
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bifunctional and regenerable molecular electrode for water electrolysis at neutral pH†
Biswanath
Das‡
 *a,
Esteban A.
Toledo-Carrillo‡
b,
Guoqi
Li
c,
Jonas
Ståhle
a,
Thomas
Thersleff
d,
Jianhong
Chen
d,
Lin
Li
c,
Fei
Ye
b,
Adam
Slabon
e,
Mats
Göthelid
f,
Tsu-Chien
Weng
c,
Jodie A.
Yuwono
g,
Priyank V.
Kumar
g,
Oscar
Verho
*h,
Markus D.
Kärkäs
i,
Joydeep
Dutta
b and
Björn
Åkermark
*a
*a,
Esteban A.
Toledo-Carrillo‡
b,
Guoqi
Li
c,
Jonas
Ståhle
a,
Thomas
Thersleff
d,
Jianhong
Chen
d,
Lin
Li
c,
Fei
Ye
b,
Adam
Slabon
e,
Mats
Göthelid
f,
Tsu-Chien
Weng
c,
Jodie A.
Yuwono
g,
Priyank V.
Kumar
g,
Oscar
Verho
*h,
Markus D.
Kärkäs
i,
Joydeep
Dutta
b and
Björn
Åkermark
*a
aDepartment of Organic Chemistry, Arrhenius Laboratory Stockholm University, Svante Arrhenius väg 16C, 10691 Stockholm, Sweden. E-mail: das.biswanath85@gmail.com; biswanath.das@su.se; bjorn.akermark@su.se
bDepartment of Applied Physics, KTH Royal Institute of Technology, Hannes Alfvéns väg 12, 114 19 Stockholm, Sweden
cSchool of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China
dDepartment of Materials and Environmental Chemistry, Stockholm University, Svante Arrhenius väg 16C, 10691 Stockholm, Sweden
eInorganic Chemistry, University of Wuppertal Gaußstr. 20, 42119 Wuppertal, Germany
fMaterial and Nanophysics, KTH Royal Institute of Technology, Hannes Alfvéns väg 12, 114 19 Stockholm, Sweden
gSchool of Chemical Engineering, University of New South Wales, Sydney 2052, Australia
hDepartment of Medicinal Chemistry, Biomedicinskt Centrum BMC, Uppsala University, SE-75123 Uppsala, Sweden. E-mail: oscar.verho@ilk.uu.se
iDepartment of Chemistry, KTH Royal Institute of Technology, SE-10044 Stockholm, Sweden
First published on 6th April 2023
Abstract
The instability of molecular electrodes under oxidative/reductive conditions and insufficient understanding of the metal oxide-based systems have slowed down the progress of H2-based fuels. Efficient regeneration of the electrode's performance after prolonged use is another bottleneck of this research. This work represents the first example of a bifunctional and electrochemically regenerable molecular electrode which can be used for the unperturbed production of H2 from water. Pyridyl linkers with flexible arms (–CH2–CH2–) on modified fluorine-doped carbon cloth (FCC) were used to anchor a highly active ruthenium electrocatalyst [RuII(mcbp)(H2O)2] (1) [mcbp2− = 2,6-bis(1-methyl-4-(carboxylate)benzimidazol-2-yl)pyridine]. The pyridine unit of the linker replaces one of the water molecules of 1, which resulted in RuPFCC (ruthenium electrocatalyst anchored on –CH2–CH2–pyridine modified FCC), a high-performing electrode for oxygen evolution reaction [OER, overpotential of ∼215 mV] as well as hydrogen evolution reaction (HER, overpotential of ∼330 mV) at pH 7. A current density of ∼8 mA cm−2 at 2.06 V (vs. RHE) and ∼−6 mA cm−2 at −0.84 V (vs. RHE) with only 0.04 wt% loading of ruthenium was obtained. OER turnover of >7.4 × 103 at 1.81 V in 48 h and HER turnover of >3.6 × 103 at −0.79 V in 3 h were calculated. The activity of the OER anode after 48 h use could be electrochemically regenerated to ∼98% of its original activity while it serves as a HE cathode (evolving hydrogen) for 8 h. This electrode design can also be used for developing ultra-stable molecular electrodes with exciting electrochemical regeneration features, for other proton-dependent electrochemical processes.
1. Introduction
The electrochemical generation of hydrogen (H2) is evolving into a reliable, commercially viable solution for resolving fossil-fuel shortages and global warming.1,2 In this area, much of the recent research efforts have been directed toward the development of durable and efficient systems for electrocatalytic water oxidation (WO) and proton reduction (PR).3–13Molecular catalysts offer an attractive alternative to metal oxide and hydroxide-based electrodes due to their improved product selectivity, and the possibility of systematic improvement.9,10,14 As has been demonstrated lately, the instability of the molecular electrocatalysts, under oxidative–reductive environments, can be overcome by immobilizing or anchoring them onto conducting solid surfaces through covalent bonding or π–π stacking interactions.15–19 Ruthenium-based complexes have so far shown the best performance in terms of turnover number (TON), turnover frequency (TOF), and overpotential.17,20–22 However, for commercial applications, further improvement of their stability, as well as strategies for catalyst regeneration are needed.
Fluorine-doped carbon cloth (FCC) has been reported and recently tested by us a as robust and durable conductive support that allows for straightforward structural modification and catalyst grafting.23,24 We recently reported the preparation of CoPFCC electrode consisting of {CoII(mcbp)(H2O)} anchored onto pyridine-modified FCC, which showed high stability and robust features under WO conditions.24 However, to enable sustainable production of H2 from water, further improvement of overpotential and TO (turnover) values is needed. We were only able to regenerate nearly about 78% of the original WO activity with the CoPFCC electrode following its first use (for 72 h) as an anode, which highlights further scopes for upgrading. The CoPFCC electrode could not show any appreciable proton reduction features, representing the need for another electrode (cathode) for proton reduction to avail H2 from water electrolysis.
In recent years, high performing dual functional electrodes that can be used for both OER and HER, have attracted attention due the possibility of using them as both anode and cathode in efficient electrolysis cells for overall water electrolysis.25–30 Usually the HER activity increases with decreasing the pH, while OER is accelerated at higher pH. Therefore, developing electrodes/electromaterials that simultaneously can perform both HER and OER at neutral pH is an additional challenge.31 Aiming towards a bifunctional and fully regenerable electrode setup for unhindered production of H2 from water, we decided to investigate the anchoring of an active ruthenium containing catalyst onto the pyridine (–CH2–CH2–pyridine) modified FCC (PFCC) surface. Ruthenium-containing catalysts have been extensively reported to show exciting features in both oxidations and reductions.32–37 Moreover, ruthenium(II) is known to form strong covalent bonds with N-donor ligands.9,17,22,38,39 The favourable Ru(II)–N (of pyridine from PFCC) interaction was envisioned to replace one of the aqua molecules in catalyst 1, an analogue of RuII(mcbp)(OH2)(py)2 which has previously been reported as a high-performing catalyst for electrochemical WO (TOFmax of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1 for WO at pH 9).22
000 s−1 for WO at pH 9).22
The mcbp2− framework, with its two benzimidazole carboxylate units, acts as a non-innocent ligand that can facilitate the proton-coupled electron transfer (PCET) processes. RuPFCC (ruthenium catalyst anchored onto PFCC) was designed to combine the electrocatalytic activity from the molecular counterpart (of 1), with the current density, and robust features of the conductive support.9,17,20–22,35,40 The pyridines were covalently attached to the carbon cloth separated by two –CH2 units, to allow for optimal distances for electron transfer and charge recombination.41
2. Results and discussion
2.1. Preparation, density functional theory calculation and electrochemical investigation of 1
The H2mcbp ligand was prepared following a reported procedure by Shatskiy et al.22 The aqua complex [RuII(mcbp)(H2O)2] (1) (Fig. 2a) was synthesized by heating a EtOH:H2O (4:1) solution of equimolar amounts of Ru(DMSO)4Cl2 and H2mcbp in the presence of three equiv. of Et3N. The identity of the molecular catalyst (Fig. 2a) was confirmed by 1H NMR, HRMS and UV-Vis analysis (Fig. S1–S6†). The carboxylate units of the mcbp2− ligand framework in [RuII(mcbp)(pyridine)2] played a pivotal role in the stability as well as catalytic water oxidation activity and similar impact was expected in 1.22 The temperature dependent NMR study (at 291 K, 298 K, 310 K and 323 K, Fig. S3†) confirmed the presence of two different types of water molecules attached to the ruthenium center in 1. They are assigned to a water molecule without direct intramolecular H-bonding interaction and another water molecule having H-bonding interaction with one of the carboxylate units of mcbp2− ligand (supported by DFT studies). DFT calculations further elaborated favourable energetics (negative adsorption energies) for the adsorption of two molecules at the Ru site, with adsorption energies of −0.69 and −0.42 eV for the first and second water molecule, respectively (Fig. 2b). The electrochemical features of 1 in solution were investigated in pH 7 phosphate buffer solution, which showed one quasi-reversible and one irreversible oxidation wave at around 1.05 V and 1.38 V (vs. RHE) which can be assigned to RuII/III and RuIII/IV redox couples, respectively. It was followed by a characteristic irreversible WO wave initiated at around +1.86 V (vs. RHE, overpotential of 630 mV) (Fig. 2c). In the case of the reduction, an irreversible one electron reduction wave was found at around 0.05 V which is attributed to the first electron transfer to one of the benzimidazole carboxylate units (in mcbp2−) that is closely held by the metal centre and partially shared by the central ruthenium unit as depicted by DFT calculated LUMO orientation of 1 (Fig. 2f). The second electron reduction that is very prominent and appears as multielectron wave at around −0.84 V must accompany catalytic water reduction which initiated just after −0.59 V (overpotential of 590 mV). Looking at these interesting WO and WR features, we carried out DFT calculations to assess and understand the mechanism of the OER (oxygen evolution reaction) and the HER (hydrogen evolution reaction) (see ESI† for free energy diagram and cartesian coordinates of the optimized structures) using 1 as the catalyst, when the ruthenium center is bonded to either one or two water molecules (denoted as Ru-1H2O and Ru-2H2O respectively). The OER proceeds from a vacant Ru active site to *OH, *O and *OOH intermediates, finally resulting in O2 evolution (Fig. 2d). The OER potential energy diagram is shown in Fig. 2e.
