
Design and synthesis of a covalent organic framework bridging CdS nanoparticles and a homogeneous cobalt–bipyridine cocatalyst for a highly efficient photocatalytic CO2 reduction†
Khai H.
Do
 ,
D. Praveen
Kumar
,
A. Putta
Rangappa
,
Jehee
Lee
,
Sungin
Yun
and
Tae Kyu
Kim
*
,
D. Praveen
Kumar
,
A. Putta
Rangappa
,
Jehee
Lee
,
Sungin
Yun
and
Tae Kyu
Kim
*
Department of Chemistry, Yonsei University, Seoul 03722, Republic of Korea. E-mail: tkkim@yonsei.ac.kr
First published on 20th March 2023
Abstract
The design and construction of highly efficient photocatalytic CO2 conversion systems are extremely desirable for technological, practical, and economic viability. In this study, a 2,2′-bipyridine (bpy)-based ketoenamine covalent organic framework (TpBpy; Tp: 1,3,5-triformylphloroglucinol), which can be prepared on a large scale by a facile and environmentally friendly hydrothermal method, was used to promote CO2 photoreduction processes by bridging heterogeneous CdS nanoparticles and a homogeneous [Co(bpy)3]2+ cocatalyst. The bpy units played multiple roles in the preparation of TpBpy, formation of strong interactions with CdS, and accommodation of the cocatalyst. In the CO2 reduction process, due to the flexible association/dissociation between the bpy ligand and the cocatalyst, the active [Co(bpy)x]+ may separate from heterogeneous CdS/TpBpy and make spaces for other unactive [Co(bpy)3]2+ species, thereby maintaining the intrinsic high activity and selectivity of the [Co(bpy)3]2+ cocatalyst. The combination of CdS, TpBpy, and [Co(bpy)3]2+ shows a strong solar light harvesting ability, a high surface area, a high CO2 adsorption capacity, highly efficient charge carrier transfer at the interface between CdS and TpBpy, and subsequent rapid photoelectron injection into the [Co(bpy)3]2+ cocatalyst. These synergistic effects lead to a robust CO production rate of 35.2 mmol g−1 with 85.0% selectivity over the first four hours of the reaction. Moreover, the quantum efficiencies (AQEs) of the reaction system, with 2 mg of CdS/TpBpy-20%, are 4.75 and 3.65% at 400 and 450 nm, respectively. Finally, the possible mechanism of the photocatalytic CO2 conversion over CdS/TpBpy is proposed and discussed here. This study on the heterostructure and photocatalytic system design might serve as a model for the development of solar-driven CO2 reduction.
1. Introduction
The depletion of fossil fuels and high anthropogenic greenhouse gas levels are the two fundamental global environmental issues.1,2 Therefore, sustained efforts have been devoted to developing sustainable energy conversion and storage alternatives, which can meet the increasing energy demand and reduce emissions into the environment.3–10 In light of the above-mentioned problems and requirements, conversion of CO2 to carbon-based fuels, using various catalytic transformation approaches, is considered a promising strategy.11–14 To this end, the solar-driven reduction of CO2 has garnered significant attention because solar energy and water are abundant and the reaction conditions are mild.15–19 Despite the numerous available reports on CO2 photoreduction using different semiconductor materials, the development of highly efficient, selective, and durable catalytic systems is still a challenging task.Polypyridine-based molecular cocatalysts containing metals such as Co, Ni, Re, and Fe attract considerable research interest, because they can effectively and selectively convert CO2 to CO.20–27 However, the complicated synthesis processes, instability of molecular catalysts during prolonged reactions, and problematic cycling tests are the major drawbacks of using such cocatalysts. Recently, the heterogenization of photocatalytic molecules, using supports such as metal–organic frameworks (MOFs),28,29 covalent organic frameworks (COFs),30,31 metal–organic polyhedra (MOPs),32 and so on,33 has been expected to increase the stability of cocatalysts owing to confinement effects. However, it has been reported that metal leaching occurs during the catalytic reaction and recycling procedures, resulting in a decrease in efficiency and selectivity.33–35 Furthermore, immobilization of homogeneous molecular complexes on/in bulky support materials reduced the flexibility and accessibility of the complexes, thereby lowering their catalytic activities.32 As a result, a new strategy is urgently required to simplify photocatalytic systems without degrading their intrinsic high activity and selectivity.
Considering unavoidable time-consuming multistep synthesis, purification process and degradation of these complexes, in situ assembly of earth-abundant metal complexes is a promising technique, wherein significant progress has been made to date.36–39 The in situ formation of [Co(bpy)3]2+ (bpy: 2,2′-bipyridine) for heterogeneous CO2 photoreduction has attracted substantial attention, because [Co(bpy)3]2+ can effectively and selectively convert CO2 to CO.40,41 Nevertheless, the interaction between photocatalysts and homogeneous [Co(bpy)3]2+ is still rarely evaluated and reported.42 Using porous materials containing bpy moieties as “bridges” enables killing two birds with one stone: first, bpy moieties can strongly interact with [Co(bpy)3]2+via coordinate bonds,30 thereby easing the electron transfer to the cocatalyst, while active [Co(bpy)x]+ separates from heterogeneous photocatalysts and makes spaces for other unactive [Co(bpy)3]2+ species,43,44 maintaining intrinsic high activity and selectivity of [Co(bpy)3]2+ solution; second, the incorporation of these materials with other semiconductors can promote charge carrier separation, suppress charge recombination, and improve solar light utilization.45 Compared to other porous materials, COFs exhibit unique advantages for photocatalysis. Their extensive π-conjugated layers offer high electron and hole mobilities, while their long-range order structures provide high surface area and continuous pores for mass transfer.31,46 Among them, the bpy-based ketoenamine COF (TpBpy; Tp: 1,3,5-triformylphloroglucinol) can be considered a prominent candidate due to its high stability, strong absorption in the visible region, good electrical properties, large surface area, and high CO2 adsorption capacity.30 Moreover, the high solubility of 5,5′-diamino-2,2′-bipyridine in acidic solutions allows the preparations of TpBpy with a high degree of crystallinity and high surface area under hydrothermal conditions using water as a green solvent, indicating great application potential of TpBpy.47
By combining these aforementioned advantages, in this work, we employed the TpBpy COF as a “bridge” material to combine cadmium sulfide (CdS) and the [Co(bpy)3]2+ cocatalyst to obtain a highly stable, efficient, and selective photocatalytic CO2 reduction system. CdS was adopted as the photo-harvester, because it possesses an excellent visible-light response and appropriate conduction band (CB) edge position.48,49 The superior activities of CdS/TpBpy stem from the synergistic effects, including a high surface area, a high CO2 adsorption capacity, a strong solar light harvesting ability, and highly efficient charge carrier transfer at the interface between CdS and TpBpy as well as that between the photocatalysts and the [Co(bpy)3]2+ cocatalyst. Additionally, during CO2 conversion, [Co(bpy)3]2+ must maintain its flexibility and accessibility because of the flexible association/dissociation between TpBpy and the cocatalyst. Consequently, in the presence of the cocatalyst and triethanolamine (TEOA; electron donor), the optimized CdS/TpBpy shows an extraordinary CO production rate of 35.2 mmol g−1 with 85.0% selectivity over the first four hours of the reaction. Furthermore, the reaction system containing 2 mg of CdS/TpBpy-20% exhibited apparent quantum efficiencies (AQEs) of 4.45 and 3.35% at 400 and 450 nm, respectively. Based on these results, the possible mechanism of photocatalytic CO2 conversion over CdS/TpBpy is proposed and discussed in this paper.
2. Materials and methods
2.1 Raw materials
2,2′-Bipyridine (bpy) and nickel(II) chloride hexahydrate (NiCl2·6H2O) were purchased from Sigma-Aldrich. 2,2′-Bipyridine 4,4′-diamine (Bpy-NH2), 1,3,5-triformylphloroglucinol (Tp), and biphenyl-4,4′-diamine (BD) were purchased from J&H Chemical Co., Ltd, China. Dimethyl sulfoxide (DMSO) and triethanolamine (TEOA) were obtained from Duksan Pharmaceutical Co., Ltd, Korea. Acetone, acetonitrile (MeCN), acid acetic, cadmium acetate dihydrate (Cd(OAc)2·2H2O), cobalt(II) chloride hexahydrate (CoCl2·6H2O), dichloromethane (DCM), dimethylacetamide (DMAc), ethanol (EtOH), iron(II) chloride tetrahydrate (FeCl2·4H2O), and tetrahydrofuran (THF) were purchased from Daejung Chemicals & Metals Co. Ltd, Korea. Iron(III) chloride hexahydrate (FeCl3·6H2O) was obtained from Alfa Aesar. Manganese(II) chloride tetrahydrate (MnCl2·4H2O) was purchased from Junsei Chemical Co., Ltd, Japan.2.2 Synthesis of TpBpy COF
TpBpy was prepared by the hydrothermal method in an acidic medium, according to a reported method with some modifications.47 To a suspension of 2,2′-bipyridine 4,4′-diamine (Bpy) (628.46 mg, 3.375 mmol) and 1,3,5-triformylphloroglucinol (Tp) (472.82 mg, 2.25 mmol) in 27.0 ml of deionized water, 13.5 ml of acid acetic was added under vigorous stirring for 15 minutes. The subsequent mixture was transferred to a Teflon-lined stainless-steel autoclave of 50 mL capacity and heated at 120 °C for 72 hours in an electric oven. A red solid precipitate was obtained after the mixture was cooled normally to room temperature. The precipitate was cleaned with a copious amount of distilled water and ethanol and then dried at 80 °C for 12 hours (isolated yield ∼ 83%).2.3 Synthesis of CdS/TpBpy composites
CdS nanoparticle loaded TpBpy was synthesized using the solvothermal method. 426.45 mg (1.6 mmol) of Cd(OAc)2·2H2O and the requisite amounts of TpBpy were vigorously stirred in 160 mL of DMSO for 1 hour. The reaction was carried out in a 200 ml Teflon-lined autoclave at 180 °C for 12 hours. Finally, the final product was collected by centrifugation and sequentially washed with water and ethanol and dried at 80 °C for 12 hours. A series of CdS/TpBpy-x% samples, where x indicated the weight percentage of TpBpy, were prepared by adjusting the amounts of the as-synthesized COF. CdS/TpBpy-10%, CdS/TpBpy-20%, CdS/TpBpy-30%, and CdS/TpBpy-40% were synthesized by using 25.68, 57.78, 99.06, and 154.09 mg of TpBpy, respectively.2.4 Photocatalytic CO2 reduction tests
The photocatalytic CO2 reduction experiments were evaluated in a 150 mL quartz reactor under ambient conditions. Typically, 2.00 mg of catalyst, 250.0 μmol of 2,2′-bipyridine (bpy), 7.0 mL of acetonitrile, 3.0 mL of H2O, 5.0 mL of triethanolamine (TEOA), and 5.00 μmol of CoCl2 were added into the quartz reactor. The reactor was sealed with a silicone rubber septum and degassed for 4 minutes. Subsequently, the reaction mixture was filled with CO2 for 1 hour to saturate the solutions. A solar simulator equipped with an AM 1.5 G filter and a 150 W Xe lamp (Abet Technologies) was used as the irradiation source. After each reaction, the generated products were quantified using an off-line Model 4900 gas chromatograph (Young Lin) with Autochro-3000 software. Both a thermal conductivity detector (TCD) and a flame ionization detector (FID) were used to detect H2 and CO gases, respectively. The liquid after the reaction was monitored by proton nuclear magnetic resonance (1H NMR) (Bruker Avance III HD 400).The 13CO2-labeling experiment was performed under the same photocatalytic conditions, except that 12CO2 was replaced with 13CO2 (99%). The products were analysed using a gas chromatography-mass spectrometry system (GC 2010 Plus, Shimadzu).
