
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActive BaTaO2N photocatalysts prepared from an amorphous Ta2O5 precursor for overall water splitting under visible light†
Shinji
Nishimae
a,
Junie Jhon M.
Vequizo
 b,
Yasunobu
Inoue
a,
Akira
Yamakata
c,
Mamiko
Nakabayashi
d,
Tomohiro
Higashi
e and
Kazunari
Domen
*bf
b,
Yasunobu
Inoue
a,
Akira
Yamakata
c,
Mamiko
Nakabayashi
d,
Tomohiro
Higashi
e and
Kazunari
Domen
*bf
aJapan Technological Research Association of Artificial Photosynthetic Chemical Process (ARPChem), 2-11-16 Yayoi, Bunkyo-ku, Tokyo 113-8656, Japan
bResearch Initiative for Supra-Materials, Interdisciplinary Cluster for Cutting Edge Research, Shinshu University, Nagano-shi, Nagano 380-8553, Japan. E-mail: domen@chemsys.t.u-tokyo.ac.jp
cDepartment of Chemistry, Faculty of Science Okayama University, 3-1-1 Tsusimanaka Kita-ku Okayama-shi, Okayama 700-8530, Japan
dInstitute of Engineering Innovation and Department of Chemical System Engineering, School of Engineering, The University of Tokyo, Tokyo 113-8656, Japan
eInstitute for Tenure Track Promotion, University of Miyazaki, Nishi 1-1 Gakuen-kibanadai, Miyazaki 889-2192, Japan
fOffice of University Professors, The University of Tokyo, 2-11-16 Yayoi, Bunkyo-ku, Tokyo 113-8656, Japan
First published on 17th February 2023
Abstract
Barium tantalum oxynitride (BaTaO2N), a photocatalyst active during one-step-excitation overall water splitting under visible light, was synthesized by NH3-based nitridation of a mixture of BaCO3 and amorphous Ta2O5·3H2O. H2 and O2 were generated from water in stoichiometric amounts and in a stable manner using this material modified with Rh (or Ru), Cr2O3 and IrO2 cocatalysts. This photocatalyst was responsive to visible light up to a wavelength of 540 nm for the water splitting reaction. Trials using nitridation temperatures from 1023 to 1273 K and durations from 0.5 to 20 h indicated that mild nitridation conditions such as 1123 K and 5 h provided high-performance specimens, whereas prolonged nitridation (15–20 h) dramatically decreased the photocatalytic activity. Transient absorption spectroscopy showed that BaTaO2N generated abundant photoexcited free electrons with long lifetimes due to a low concentration of defects such as oxygen vacancies, but was deactivated after prolonged nitridation. X-ray photoelectron spectra indicated that overly long nitridation generated recombination sites such as Ta3+. The mechanism by which active BaTaO2N was formed was examined, and the high reactivity of the amorphous Ta2O5·3H2O nanoparticles was determined to be important. The use of highly reactive nanoparticles under mild nitridation conditions could allow the future development of high-performance oxynitride photocatalysts for overall water splitting.
Introduction
Overall water splitting via single-step photoexcitation using particulate photocatalysts is recognized as one of the most useful means of achieving sustainable solar hydrogen production, partly because this process can be readily scaled up.1,2 As an example, solar hydrogen production from water using Al-doped SrTiO3 as an ultraviolet (UV) photoresponsive powder photocatalyst has recently been demonstrated in a 100 m2 system.3 Even so, the development of cost-effective systems will require particulate photocatalysts capable of driving one-step-excitation water splitting in response to visible light wavelengths of 400 to 700 nm. To date, studies regarding visible-light-driven photocatalysts have been primarily performed using (oxy)nitrides4–7 and oxysulfides. Unfortunately, only a limited number of such compounds, including TaON, Zr-doped TaON,8 Ta3N5 grown on KTaO3 (ref. 9) and Y2Ti2O5S2,10 have been reported to be photocatalytically active for overall water splitting.Barium tantalum oxynitride, BaTaO2N, has a narrow bandgap energy of 1.9 eV along with a robust perovskite structure, and is highly stable in water. Thus, this material is considered a promising catalyst capable of utilizing long-wavelength visible light. Significant work has been devoted over the past two decades to tuning the morphology of BaTaO2N to obtain a material that is active for water splitting. Complete nitridation based on reaction durations longer than 10 h under a flow of gaseous NH3 at temperatures higher than 1123 K, both in the absence and presence of a flux, has been employed. BaTaO2N obtained from these processes has been found to promote half-reactions evolving either H2 or O2 when using sacrificial reagents.6,7,11–13 This material has also been applied to Z-scheme water splitting as a particulate photocatalyst for hydrogen evolution14,15 in combination with WO3. Very recently, Mg-doped BaTaO2N was reported to be active for water splitting after having Cr2O3 and Rh deposited on its surface as cocatalysts.16 However, only minimal activity was observed without Mg addition. Overall water splitting has not yet been realized on pristine BaTaO2N particulate photocatalysts, and the successful achievement of this process without the incorporation of dopants remains an important challenge.