Importantly, we found that the overpotential for OER is significantly reduced at Ru-2H2O (an overpotential of 0.33 eV) compared to that at Ru-1H2O (0.67 eV). Further, we found that the rate-limiting step is *OOH to O2 evolution for Ru-1H2O, while it is *OH to *O formation for Ru-2H2O. Our HER calculations showed that the free energy of H adsorption (H*) is pushed closer to thermoneutral for Ru-2H2O 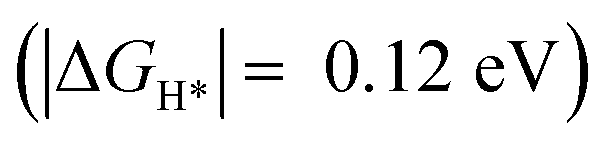 compared to that at Ru-1H2O
compared to that at Ru-1H2O 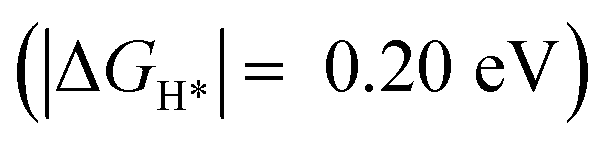 , suggesting that it is also easier for HER to proceed at Ru-2H2O.
, suggesting that it is also easier for HER to proceed at Ru-2H2O.
Finding this additional HER capability, i.e., understanding the dual functionality of [RuII(mcbp)(H2O)2] is one of the novelties of this work. In terms of Ru–N coordination and bond length variations, 1 followed very similar reactive intermediate structures as discussed by us with [RuII(mcbp)(py)2] catalyzed oxygen evolution.22 It is important to note that an increase in the number of water molecules attached to the active site makes the ligand (mcbp2−) less firmly bound and the catalyst becomes more susceptible to decomposition while turning over. Anchoring the catalyst onto a conductive solid surface through a suitable linker can improve both the stability and the overall reactivity, leading to the enhancement of the current density.17,20
2.2. Construction and characterization of RuPFCC
A stepwise procedure (Fig. S7†) was followed to prepare the RuPFCC (see ESI†). FCC and PFCC were prepared following a previously reported procedure (Fig. 1).23,24 The PFCC was dipped into a degassed MeOH:H2O solution of 1 for 5 h at 50 °C, followed by thorough washing with water several times to remove the loosely bound catalysts from the surface. The resulting electrode was dried overnight to furnish the target RuPFCC. From the overall weight percentage analysis, around 0.04 wt% of ruthenium on the surface of RuPFCC was calculated (see ESI†). The electrodes (RuPFCC and PFCC) were characterized by Scanning Electron Microscopy (SEM; Fig. 3a–c), Transmission Electron Microscopy (TEM; Table S1, Fig. S9†), X-ray Photoelectron Spectroscopy (XPS; Fig. 3d, S10†), thermogravimetric analysis (TGA; Fig. 3e, S13†), Electrochemical Impedance Spectroscopy (EIS; Fig. 3f), Fourier-transform Infrared Spectroscopy (FTIR) (Fig. S11†), Raman spectroscopy (Fig. S12, Table S2†), and Tafel diagram (Fig. S14†). The SEM images of RuPFCC clearly showed the presence of the catalyst aggregates (Fig. 3c) with an average size of ∼94 nm (Fig. S8†). The formation of the aggregates is directly related to the inhomogeneous grafting (electronic and steric factors) of the –CH2–CH2–pyridine units onto the FCC, which is followed by the formation of the covalent bonding interaction between pyridine (of PFCC) and ruthenium (of 1) and a strong H-bonding network around the hydrophilic centers (especially Ru–OH2 unit) of the complex. The elemental composition of these aggregates was investigated by STEM EDX mapping, which shows uniform distribution of C, O, N, and Ru (Fig. S9a–e, TEM discussion in ESI†).