3. Results and discussion
3.1 Structural and morphological characterization
The synthesis procedures of the TpBpy and CdS/TpBpy nanohybrids are illustrated in Fig. 1a. First, highly stable TpBpy was fabricated via the hydrothermal polymerization of 1,3,5-triformylphloroglucinol (Tp) and 2,2′-bipyridine-5,5′-diamine (Bpy-NH2) in an acetic acid medium.47 Then, CdS was anchored to TpBpy under solvothermal conditions using Cd(CH3COO)2 as a Cd2+ source and dimethyl sulfoxide (DMSO) as a solvent and S2− source.49 The coordination of the bpy and phenol–imine moieties with Cd2+ can result in strong interactions between the two components and prevent the agglomeration of CdS. According to the inductively coupled plasma optical emission spectrometry (ICP-OES) analysis, the CdS loadings in the composites are near the theoretical one (Table S1†).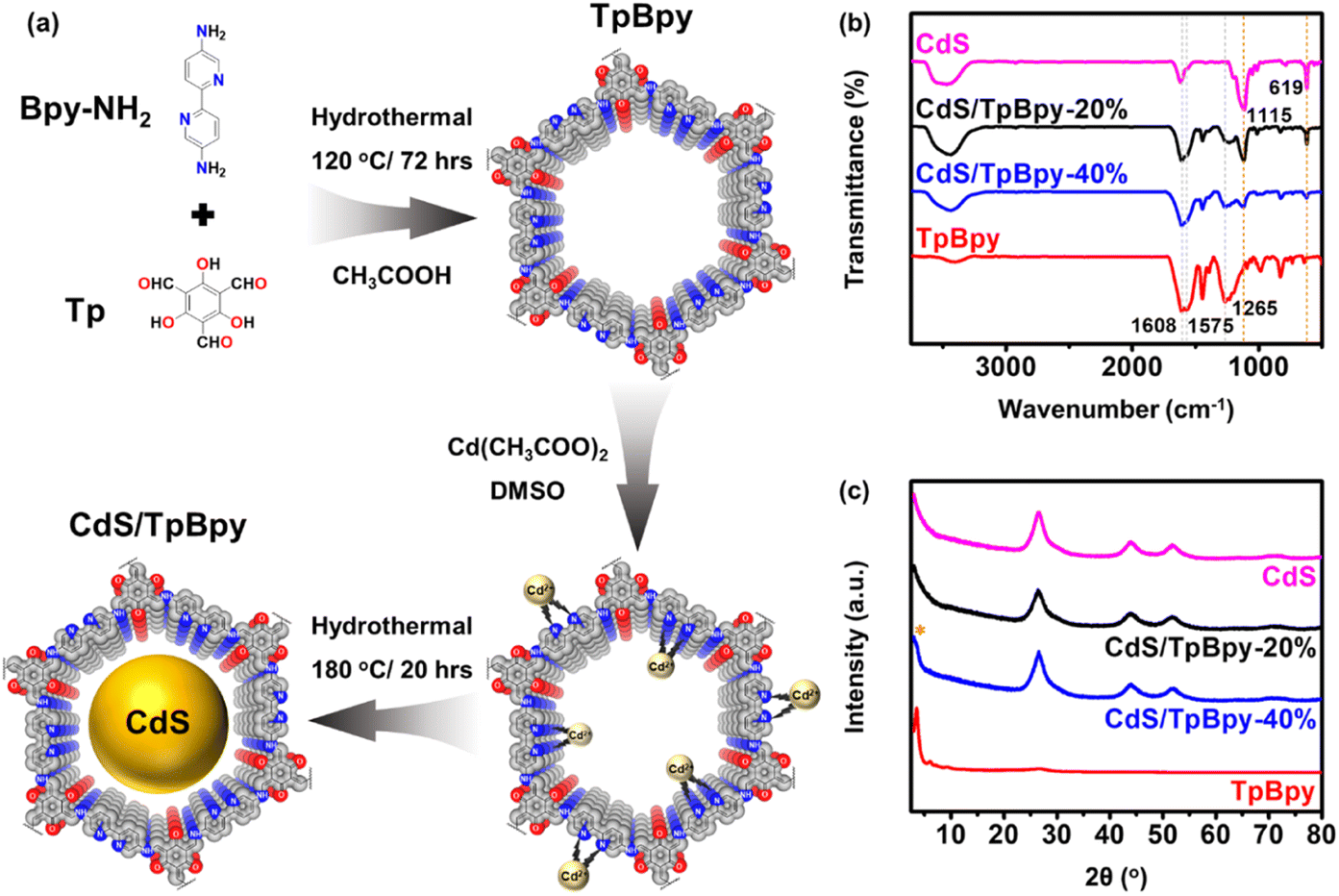 | ||
| Fig. 1 (a) Schematic illustration of TpBpy and CdS/TpBpy. (b) FTIR spectra of CdS, TpBpy and CdS/TpBpy composites. (c) XRD patterns of the as-synthesized materials (* denotes the TpBpy singal). | ||
The functional groups of the TpBpy and CdS/TpBpy nanohybrids were studied using Fourier transform infrared (FT-IR) spectroscopy (Fig. 1b). In the TpBpy FT-IR spectrum, the absence of –NH2 stretching bands (3112 and 3317 cm−1) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency (1637 cm−1) exclude the presence of Bpy-NH2 and Tp, respectively.50 Furthermore, the strong peaks at 1608, 1575, and 1265 cm−1 are ascribed to the stretching frequencies of –CO, –CC–, and –C–N–, respectively.51 The successful formation of TpBpy under hydrothermal conditions can also be validated by the 13C nuclear magnetic resonance (NMR) result shown in Fig. S1.†52 The FT-IR spectra of the composites contain additional characteristic signals of CdS at 1115 and 619 cm−1,53–55 suggesting that TpBpy is highly stable under hydrothermal conditions. The powder X-ray diffraction (XRD) patterns of TpBpy reveal a highly crystalline eclipsed stacking framework. The intense peak at 3.6° and the broad peak at 25.1–28.6° can be assigned to the (100) facet and the π–π stacking between the TpBpy COF layers, respectively.56 The XRD result of CdS exhibits a series of signals at 26.6°, 44.0°, and 52.1°, matching a cubic phase structure.57 The intense diffraction signals of CdS dominate the XRD profiles of CdS/TpBpy composites because compared to CdS, the amount of TpBpy is smaller, and its diffraction peak intensities are inferior. Moreover, the broad diffraction peaks of the hybrids reflect a decrease in the particle size of CdS (Fig. S2†).49,58
O stretching frequency (1637 cm−1) exclude the presence of Bpy-NH2 and Tp, respectively.50 Furthermore, the strong peaks at 1608, 1575, and 1265 cm−1 are ascribed to the stretching frequencies of –CO, –CC–, and –C–N–, respectively.51 The successful formation of TpBpy under hydrothermal conditions can also be validated by the 13C nuclear magnetic resonance (NMR) result shown in Fig. S1.†52 The FT-IR spectra of the composites contain additional characteristic signals of CdS at 1115 and 619 cm−1,53–55 suggesting that TpBpy is highly stable under hydrothermal conditions. The powder X-ray diffraction (XRD) patterns of TpBpy reveal a highly crystalline eclipsed stacking framework. The intense peak at 3.6° and the broad peak at 25.1–28.6° can be assigned to the (100) facet and the π–π stacking between the TpBpy COF layers, respectively.56 The XRD result of CdS exhibits a series of signals at 26.6°, 44.0°, and 52.1°, matching a cubic phase structure.57 The intense diffraction signals of CdS dominate the XRD profiles of CdS/TpBpy composites because compared to CdS, the amount of TpBpy is smaller, and its diffraction peak intensities are inferior. Moreover, the broad diffraction peaks of the hybrids reflect a decrease in the particle size of CdS (Fig. S2†).49,58
To further evaluate the chemical states and the elemental compositions of materials, X-ray photoelectron spectroscopy (XPS) analysis was employed. The XPS spectra of CdS/TpBpy confirm the presence of Cd, S, C, and O (Fig. 2c and d and S3†). No N signals appear because of the high intensities of the Cd signals and the overlapping between the Cd and N peaks. The XPS spectrum of Cd 3d consists of Cd 3d3/2 at 411.9 eV and Cd 3d5/2 at 405.2 eV (Fig. 2a), indicating that the Cd is in a +2 oxidation state.59 Furthermore, the binding energies of S 2p1/2 and S 2p3/2 are 162.7 and 161.6 eV, respectively, corresponding to S2− species (Fig. S3b†).43 Notably, no differences are observed between the XPS spectra of CdS and those of surface CdS/TpBpy (Fig. 2a and S3b†), indicating the similar chemical state of both surface materials. However, after etching with Ar+ ions to remove the outer CdS layers, the binding energies of Cd and S species in CdS/TpBpy show positive shifts relative to that of pristine CdS (Fig. 2b–d and S3c†). These observations can be explained by the formation of tiny CdS particles in the interior cavities of TpBpy as well as strong interactions between CdS and TpBpy.43,60–62 Moreover, as depicted in Fig. S3d,† the atomic contents of Cd and S in CdS/TpBpy diminish, while the C content increases with the increased etching time, implying that CdS distributes throughout TpBpy, possibly including pores of TpBpy. To validate this, the porous structures and specific surface areas of the as-synthesized materials are investigated by nitrogen sorption at 77 K (Fig. S4–S6†). The presence of micropores in TpBpy leads to a high specific surface area (1468.73 m2 g−1) and a high pore volume (1.089 cm3 g−1), which are comparable to those of its solvothermal counterpart.30,47,51,63 The 11-fold lower specific surface area (136.4 m2 g−1) and inferior pore volume (0.318 cm3 g−1) of CdS/TpBpy indicate that CdS deposition occurs not only on the surfaces but also inside the micropores of TpBpy.64 In contrast, the specific surface area of CdS/TpBpy is still higher than that of bare CdS (89.7 m2 g−1), and this high surface area facilitates a cocatalyst-mediated CO2 capture (Fig. S8†).