The methods used to synthesize BaTaO2N are now commonly based on direct nitridation of mixtures of various materials, such as barium and tantalum oxides, rather than using a barium tantalate precursor such as Ba5Ta4O15. During the nitridation of Ba5Ta4O15 by gaseous NH3, lattice O atoms are replaced by N atoms in conjunction with reduction by hydrogen obtained from the NH3. However, this reaction proceeds from the surface to the interior of the catalyst particles, resulting in inhomogeneous phase formation with variations in the extent of nitridation. In contrast, the nitridation of a mixture involves a solid-phase reaction between barium and tantalum oxides that accompanies the nitridation process and generates a more homogeneous product. Consequently, BaTaO2N exhibiting improved photocatalytic performance can be obtained. As an example, in prior work a mixture of BaCO3, MgO and Ta2O5 was directly nitrided in a RbCl flux to produce active Mg-BaTaO2N.16
Our own group previously demonstrated that amorphous Ta2O5·3H2O nanoparticles can be used as a starting material for the synthesis of TaON and Zr-doped TaON exhibiting very high activity for overall water splitting compared with materials obtained from large crystalline Ta2O5 particles.8 There are two advantages to the use of amorphous Ta complexes. Firstly, the high reactivity of these materials allows rapid nitridation at relatively low temperatures and so may prevent or at least minimize the formation of reduced Ta3+/Ta4+ species. Secondly, smaller catalyst particles can be obtained. Thus, the use of amorphous Ta2O5 is also an interesting approach to the synthesis of BaTaO2N photocatalysts for overall water splitting. On this basis, in the present study, BaTaO2N was prepared by direct nitridation of amorphous Ta2O5 with BaCO3via the reaction
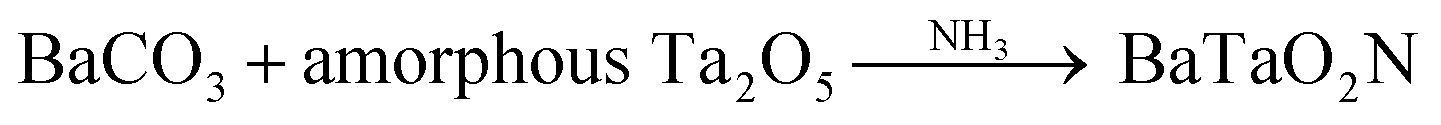 | (1) |
Results and discussion
Characterization of BaTaO2N
Details regarding the preparation of BaTaO2N are provided in the Methods section. Briefly, a white precipitate comprising amorphous Ta2O5·nH2O (n = 3; referred to herein as a-Ta2O5) was obtained by H2SO4 neutralization of an alkaline solution of Na3TaO4 in H2O. This compound was subsequently combined with BaCO3 and then nitrided in an NH3 flow at various temperatures between 1073 and 1273 K, employing nitridation durations between 0.5 and 20 h. Fig. 1 shows X-ray diffraction (XRD) patterns for BaTaO2N specimens prepared at 1123 K with durations from 3 to 20 h. After 3 h of nitridation, intense peaks appeared at 2θ = 30.7° and 43.9° that were attributed to the (110) and (200) planes of BaTaO2N having a characteristic perovskite structure. The XRD pattern for this sample also contained less intense peaks assigned to Ta3N5 and Ba5Ta4O15 phases. After a 5 h nitridation, the peaks due to the oxide disappeared whereas those related to Ta3N5 remained. With increasing nitridation time from 15 to 20 h, the BaTaO2N peaks became more intense and sharp, indicating a higher degree of crystallinity, while Ta3N5 peaks were still observed. Fig. 2 presents UV-visible diffuse reflectance spectra acquired from BaTaO2N samples prepared at 1123 K with different nitridation times. The sample treated for 3 h showed absorption that began at approximately 620 nm and increased with decreasing wavelength. Upon increasing the nitridation time to 7 h or longer, the absorption underwent a red shift. The samples nitrided for 3 and 5 h exhibited strong absorption beyond 650 nm but this absorption was reduced with increasing nitridation time. This was also associated with enhanced crystallinity of BaTaO2N.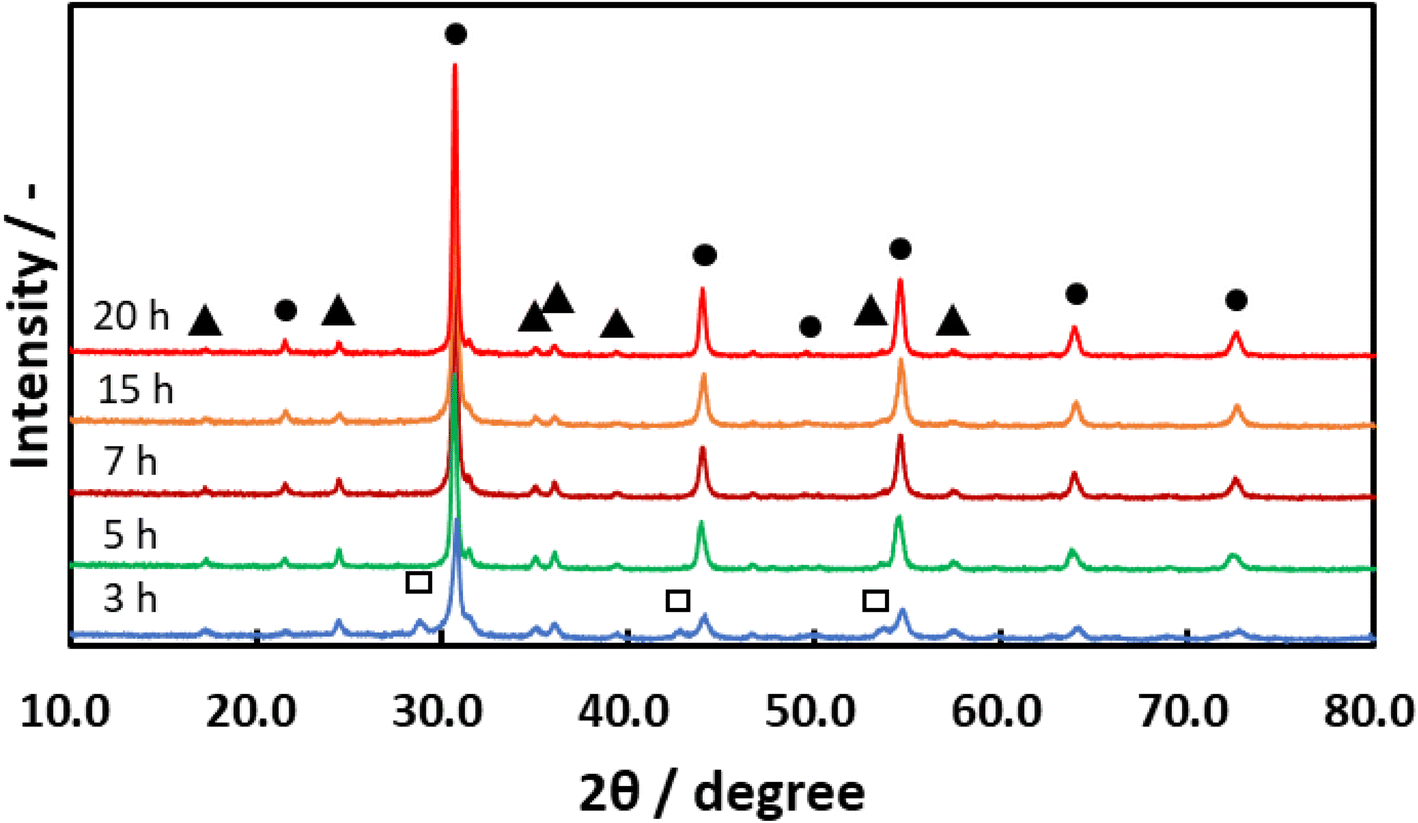 | ||
| Fig. 1 X-ray diffraction patterns for BaTaO2N prepared in a flow of NH3 at 1123 K using durations of 3–20 h. ●: BaTaO2N, ▲: Ta3N5, and □: Ba5Ta4O15. | ||
 | ||
| Fig. 2 UV-visible diffuse reflectance spectra obtained from BaTaO2N prepared in a flow of NH3 at 1123 K using durations of 3–20 h. | ||
Fig. 3 provides the SEM images of various specimens. The original amorphous Ta2O5 used as a raw material evidently was made of particles having a fluffy, cloud-like shape with sizes of 20–30 nm. The nitridation of this compound with BaCO3 at 1123 K produced aggregates of irregularly shaped particles roughly 50–150 nm in size. With increasing nitridation duration to 15–20 h, these irregularly shaped particles partly grew into large aggregates and connected with one another. Raising the nitridation temperature to 1173–1273 K caused the particle shape to vary and produced sintered structures (Fig. S1†). Fig. S2(a) and (b)† show the HR-TEM images of a-Ta2O5 and BaTaO2N, respectively. In contrast to a-Ta2O5 with amorphous structural characteristics, BaTaO2N exhibits crystalline lattice fringes.