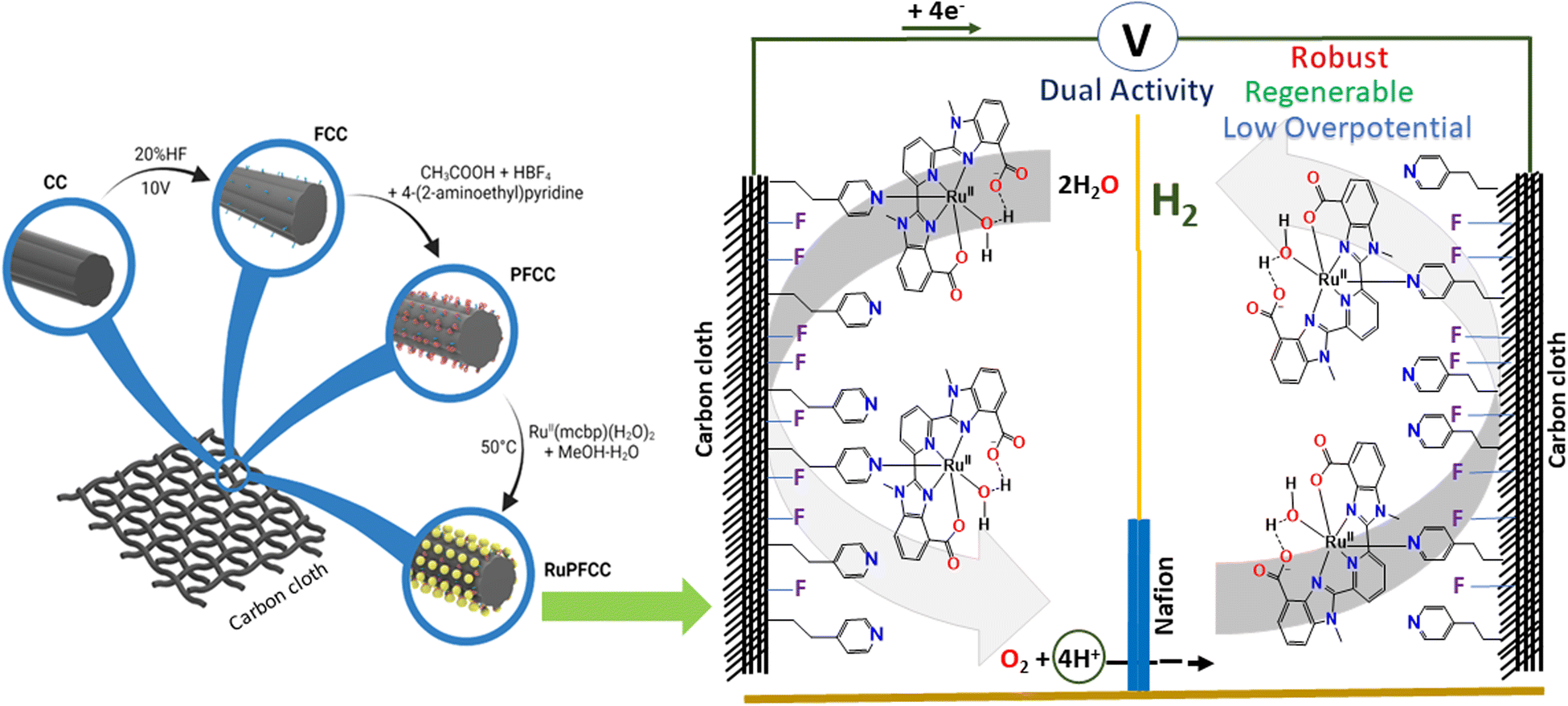 | ||
| Fig. 1 Left: A schematic representation of the preparative procedure for the RuPFCC electrode and right: graphical depiction of the RuPFCC surface and of the potential use of RuPFCC as both anode and cathode for producing H2 from water. | ||
FCC and PFCC showed very similar high-resolution XPS spectra (C 1s and Ru 3d, Fig. 2d, S10†) with two peaks at about 284 and 292 eV, which arises from sp2 carbons in the graphitic structure and the CF2 groups incorporated upon fluorination, respectively.42 In contrast, RuPFCC shows a higher peak intensity of the band located at 284 eV and an extra Ru 3d signal, which is a composition of three different Ru peaks, attributed to N(pyridine)-Ru, N(mcbp ligand)-Ru, and O-Ru.42 Impedance results are shown as Nyquist plots (Fig. 3f), where the semicircles in the high-frequency regime are assigned to a charge transfer process (Mechanism I), a linear region in the intermediate frequencies is associated to mass transport processes (Mechanism II), and a final linear region with a higher slope in the low frequency region is assigned to capacitive behaviour (Mechanism III). The diameter of the semicircle corresponds to the charge transfer resistance (Rct).43,44 A clear reduction in the Rct is observed for RuPFCC, demonstrating an enhancement in the electrochemical activity. The appearance of capacitive behavior at higher frequencies for RuPFCC, is associated to a faster mass transport to the metal centers. In other words, the active sites in the molecular catalyst are easily accessible for substrate (H2O) molecules.
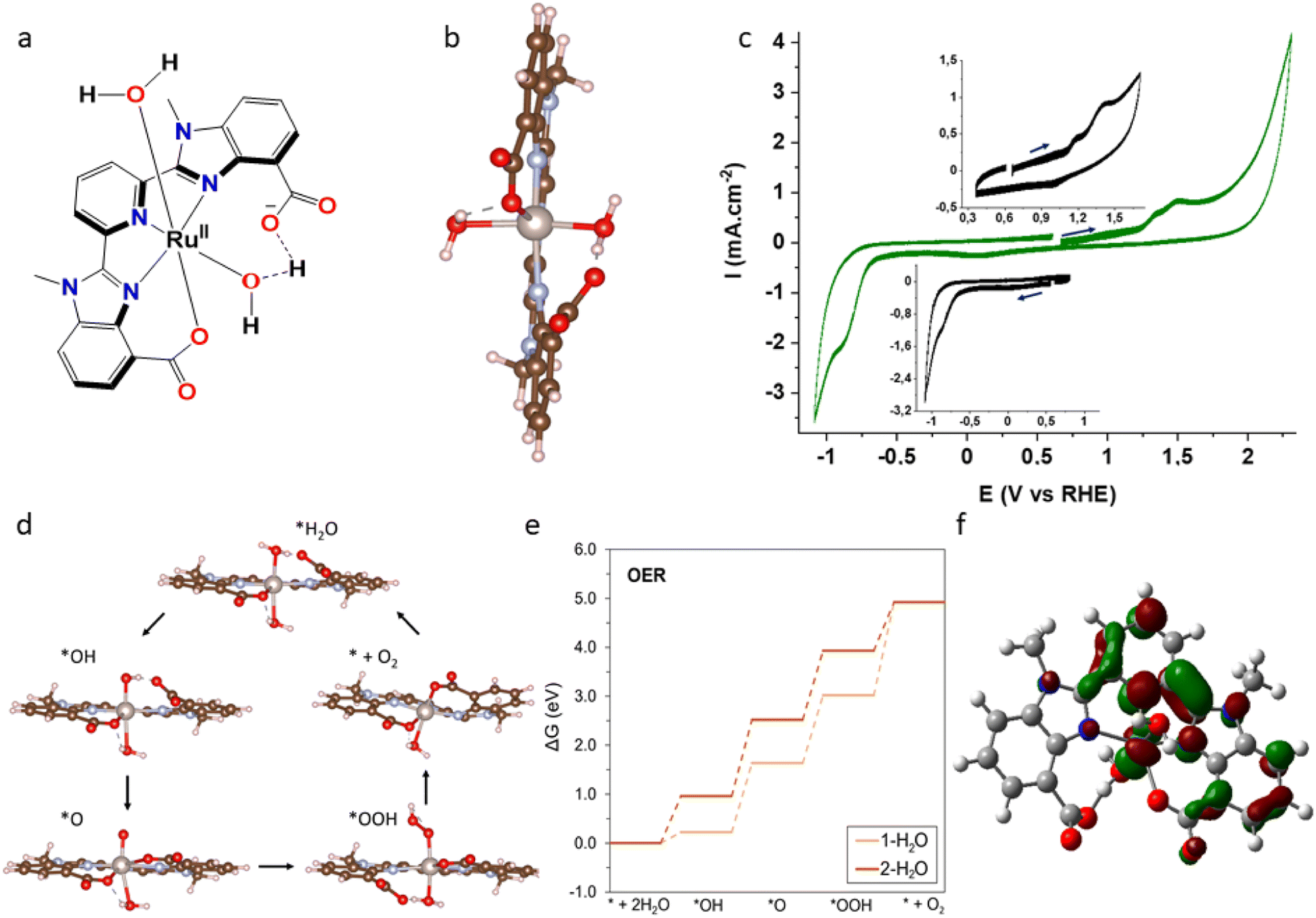 | ||
| Fig. 2 (a) Graphical representation of 1 [RuII(mcbp)(H2O)2], (b) DFT optimized structure of 1 highlighting plausible H-bonding interactions and coordinated aqua molecules. The ruthenium, carbon, oxygen, nitrogen, and hydrogen atoms are represented as grey, brown, red, blue and pinkish-white spheres, respectively, and (c) CVs (–1.1 V to +2.3 V, vs. RHE) of a pH 7.0 phosphate buffer (0.1 M) solution using a glassy carbon (d = 3 mm) as the working electrode (WE) at a scan rate of 100 mV s−1, in presence (green) of 1 mM of 1 (homogeneous conditions). Pt wire and an Ag/AgCl (3.0 M KCl) electrodes were used as counter electrode and reference electrode respectively. Insets show first two oxidations (+0.35 v to +1.70 V) and reductions (between 0.8 to −1.1 V), (d) structural models of the different OER intermediates used in our DFT calculations. The example of Ru-2H2O is shown. (e) The OER potential energy diagram comparing the energetics at the Ru-1H2O and Ru-2H2O active site, (f) LUMO representation of the DFT optimized (unrestricted, using SMD water solvation model and b97d3 function) structure of 1. | ||
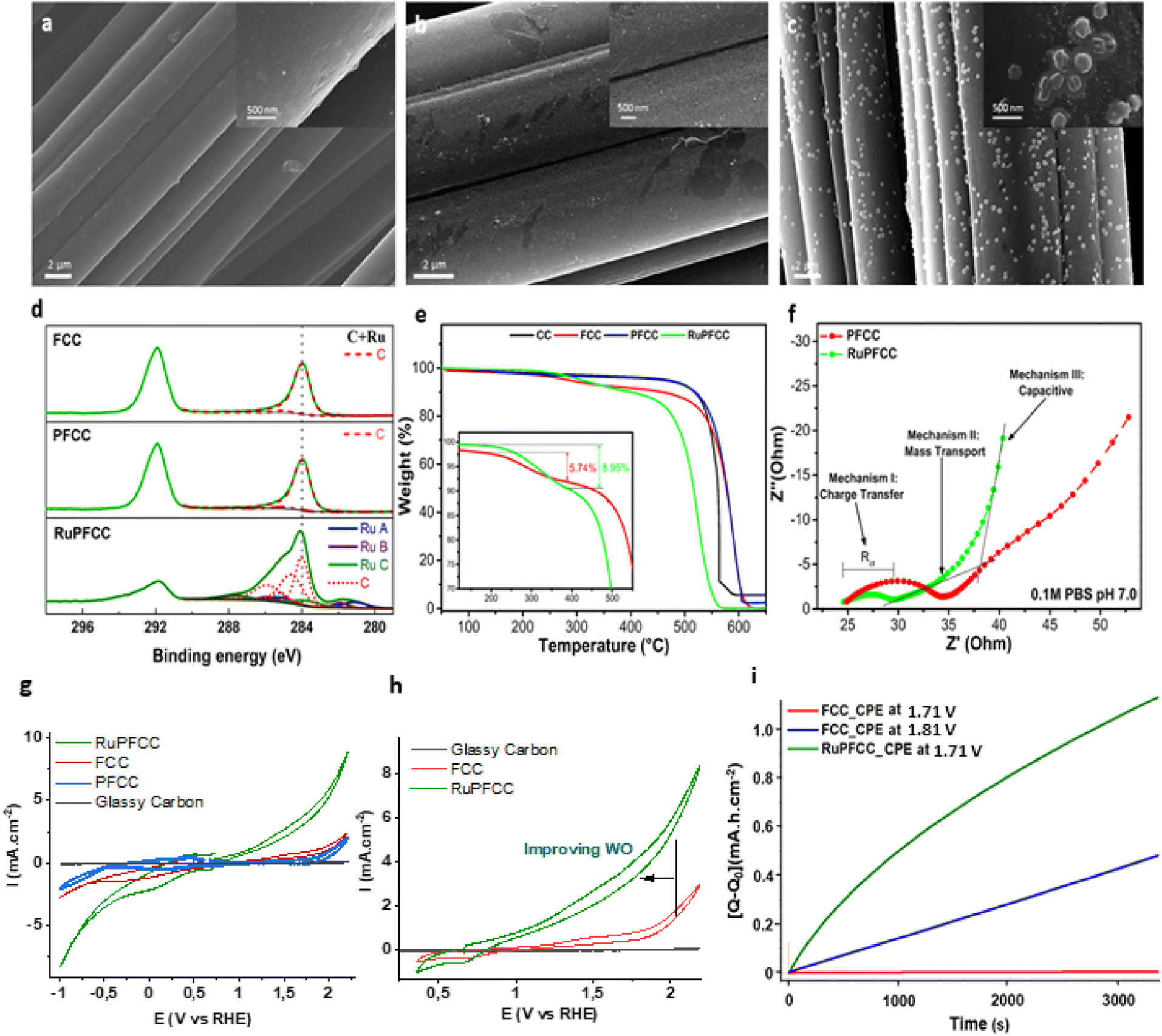 | ||
| Fig. 3 SEM images for (a) FCC (b) PFCC and (c) RuPFCC. (d) XPS spectra of C 1s of FCC, PFCC and RuPFCC, (e) TGA of CC, FCC, PFCC and RuPFCC (discussion in ESI†), (f) EIS of PFCC and RuPFCC, (g) CVs (−1.05 V to +2.2 V, vs. RHE) of a pH 7 phosphate buffer (0.1 M) solution using 1 cm × 0.5 cm RuPFCC (green trace), FCC (red trace), PFCC (blue trace) and a glassy carbon (d = 1 mm) (grey trace) as the working electrodes (WE) at scan rate of 100 mV s−1. Pt wire and Ag/AgCl (3.0 M KCl) electrodes were used as counter electrode and reference electrode respectively, (h) CVs (0.35 V to +2.2 V, vs. RHE) using 1 cm × 0.5 cm RuPFCC (green trace), FCC (red trace), and a glassy carbon (d = 1 mm) (grey trace) as the working electrodes (WE), (i) CPE results focusing over first 3.4k seconds performed at 1.71 V [using 1 cm × 0.5 cm RuPFCC (green)] and at 1.81 V and 1.71 V [using 1 cm × 0.5 cm FCC (blue and red trace respectively)]. | ||