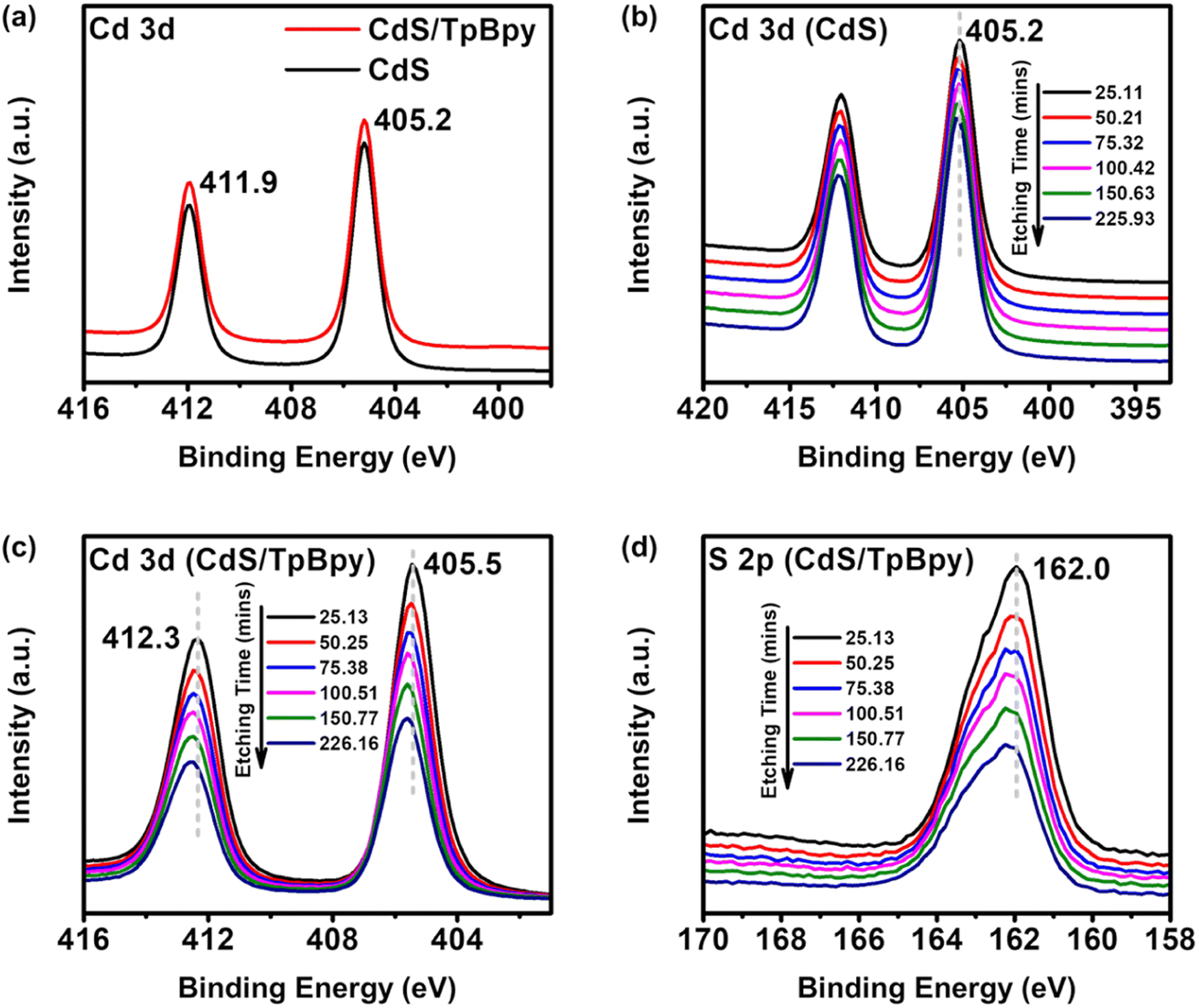 | ||
| Fig. 2 (a) Cd 3d XPS spectra at the surface of CdS/TpBpy-20% and CdS. (b) Cd 3d high resolution XPS spectra of CdS at different etching times. (c) Cd 3d and (d) S 2p high resolution XPS spectra of CdS/TpBpy-20% at different etching times. | ||
Field-emission scanning electron microscopy (FESEM) images of TpBpy show a large particle with a three-dimensional (3D) fiber network (Fig. 3a and b and S9†). Without TpBpy incorporation, the CdS particles agglomerate to form large particles (200–400 nm) (Fig. S10†). However, using TpBpy as a supporting material leads to a uniform distribution of CdS on the sample surface along with a reduction in the particle size (100–130 nm), which is confirmed by the SEM and transmission electron microscopy (TEM) images shown in Fig. 3c and d, S11, and S12.† Furthermore, Fig. S11e and f† show that smaller CdS particles anchor on TpBpy fibers at some positions where the external CdS particles detach, demonstrating the strong interfacial contact between CdS and TpBpy and well-preserved morphology structures of TpBpy. In order to confirm the formation of CdS particles on/in TpBpy fibers, the surface of CdS/TpBpy was sputtered using a gallium (Ga) focused ion beam (FIB). As shown in the FIB SEM and FIB aberration-corrected annular dark-field scanning transmission electron microscopy (FIB AC-ADF-STEM) images (Fig. 3e and f, and S13†), tiny CdS particles with a size down to 1 nm spread over the COF, proving that CdS particles are possibly anchored inside the pores of TpBpy (2.1 nm).65,66 The decrease in CdS size and the strong interfacial interaction between CdS and TpBpy are supposed to suppress the bulk electron–hole recombination of CdS and facilitate the interfacial charge transfer.14,67 From the high-resolution TEM (HRTEM) and STEM images of the hybrids (Fig. S12d and S13e†), the interplanar spacings of the dispersed CdS particles and TpBpy are determined to be 0.33 and 0.34 nm, corresponding to the (111) plane and the π–π stacking between the COF layers. Energy-dispersive X-ray (EDX) elemental mapping images (Fig. 3g, S13g, and S14†) reveal the homogeneous dispersion of CdS over TpBpy.
 | ||
| Fig. 3 (a) and (b) SEM images of TpBpy. (c) SEM and (d) TEM images of CdS/TpBpy. (e) FIB SEM and (f) FIB AC-ADF-STEM images of CdS/TpBpy. (g) TEM EDX elemental mapping images of CdS/TpBpy. | ||
3.2 Photocatalytic performances
Typically, the photocatalytic CO2 reduction experiments were carried out in a CO2-saturated MeCN/water mixed solution using [Co(bpy)3]2+ as the cocatalyst and TEOA as the sacrificial donor under simulated solar light (AM 1.5G). During the photocatalytic reaction, CO is the main product and no liquid CO2 reduction products (e.g., HCOOH, CH3OH, and C2H5OH) are produced (Fig. S15 and S16†). Light, CO2, TEOA, [Co(bpy)3]2+, and the composites play important roles in obtaining an efficient CO2 reduction performance (Fig. S17†). Only H2 is generated under an Ar atmosphere, suggesting that CO might be formed from CO2 reduction (Fig. S15†). To verify the carbon source of CO, an isotope 13CO2 photoconversion experiment was performed (Fig. S18†). According to the mass spectrum, an intense signal of 13CO with a mass-to-charge (m/z) value of 29 indicates that CO is indeed derived from CO2. MeCN/water is the most suitable solvent for photocatalytic CO2 reduction systems comprising [Co(bpy)3]2+ and TEOA. Furthermore, changing the water-to-MeCN ratio affects the efficiency and selectivity of the reaction system (Fig. S19a†). Compared to water, MeCN absorbs a higher amount of CO2, while water acts as a hydrogen source.68 An appropriate ratio of water to MeCN produces the highest performance and selectivity.69 There is an increase in CO and H2 evolution rates, when the TEOA concentration is increased up to 5 mL (Fig. S19b†). The further addition of TEOA has adverse effects on the CO2 photoreduction performance due to the high viscosity of the reaction medium and reduced light-harvesting ability of the photocatalysts.70 The gas evolution rates increase with the addition of [Co(bpy)3]2+ up to 5 μmol (Fig. S19c†). Additionally, the amount of bpy also profoundly influences the selectivity and activity (Fig. S19d†). Without bpy, H2 dominates the CO2 reduction products. Adding bpy to the reaction system converts the major product from H2 to CO. The excess amount of bpy can avoid the precipitation of [Co(bpy)3]2+ and suppress the competition between the coordination of bpy and solvents with Co2+, leading to enhanced performance and selectivity.42,71 Polypyridine-based molecular cocatalysts containing metals such as Ni, Fe, and Mn have been widely reported for efficient CO2-to-CO conversion,30,72–74 so various metal salts such as NiCl2, FeCl2, FeCl3, and MnCl2 were also evaluated for photoreduction of CO2 (Fig. S19e†). The highest activity and selectivity of CoCl2 imply that Co2+ is indispensable to the reaction system. An excessive amount of the cocatalyst reduces the light-harvesting ability of the materials in the ultraviolet (UV) region75 and decreases the probability of interaction between the active [Co(bpy)x]+ species and CO2, leading to a slight drop in efficiency.27 The photocatalytic activity remains unchanged at photocatalyst doses > 2 mg due to the light shielding effect (Fig. S19f†).76As shown in Fig. 4a, the combination of CdS and TpBpy can greatly increase the photocatalytic efficiencies. Because of the fast electron–hole pair recombination, pristine CdS has a low CO evolution rate of 3.70 mmol h−1 g−1 and a H2 generation rate of 0.974 mmol h−1 g−1, while TpBpy yields 0.799 mmol h−1 g−1 of CO with a selectivity of 84.0% over H2. After TpBpy is loaded with the CdS nanoparticles, a drastic increase in the CO2-to-CO conversion efficiency is observed; a maximum of 8.80 mmol h−1 g−1 of CO with a high CO selectivity of 83.7% is observed for CdS/TpBpy-20%. The bpy units of TpBpy guarantee the fast photogenerated electron transfer from CdS to [Co(bpy)3]2+, leading to the superior activities of CdS/TpBpy. The AQE remarkably depends on the light wavelength, indicating that the CO2 photoreduction reaction is induced by the light photon harvesting over the optimized composite (Fig. S20†). CdS/TpBpy-20% exhibits high AQEs of 4.75% and 3.65% under monochromatic light of wavelengths 400 and 450 nm, respectively.