 | ||
| Fig. 3 SEM images of (a) a-Ta2O5 and BaTaO2N prepared in a flow of NH3 at 1123 K with nitridation durations of (b) 3, (c) 5, (d) 7, (e) 15 and (f) 20 h. | ||
Water splitting reaction
Fig. 4 summarizes the extent of H2O decomposition in response to Xe lamp illumination of a BaTaO2N specimen nitrided at 1123 K for 5 h and loaded with Rh, Cr2O3 and IrO2 as cocatalysts. Both H2 and O2 were generated in stoichiometric amounts during the initial stage of the reaction and the quantities of these gases increased linearly during the reaction. During the second and third runs following the evacuation of gaseous products, the gas amounts produced were nearly the same as those in the first run. The stoichiometric and constant production of H2 and O2 in these trials clearly indicates that one-step-excitation overall water splitting took place. Fig. 5 plots H2 and O2 production as functions of nitridation time as a means of assessing photocatalytic activity. The activity is seen to have increased sharply over the range of 3–5 h, passed through a maximum at 5 h, and then decreased over the range of 7–15 h. The H2 evolution for BaTaO2N heated for 20 h was lower than 6% of that for 5 h and little O2 evolution was observed with this sample. The effect of the nitridation duration on the activity of BaTaO2N after processing at 1173 or 1073 K is presented in Fig. S3 and S4,† respectively. The maximum activity appeared after 2 and 15 h at 1173 and 1073 K, respectively. Fig. S5† summarizes the changes in activity over a wide variety of nitridation times and temperatures in the form of a three-dimensional map. It is evident that lower nitridation temperatures required longer durations to achieve suitable water splitting activity. Out of this wide range of times and temperatures, the combination of 1123 K and 5 h provided the highest photocatalytic activity during overall water splitting.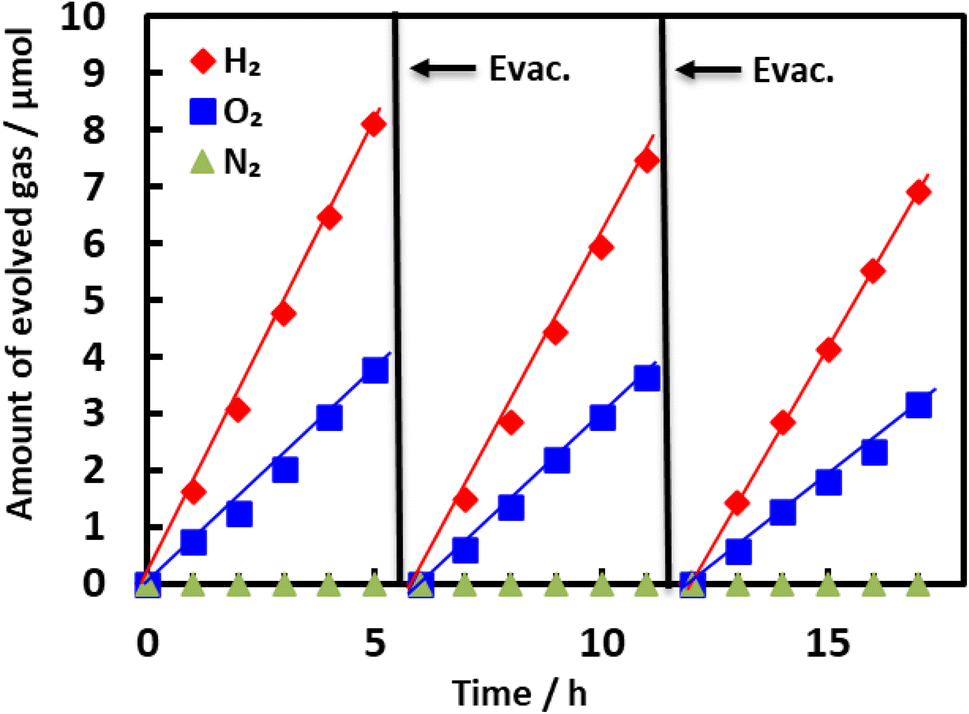 | ||
| Fig. 4 Evolution of various gases over time during the water splitting reaction over Rh/Cr2O3/IrO2-loaded BaTaO2N under visible light (λ > 420 nm). BaTaO2N was prepared by nitridation at 1123 K for 5 h and was loaded with 2 wt% Rh, 1 wt% Cr2O3 and 0.3 wt% IrO2 as cocatalysts. Reaction conditions: lamp source: 300 W Xe lamp, reaction temperature: 288 K, and background pressure: Ar at 5 kPa. | ||
 | ||
| Fig. 5 Effects of nitridation time on the photocatalytic activity of BaTaO2N during the water splitting reaction. BaTaO2N was prepared by nitridation at 1123 K for varying durations and was loaded with 2 wt% Rh, 1 wt% Cr2O3 and 0.3 wt% IrO2 as cocatalysts. Reaction conditions: lamp source: 300 W Xe lamp (λ > 420 nm), reaction temperature: 288 K, and background pressure: Ar at 5 kPa. | ||
As shown in Fig. 1, BaTaO2N contained a small amount of Ta3N5. To avoid or at least suppress the formation of the Ta3N5 phase, the Ba/Ta ratio for the initial mixture of BaCO3 and a-Ta2O5 was varied between 0.9 and 1.2. Fig. 6(a) shows XRD patterns for samples prepared by nitridation at 1123 K for 5 h. Peaks related to the Ta3N5 phase remained present at a Ba/Ta ratio of 1.1 but nearly disappeared at Ba/Ta = 1.2, while peaks due to Ba5Ta4O15 appeared. The ratio of the intensity of the Ta3N5 peaks to the BaTaO2N peaks is plotted against the Ba/Ta ratio in Fig. 6(b), and this ratio is seen to decrease with increasing ratio. The photocatalytic activity of the catalyst (as assessed based on H2 and O2 evolution) increased beginning at Ba/Ta = 0.9 and peaked at Ba/Ta = 1, with dramatic decreases as the ratio was further increased to a value of 2 (Fig. 6(c)). A Ba/Ta ratio of 1 was determined to provide the highest activity regardless of the presence of Ta3N5. Although it is possible that the Ta3N5 phase contributed to photocatalysis, the poor correlation between the amount of Ta3N5 phase and the activity of the material suggests that this phase had only a minimal effect on the photocatalytic activity of BaTaO2N. Water splitting trials were also carried out under argon at pressures of 5, 20 or 50 kPa. As shown in Fig. S6,† at least 80% of the original activity was maintained even at 50 kPa, indicating that the effect of background pressure on the photocatalytic activity was not especially strong. Fig. S7† shows changes in the amounts of H2 and O2 in the gas phase after turning the light irradiation off (in the dark). Their decreases were 4% for 3 h, indicating that the extent of the backward reaction from H2/O2 to H2O is low.
 | ||
| Fig. 6 (a) X-ray diffraction patterns for BaTaO2N prepared using various Ba/Ta ratios in the starting mixture, (b) the ratio of the intensity of the Ta3N5 XRD peak to the sum of the intensities of the BaTaO2N and Ta3N5 peaks as a function of the Ba/Ta ratio, and (c) the photocatalytic activity of Rh/Cr2O3/IrO2-loaded BaTaO2N (as reflected in the evolution of gaseous products) during the water splitting reaction as a function of the Ba/Ta ratio. BaTaO2N was prepared by nitridation at 1123 K for 5 h and was loaded with 2 wt% Rh, 1 wt% Cr2O3 and 0.3 wt% IrO2 as cocatalysts. Reaction conditions: lamp source: 300 W Xe lamp (λ > 420 nm), reaction temperature: 288 K, and background pressure: Ar at 5 kPa. | ||
Effects of cocatalysts
The photocatalytic activity of the catalyst during water splitting was found to vary with changes in the amounts of Rh, Cr2O3 and IrO2 that were deposited. Specifically, the activity increased with increasing Rh loading up to 4 wt% (Fig. S8†). Small amounts of Cr2O3 (meaning concentrations in the range of 0.5 to 1.0 wt%) were also found to lead to high photocatalytic activity (Fig. S9†). The concentration of IrO2 did not have a large effect, such that even a low concentration of 0.3 wt% was sufficient to promote oxygen evolution during water splitting. Annular dark-field STEM images and STEM-EDS elemental maps were obtained to assess the states of Rh, Cr2O3 and IrO2 deposited on BaTaO2N. As shown in Fig. 7, the BaTaO2N particles were found to have uniform distributions of Ba, Ta, O and N. However, Rh particles on Rh/Cr2O3/IrO2–BaTaO2N were determined to form aggregates on the BaTaO2N particles while Cr2O3 loaded by photodeposition were concentrated on the Rh particles. This last result was indicative of a core–shell structure that has been previously proposed for this type of cocatalyst.1,17–19 IrO2 was likely to have been uniformly distributed over the BaTaO2N surface, but the evidence for this was not as clear because of the poor color pattern of the associated images, possibly owing to the low concentration of this cocatalyst. | ||
| Fig. 7 Annular dark-field STEM images and STEM-EDS elemental maps for Rh/Cr2O3/IrO2-loaded BaTaO2N. BaTaO2N was prepared by nitridation at 1123 K for varying durations and was loaded with 4 wt% Rh, 1 wt% Cr2O3 and 0.3 wt% IrO2 as cocatalysts. | ||
To see the role of the cocatalysts, the water splitting reaction was tested for Rh/Cr2O3- and IrO2-loaded BaTaO2N, separately. As shown in Fig S10(a),† Rh/Cr2O3-loaded BaTaO2N was able to produce H2 and O2, indicating the effectiveness of Rh/Cr2O3 cocatalysts for water splitting. However, the evolution of O2 was slightly low, compared to the case of Rh/Cr2O3/IrO2-loading. Fig. S10(b)† shows no activity of IrO2-loaded BaTaO2N for the water splitting reaction. From these results, it is concluded that IrO2 has the ability of promoting O2 evolution.