2.3. Electrochemical response and oxygen evolution experiments using RuPFCC as anode
The cyclic voltammograms of FCC, PFCC and RuPFCC as working electrodes were recorded in pH 7 phosphate buffer (0.1 M) from +2.21 V to −1.0 V (vs. RHE) (Fig. 3g). In the case of the FCC, an irreversible oxidation was observed at around +1.41 V (vs. RHE), which was ca. 200 mV shifted relative to that recorded by Dutta and co-workers in 5 M NaCl solution using FCC.45 Compared to FCC and PFCC, RuPFCC showed very different oxidation and reduction features throughout the redox window (+2.25 V to −1.0 V, vs. RHE). An irreversible oxidation wave at around 1.26 V was observed, followed by characteristic WO wave. The redox waves observed at 1.05 V and 1.38 V (vs. RHE) in the homogeneous conditions (discussed above) are not distinguishable on the RuPFCC surface, possibly due to the much higher current on the conductive surface and improved overpotentials for WO and WR. In the reduction window, a prominent irreversible reduction at around 0.06 V (attributed to the mcbp2− centred reduction as discussed above) was observed followed by a characteristic water reduction (WR) wave starting from at around −0.34 V. No such distinct features for WO or WR were observed between 1.9 V and −0.8 V with PFCC. Instead PFCC showed a small positive shift (∼50 mV) of the irreversible reduction wave, with associated diminution of both the catalytic oxidation and reduction behaviour. In comparison to these two electrodes, RuPFCC showed higher activity for both oxidation and reduction (Fig. 3g). Almost 500 mV improvement of the WO and WR overpotentials (compared to FCC and PFCC) accompanied with a nearly five-fold increment in the current at +1.71 V and a four-fold increment of the current at −0.81 V (Fig. 3g and h) could be observed.Controlled potential electrolysis (CPE) experiments were performed at 1.71 V (8 h and 20 h), and −0.81 V to confirm WO (through oxygen detection) and WR (through hydrogen detection) and to investigate electrode performance (charge accumulation and current density) and stability. No drastic changes in the charge or current were observed, however, a continuous electron transfer could be seen in both the cases. For WO, RuPFCC yielded a TO of >1.7 × 103 over a period of 8 h at 1.71 V. During the first two hours of the first CPE experiment, 12.5 μmol of oxygen was evolved with 82% of Faraday efficiency (Table 1, entry 1 and Fig. S15†). Yagi et al., recently reported 91% Faraday efficiency of oxygen evolution with the RuCP electrode {RuCP = immobilized proximal,proximal-[Ru2L(C8Otpy)2(OH)(OH2)]3+ on carbon paper, where C8Otpy = 4′-octyloxy-2,2′; 6′,2′′-terpyridine and L = 5-phenyl-2,8-di(2-pyridyl)-1,9,10-anthyridine} for the first one hour of electrolysis at 2.01 V at pH 7.20 The Faraday efficiency with RuCP electrode over the first 3 h decreases to 45% when the applied potential was decreased by 450 mV. In our study, when the CPE using RuPFCC was repeated at 1.81 V for 48 h a TON of >7.4 × 103 could be achieved. The efficiency of the resulting electrode after CPE experiments were checked before and after washing with water in a fresh buffer (pH 7) solution. When the electrode was reused as a WO anode under the same reaction conditions after 48 h of continuous oxygen evolution, it evolved 3.5 μmol of oxygen over the first two hours of CPE with a 44% Faraday efficiency (Table 1, entry 2). Compared to the first CPE, a clear difference (Fig. 4a) in the current density was also observed this time.