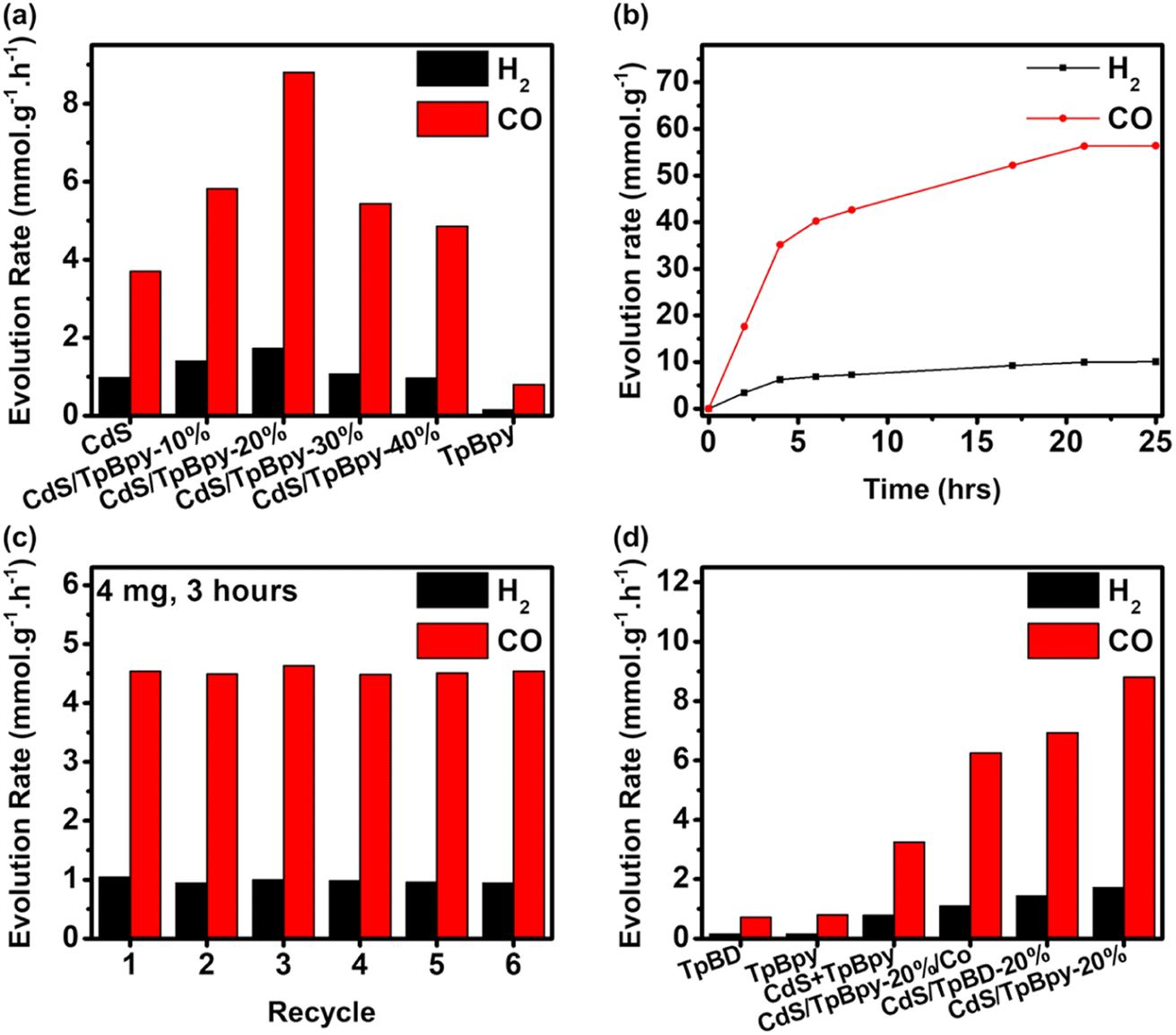 | ||
| Fig. 4 (a) Influence of the amount of TpBpy integrated with CdS on the CO and H2 evolution rates. (b) Product yield activity over time for CdS/TpBpy-20%. (c) Catalyst recycling experiments of the optimized nanohybrid. (d) Comparison of activities of various samples. | ||
The CO production rate of the optimized composite is 35.2 mmol g−1 with 85.0% selectivity over the first four hours of reaction and then increases steadily during the next 17 hours (Fig. 4b). The saturation of the products and consumption of the reactants result in reduced activity after four-hour irradiation. Moreover, the stability of CdS/TpBpy-20% surpasses that of CdS due to the confinement effect (Fig. S21†). As observed in the recycling experiments (Fig. 4c), the unchanged activity up to 6 cycles highlights the stability and reusability of the catalysts. After the cycling experiments, there are no noticeable changes in the structure, as evidenced by the XRD patterns, SEM images, and TEM images shown in Fig. S22–S24.† The FT-IR and XPS results of the recovered composite also confirm the stability of the photocatalysts (Fig. S25 and S26†).
The bpy moieties play a vital role in synthesizing the COF and in improving the CO2 photoreduction performance of the CdS/TpBpy nanohybrids. TpBD (BD: benzidine), an analogue of TpBpy, was prepared under identical conditions for the CO2 photoreduction. The poor solubility of benzidine in acidic solutions leads to a complicated purification process and low yield, indicating the important role of the bpy units under hydrothermal conditions using water as a green solvent (Fig. S27 and S28†). In addition, TpBD and its integration with CdS (Fig. S29 and Table S1†) show lower activities, in comparison with TpBpy and CdS/TpBpy, due to a lower surface area and lack of connection with the cocatalyst. The higher activity of CdS/TpBpy compared to that of CdS/TpBpy-Co (Fig. S30–S32†) can be ascribed to the deficient amount of the cocatalyst (Fig. S19c and Table S1†). The photocatalytic activity of the mechanically mixed pure COF and CdS system (CdS + TpBpy: 3.25 mmol h−1 g−1 of CO and 0.781 mmol h−1 g−1 of H2) is inferior to those of CdS/TpBpy and CdS, highlighting the importance of the in situ growth of CdS within the COF structure.