To better understand the effect of the cocatalysts on the activity, Ru was used in place of Rh, together with Cr2O3 and IrO2. Fig. 8(a) shows the results for the water splitting reaction on Ru/Cr2O3/IrO2–BaTaO2N. These data demonstrate that both H2 and O2 were evolved and that the amounts of these gases increased in an essentially linear fashion as the irradiation time was increased. As shown in Fig. 8(b), Ru metal provided almost the same level of activity as Rh although the highest activities were obtained at 1 to 2 wt% Ru whereas 4 wt% Rh was required for optimal performance (Fig. S11†). Fig. S12† provides an annular dark-field STEM image and STEM-EDS elemental maps obtained from a cross-sectional sample of Ru/Cr2O3/IrO2–BaTaO2N. These results confirm that Ru metal was uniformly dispersed over BaTaO2N in the form of small particles. Thus, the difference in the amount of metal required to obtain high activity between Ru and Rh was likely related to the higher degree of dispersion of the former. Fig. S13† shows a HR-TEM image and its EDS mappings. The oval shaped Ru metal particles are attached on BaTaO2N and surrounded by a thin Cr2O3 layer, which indicates the formation of a core–shell structure.
 | ||
| Fig. 8 (a) Evolution of gaseous products during the water splitting reaction over Ru-loaded BaTaO2N under visible light. Cocatalysts: Ru at 4 wt%, Cr2O3 at 1 wt% and IrO2 at 0.3 wt%. (b) Comparison of the photocatalytic activities during the water splitting reaction using Ru and Rh cocatalysts. Cocatalysts: Rh at 2 wt% or Ru at 1 wt%, Cr2O3 at 0.3 wt% and IrO2 at 0.3 wt%. BaTaO2N was prepared by nitridation at 1123 K for 5 h. Reaction conditions: lamp source: 300 W Xe lamp (λ > 420 nm), reaction temperature: 288 K, and background pressure: Ar at 5 kPa. | ||
Prior to the water splitting trials, a Cr2O3 layer was applied to Rh by photo-reduction of Cr6+ to Cr3+ using a K2CrO4 solution containing 10% CH3OH as a sacrificial reagent. After Rh-loaded BaTaO2N was illuminated in an aqueous K2CrO4 solution without CH3OH, water splitting was found to proceed with the generation of stoichiometric amounts of H2 and O2 (Fig. S14(a)†). It should also be noted that Cr2O3 could be loaded on Rh without the aid of a sacrificial reagent to produce a Cr2O3/Rh core–shell structure, after which the water splitting reaction would take place. As shown in Fig. 9, the activity increased with increasing K2CrO4 concentration, and the highest activity was obtained at approximately 0.22 mM.
 | ||
| Fig. 9 Effects of the Cr concentration in aqueous solution on the photocatalytic activity of Rh-loaded BaTaO2N during the water splitting reaction. In these trials, 2 wt% Rh-loaded BaTaO2N was dispersed in an aqueous solution of K2CrO4. A concentration of 0.22 mM K2CrO4 corresponded to a loading of 1 wt% Cr on BaTaO2N. BaTaO2N was prepared by nitridation at 1123 K for 5 h. Reaction conditions: lamp source: 300 W Xe lamp (λ > 420 nm), reaction temperature: 288 K, and background pressure: Ar at 5 kPa. | ||
The effect of pH on the photocatalytic activity of Rh-loaded BaTaO2N in aqueous K2CrO4 solutions was also assessed. The data in Fig. S14(b)† establish that the photocatalytic activity increased with increasing pH from 7 to 9. However, a sharp drop in activity occurred at pH 10 because Cr2O3 was highly soluble in the alkaline medium. The activity enhancements observed at pH 8 and 9 were possibly related to reductions in the energy barrier to oxygen evolution with increasing OH− concentration. This outcome is in agreement with the results obtained from previous photoelectrochemical analyses of BaTaO2N electrodes, in which oxygen evolution was enhanced at higher pH values.20
Fig. 10 plots the apparent quantum yield (AQY) obtained from Rh/Cr2O3/IrO2-loaded BaTaO2N, together with the optical absorption of the material. The AQY at 400 nm was 0.1% but this value gradually decreased beginning at 420 nm and then dropped sharply at around 500 nm and exhibited a slower decline above 540 nm. These changes in the AQY were roughly analogous to those in optical absorption. Even though the AQY for this material was relatively low, it is noteworthy that the overall water splitting reaction proceeded at a long wavelength of 540 nm. Fig. 11 summarizes the results of the water splitting reaction on Rh/Cr2O3/IrO2-BaTaO2N when irradiated using a solar simulator. Constant generation of both H2 and O2 from H2O was observed during prolonged irradiation and the solar-to-hydrogen (STH) yield was estimated to be 5 × 10−4%. This value is slightly higher than 4 × 10−4% reported for Mg-doped BaTaO2N.16
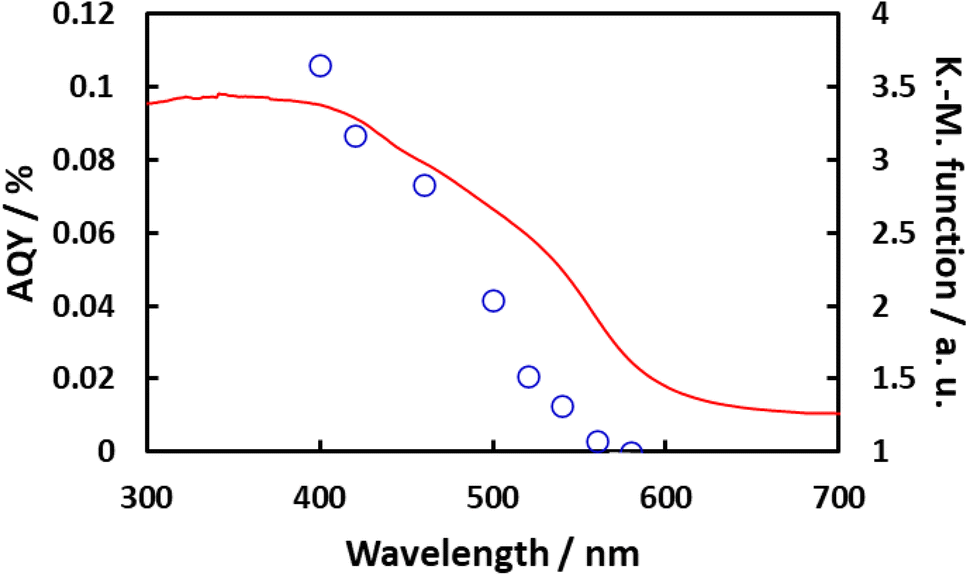 | ||
| Fig. 10 AQY (○) and absorption (solid line) data for Rh/Cr2O3/IrO2-loaded BaTaO2N as a function of wavelength. BaTaO2N was prepared by nitridation at 1123 K for 5 h and was loaded with 2 wt% Rh, 1 wt% Cr2O3 and 0.3 wt% IrO2 as cocatalysts. Reaction conditions: lamp source: 300 W Xe lamp, reaction temperature: 288 K, and background pressure: Ar at 5 kPa. | ||
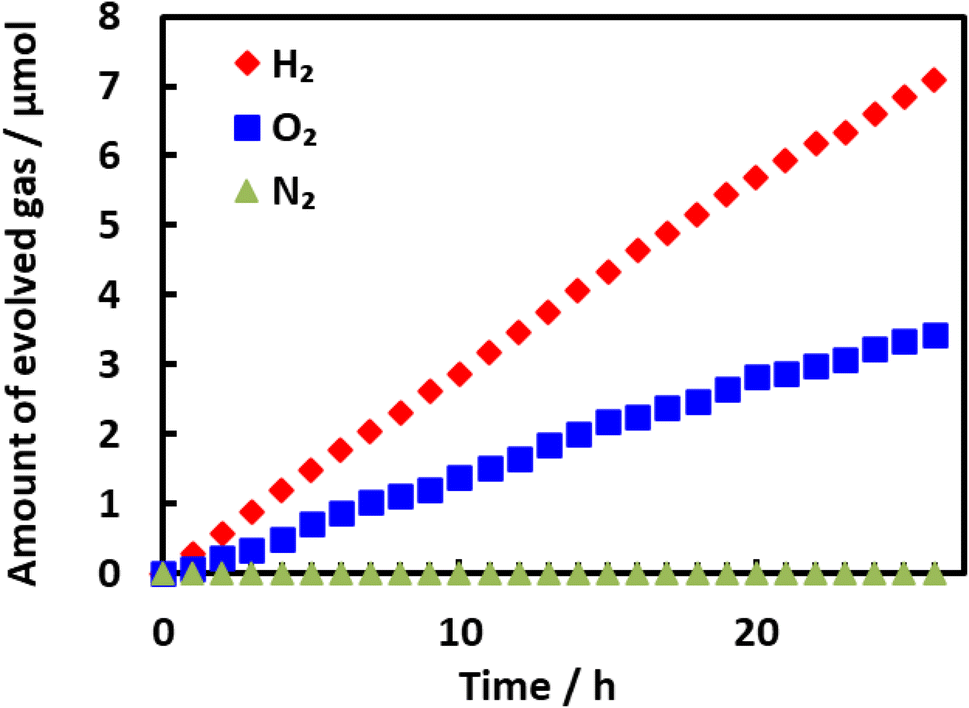 | ||