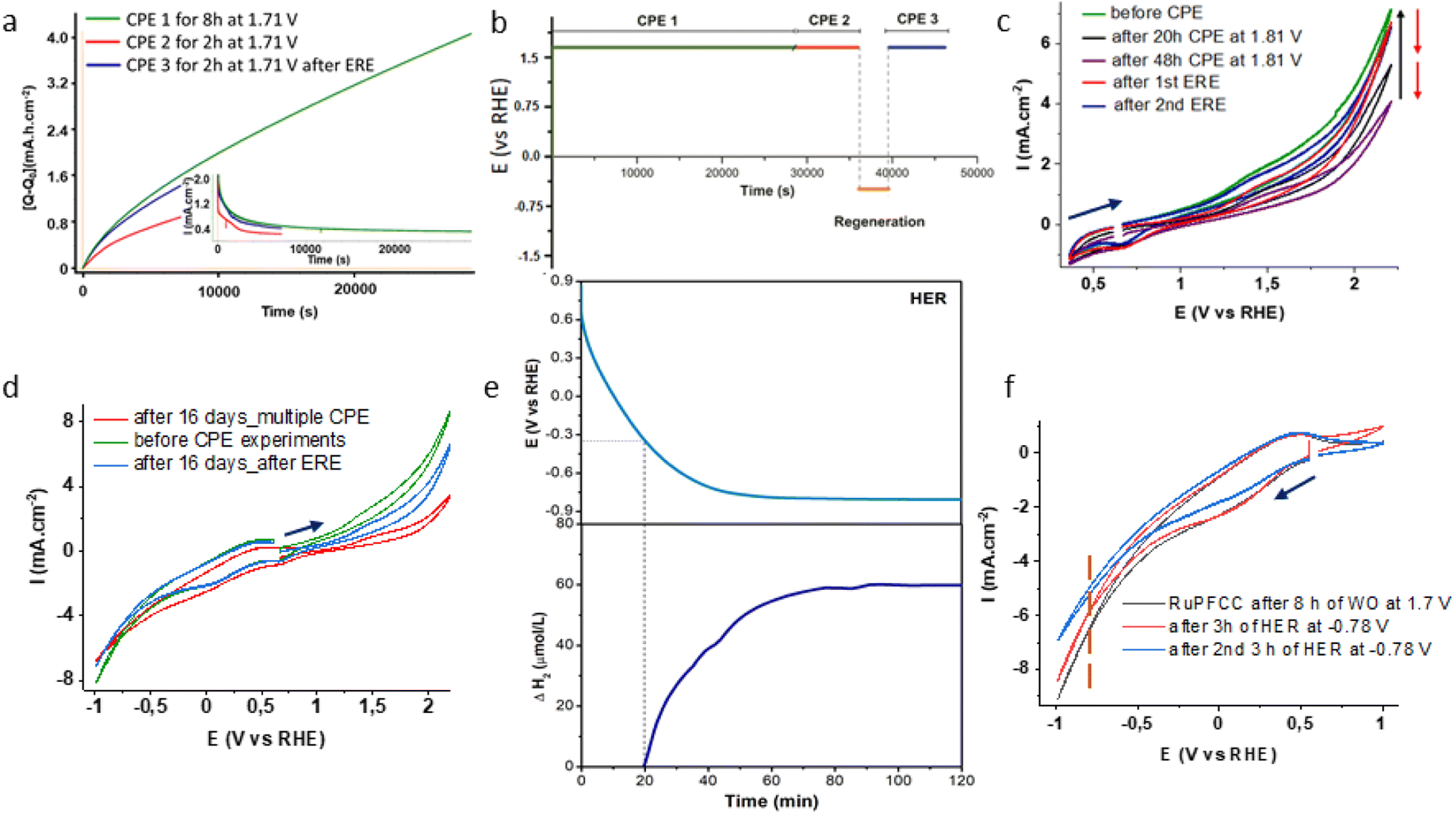 | ||
| Fig. 4 (a) Positive charge (Q) vs. time (s) plot for CPE over 8 h, at 1.71 V (green trace, using 1 cm × 0.5 cm RuPFCC as working electrode), next day over 2 h (red trace) and after the electrochemical regeneration of the electrode (ERE, see ESI†) over 1 h. Inset shows corresponding current (mA) vs. time (s) plot. For first 1k seconds 98% recovery was possible, (b) overall oxidation and ERE (electrochemical regeneration experiment) timeline; (c) CVs (from +0.35 V to +2.21 V) before starting CPE experiment (dark green), after 20 h (black trace) and 48 h (purple trace) of CPE experiments at +1.81 V. Red and blue traces show regeneration of the electrocatalytic activity after 68 h of oxidation; (d) CVs (from –1.0 V to +2.21 V) of RuPFCC before CPE experiment (dark green), after 16 days (red) kept in the same buffer solution under N2 and multiple CPE experiments (4 oxidations of 4 h, 8 h, 20 h and 48 h at +1.81 V and 3 ERE of 30 min, 1 h and 2.5 h at –0.59 V) and after last ERE (6 h) (blue trace); (e) potential (V) vs. time (min) (above) and corresponding H2 detection (below) plots of a chronopotentiometric experiment at a constant current of –5 mA cm−2 in pH 7 phosphate buffer electrolyte over 2 h and (f) electrochemical response of RuPFCC under the HER conditions. In both the HER related experiments, RuPFCC (1 cm × 0.5 cm) after 8 h of WO was used as working electrode. Carbon paper (1 cm × 0.5 cm) and Ag/AgCl (3.0 M KCl) electrodes were used as counter electrode and reference electrode respectively. | ||
A small positive shift in WO oxidation potential and a decrease in the current density were observed after every oxidation (CPE) process (Fig. 4c and d). A longer CPE process caused a more prominent change. 1H and 19F NMR (500 MHz) of the solution after the electrolysis confirmed that no catalyst and fluoride leaching happened during the CPE process. In an additional experiment, the Pt wire was replaced by a carbon rod as counter electrode for 48 h to investigate whether Pt corrosion is impacting the overall reactivity. No appreciable difference (<5% at 1.75 V) in the cyclic voltammetric response was observed.
In a separate experiment, dissolved oxygen was monitored by chronopotentiometry keeping a constant current density at 2.6 mA cm−2 (Fig. S16†). A sharp increase of oxygen close to the working electrode at a potential of 1.44 V (oxygen evolution rate of 1.048 μmol min−1 L−1 for first 30 min) was seen, corresponding to an overpotential of ∼215 mV. This indicates an appreciable shift (∼415 mV) in the overpotential of WO upon coordination of the electrocatalyst onto the PFCC through the pyridine linkers.39
2.4. Electrochemical regeneration of RuPFCCox anode and plausible mechanism
In terms of the overpotential for OER and turnovers at minimum applied potential (Table 2) as a molecular anode, RuPFCC can be considered a state-of-the-art WO electrode. Envisioning that the prolonged oxidation might be a reason for partial deactivation and looking at the exciting electrochemical behaviour of RuPFCC in the negative potentials (Fig. 4d), we performed a CPE experiment at –0.59 V (see ESI†) for 60 min after the 8 h CPE at +1.71 V.24 This reduction recovered the potential of WO. The next CPE at +1.71 V using the regenerated electrode as anode showed as high as 98% of the current density for the first 1k seconds (Fig. 4a and b) and evolution of 9.6 μmol of oxygen over the first two hours of CPE with 77% Faraday efficiency (Table 1, entry 3) was observed. After 68 h (20 h and 48 h) of oxidation at 1.81 V, up to 94% of the current density (measured at 2.21 V) could be recovered following two consecutive reductions at –1.0 V for 4 h (Fig. 4c). The electrode's performance after 16 days (under nitrogen) of multiple oxidations and reductions and an electrochemical regeneration of the electrode (ERE) at –0.59 V for 6 h, recovered 74% of the corresponding current density (Fig. 4d).| Entry | Catalyst | Conductive support | pH | η (mV) | TO (h, AP) |
|---|---|---|---|---|---|
| a Calculated value from the reported electrochemical response. | |||||
| 1 | [Ru(mcbp)(H2O)2] (1) | PFCC (this work) | 7 | 215 | 7430 (48, 1.40) |
| 2 (ref. 20) | Ru2L(C8Otpy)2(OH)(OH2)]+ | Carbon paper | 7 | 260 | 4080 (35, 1.64) |
| 3 (ref. 46) | Ru2L(cptpy)2(OH)(OH2)]+ | Nano-TiO2/ITO | 7 | 530 | 416 (3, 1.60) |
| 4 (ref. 46) | [Ru2(Hcptpy)2L(μ-Cl)]3+ | Nano-TiO2/ITO | 7 | 830a | 22 (3, 1.60) |
| 5 (ref. 17) | Ru(tda)(4, 4′-bpy)]15(4, 4′-bpy) | MWCNT/GC | 7 | 400 | 2 × 105 (12, 1.86) |
| 6 (ref. 39) | Poly-[Ru(tda)(pyrS)2] | Carbon paper | 7 | 385a | 5000 (0.5, 1.40) |
An increase in the broad irreversible reduction wave was observed at around −0.19 V after 72 h of continuous OER at 1.71 V, which can be attributed to the pyridinium(py+)/pyridine(py) redox wave, (Fig. S17†).31 This disappeared after 2 h of ERE at −0.59 V (Fig. S17†). Moreover, pH changes of the bulk solution from 7.1 to 6.6 after 72 h of continuous OE was observed. An increase in the protonation of the free pyridyl units of the RuPFCC surface upon prolonged WO experiments leads to a decrease of the electrocatalytic WO activity due to the lesser availability of the proton acceptors on the electrode surface. These finding allowed us to propose a mechanism (Figure S18†) where the ERE experiment allows deprotonation of the pyridinium units and regeneration of the WO efficiency.