3.3 Band structures
UV-Vis diffuse reflectance spectroscopy (UV-Vis DRS) was employed to investigate the light-harvesting abilities of all the materials (Fig. 5a). The light absorption edge of CdS is ∼530 nm, and a blue shift in the band-edge position of CdS is observed for the CdS nanoparticles grown on/within TpBpy. This band-edge blueshift can be explained by the quantum size effect of the CdS nanocrystals in the composites, in agreement with the XRD, XPS, SEM, and TEM results. In addition, the introduction of TpBpy results in the enhancement of the absorption in the visible regime, leading to a superior photocatalytic performance of the composites. The bandgaps of CdS and TpBpy are determined to be 2.35 and 1.65 eV, respectively (Fig. S33b†). To analyze the energy band structures of the as-synthesized materials, Mott–Schottky (MS) measurements were carried out (Fig. 5b and c and S34†). The flat band potential (EFB) values of CdS, TpBpy, and CdS/TpBpy are −1.08, −0.950, and −1.00 V vs. EAg/AgCl, respectively, corresponding to −0.883, −0.753, and −0.803 V vs. NHE (NHE: normal hydrogen electrode), respectively. Because the flat band potentials are approximately close to the CB potentials for n-type semiconductors, the CB potentials (ECB) of CdS and TpBpy are −0.883 and −0.753 V vs. NHE, respectively, which are more negative than the redox potentials of [Co(bpy)3]2+/[Co(bpy)3]+ (−0.45 V vs. NHE)77,78 and CO2/CO (−0.104 V vs. NHE).79 Based on the bandgaps and CBs, the valence band (VB) potentials (EVB) of CdS and TpBpy are calculated to be 1.47 and 0.897 V vs. NHE, respectively. The energy gaps between the VB and Fermi levels are found to be 1.55 and 1.42 eV from the VB-XPS spectra of CdS and TpBpy, respectively (Fig. 5d).80,81 According to the EVB values, the Fermi levels (Ef) are estimated to be −0.083 and −0.523 V vs. NHE for CdS and TpBpy, respectively. Thus, the electrons transfer from TpBpy to CdS until Ef at the interface reaches an equilibrium, forming a straddling gap (type I) CdS/TpBpy heterojunction and causing the band bending shown in Scheme 1. The charge transfer and CO2 photoreaction mechanism are further discussed in the next section.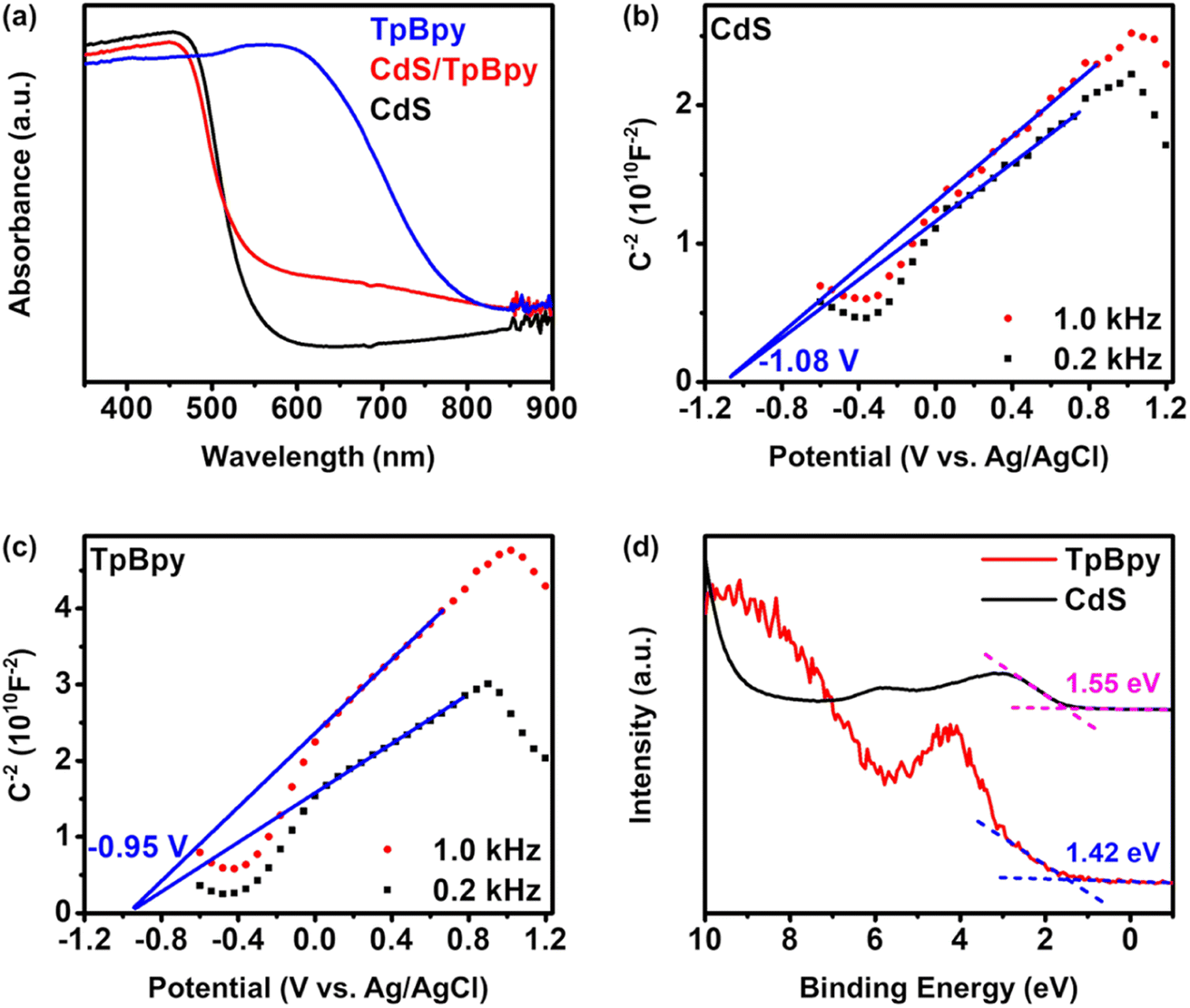 | ||
| Fig. 5 (a) UV-Vis diffuse reflectance spectra of the as-prepared materials. Mott–Schottky analyses of (b) CdS and (c) TpBpy. (d) VB-XPS spectra of CdS and TpBpy. | ||
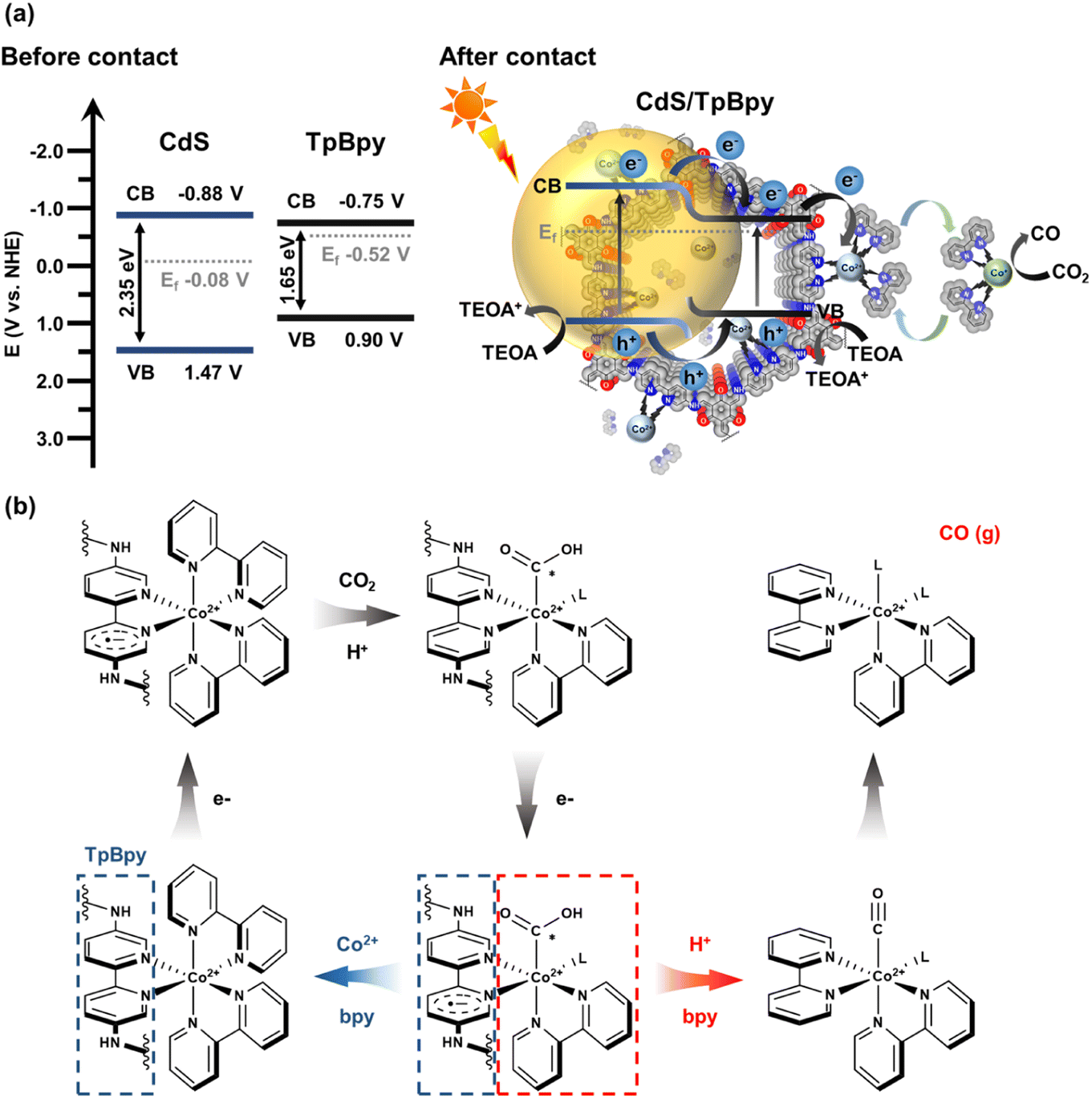 | ||
| Scheme 1 (a) Schematic representation of the reaction mechanism of photocatalytic CO2 reduction using CdS/TpBpy as the catalyst, [Co(bpy)3]2+ as the cocatalyst, and TEOA as the hole scavenger, under sunlight irradiation. (b) Proposed mechanism of CO2 photoreduction to CO by [Co(bpy)3]2+. | ||
3.4 Discussion of the mechanism of activity enhancement
To examine the transfer kinetics of the photogenerated carriers, photo/electrochemical measurements were conducted. The transient photocurrent responses recorded over 420 seconds of 30 s on/off show the highest intensity of CdS/TpBpy, implying that the migrated charge carriers are effectively separated over the composites (Fig. 6a). In addition, according to the Nyquist plots of CdS, TpBpy, and CdS/TpBpy, the CdS/TpBpy composite exhibits the smallest semicircle (Fig. S35†), illustrating a lower interfacial charge-transfer resistance. Two strong emission bands at 423 and 614 nm are observed in the emission spectrum of TpBpy (Fig. S36a†), consistent with the results of a previous report.82 As shown in Fig. 6b, CdS exhibits a strong band-edge emission peak at 520 nm. By contrast, the much lower fluorescence intensity of CdS/TpBpy suggests that the charge carrier recombination is strongly suppressed. The redshift of the photoluminescence (PL) maximum is associated with the decreased band gap energy of CdS in the composites. Time-resolved PL (TRPL) measurements were carried out at a laser excitation wavelength of 375 nm and the CdS band-edge emission of the as-prepared materials (∼520 nm) was monitored (Fig. 6c and Table S2†). The TRPL decay curves were fitted with bi-exponential functions, where short lifetime components (A1, τ1, and B1) correspond to the radiative recombination and long lifetime components (A2, τ2, and B2) reflect the non-radiative process. When compared to bare CdS, the incorporation between CdS and TpBpy leads to decreases in the proportion (A1) and fractional contribution (B1), and increases in the proportion (A2) and fractional contribution (B2), implying that CdS/TpBpy has more favourable non-radiative decay pathways.83,84 Additionally, the charge carrier transfers at the interface shorten the non-radiative lifetimes τ2, while the lower concentrations of photogenerated charges in CdS can lengthen the radiative carrier lifetime τ1. Overall, the average lifetime of CdS (0.662 ns) is shorter than that of CdS/TpBpy (0.762 ns) due to the efficient transfer and separation of photogenerated electrons and holes.85,86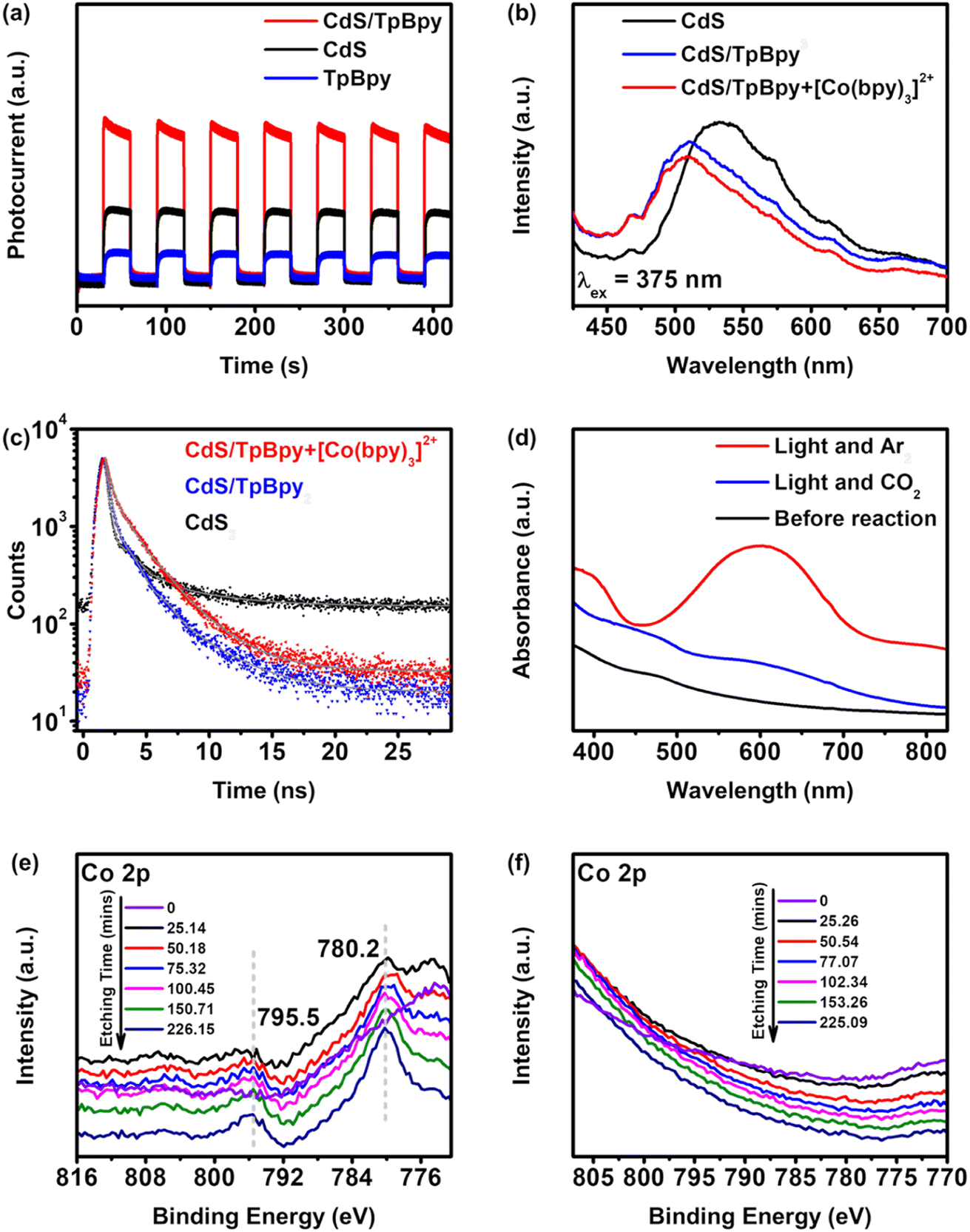 | ||