| Fig. 11 Steady evolution of gaseous products during the water splitting reaction on Rh/Cr2O3/IrO2-loaded BaTaO2N under simulated light. BaTaO2N was prepared by nitridation at 1123 K for 5 h and was loaded with 2 wt% Rh, 1 wt% Cr2O3 and 0.3 wt% IrO2 as cocatalysts. Reaction conditions: lamp source: AM 1.5G solar radiation, reaction temperature: 288 K, and background pressure: Ar at 5 kPa. | ||
Mechanism of active BaTaO2N formation
It is important to have an understanding of the mechanism responsible for the formation of photocatalytically active BaTaO2N. The generation of BaTaO2N from a mixture of BaCO3 and a-Ta2O5 in a flow of NH3 involves several parallel reaction paths, but these can be simplified by dividing them into several separate processes, as summarized in Fig. S15(a).† Here, reaction path (1) involves the rapid formation of an oxide precursor having the general formula BaTaOx (but most likely Ba5Ta4O15) at increasing temperatures, followed by nitridation of this oxide. In contrast, path (2) proceeds via the selective formation of an oxynitride precursor such amorphous BaTaOxNy at low temperatures followed by complete nitridation. In path (3), the nitridation of a-Ta2O5 to TaON proceeds preferentially after which reaction with BaCO3 occurs to produce BaTaO2N in a flow of NH3. The nitridation of TaON to Ta3N5 readily proceeds, as demonstrated in prior work, and the reaction between Ta3N5 and BaCO3 is a well-known oxynitride reaction in N2.20 In this process, some unreacted Ta3N5 also remains in the product. There have been several reports that BaCN2 is formed by the nitridation of BaCO3, but XRD peaks related to BaCN2 were not observed and so the contribution of this reaction to BaTaO2N formation was likely negligible.21–23 Reaction paths (1) to (5) were examined by synthesizing BaTaO2N via each path, with the results presented in Fig. S15(b).† Both reaction paths (1) and (2) were found to generate BaTaO2N that was active for overall water splitting. However, the latter showed higher activity than the former, indicating that path (2) is superior. In contrast, the processes from path (3) to (5) resulted in the formation of inactive BaTaO2N. The generation of highly active BaTaO2N via reaction path (2) clearly demonstrates the importance of the reactivity of a-Ta2O5, which allows rapid nitridation even at low temperatures. To confirm the validity of this concept, large crystalline Ta2O5 particles were used instead of a-Ta2O5 for BaTaO2N preparation using the same nitridation conditions as were applied in trials with a-Ta2O5. The photocatalytic activity of the resulting BaTaO2N after Rh/Cr2O3/IrO2 loading was negligibly small, confirming the importance of amorphous Ta2O5. One of the functions of amorphous a-Ta2O5 particles is that it has higher reactivity than crystalline Ta2O5, which enables BaTaO2N synthesis at low temperatures and thus small particle formation. In addition, it is likely that the reaction with BaCO3 will proceed uniformly, compared to crystalline Ta2O5 that consists of crystalline planes with different reactivity. These functions of amorphous a-Ta2O5 are thought to result in the formation of high-quality BaTaO2N.The plot of photocatalytic activity against nitridation time in Fig. 5 indicates that the high activity of BaTaO2N nitrided for 5 h was greatly decreased after 15 to 20 h of nitridation. Transient absorption spectroscopy (TAS) was employed to analyze the photoexcited states of active and inactive BaTaO2N prepared by nitridation at 5 and 20 h, respectively. The TAS data in Fig. 12(a) and (b) contain two absorption features in the range 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–5000 cm−1 and below 4000 cm−1. These can be assigned to deeply trapped electrons and free electrons, respectively.24,25 Increasing the nitridation time from 5 to 20 h drastically altered the shape of the spectrum. Specifically, the intensity of the peak related to deeply trapped electrons was significantly increased whereas that of the free electron peak decreased. These results suggest that the concentration of mid-gap states capable of deeply trapping photogenerated electrons was increased by extending the nitridation time. Furthermore, the absorption in the region of 5000–16,000 cm−1 was very low in the case of the specimen nitrided for 5 h but very high for the 20 h specimen. It is therefore evident that the number of electrons trapped in vacancies was much smaller in the former compared with the latter. Fig. 12(c) shows the TAS data obtained at 5000 nm (2000 cm−1) from samples nitrided for 5 and 20 h. The signal generated by the former material decayed very slowly and maintained a high level of intensity whereas that for the latter decayed more rapidly to a very low level. The intensity of the TA signal is correlated with the population of free electrons and so these results indicate that the 5 h specimen was able to generate long-lived free electrons. It is apparent that the active 5 h sample had a very low density of trap sites induced by defects (such as oxygen vacancies) and a high level of long-lived free electrons, both of which contributed to its excellent photocatalytic performance. Prolonged nitridation (15–20 h) increased the concentration of oxygen vacancies and other defects capable of deeply trapping photogenerated electrons.