2.5. Hydrogen evolution experiments
Interestingly, gas bubbles around the working RuPFCCox electrode were seen while performing this ERE process, i.e., during the WO recovery. The hydrogen detector confirmed the identity of the gas bubbles as H2 throughout this process. In our previous work, we could not see any such gas bubbles around CoPFCCox while performing ERE process at −0.59 V and −0.79 V.24 The production of H2 at around −0.59 V is an additional characteristic of this ruthenium electrocatalyst anchored electrode, RuPFCC. Understanding that one of the major challenges on the way of producing H2 at neutral pH and aqueous solution is still the lack of efficient electrodes for HER, a series of new experiments using RuPFCC as cathode at the pH 7 phosphate buffer solution were carried out.47,48 Chronopotentiometric testing was conducted (Fig. 4e) in order to assess the stability of the electrocatalytic properties and confirm the evolution of H2 gas. A constant current of −5 mA cm−2 was applied over 120 min, while the dissolved gas was detected using a microsensor (Unisense) immersed in the electrolyte (pH 7, 0.1 M PBS). During cathodic polarization, a delay between the current applied and the hydrogen production was observed, which was ascribed to the capacitive charging of the carbon substrate. Thus, after 20 min of polarization the onset potential for the hydrogen evolution reaction is reached (−0.34 V vs. RHE) and a sharp increase in the hydrogen content is detected with an overpotential of ∼330 mV.The electrode potential reached a stable value of −0.79 V vs. RHE after 55 min, showing no sign of degradation (Fig. 4e). The hydrogen content reached a stable value after 70 min, which could be associated to the release of hydrogen into the gas phase. Another set CPE experiments at −0.79 V (vs. RHE) reveals that RuPFCC loses its electrocatalytic proton reduction activity by 8% (measured at −1.0 V) after 3 h of constant PR and by 24% after 20 h of PR (Fig. 4f). During the 3 h PR experiment at applied potential of −0.79 V, a total TOPR of 3680 was calculated from the current density plot (see ESI†). In another experiment, when a freshly prepared RuPFCC was tested under HER conditions, at −0.79 V, it showed nearly 18% lower current density in the first 2 h of the experiment (Fig. S19†) than RuPFCCox. This supports our proposed mechanism (Fig. S18†) where the formation of the pyridinium units on RuPFCC surface (RuPFCCox) during the WO experiment makes it a better electrode for HER. While releasing H2 during HER process, pyridinium units transform back to pyridines which makes it again suitable for WO, i.e., regeneration. This simple but effective design strategy can potentially be used for electrode preparation in various proton dependent electrochemical processes.
The overall water electrolysis at pH 7 and quantification of the evolved gas from the anode and cathode compartments were performed using a commercial H-cell and a home-made volumetric extension (Fig. S15†). It confirmed the anodic production of O2 and cathodic production of H2 while using RuPFCC as electrodes in both the compartments. An overall cell voltage of 1.78 V at pH 7 for anodic OER (starts at 1.44 V) and cathodic HER (starts at −0.34 V) was calculated. Although these values (overpotential and overall cell voltage) are on the higher side compared to reported state-of-the-art RuO2/IrO2 cluster and nanostructured transition metal electrocatalysts (overall cell voltages are around 1.55 V), low loading (∼0.04 wt% ruthenium) of the ruthenium centres on RuPFCC, together with its regeneration feature can make the water electrolysis cost-effective, as well environmentally more benign.49,50
Electron microscopy studies were carried out to examine the chemical stability of the RuPFCC electrode after continuous electrolysis for 12 h of OER (∼98% regenerable), 16 days (∼74% regenerable), 3 and 4 months (<60% regenerable), of multiple OER and HER experiments. It can be observed that the extent of over-oxidation of the molecular catalysts gradually increases with the number of OER and HER. As indicated in the TEM images (Fig. S21–S23†), the oxidation product, RuO2 nanoparticles were formed on the molecular electrode and the amount increases with the time of electrolysis.
As previously reported by our group, to be effective, the ruthenium center of 1 must proceed RuIV/RuV oxidation, which is involved in the rate-limiting oxygen–oxygen bond formation step through water nucleophilic attack.22 For this, it is pivotal to have the mcbp2− type ligand system remain attached to the ruthenium center. Over-oxidation can produce different kinds of ruthenium oxides, among which (i) mesoporous RuO2 (MP-RuO2), (ii) commercially available RuO2 nanoparticles (C-RuO2) and (iii) high surface area containing RuO2, also known as Ad-RuO2 are extensively discussed.51,52 While MP-RuO2 showed interesting OER activities, others have shown less to almost no reactivity. Most likely, the molecular catalysts on PFCC after more than 65 h of continuous OER experiment transform into appreciable amount of nonreactive/unreactive ruthenium oxides that reflects in incomplete (<95%) regeneration of the electrode's activity. The formed RuO2 nanoparticles on the PFCC surface seem to be less active than 1. Although no catalyst leaching could be observed (by high resolution NMR with 10% D2O insert) in the resulting electrolyte solution after more than 65 h of continuous electrolysis, after 4 months of multiple OER and HER experiments fragmented H2mcbp ligands (Fig. S25†) could be seen, indicating a slow decomposition of the electrocatalyst.
3. Experimental section
3.1. Preparation of fluorine doped carbon cloth (FCC) and pyridine modified fluorine doped carbon cloth (PFCC)
FCC and PFCC were prepared following a previously reported procedure from our group.243.2. Synthesis of [RuII(mcbp)(H2O)2] (1)
The H2mcbp ligand for the catalyst [RuII(mcbp)(H2O)2] (1) was prepared following another reported procedure from us.21 A EtOH/H2O (4:1) solution (25 mL) of H2mcbp (0.2 g, 0.47 mmol) and of Ru(DMSO)4Cl2 (0.23 g, 0.47 mmol, 1 equiv.) was degassed (by N2) for a period of 5 min before adding degassed Et3N (1.41 mmol, 0.2 mL, 3 equiv.). The resulting solution was refluxed at 80 °C for a period of 5 h. The color change from yellow to dark brown upon heating indicated complexation. The solution was filtered and kept in a fridge overnight to get a brown precipitate. The precipitate was collected and washed with ice-cold ethanol and diethyl ether to get a brown powder of 1. It was dried overnight under vacuum (0.16 g, 61% yield) and characterized by HRMS and NMR spectroscopy before anchoring to PFCC. The existence of water molecules in the structure is supported by the temperature dependent NMR, ESI-MS and UV-Vis spectroscopic analysis (Fig. S2, S3, S5, and S6†). 1H NMR (CD3OD): δ 8.54 (d, J = 8.2 Hz, 2H), 8.20 (t, J = 8.2 Hz, 1H), 8.03 (dd, J = 8.4 and 0.7 Hz, 2H), 8.01 (dd, J = 7.4 Hz, 0.7 Hz, 2H), 7.71 (dd, J = 8.4 Hz, 7.4 Hz, 2H), 6.90 (s, 1H), 6.61 (s, 1H), 4.54 (s, 6H, –CH3 protons). ESI-HRMS in MeOH: calculated for [RuII(mcbp)Na]+m/z 550.0066 (C23H15N5O4RuNa+), and experimental m/z 550.0039.
