko, S. K. Mohapatra, J. Pei, S. Barlow and S. R. Marder, Reactivity of an Air-Stable Dihydrobenzoimidazole n-Dopant with Organic Semiconductor Molecules, Chem, 2021, 7(4), 1050–1065, DOI:10.1016/j.chempr.2021.01.020.
- R. A. Schlitz, F. G. Brunetti, A. M. Glaudell, P. L. Miller, M. A. Brady, C. J. Takacs, C. J. Hawker and M. L. Chabinyc, Solubility-Limited Extrinsic n-Type Doping of a High Electron Mobility Polymer for Thermoelectric Applications, Adv. Mater., 2014, 26(18), 2825–2830, DOI:10.1002/adma.201304866.
- H. Guo, C.-Y. Yang, X. Zhang, A. Motta, K. Feng, Y. Xia, Y. Shi, Z. Wu, K. Yang, J. Chen, Q. Liao, Y. Tang, H. Sun, H. Y. Woo, S. Fabiano, A. Facchetti and X. Guo, Transition Metal-Catalysed Molecular n-Doping of Organic Semiconductors, Nature, 2021, 599(7883), 67–73, DOI:10.1038/s41586-021-03942-0.
- B. D. Naab, S. Guo, S. Olthof, E. G. B. Evans, P. Wei, G. L. Millhauser, A. Kahn, S. Barlow, S. R. Marder and Z. Bao, Mechanistic Study on the Solution-Phase n-Doping of 1,3-Dimethyl-2-Aryl-2,3-Dihydro-1H-Benzoimidazole Derivatives, J. Am. Chem. Soc., 2013, 135(40), 15018–15025, DOI:10.1021/ja403906d.
- H.-I. Un, S. A. Gregory, S. K. Mohapatra, M. Xiong, E. Longhi, Y. Lu, S. Rigin, S. Jhulki, C.-Y. Yang, T. V. Timofeeva, J.-Y. Wang, S. K. Yee, S. Barlow, S. R. Marder and J. Pei, Understanding the Effects of Molecular Dopant on N-Type Organic Thermoelectric Properties, Adv. Energy Mater., 2019, 9(24), 1900817, DOI:10.1002/aenm.201900817.
- L. H. Doerrer and M. L. H. Green, Oxidation of [M(η-C5H5)2], M = Cr, Fe or Co, by the New Brønsted Acid H2O·B(C6F5)3 Yielding the Salts [M(η-C5H5)2]+A−, Where A− = [(C6F5)3B(μ-OH)B(C6F5)3]− or [(C6F5)3BOH·H2OB(C6F5)3]−, J. Chem. Soc., Dalton Trans., 1999, 24, 4325–4329, 10.1039/A905892C.
- P. S. Marqués, G. Londi, B. Yurash, T.-Q. Nguyen, S. Barlow, S. R. Marder and D. Beljonne, Understanding How Lewis Acids Dope Organic Semiconductors: A “Complex” Story, Chem. Sci., 2021, 12(20), 7012–7022, 10.1039/D1SC01268A.
- M. Cassinelli, S. Cimò, T. Biskup, X. Jiao, A. Luzio, C. R. McNeill, Y.-Y. Noh, Y.-H. Kim, C. Bertarelli and M. Caironi, Enhanced N-Type Doping of a Naphthalene Diimide Based Copolymer by Modification of the Donor Unit, Adv. Electron. Mater., 2021, 7(12), 2100407, DOI:10.1002/aelm.202100407.
- D. Kiefer, A. Giovannitti, H. Sun, T. Biskup, A. Hofmann, M. Koopmans, C. Cendra, S. Weber, L. J. Anton Koster, E. Olsson, J. Rivnay, S. Fabiano, I. McCulloch and C. Müller, Enhanced N-Doping Efficiency of a Naphthalenediimide-Based Copolymer through Polar Side Chains for Organic Thermoelectrics, ACS Energy Lett., 2018, 3(2), 278–285, DOI:10.1021/acsenergylett.7b01146.
- R. A. Schlitz, F. G. Brunetti, A. M. Glaudell, P. L. Miller, M. A. Brady, C. J. Takacs, C. J. Hawker and M. L. Chabinyc, Solubility-Limited Extrinsic n-Type Doping of a High Electron Mobility Polymer for Thermoelectric Applications, Adv. Mater., 2014, 26(18), 2825–2830, DOI:10.1002/adma.201304866.
- S. Zhang, B. D. Naab, E. V. Jucov, S. Parkin, E. G. B. Evans, G. L. Millhauser, T. V. Timofeeva, C. Risko, J.-L. Brédas, Z. Bao, S. Barlow and S. R. Marder, N-Dopants Based on Dimers of Benzimidazoline Radicals: Structures and Mechanism of Redox Reactions, Chem.–Eur. J., 2015, 21(30), 10878–10885, DOI:10.1002/chem.201500611.
- F. Li, A. Werner, M. Pfeiffer, K. Leo and X. Liu, Leuco Crystal Violet as a Dopant for N-Doping of Organic Thin Films of Fullerene C60, J. Phys. Chem. B, 2004, 108(44), 17076–17082, DOI:10.1021/jp0478615.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta00231d |
| This journal is © The Royal Society of Chemistry 2023 |