000–5000 cm−1 and below 4000 cm−1. These can be assigned to deeply trapped electrons and free electrons, respectively.24,25 Increasing the nitridation time from 5 to 20 h drastically altered the shape of the spectrum. Specifically, the intensity of the peak related to deeply trapped electrons was significantly increased whereas that of the free electron peak decreased. These results suggest that the concentration of mid-gap states capable of deeply trapping photogenerated electrons was increased by extending the nitridation time. Furthermore, the absorption in the region of 5000–16,000 cm−1 was very low in the case of the specimen nitrided for 5 h but very high for the 20 h specimen. It is therefore evident that the number of electrons trapped in vacancies was much smaller in the former compared with the latter. Fig. 12(c) shows the TAS data obtained at 5000 nm (2000 cm−1) from samples nitrided for 5 and 20 h. The signal generated by the former material decayed very slowly and maintained a high level of intensity whereas that for the latter decayed more rapidly to a very low level. The intensity of the TA signal is correlated with the population of free electrons and so these results indicate that the 5 h specimen was able to generate long-lived free electrons. It is apparent that the active 5 h sample had a very low density of trap sites induced by defects (such as oxygen vacancies) and a high level of long-lived free electrons, both of which contributed to its excellent photocatalytic performance. Prolonged nitridation (15–20 h) increased the concentration of oxygen vacancies and other defects capable of deeply trapping photogenerated electrons.
 | ||
| Fig. 12 (a) and (b) Time-resolved spectra and (c) decay kinetics of BaTaO2N prepared by nitridation at 1123 K for 5 or 20 h. Active and inactive BaTaO2N specimens were obtained by nitridation at 1123 K for 5 and 20 h, respectively (see Fig. 5). The TA spectra were obtained by scanning the probe energy from 17000–1200 cm−1 (0.15–2.1 eV). In the mid-IR region (6000–1200 cm−1), an IR beam emitted by a MoSi2 coil was used, while, in the visible to near-IR region (17000–6000 cm−1), a probe beam generated by a halogen lamp was focused on the sample. During the acquisition of the transient decay profile of photoexcited free electrons, the absorption signal was monitored at 2000 cm−1 (5000 nm) after irradiation with 470 nm laser pulses (fluence: 1 mJ per pulse) at a frequency of 1 Hz to achieve bandgap excitation of photocarriers in BaTaO2N. Measurements were carried out in a N2 atmosphere at room temperature. | ||
The effect of oxygen vacancies was investigated by producing inactive BaTaO2N via a 15 h nitridation and then exposing this material to a N2 atmosphere containing 100 ppm O2 at 823 K for 2 h. As shown in Fig. S16,† the initial low photocatalytic activity was increased by a factor of 5 and H2 and O2 were generated in a nearly stoichiometric ratio. The incorporation of oxygen in the specimen evidently recovered its original activity. This finding provides evidence that the oxygen vacancies formed by prolonged nitridation deactivated BaTaO2N. Analyses by X-ray photoelectron spectroscopy (XPS) as presented in Fig. S17(a) and (b)† demonstrated that the intensities of the Ta 4f peaks (associated with Ta3+) were much higher in the case of BaTaO2N nitrided for 20 h compared with the 5 h specimen. Because Ta3+ sites can act as recombination centers for photoexcited electrons and holes, increasing the number of these sites decreased the photocatalytic activity. Together, these TAS and XPS results confirm the exceptional performance of the photocatalytically active BaTaO2N obtained by 5 h nitridation and explain the enhanced performance of this material during the water splitting reaction.
Conclusions
To the best of our knowledge, this work represents the first-ever demonstration of one-step-excitation overall water splitting using pristine BaTaO2N. The direct nitridation of a mixture of amorphous Ta2O5 (Ta2O5·3H2O) nanoparticles and BaCO3 produced a BaTaO2N photocatalyst active for overall water splitting under visible light extending to a wavelength of 540 nm following modification with Rh (or Ru), Cr2O3 and IrO2 as cocatalysts. The photocatalytic activity was found to be greatly affected by the concentrations of the cocatalysts, and the optimal amounts were determined to be 4 wt% (2 wt% for Ru), 1 wt% and 0.3 wt%, respectively. A wide range of nitridation conditions was examined with temperatures from 1023 to 1273 K and times from 0.5 to 20 h, and the highest activity was obtained for mild NH3-based nitridation at 1123 K for 5 h. Even though a low nitridation temperature was employed, extending the duration to 15–20 h produced BaTaO2N with negligible activity. Analyses by TAS clearly showed that the active BaTaO2N contained the largest population of photoexcited free electrons, and that these electrons had long lifetimes owing to a small number of defects. The mechanism by which BaTaO2N was formed during NH3 nitridation from the present mixture of starting materials was assessed, and the elevated reactivity of amorphous Ta2O5 nanoparticles is thought to be an important aspect of this process. Although the efficiency of the photocatalysis remains very low, the concept of employing highly reactive nanoparticles shows promise and could be used in future to develop high-quality oxynitride photocatalysts.Methods
Synthesis of BaTaO2N
Na3TaO4 was initially prepared as a starting material for the synthesis of amorphous tantalum oxides. In this process, Ta2O5 (99.99%, Rare Metal Co.) and NaOH (98%, Fujifilm Wako Pure Metal Co.) were thoroughly mixed at a Na:Ta molar ratio of 5:1. The mixture was then transferred to an alumina crucible and heated in air at 623 K for 12 h. The resulting solid generated an XRD pattern containing peaks attributable to Na3TaO4 and to excess NaOH. This product was subsequently dissolved in H2O by sonication and stirring to produce an aqueous solution of Na8[Ta6O19] with a pH close to 13.26–28 This alkaline solution was neutralized by adding H2SO4 until a white precipitate was observed at pH 7 based on the protonation of [Ta6O19]8− anions followed by decomposition to produce amorphous hydrous tantalum, Ta2O5·nH2O. The precipitate was washed five times with pure water to remove residual Na2SO4 and then dried overnight under vacuum at 313 K. The XRD pattern of the product confirmed its amorphous structure and the material was then converted to crystalline Ta2O5 by heating in air at 1023 K. Thermogravimetric/differential thermal analysis data showed that the amorphous hydrous tantalum oxide had the formula Ta2O5·3H2O. This material is referred to herein as a-Ta2O5.
BaTaO2N was prepared by a solid state reaction. In this synthesis, BaCO3 (99.99%, Rare Metallic Co.) and a-Ta2O5 were thoroughly mixed in an agate mortar containing a drop of C2H5OH. The resulting mixture was placed in an alumina crucible and directly nitrided under a flow of NH3 at a rate of 500 mL min−1 and at various temperatures from 1023 to 1273 K with durations ranging from 0.5 to 20 h. The resulting BaTaO2N was washed with dilute HCl and water in conjunction with sonication, removed by filtration and then vacuum dried at 313 K for 2 h.