3.3. Anchoring of [RuII(mcbp)(H2O)] onto PFCC
A 1.5 cm × 3 cm foil of PFCC was dipped into a degassed 10 mL of MeOH:H2O (4:1) solution of 0.05 M of 1 and left in the solution for 5 h at 50 °C under continuous stirring, followed by washing with cold water (2 × 5 mL) to remove the loosely bound catalysts from the PFCC surface. The resulting electrode was dried overnight in a 65 °C oven to get RuPFCC (ruthenium-electrocatalyst anchored pyridine modified fluorine doped carbon cloth).
3.4. Techniques used for characterization of PFCC and RuPFCC electrodes' surface
The surface of PFCC and RuPFCC were analyzed by several spectroscopic techniques, including scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), Raman spectral analysis, thermogravimetric analysis (TGA), electrochemical impedance spectroscopy (EIS).The morphology and roughness of PFCC and RuPFCC were determined using SEM using Zeiss GEMINI® Ultra 55. Transmission electron microscopy analysis was performed using an aberration-corrected Themis Z instrument from Thermo Fisher. TEM was operated at 300 kV in Scanning TEM (STEM) mode. A TEM grid was prepared by immersing RuPFCC in 1 M aqueous KOH and ultrasonicating it for 15 minutes, after which it was neutralized with 1 M HCl and dispersed onto a thin carbon support film. This allowed detaching some of the aggregates from RuPFCC (Fig. 3c), enabling a detailed TEM analysis (Fig. S9†). XPS was performed using Thermo Scientific ESCALAB 250Xi with a monochromatic Al Kα source (hν = 1486.6 eV). The Thermo Scientific™ Avantage software was used for data analysis and curve fitting. Raman spectral analysis was performed with an iHR550 Spectrometer (Horiba Jobin Yvon) in the range between 200 to 4000 cm−1 with a 633 nm laser light for excitation. A grating of 1200, a pinhole of 130 with 200 acquisition and a time of 1 s were used to see the changes between FCC, PFCC, and RuPFCC (Fig. S10†). TGA was performed using a TGA Q500 equipment (TA Instrument) under airflow to determine weight losses due to the elimination of surface functionalities and degradation of carbon material. Electrochemical measurements were performed in a conventional three-electrode setup using a Potentiostat/Galvanostat/ZRA Gamry Interface 1010E. EIS was performed in potentiostatic mode at open circuit potential (bias potential) within a frequency range from 100 kHz to 10 MHz. The amplitude of the sinusoidal was 10 mV rms.
3.5. Detection techniques for oxygen and hydrogen
Control potential electrolysis experiments were conducted to confirm the oxidation of water, and reduction of protons. These experiments also allowed evaluation the stability of the electrode surface. For oxygen evolution, a constant current of 2.6 mA cm−2 and for hydrogen production, a constant current of −5 mA cm−2 was applied over 120 min to a RuPFCC working electrode (1 cm × 1 cm). The experiments were performed in a 0.1 M phosphate buffer Solution (pH 7), with a carbon counter electrode and Ag/AgCl as reference electrode. The dissolved gas was detected in a closed system using an O2 and H2 measurement system (Unisense) comprising a needle sensor of 2.1 × 80 mm connected to an O2 and H2 UniAmp units. Prior to starting the CPE experiments, the system was purged with N2 for 30 min.4. Conclusions
We have demonstrated an electrochemically regenerable and highly efficient molecular electrode that was prepared by anchoring a Ru-electrocatalyst onto a conductive PFCC surface. The catalyst [RuII(mcbp)(H2O)2] (1) with labile coordination (H2O) sites was attached onto the PFCC through favorable Ru–N interactions. The resulting RuPFCC electrode (with ∼0.04 wt% ruthenium) initiated WO at ∼215 mV overpotential and HER at ∼330 mV overpotential in a pH 7 phosphate buffer. A TO of >7 × 103 over a period of 48 h of continuous WO at 1.81 V and TO of >3.6 × 103 at −0.79 V for HER in the first 3 h was observed. Around 82% Faraday efficiency during the first 2 h of WO-CPE experiment and up to 94% electrochemical regeneration of the electrocatalytic WO activity after 68 h of WO were observed. Moreover, after 16 days of multiple electrolysis experiments, 74% of the original activity could be recovered. Overall, this work provides an efficient strategy for developing ultra-stable, bifunctional (OER and HER) molecular electrodes with exciting electrochemical regeneration originating from the unique design and highly active ruthenium centre encapsulated by a non-innocent benzimidazole-carboxylate ligand framework. Being a molecular electrode, it also allows us to improve the activity further, through strategic modifications of the ligand framework as well as the linker.Author contributions
BD, OV and BÅ designed and supervised the project; BD and ET performed the majority of the experiments. GL, JS, TT, JC, LL, FY, AS, MG, TW, MK, JD helped in characterizing the electrode material and analysing the results. JAY, PVK, and BD did the theoretical calculations. All the authors participated in the discussion and writing of this manuscript.Conflicts of interest
The authors declare no known competing interest.Acknowledgements
BD, OV, JD, BÅ would like to thank Futura foundation for the financial support. A. S. acknowledges funding from Vinnova, Sweden's Innovation Agency (project: C1Bio, reference number 2019-03174). B. D. and B. Å. acknowledge the computational facilities from Swedish National Infrastructure [Computational time (SNIC 2020/13-64) awarded to B. D.]. E. T. thanks the National Research and Development Agency of Chile – ANID (former CONICYT) for Doctoral scholarship “Beca Chile” 2018-72190682. T. T. acknowledges support from the Electron Microscopy Center (EMC) at Stockholm University. MG will like to acknowledge economic support from VR (Sweden) for the Raman spectrometer. J. A. Y. and P. V. K. acknowledge the resources and services from the National Computational Infrastructure (NCI), which is supported by the Australian Government. P. V. K. acknowledges the Scientia Fellowship scheme at The University of New South Wales (UNSW) and the ARC for financial support through the Discovery Early Career Researcher Award (DE210101259).References
- W. Lubitz and W. Tumas, Chem. Rev., 2007, 107, 3900–3903 CrossRef CAS PubMed.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed.
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef CAS PubMed.
- B. Zhang, J. Shan, W. Wang, P. Tsiakaras, Y. Li, B. Zhang, J. Shan, W. Wang, Y. Li and P. Tsiakaras, Small, 2022, 18, 2106012 CrossRef CAS PubMed.
- P. Garrido-Barros, I. Funes-Ardoiz, S. Drouet, J. Benet-Buchholz, F. Maseras and A. Llobet, J. Am. Chem. Soc., 2015, 137, 6758–6761 CrossRef CAS PubMed.
- R. Pokhrel, M. K. Goetz, S. E. Shaner, X. Wu and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 8384–8387 CrossRef CAS PubMed.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, S. Trudel and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 11580–11586 CrossRef CAS PubMed.
- B. Das, A. Rahaman, A. Shatskiy, O. Verho, M. D. Kärkäs and B. Åkermark, Acc. Chem. Res., 2021, 54, 3326–3337 CrossRef CAS PubMed.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed.
- T. J. Meyer, M. V. Sheridan and B. D. Sherman, Chem. Soc. Rev., 2017, 46, 6148–6169 RSC.
- J. Xie, S. A. Shevlin, Q. Ruan, S. J. A. Moniz, Y. Liu, X. Liu, Y. Li, C. C. Lau, Z. X. Guo and J. Tang, Energy Environ. Sci., 2018, 11, 1617–1624 RSC.
- J. L. Boyer, D. E. Polyansky, D. J. Szalda, R. Zong, R. P. Thummel and E. Fujita, Angew. Chem., Int. Ed., 2011, 50, 12600–12604 CrossRef CAS PubMed.