| Fig. 6 (a) Transient photocurrent responses, (b) photoluminescence (PL) spectra, and (c) time-resolved photoluminescence (TRPL) spectra of the as-synthesized materials. (d) UV-Vis absorption spectra of the photocatalytic system before and after illumination. (e) Co 2p high resolution XPS spectra of CdS/TpBpy-20%/Co. (f) Co 2p high resolution XPS spectra of the optimized composite after the reaction. | ||
To identify the role of [Co(bpy)3]2+ in improving the efficient and selective photoreduction of CO2 to CO, UV-vis absorption measurements were employed. As shown in Fig. 6d, in an Ar atmosphere and under irradiation, a new peak in the 500–700 nm region is recorded, which is ascribed to the generation of [Co(bpy)x]+.37 The absorption band intensity reduces when CO2 replaces Ar, implying that [Co(bpy)x]+ is oxidized to [Co(bpy)3]2+ by CO2. This kinetic behavior is also proved via the photochromic phenomenon (Fig. S37 and S38†). The color of the suspension changes from yellow to green under irradiation.71 However, after turning off the light, the green suspension is gradually restored to yellow in a CO2 atmosphere, while the color of the suspension remains unchanged in an Ar atmosphere. The addition of the cocatalyst suppresses the photoinduced recombination and accelerates the migration and separation of charge carriers (Fig. 6b and c).
Further characterization of CdS/TpBpy-20%/Co was performed to understand the interaction between CdS/TpBpy and Co2+/[Co(bpy)3]2+. The XPS spectra and ICP-OES result confirm the presence of Co2+ in CdS/TpBpy-20%/Co (Fig. 6e, S40 and Table S1†). Moreover, in the XPS profile of the CdS/TpBpy/Co surface, no signal of Co 2p is observed, whereas the Cd and S signals show positive shifts, compared to those of CdS/TpBpy, after etching with Ar+ ions. These observations indicate that the unsaturated S atoms weakly interact with the metal ions87 but can coordinate to Co2+ in the presence of the bpy moieties. As shown in Fig. S33a,† the UV-Vis DRS spectrum of CdS/TpBpy-20%/Co shows an enhancement of absorption in the visible region, which is ascribed to the coordination of bpy units to Co2+.45,56 Although implanting Co2+ into CdS/TpBpy leads to a reduction in the surface area (Fig. S7†), the CO2 adsorption ability of this material is unaffected (Fig. S8†). Finally, the morphology of CdS/TpBpy-20%/Co is found to be similar to that of CdS/TpBpy-20%. In summary, the coordination of the bpy units to Co2+ is advantageous to CO2 photoreduction. Notably, this coordination is also the reason for the lower activity of the mechanically mixed pure CdS and TpBpy system, compared to that of CdS (Fig. 4d). The high affinity between TpBpy and Co2+ decreases the probability of interaction between [Co(bpy)3]2+ and CdS. Besides, the bulk charge carrier recombination of CdS and the weak interfacial contact between bulk CdS and TpBpy results in an inefficient charge carrier transfer and a strong carrier recombination. On the other hand, no signal of Co is found in the Co 2p spectra of the recovered CdS/TpBpy (Fig. 6f) and a drastic decrease in photocatalytic activity of CdS/TpBpy-20%/Co is observed (Fig. S32b and S39†). These results unambiguously imply that the cocatalyst easily accepts electrons from the photocatalyst via the coordination of nitrogen atoms to the Co2+ ions. Due to the dissociation of a bpy ligand in the CO2 reduction process,43,44 the active [Co(bpy)x]+ can separate from the heterogeneous photocatalysts and make spaces for other unactive [Co(bpy)3]2+ species, and the intrinsic high activity and selectivity of the [Co(bpy)3]2+ solution are preserved. In addition, the active [Co(bpy)x]+ can also be anchored to the bpy units of TpBpy, thereby stabilizing the cocatalyst to some extent during the CO2 photoconversion (Fig. S21†).
A plausible mechanism for the sunlight-driven CO2 conversion catalyzed by CdS/TpBpy using TEOA and [Co(bpy)3]2+ as the hole scavenger and cocatalyst, respectively, is depicted in Scheme 1. Both CdS and TpBpy produce excited electrons and holes when exposed to sunlight. The type I band alignment and energy differences assist the photoexcited charge carrier transportation and separation. The photoinduced electrons migrate directly from the CB of CdS to that of TpBpy. After receiving electrons from CdS/TpBpy, the metal center of [Co(bpy)3]2+ (Co2+) converts to [Co(bpy)x]+ (Co+) through ligand metal charge transfer (LMCT), and then loses a bpy ligand to coordinate to CO2, thereby reducing CO2 into CO (Scheme 1b). Due to the flexible bpy ligand association/dissociation, the active [Co(bpy)x]+ can dissociate from the heterogeneous photocatalysts and provides spaces for freely inactive [Co(bpy)3]2+ species. This preserves the [Co(bpy)3]2+ solution's inherently high activity and selectivity. Besides, the slower migration of holes and the band bending lead to the efficient separation of charge carriers.88–90 The photogenerated holes in the VB of the CdS can slowly transfer to the VB of TpBpy or remain in the VB of CdS, and then holes oxidize the sacrificial electron donor TEOA.