Loading of cocatalysts on BaTaO2N
The cocatalysts were typically loaded onto BaTaO2N in the order Rh, Cr2O3 and IrO2. The Rh was deposited using an impregnation method. In this process, a solution of RhCl3·3H2O (99.9%, Wako Pure Chemical Co. Ltd.) in water was added dropwise to BaTaO2N powder until incipient wetness was obtained. After drying, the powder was heated under a flow of H2 (10%)/N2 (90%) at 573 K for 1 h so as to convert the Rh compound to Rh metal particles on BaTaO2N. The Rh loadings on these specimens were in the range 1 to 4 wt%. A Cr2O3 layer was applied by dispersing a quantity of Rh-loaded BaTaO2N in an aqueous K2CrO4 solution containing 10 vol% CH3OH as a sacrificial reagent with subsequent illumination (>350 nm) for 4 h. This procedure reduced Cr6+ to Cr3+ on the Rh metal surfaces to produce a core–shell structure in which Rh metal particles were covered with Cr2O3. This structure helped to suppress backward reactions involving H2 and O2, as has been reported previously.17,18 Following this, colloidal IrO2 nanoparticles obtained from the hydrolysis of Na2IrCl6 in an aqueous alkaline solution29,30 were loaded onto the catalyst at a concentration of 0.3 wt%. The resulting photocatalysts are referred to herein as IrO2/Cr2O3/Rh-BaTaO2N. In the case of Ru impregnation, RuCl3·3H2O (Kanto Ltd, >95%) was used, and nearly the same procedure as that described above was applied except that the loading was in the range 0.5 to 2 wt% and the reduction temperature was 623 K.Water splitting reaction
Overall water splitting was performed using a closed gas circulation apparatus equipped with a gas chromatograph (GC 8 A, Shimadzu, with a thermal conductivity detector, molecular sieve 5 A column and Ar as the carrier gas). A Pyrex top-illuminated reaction vessel was connected to the reaction apparatus and used as the reactor. In each trial, a quantity of the Rh/Cr2O3/IrO2-loaded BaTaO2N photocatalyst (0.2 g) was dispersed in ultrapure water (150 mL) having a pH of approximately 7 unless otherwise specified. After complete removal of dissolved gases in the water, the reactor was filled with Ar to a background pressure of 5 kPa and then illuminated with a 300 W xenon lamp coupled with a cold mirror and a cut-off filter (L42, λ > 420 nm) or a solar simulator (SAN-EI electronic, XRS40S1, AM 1.5G, 100 mW cm−2) with continuous agitation using a magnetic stirrer. The temperature of the reactant solution was maintained at 288 K with a flow of cooling water.Characterization
XRD patterns for the various samples were acquired using a Rigaku RINT-Ultima III diffractometer with a Cu Kα radiation source. The absorption characteristics of the specimens were assessed using UV-visible diffuse reflectance spectroscopy employing a spectrophotometer (JASCO V-670DS), with Kubelka–Munk conversion of the data. Scanning transmission electron microscopy (STEM) images were obtained with a JEOL JEM-2800 instrument. Energy dispersive X-ray spectroscopy (EDS) data together with STEM images were obtained with a JEOL JEM-2800 system equipped with an Oxford Instruments X-MAX 100TLE SDD detector. XPS data were acquired using powder samples fixed on conductive carbon adhesive tape without a conductive Al pillar. Spectra were acquired with a KRATOS ULTRA2 instrument having an Al Kα source and equipped with a charge neutralizer. The binding energy values for these spectra were calibrated based on the C 1s peak at 284.4 eV.Microsecond-millisecond TAS analyses were performed using a pump-probe Nd:YAG laser system (Continuum, Surelite I; duration: 6 ns) equipped with custom-built spectrometers.31,32 The TA spectra were obtained by scanning with probe energy from 17000 to 1200 cm−1 (0.15–2.1 eV). Photoexcited carriers in the mid-infrared (IR) region (6000–1200 cm−1) were probed using an IR beam emitted by a MoSi2 coil that was applied to a film sample. The transmitted IR beam was then passed through a monochromatic grating spectrometer and to a mercury-cadmium-telluride detector (Kolmar). Photocarriers in the visible to near-IR region (17000–6000 cm−1) were analyzed using a probe beam generated from a halogen lamp that was focused on the sample. Reflected light from the sample entered the grating spectrometer and was sent to a Si photodetector. The output electric signal was processed using an alternating current coupled amplifier (Stanford Research Systems, SR560, bandwidth: 1 MHz), and the time resolution of the spectrometer was limited to 1 μs by the bandwidth of the amplifier. The transient decay profile for photoexcited free electrons was captured by monitoring the absorption signal at 2000 cm−1 (5000 nm). The bandgap excitation of photocarriers in BaTaO2N was assessed using 470 nm laser pulses (fluence: 1 mJ per pulse) at a frequency of 1 Hz. In preparation for these analyses, a portion of the BaTaO2N powder was dispersed in water, drop-cast onto a CaF2 substrate, and dried naturally in air overnight to obtain a powder film with a density of approximately 1.1 mg cm−2. Analyses were carried out in a N2 atmosphere at room temperature.
Apparent quantum yield
The AQY for one step photoexcitation overall water splitting was obtained from trials under illumination of a 300 W Xe lamp (Asahi Spectra, MAX-303) using a bandpass filter with a full width at half maximum of 15 nm. The center wavelength was varied from 400 to 480 nm in intervals of 20 nm and the number of incident photons was monitored using a diffraction grating spectroradiometer (EKO Instr. Co., LS-100). The AQY value was calculated as| AQY (%) = [2 × N(H2)]/N(photon) × 100, |
Solar-to-hydrogen (STH) energy conversion efficiency
Water splitting reactions were conducted under simulated sunlight generated using a solar simulator (Sanei Electric Co., XES-40S1). The STH values were calculated as| STG (%) = (R(H2) × ΔG)/(I × A) × 100 |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the Artificial Photosynthesis Project (ARPChem) of the New Energy and Industrial Technology Development organization (NEDO).References
- S. Chen, T. Takata and K. Domen, Particulate photocatalysts for overall water splitting, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- T. Hisatomi and K. Domen, Reaction systems for solar hydrogen production via water splitting with particulate semiconductor photocatalysts, Nat. Catal., 2019, 2, 387–399 CrossRef CAS.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanaba, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Photocatalytic solar hydrogen production from water on a 100-m2 scale, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- C. Pan, T. Takata and K. Domen, Overall Water Splitting on the Transition-Metal Oxynitride Photocatalyst LaMg1/3Ta2/3O2N over a Large Portion of the VisibleLight Spectrum, Chem.–Eur. J., 2016, 22, 1854–1862 CrossRef CAS PubMed.
- C. Pan, T. Takata, M. Nakabayashi, T. Masumoto, N. Shibata, Y. Ikuhara and K. Domen, A Complex Perovskite-Type Oxynitride: The First Photocatalyst for Water Splitting Operable at up to 600 nm, Angew. Chem., Int. Ed., 2015, 54, 2955–2959 CrossRef CAS PubMed.
- K. Ueda, T. Minegishi, J. Clune, M. Nakabayashi, T. Hisatomi, H. Nishiyama, M. Katayama, N. Shibata, J. Kubota, T. Yamada and K. Domen, Photoelectrochemical Oxidation of Water Using BaTaO2N Photoanodes Prepared by Particle Transfer Method, J. Am. Chem. Soc., 2015, 137, 2227–2230 CrossRef CAS PubMed.
- H. Okamoto, M. Kodera, T. Hisatomi, M. Katayama, T. Minegishi and K. Domen, Effects of annealing conditions on the oxygen evolution activity of a BaTaO2N photocatalyst loaded with cobalt species, Catal. Today, 2020, 354, 204–210 CrossRef CAS.
- J. Xiao, S. Nishimae, J. Vequizo, M. Nakabayashi, T. Hisatomi, H. Li, L. Lin, N. Shibata, A. Yamakata, Y. Inoue and K. Domen, Enhanced Overall Water Splitting by a Zirconium-Doped TaON-Based Photocatalyst, Angew. Chem., Int. Ed., 2022, 61, e2021165 Search PubMed.
- Z. Wang, Y. Inoue, T. Hisatomi, R. Ishikawa, Q. Wang, T. Takata, S. Chen, N. Shibata, Y. Ikuhara and K. Domen, Overall water splitting by Ta3N5 nanorod single crystals grown on the edges of KTaO3 particles, Nat. Catal., 2018, 1, 756–763 CrossRef CAS.
- Q. Wang, M. Nakabayashi, T. Hisatomi, S. Sun, S. Akiyama, Z. Wang, Z. Pan, X. Xiao, T. Watanabe, T. Yamada, N. Shibata, T. Takata and K. Domen, Oxysulfide photocatalyst for visible-lightdriven overall water splitting, Nat. Mater., 2019, 18, 827–832 CrossRef CAS PubMed.