- B. Das, A. Thapper, S. Ott and S. B. Colbran, Sustainable Energy Fuels, 2019, 3, 2159–2175 RSC.
- K. Torbensen, D. Joulié, S. Ren, M. Wang, D. Salvatore, C. P. Berlinguette and M. Robert, ACS Energy Lett., 2020, 5, 1512–1518 CrossRef CAS.
- J. Soriano-López and C. Casadevall, Water, 2022, 14, 371 CrossRef.
- M. A. Hoque, M. Gil-Sepulcre, A. de Aguirre, J. A. A. W. Elemans, D. Moonshiram, R. Matheu, Y. Shi, J. Benet-Buchholz, X. Sala, M. Malfois, E. Solano, J. Lim, A. Garzón-Manjón, C. Scheu, M. Lanza, F. Maseras, C. Gimbert-Suriñach and A. Llobet, Nat. Chem., 2020, 12, 1060–1066 CrossRef CAS PubMed.
- C. Jia, K. Ching, P. V. Kumar, C. Zhao, N. Kumar, X. Chen and B. Das, ACS Appl. Mater. Interfaces, 2020, 12, 41288–41293 CrossRef CAS PubMed.
- H.-Y. Liu, C. C. Cody, J. A. Jacob-Dolan, R. H. Crabtree and G. W. Brudvig, ACS Energy Lett., 2020, 5, 3195–3202 CrossRef CAS.
- Y. Tsubonouchi, T. Hayasaka, Y. Wakai, E. A. Mohamed, Z. N. Zahran and M. Yagi, ACS Appl. Mater. Interfaces, 2022, 14, 15154–15164 CrossRef CAS PubMed.
- J. Yang, L. Wang, S. Zhan, H. Zou, H. Chen, M. S. G. Ahlquist, L. Duan and L. Sun, Nat. Commun., 2021, 12, 1–10 CrossRef PubMed.
- A. Shatskiy, A. A. Bardin, M. Oschmann, R. Matheu, J. Benet-Buchholz, L. Eriksson, M. D. Kärkäs, E. V. Johnston, C. Gimbert-Suriñach, A. Llobet and B. Åkermark, ChemSusChem, 2019, 12, 2251–2262 CrossRef CAS PubMed.
- E. Toledo-Carrillo, X. Zhang, K. Laxman and J. Dutta, Electrochim. Acta, 2020, 358, 136939 CrossRef CAS.
- B. Das, E. A. Toledo-Carrillo, L. Li, F. Ye, J. Chen, A. Slabon, O. Verho, L. Eriksson, M. Göthelid, J. Dutta and B. Åkermark, ChemCatChem, 2022, e202200538 CAS.
- S. Sanati, A. Morsali and H. García, Energy Environ. Sci., 2022, 15, 3119–3151 RSC.
- H. J. Kim, H. Y. Kim, J. Joo, S. H. Joo, J. S. Lim, J. Lee, H. Huang, M. Shao, J. Hu, J. Y. Kim, B. J. Min, S. W. Lee, M. Kang, K. Lee, S. Choi, Y. Park, Y. Wang, J. Li, Z. Zhang, J. Ma and S. Il Choi, J. Mater. Chem. A, 2022, 10, 50–88 RSC.
- H. Zeng, X. Liu, F. Chen, Z. Chen, X. Fan and W. Lau, ACS Appl. Mater. Interfaces, 2020, 12, 52549–52559 CrossRef CAS PubMed.
- N. Mushtaq, C. Qiao, H. Tabassum, M. Naveed, M. Tahir, Y. Zhu, M. Naeem, W. Younas and C. Cao, Sustainable Energy Fuels, 2020, 4, 5294–5300 RSC.
- R. Prasannachandran, T. V. Vineesh, M. B. Lithin, R. Nandakishore and M. M. Shaijumon, Chem. Commun., 2020, 56, 8623–8626 RSC.
- R. Souleymen, Z. Wang, C. Qiao, M. Naveed and C. Cao, J. Mater. Chem. A, 2018, 6, 7592–7607 RSC.
- S. Wang, A. Lu and C. J. Zhong, Nano Convergence, 2021, 8, 1–23 CrossRef CAS PubMed.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- J. An, L. Duan and L. Sun, Faraday Discuss., 2012, 155, 267–275 RSC.
- B. Das, C. Jia, K. Ching, M. Bhadbhade, X. Chen, G. E. Ball, S. B. Colbran and C. Zhao, ChemCatChem, 2020, 12, 1292–1296 CrossRef CAS.
- B. Das, L. Ezzedinloo, M. Bhadbhade, M. P. Bucknall and S. B. Colbran, Chem. Commun., 2017, 53, 10006–10009 RSC.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2008, 38, 89–99 RSC.
- C. Bachmann, B. Probst, M. Oberholzer, T. Fox and R. Alberto, Chem. Sci., 2015, 7, 436–445 RSC.
- L. Li, B. Das, A. Rahaman, A. Shatskiy, F. Ye, P. Cheng, C. Yuan, Z. Yang, O. Verho, M. D. Kärkäs, J. Dutta, T. C. Weng and B. Åkermark, Dalton Trans., 2022, 51, 7957–7965 RSC.
- M. Ventosa, M. Gil-Sepulcre, J. Benet-Buchholz, C. Gimbert-Suriñach and A. Llobet, ACS Appl. Energy Mater., 2021, 4, 9775–9782 CrossRef CAS.
- S. Neudeck, S. Maji, I. López, S. Meyer, F. Meyer and A. Llobet, J. Am. Chem. Soc., 2013, 136, 24–27 CrossRef PubMed.
- J. Wiberg, L. Guo, K. Pettersson, D. Nilsson, T. Ljungdahl, J. Mårtensson and B. Albinsson, J. Am. Chem. Soc., 2007, 129, 155–163 CrossRef CAS PubMed.
- X. Liang, M. Lao, D. Pan, S. Liang, D. Huang, W. Zhou and J. Guo, Appl. Surf. Sci., 2017, 400, 339–346 CrossRef CAS.
- B. A. Mei, O. Munteshari, J. Lau, B. Dunn and L. Pilon, J. Phys. Chem. C, 2018, 122, 194–206 CrossRef CAS.
- T. W. Lin, C. S. Dai and K. C. Hung, Sci. Rep., 2014, 4, 1–10 Search PubMed.
- M. I. Alvarado Ávila, E. Toledo-Carrillo and J. Dutta, Clean. Eng. Technol., 2020, 1, 100016 CrossRef.
- Y. Tanahashi, S. Nagai, Y. Tsubonouchi, M. Hirahara, T. Sato, E. A. Mohamed, Z. N. Zahran, K. Saito, T. Yui and M. Yagi, ACS Appl. Energy Mater., 2020, 3, 12172–12184 CrossRef CAS.
- Z. Zhou, Z. Pei, L. Wei, S. Zhao, X. Jian and Y. Chen, Energy Environ. Sci., 2020, 13, 3185–3206 RSC.
- X. Xie, M. Song, L. Wang, M. H. Engelhard, L. Luo, A. Miller, Y. Zhang, L. Du, H. Pan, Z. Nie, Y. Chu, L. Estevez, Z. Wei, H. Liu, C. Wang, D. Li and Y. Shao, ACS Catal., 2019, 9, 8712–8718 CrossRef CAS.
- H. Wu, C. Feng, L. Zhang, J. Zhang and D. P. Wilkinson, Electrochem. Energy Rev., 2021, 4, 473–507 CrossRef CAS.
- A. Ali, F. Long and P. K. Shen, Electrochem. Energy Rev., 2022, 5(1) DOI:10.1007/s41918-022-00136-8.
- M. N. Iqbal, A. F. Abdel-Magied, H. N. Abdelhamid, P. Olsén, A. Shatskiy, X. Zou, B. Åkermark, M. D. Kärkäs and E. V. Johnston, ACS Sustainable Chem. Eng., 2017, 5, 9651–9656 CrossRef CAS.
- S. W. Lee, C. Baik and C. Pak, Catal. Today, 2020, 358, 203–209 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta00071k |
| ‡ BD and ET equally contributed to the experimental work. |
| This journal is © The Royal Society of Chemistry 2023 |