4. Conclusion
A simple and eco-friendly approach was used to synthesize a bpy-based ketoenamine COF (TpBpy) on a large scale. The bpy units aid in the synthesis of TpBpy, produce strong interactions with CdS, and accommodate the [Co(bpy)3]2+ cocatalyst, thereby bridging the heterogeneous CdS nanoparticles and the homogeneous cocatalyst. A robust CO production rate of 35.2 mmol g−1 with an 85.0% selectivity over the first four hours of the reaction is obtained owing to synergistic effects, i.e., a high surface area, a high CO2 adsorption capacity, a strong solar light harvesting ability, highly efficient charge transfer at the CdS/TpBpy interface, and subsequent rapid photoelectron injection into the [Co(bpy)3]2+ cocatalyst. Specifically, during CO2 conversion, [Co(bpy)3]2+ must preserve its flexibility and accessibility because of the flexible association/dissociation between TpBpy and the cocatalyst, thereby maintaining the intrinsic high activity and selectivity of the [Co(bpy)3]2+ cocatalyst. Furthermore, the reaction system comprising 2 mg of CdS/TpBpy-20% exhibits 4.75% and 3.65% AQE at 400 and 450 nm, respectively. Based on these observations, a possible mechanism of the photocatalytic CO2 conversion over CdS/TpBpy is developed. The presented rational design and construction of a heterostructure and photocatalytic system will motivate further research on the photoconversion of CO2 in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Research Foundation of Korea (NRF), funded by the Ministry of Science and ICT of Korea (2020H1D3A1A02081461 and 2022R1A2C3003081). Khai H. Do is greatly thankful to the Hyundai Motor Chung Mong-Koo Foundation for providing the financial support.References
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- L. P. Duan, D. Walter, N. T. Chang, J. Bullock, D. Kang, S. P. Phang, K. Weber, T. White, D. Macdonald, K. Catchpole and H. P. Shen, Nat. Rev. Mater., 2023 DOI:10.1038/s41578-022-00521-1.
- K. Zheng, Y. Wu, Z. X. Hu, S. M. Wang, X. C. Jiao, J. C. Zhu, Y. F. Sun and Y. Xie, Chem. Soc. Rev., 2023, 52, 8–29 RSC.
- G. Yang, X. T. Zhu, G. Cheng, R. Chen, J. Y. Xiong, W. J. Li and Y. C. Wei, J. Mater. Chem. A, 2021, 9, 22781–22809 RSC.
- Y. J. Wang and T. He, J. Mater. Chem. A, 2021, 9, 87–110 RSC.
- T. Q. Zhang, Z. F. Mao, X. J. Shi, J. Jin, B. B. He, R. Wang, Y. S. Gong and H. W. Wang, Energy Environ. Sci., 2022, 15, 158–168 RSC.
- D. V. Lam, U. N. T. Nguyen, E. Roh, W. Choi, J. H. Kim, H. Kim and S. M. Lee, Small, 2021, 17, 2100670 CrossRef CAS PubMed.
- U. N. T. Nguyen, D. V. Lam, H. C. Shim and S. M. Lee, Renewable Energy, 2021, 171, 116–123 CrossRef.
- B. H. Ko, B. Hasa, H. Shin, Y. R. Zhao and F. Jiao, J. Am. Chem. Soc., 2022, 144, 1258–1266 CrossRef CAS PubMed.
- X. F. Qiu, H. L. Zhu, J. R. Huang, P. Q. Liao and X. M. Chen, J. Am. Chem. Soc., 2021, 143, 7242–7246 CrossRef CAS PubMed.
- Z. H. Li, J. J. Liu, R. Shi, G. I. N. Waterhouse, X. D. Wen and T. R. Zhang, Adv. Energy Mater., 2021, 11, 2002783 CrossRef CAS.
- J. Zhang, B. An, Z. Li, Y. Cao, Y. Dai, W. Wang, L. Zeng, W. Lin and C. Wang, J. Am. Chem. Soc., 2021, 143, 8829–8837 CrossRef CAS PubMed.
- D. P. Kumar, A. P. Rangappa, H. S. Shim, K. H. Do, Y. Hong, M. Gopannagari, K. A. J. Reddy, P. Bhavani, D. A. Reddy, J. K. Song and T. K. Kim, Mater. Today Chem., 2022, 24, 100827 CrossRef CAS.
- S. Zhu, X. Li, X. Jiao, W. Shao, L. Li, X. Zu, J. Hu, J. Zhu, W. Yan, C. Wang, Y. Sun and Y. Xie, Nano Lett., 2021, 21, 2324–2331 CrossRef CAS PubMed.
- G. Wang, C. T. He, R. Huang, J. Mao, D. Wang and Y. Li, J. Am. Chem. Soc., 2020, 142, 19339–19345 CrossRef CAS PubMed.
- N. Sun, Y. X. Zhu, M. W. Li, J. Zhang, J. N. Qin, Y. X. Li and C. Y. Wang, Appl. Catal., B, 2021, 298, 120565 CrossRef CAS.
- L. Wang, B. H. Zhao, C. H. Wang, M. Y. Sun, Y. F. Yu and B. Zhang, J. Mater. Chem. A, 2020, 8, 10175–10179 RSC.
- J. Xu, Z. Ju, W. Zhang, Y. Pan, J. Zhu, J. Mao, X. Zheng, H. Fu, M. Yuan, H. Chen and R. Li, Angew. Chem., Int. Ed., 2021, 60, 8705–8709 CrossRef CAS PubMed.
- Y. Tamaki and O. Ishitani, Faraday Discuss., 2017, 198, 319–335 RSC.
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS.
- Y. Kou, Y. Nabetani, D. Masui, T. Shimada, S. Takagi, H. Tachibana and H. Inoue, J. Am. Chem. Soc., 2014, 136, 6021–6030 CrossRef CAS PubMed.
- J. Agarwal, E. Fujita, H. F. Schaefer 3rd and J. T. Muckerman, J. Am. Chem. Soc., 2012, 134, 5180–5186 CrossRef CAS PubMed.
- N. Elgrishi, M. B. Chambers, V. Artero and M. Fontecave, Phys. Chem. Chem. Phys., 2014, 16, 13635–13644 RSC.
- N. Elgrishi, M. B. Chambers and M. Fontecave, Chem. Sci., 2015, 6, 2522–2531 RSC.
- J. L. Lin, B. Qin and Z. X. Fang, Catal. Lett., 2019, 149, 25–33 CrossRef CAS.
- Z. Guo, S. Cheng, C. Cometto, E. Anxolabehere-Mallart, S. M. Ng, C. C. Ko, G. Liu, L. Chen, M. Robert and T. C. Lau, J. Am. Chem. Soc., 2016, 138, 9413–9416 CrossRef CAS PubMed.
- K. M. Choi, D. Kim, B. Rungtaweevoranit, C. A. Trickett, J. T. Barmanbek, A. S. Alshammari, P. Yang and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 356–362 CrossRef CAS PubMed.
- T. C. Zhuo, Y. Song, G. L. Zhuang, L. P. Chang, S. Yao, W. Zhang, Y. Wang, P. Wang, W. Lin, T. B. Lu and Z. M. Zhang, J. Am. Chem. Soc., 2021, 143, 6114–6122 CrossRef CAS PubMed.
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS PubMed.
- Y. G. Xiang, W. B. Dong, P. Wang, S. Y. Wang, X. Ding, F. Ichihara, Z. Wang, Y. Wada, S. B. Jin, Y. X. Weng, H. Chen and J. H. Ye, Appl. Catal., B, 2020, 274, 119096 CrossRef CAS.
- H. S. Lee, S. Jee, R. Kim, H. T. Bui, B. Kim, J. K. Kim, K. S. Park, W. Choi, W. Kim and K. M. Choi, Energy Environ. Sci., 2020, 13, 519–526 RSC.
- B. Yu, L. Li, S. Liu, H. Wang, H. Liu, C. Lin, C. Liu, H. Wu, W. Zhou, X. Li, T. Wang, B. Chen and J. Jiang, Angew. Chem., Int. Ed., 2021, 60, 8983–8989 CrossRef CAS PubMed.
- Y. Wei, L. Chen, H. Chen, L. Cai, G. Tan, Y. Qiu, Q. Xiang, G. Chen, T. C. Lau and M. Robert, Angew. Chem., Int. Ed., 2022, 61, e202116832 CAS.
- S. Kim, S. Jee, K. M. Choi and D.-S. Shin, Nano Res., 2020, 14, 486–492 CrossRef.
- X. Chen, Q. Dang, R. Sa, L. Li, L. Li, J. Bi, Z. Zhang, J. Long, Y. Yu and Z. Zou, Chem. Sci., 2020, 11, 6915–6922 RSC.
- K. H. Do, D. P. Kumar, A. P. Rangappa, J. M. Wang, Y. Hong, E. Kim, D. A. Reddy and T. K. Kim, Mater. Today Chem., 2021, 22, 100589 CrossRef CAS.
- B. P. Biswal, H. A. Vignolo-Gonzalez, T. Banerjee, L. Grunenberg, G. Savasci, K. Gottschling, J. Nuss, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2019, 141, 11082–11092 CrossRef CAS PubMed.
- Z. Xu, Y. Cui, D. J. Young, J. X. Wang, H. Y. Li, G. Q. Bian and H. X. Li, J. CO2 Util., 2021, 49, 101561 CrossRef CAS.
- L. L. Li, D. K. Ma, Q. L. Xu and S. M. Huang, Chem. Eng. J., 2022, 437, 135153 CrossRef CAS.
- E. Kim, K. H. Do, J. Wang, Y. Hong, A. Putta Rangappa, D. Amaranatha Reddy, D. Praveen Kumar and T. K. Kim, Appl. Surf. Sci., 2022, 587, 152895 CrossRef CAS.
- Q. Q. Mu, Y. H. Su, Z. H. Wei, H. Sun, Y. B. Lian, Y. Y. Dong, P. W. Qi, Z. Deng and Y. Peng, J. Catal., 2021, 397, 128–136 CrossRef CAS.
- F. Wang, T. Hou, X. Zhao, W. Yao, R. Fang, K. Shen and Y. Li, Adv. Mater., 2021, 33, e2102690 CrossRef PubMed.
- M. F. Kuehnel, K. L. Orchard, K. E. Dalle and E. Reisner, J. Am. Chem. Soc., 2017, 139, 7217–7223 CrossRef CAS PubMed.
- C. Chen, T. Wu, H. Wu, H. Liu, Q. Qian, Z. Liu, G. Yang and B. Han, Chem. Sci., 2018, 9, 8890–8894 RSC.
- F. M. Zhang, J. L. Sheng, Z. D. Yang, X. J. Sun, H. L. Tang, M. Lu, H. Dong, F. C. Shen, J. Liu and Y. Q. Lan, Angew. Chem., Int. Ed., 2018, 57, 12106–12110 CrossRef CAS PubMed.
- J. Thote, H. Barike Aiyappa, R. Rahul Kumar, S. Kandambeth, B. P. Biswal, D. Balaji Shinde, N. Chaki Roy and R. Banerjee, IUCrJ, 2016, 3, 402–407 CrossRef CAS PubMed.