- M. Hojamberdiev, K. Yabuta, J. J. M. Vequizo, A. Yamakata, S. Oishi, K. Domen and K. Teshima, NH3-Assisted Flux Growth of Cube-like BaTaO2N Submicron Crystals in a Completely Ionized Nonaqueous High-Temperature Solution and Their Water Splitting Activity, Cryst. Growth Des., 2015, 15, 4663–4671 CrossRef CAS.
- H. Li, D. Lu, S. Chen, T. Hisatomi, J. Vequizo, J. Xiao, Z. Wang, L. Lin, Q. Xiao, Y. Sun, Y. Miseki, K. Sayama, A. Yamakata, T. Takata and K. Domen, A Na-containing Pt cocatalyst for efficient visible-light-induced hydrogen evolution on BaTaO2N, J. Mater. Chem. A, 2021, 9, 13851–13854 RSC.
- M. Higashi, Y. Yamanaka, O. Tomita and R. Abe, Fabrication of cation-doped BaTaO2N photoanodes for efficient photoelectrochemical water splitting under visible light irradiation, APL Mater., 2015, 3, 104418 CrossRef.
- Z. Wang, Y. Luo, T. Hisatomi, J. Vequizo, S. Suzuki, S. Chen, M. Nakabayashi, L. Lin, Z. Pan, N. Kariya, A. Yamakata, N. Shibata, T. Takata, K. Teshima and K. Domen, Sequential cocatalyst decoration on BaTaO2N towards highly-active Z-scheme water splitting, Nat. Commun., 2021, 12, 1005 CrossRef CAS PubMed.
- M. Higashi, R. Abe, T. Takata and K. Domen, Photocatalytic Overall Water Splitting under Visible Light Using ATaO2N (A=Ca, Sr, Ba) and WO3 in a IO3−/I− Shuttle Redox Mediated System, Chem. Mater., 2009, 21, 1543–1549 CrossRef CAS.
- H. Li, J. Xiao, J. J. M. Vequizo, T. Hisatomi, M. Nakabayashi, Z. Pan, N. Shibata, A. Yamakata, T. Takata and K. Domen, One-Step Excitation Overall Water Splitting over a Modified Mg-Doped BaTaO2N Photocatalyst, ACS Catal., 2022, 12, 10179–10185 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Roles of Rh/Cr2O3 (Core/Shell) Nanoparticles Photodeposited on Visible-Light-Responsive (Ga1−xZnx)(N1−xOx) Solid Solutions in Photocatalytic Overall Water Splitting, J. Phys. Chem. C, 2007, 111, 7554–7560 CrossRef CAS.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Photocatalytic water splitting with a quantum efficiency of almost unity, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- M. Yoshida, K. Takanabe, K. Maeda, A. Ishikawa, J. Kubota, Y. Sakata, Y. Ikezawa and K. Domen, Role and Function of Noble-Metal/Cr-Layer Core/Shell Structure Cocatalysts for Photocatalytic Overall Water Splitting Studied by Model Electrodes, J. Phys. Chem. C, 2009, 113, 10151–10157 CrossRef CAS.
- S. Nishimae, Y. Mishima, H. Nishiyama, Y. Sasaki, M. Nakabayashi, Y. Inoue, M. Katayama and K. Domen, Fabrication of BaTaO2N Thin Films by Interfacial Reactions of BaCO3/Ta3N5 Layers on a Ta Substrate and Resulting High Photoanode Efficiencies During Water Splitting, Sol. RRL, 2020, 4, 1900542 CrossRef CAS.
- Y. Masubuchi, S. Nishitani, A. Hosono, Y. Kitagawa, J. Ueda, S. Tanabe, H. Yamane, M. Higuchi and S. Kikkawa, Red-emission over a wide range of wavelength at various temperatures from tetragonal BaCN2:Eu2+, J. Mater. Chem. C, 2018, 6, 6370 RSC.
- A. Hosono, Y. Masubuchi, T. Endo and S. Kikkawa, Molten BaCN2 for the sintering and crystal growth of dielectric oxynitride perovskite Sr1−xBaxTaO2N (x = 0.04–0.23), Dalton Trans., 2017, 46, 16837 RSC.
- A. Hosono, R. P. Stoffel, Y. Masubuchi, R. Dronskowski and S. Kikkawa, Melting Behavior of Alkaline-Earth Metal Carbodiimides and Their Thermochemistry from First-Principles, Inorg. Chem., 2019, 58, 8938–8942 CrossRef CAS PubMed.
- A. Yamakata, M. Yoshida, J. Kubota, M. Osawa and K. Domen, Potential-Dependent Recombination Kinetics of Photogenerated Electrons in n- and p- Type GaN Photoelectrodes Studied by Time-Resolved IR Absorption Spectroscopy, J. Am. Chem. Soc., 2011, 133, 11351–11357 CrossRef CAS PubMed.
- J. J. Vequizo, S. Nishioka, J. Hyodo, Y. Yamazaki, K. Maeda and A. Yamakata, Crucial impact of reduction on the photocarrier dynamics of SrTiO3 powders studied by transient absorption spectroscopy, J. Mater. Chem. A, 2019, 7, 26139–26146 RSC.
- W. H. Nelson and R. S. Tobias, Structure of the Polyanions of the Transition Metals in Aqueous Solution: The Hexatantalate, Inorg. Chem., 1963, 2, 985–992 CrossRef CAS.
- M. Filowitz, R. K. C. Ho, W. G. Klemperer and W. Shum, Oxygen-17 nuclear magnetic resonance spectroscopy of polyoxometalates. 1. Sensitivity and resolution, Inorg. Chem., 1979, 18, 93–103 CrossRef CAS.
- S. Yamazoe, K. Shibata, K. Kato and T. Wada, Needle-like NaNbO3 Synthesis via Nb6O198− Cluster Using Na3NbO4 Precursor by Dissolution–Precipitation Method, Chem. Lett., 2013, 42, 380–382 CrossRef CAS.
- M. Hara, C. C. Waraksa, J. T. Lean, B. A. Lewis and T. E. Mallouk, Photocatalytic water oxidation in a buffered tris(2,2′-bipyridyl)ruthenium complex-colloidal IrO2 system, J. Phys. Chem. A, 2000, 104, 5275–5280 CrossRef CAS.
- A. Ishikawa, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, Oxysulfide Sm2Ti2S2O5 as a Stable Photocatalyst for Water Oxidation and Reduction under Visible Light Irradiation (λ ≤ 650 nm), J. Am. Chem. Soc., 2002, 124, 13547 CrossRef CAS PubMed.
- J. J. M. Vequizo, H. Matsunaga, T. Ishiku, S. Karnimura, T. Ohno and A. Yamakata, Trapping-Induced Enhancement of Photocatalytic Activity on Brookite TiO2 Powders: Comparison with Anatase and Rutile TiO2 Powders, ACS Catal., 2017, 7, 2644–2651 CrossRef CAS.
- J. D. Xiao, J. J. M. Vequizo, T. Hisatomi, J. Rabeah, M. Nakabayashi, Z. Wang, Q. Xiao, H. H. Li, Z. H. Pan, M. Krause, N. Yin, G. Smith, N. Shibata, A. Bruckner, A. Yamakata, T. Takata and K. Domen, Simultaneously Tuning the Defects and Surface Properties of Ta3N5 Nanoparticles by Mg-Zr Codoping for Significantly Accelerated Photocatalytic H2 Evolution, J. Am. Chem. Soc., 2021, 143, 10059–10064 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta10010j |
| This journal is © The Royal Society of Chemistry 2023 |