- K. H. Do, D. P. Kumar, A. P. Rangappa, Y. Hong, D. A. Reddy and T. K. Kim, Chemcatchem, 2020, 12, 4550–4557 CrossRef CAS.
- S. W. Cao, Y. J. Wang, B. C. Zhu, G. C. Xie, J. G. Yu and J. R. Gong, J. Mater. Chem. A, 2020, 8, 7671–7676 RSC.
- D. B. Shinde, H. B. Aiyappa, M. Bhadra, B. P. Biswal, P. Wadge, S. Kandambeth, B. Garai, T. Kundu, S. Kurungot and R. Banerjee, J. Mater. Chem. A, 2016, 4, 2682–2690 RSC.
- Z. L. Liu, Y. Q. Huang, S. Q. Chang, X. L. Zhu, Y. H. Fu, R. Ma, X. Q. Lu, F. M. Zhang, W. D. Zhu and M. H. Fan, Sustainable Energy Fuels, 2021, 5, 2871–2876 RSC.
- R. Guo, Y. Liu, Y. Huo, A. Zhang, J. Hong and Y. Ai, J. Colloid Interface Sci., 2022, 606, 1617–1626 CrossRef CAS PubMed.
- X. Y. Li, D. Wu, Q. Z. Luo, J. An, R. Yin and D. S. Wang, J. Mater. Sci., 2017, 52, 736–748 CrossRef CAS.
- J. Fu, B. B. Chang, Y. L. Tian, F. N. Xi and X. P. Dong, J. Mater. Chem. A, 2013, 1, 3083–3090 RSC.
- L. F. Zhang, F. R. Huang, C. H. Liang, L. Y. Zhou, X. G. Zhang and Q. Pang, J. Taiwan Inst. Chem. Eng., 2016, 60, 643–650 CrossRef CAS.
- H. B. Aiyappa, J. Thote, D. B. Shinde, R. Banerjee and S. Kurungot, Chem. Mater., 2016, 28, 4375–4379 CrossRef CAS.
- H. Q. Xu, S. Z. Yang, X. Ma, J. E. Huang and H. L. Jiang, ACS Catal., 2018, 8, 11615–11621 CrossRef CAS.
- J. Thote, H. B. Aiyappa, A. Deshpande, D. Diaz Diaz, S. Kurungot and R. Banerjee, Chem.–Eur. J., 2014, 20, 15961–15965 CrossRef CAS PubMed.
- A. P. Rangappa, D. P. Kumar, M. Gopannagari, D. A. Reddy, Y. Hong, Y. Kim and T. K. Kim, Appl. Surf. Sci., 2020, 508, 144803 CrossRef CAS.
- J. Li, S. Tang, L. Lu and H. C. Zeng, J. Am. Chem. Soc., 2007, 129, 9401–9409 CrossRef CAS PubMed.
- X. M. Ning, Y. L. Wu, X. F. Ma, Z. Zhang, R. Q. Gao, J. Chen, D. L. Shan and X. Q. Lu, Adv. Funct. Mater., 2019, 29, 1902992 CrossRef.
- R. J. Feng, K. W. Wan, X. Y. Sui, N. Zhao, H. X. Li, W. Y. Lei, J. G. Yu, X. F. Liu, X. H. Shi, M. L. Zhai, G. Liu, H. Wang, L. R. Zheng and M. H. Liu, Nano Today, 2021, 37, 101080 CrossRef CAS.
- M. P. Kou, W. Liu, Y. Y. Wang, J. D. Huang, Y. L. Chen, Y. Zhou, Y. Chen, M. Z. Ma, K. Lei, H. Q. Xie, P. K. Wong and L. Q. Ye, Appl. Catal., B, 2021, 291, 120146 CrossRef CAS.
- C. Q. Wang, S. H. Shi, F. Duan, S. L. Lu, H. Zhu, M. L. Du, X. Chen and M. Q. Chen, J. Mater. Chem. A, 2022, 10, 16524–16532 RSC.
- Y. Deng, Z. Zhang, P. Y. Du, X. M. Ning, Y. Wang, D. X. Zhang, J. Liu, S. T. Zhang and X. Q. Lu, Angew. Chem., Int. Ed., 2020, 59, 6082–6089 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, J. Perman, N. Nguyen and S. Q. Ma, J. Am. Chem. Soc., 2016, 138, 15790–15796 CrossRef CAS PubMed.
- A. P. Rangappa, D. P. Kumar, J. M. Wang, K. H. Do, E. Kim, D. A. Reddy, H. S. Ahn and T. K. Kim, J. Mater. Chem. A, 2022, 10, 7291–7299 RSC.
- H. Xu, S. You, Z. Lang, Y. Sun, C. Sun, J. Zhou, X. Wang, Z. Kang and Z. Su, Chem.–Eur. J., 2020, 26, 2735–2740 CrossRef CAS PubMed.
- J. Lin, R. Liao and J. Xu, RSC Adv., 2018, 8, 3798–3802 RSC.
- C. Kutal, M. A. Weber, G. Ferraudi and D. Geiger, Organometallics, 1985, 4, 2161–2166 CrossRef CAS.
- H. Zhong, Z. X. Hong, C. Yang, L. Y. Li, Y. S. Xu, X. C. Wang and R. H. Wang, Chemsuschem, 2019, 12, 4493–4499 CrossRef CAS PubMed.
- Y. A. Wang, T. Liu, L. X. Chen and D. B. Chao, Inorg. Chem., 2021, 60, 5590–5597 CrossRef CAS PubMed.
- X. Z. Wang, S. L. Meng, J. Y. Chen, H. X. Wang, Y. Wang, S. Zhou, X. B. Li, R. Z. Liao, C. H. Tung and L. Z. Wu, Angew. Chem., Int. Ed., 2021, 60, 26072–26079 CrossRef CAS PubMed.
- D. C. Grills, M. Z. Ertem, M. McKinnon, K. T. Ngo and J. Rochford, Coord. Chem. Rev., 2018, 374, 173–217 CrossRef CAS.
- J. Lin, Y. Hou, Y. Zheng and X. Wang, Chem.–Asian J., 2014, 9, 2468–2474 CrossRef CAS PubMed.
- D. P. Kumar, S. Seo, A. P. Rangappa, S. Kim, K. A. J. Reddy, M. Gopannagari, P. Bhavani, D. A. Reddy and T. K. Kim, J. Alloys Compd., 2022, 905, 164193 CrossRef CAS.
- J. L. Lin, B. Qin and G. L. Zhao, J. Photochem. Photobiol., A, 2018, 354, 181–186 CrossRef CAS.
- S. Y. Wang, X. Hai, X. Ding, S. B. Jin, Y. G. Xiang, P. Wang, B. Jiang, F. Ichihara, M. Oshikiri, X. G. Meng, Y. X. Li, W. Matsuda, J. Ma, S. Seki, X. P. Wang, H. Huang, Y. Wada, H. Chen and J. H. Ye, Nat. Commun., 2020, 11, 1149 CrossRef PubMed.
- K. Sun, Y. Qian and H. L. Jiang, Angew. Chem., Int. Ed., 2023, e202217565, DOI:10.1002/anie.202217565.
- Q. Chen, X. J. Chen, Q. R. Jiang, Z. P. Zheng, Z. J. Song, Z. Y. Zhao, Z. X. Xie and Q. Kuang, Appl. Catal., B, 2021, 297, 120394 CrossRef CAS.
- W. Li, X. Wang, Q. Ma, F. Wang, X. S. Chu, X. C. Wang and C. Y. Wang, Appl. Catal., B, 2021, 284, 119688 CrossRef CAS.
- Q. Pan, M. Abdellah, Y. Cao, W. Lin, Y. Liu, J. Meng, Q. Zhou, Q. Zhao, X. Yan, Z. Li, H. Cui, H. Cao, W. Fang, D. A. Tanner, M. Abdel-Hafiez, Y. Zhou, T. Pullerits, S. E. Canton, H. Xu and K. Zheng, Nat. Commun., 2022, 13, 845 CrossRef CAS PubMed.
- C. B. Bie, B. C. Zhu, L. X. Wang, H. G. Yu, C. H. Jiang, T. Chen and J. G. Yu, Angew. Chem., Int. Ed., 2022, 61, e2022120 CrossRef PubMed.
- K. Das, S. N. Sharma, M. Kumar and S. K. De, J. Phys. Chem. C, 2009, 113, 14783–14792 CrossRef CAS.
- W. W. Shao, S. M. Wang, J. C. Zhu, X. D. Li, X. C. Jiao, Y. Pan, Y. F. Sun and Y. Xie, Nano Res., 2021, 14, 4520–4527 CrossRef CAS.
- B. Su, L. J. Huang, Z. Xiong, Y. C. Yang, Y. D. Hou, Z. X. Ding and S. B. Wang, J. Mater. Chem. A, 2019, 7, 26877–26883 RSC.
- P. Bhavani, D. P. Kumar, H. S. Shim, P. Rangappa, M. Gopannagari, D. A. Reddy, J. K. Song and T. K. Kim, Catal. Sci. Technol., 2020, 10, 3542–3551 RSC.
- Z. Fang, Y. F. Liu, Y. T. Fan, Y. H. Ni, X. W. Wei, K. B. Tang, J. M. Shen and Y. Chen, J. Phys. Chem. C, 2011, 115, 13968–13976 CrossRef CAS.
- H. J. Li, Y. Zhou, W. G. Tu, J. H. Ye and Z. G. Zou, Adv. Funct. Mater., 2015, 25, 998–1013 CrossRef CAS.
- H. Jung, Y. Whang and S. W. Han, Bull. Korean Chem. Soc., 2021, 42, 806–809 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 13C NMR spectra, FT-IR results, XPS spectra, N2 and CO2 isotherms, TEM and SEM analyses, Tauc plots, Mott–Schottky plots, impedance measurement results, and ICP results of the as-synthesized materials. Detailed CO2 photoreduction studies and comparison of the CO2 photoreduction performance of different photocatalysts. See DOI: https://doi.org/10.1039/d3ta00079f |
| This journal is © The Royal Society of Chemistry 2023 |