
Fe, Cu dual-metal single atom catalyst on commercial carbon black for efficient oxygen reduction reaction†
Hongzhou
Yang‡
ab,
He
Huang‡
 a,
Qing
Wang
b,
Lu
Shang
*b,
Tierui
Zhang
bc and
Shouguo
Wang
*ad
a,
Qing
Wang
b,
Lu
Shang
*b,
Tierui
Zhang
bc and
Shouguo
Wang
*ad
aBeijing Advanced Innovation Center for Materials Genome Engineering, School of Materials Science and Engineering, University of Science and Technology Beijing, Beijing, 100083, China. E-mail: sgwang@ustb.edu.cn
bKey Laboratory of Photochemical Conversion and Optoelectronic Materials Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing, 100190, China. E-mail: lushang@mail.ipc.ac.cn
cCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing, 100049, China
dSchool of Materials Science and Engineering, Anhui University, Hefei, 230601, China
First published on 20th February 2023
Abstract
3d transition metal single atom catalysts (SACs) (e.g. M = Fe, Co, Cu) are considered as promising catalysts for oxygen reduction reaction (ORR), among which Fe SACs display comparable performance to commercial Pt/C. However, Fe SACs still demand significant improvements in activity and stability for further industrial deployment. Herein, a Fe, Cu dual metal SAC was synthesized using a facile ligand-mediated method. Fe and Cu atoms were atomically dispersed on commercial carbon black in the form of FeN4 and CuN4 sites. Owing to the interactions between the adjacent Fe and Cu atoms co-existing in a very tiny area, the as-synthesized Fe, Cu dual metal SAC exhibited excellent ORR performance with a high half-wave potential of 0.926 V and good stability in 0.1 M KOH, which are significantly higher than that of the Fe-SAC or Cu-SAC control samples. Density function theory calculations showed that the Cu single atom can act as an electron donator enriching the electron density of Fe sites, thus endowing the Fe sites with optimized adsorption/desorption energy for ORR intermediates and thereby facilitating the ORR activity of FeCu-SAC. Moreover, a study on three typical commercial carbon substrates indicated that carbon support with high surface areas and hierarchical pore structure was beneficial for ORR. The excellent ORR activity and durability of FeCu-SAC enabled the constructed Zn–air battery to have a high power density of 201.4 mW cm−2 and high specific energy of 827.68 W h kg−1.
Introduction
Oxygen reduction reaction (ORR) is a key electrocatalytic reaction in many next-generation energy storage and conversion devices, such as fuel cells and metal–air batteries.1–4 However, its sluggish kinetics have been the main bottleneck of these technologies.5 To date, platinum (Pt)-based materials are used as the most efficient catalysts for ORR.6–9 However, the high cost and low abundance of Pt limit its large-scale applications. Recently, single atom catalysts (SACs) have been considered promising alternatives to Pt-based ORR catalysts because metalloporphyrin active sites are intrinsically active for ORR.1,5,10–12 Among various SACs, Fe SACs have been reported to display the best ORR activity owing to the optimized adsorption binding energy (despite being strong) of Fe centres to ORR intermediates.13–15 To further improve the ORR activity of Fe SACs, dual metal SACs with secondary metal sites were developed with the expectation that the synergistic effect of two metal sites can facilitate the catalytic activity.16–25 For example, Li et al. synthesized Fe, Co dual metal SAC via pyrolyzing a metal organic framework (MOF) precursor containing two metal ions. Because of the formation of new FeCoN6 active sites, this catalyst exhibited excellent ORR performance in acidic media.26 Thereafter, many dual metal SACs for ORR were developed, such as Fe, Ni-SAC,27 Fe, Mn-SAC,28,29 and Zn, Co-SAC.30However, there are limited studies on Fe, Cu dual metal SACs, which may be mainly because the activity of Cu SACs was usually inferior to other SACs.31–36 However, a theoretical study indicated that Fe, Cu dual metal SACs may have high intrinsic activity for ORR because the Cu sites with a high d orbital electron density (d10) can act as electron donors to adjust the electron structure of Fe site and thus facilitate the electrochemical activity.37 Moreover, some experimental results indicated that the introduction of Cu sites in Fe SACs can effectively improve its activity.38–40 However, the reported Fe, Cu-SACs, similar to most SACs, were synthesized based on MOF.41 This synthetic route involved abundant evaporation of carbonic species, resulting in a low yield of the final product.42 Additionally, the ORR activity of MOF-based Fe, Cu dual metal SACs was still unsatisfactory owing to the low metal content (insufficient active sites for ORR) or improper pore size distribution of the carbon support (mainly contained micropores that cannot be exposed to electrolyte and contribute to oxygen reduction).43,44
In a previous study, we developed a facile and universal ligand-mediated method for synthesizing SACs with high metal loadings on a commercial carbon black.45 A series of single and dual SACs with high metal loadings were synthesized in a large scale. Herein, the Fe, Cu dual metal SAC (denoted as FeCu-SAC) was successfully synthesized via a similar ligand-mediated method, followed by an investigation of the influence of different typical commercial carbon black supports on their ORR performance. The synthesis involved the coordination of Fe and Cu ions with 1,10-phenanthroline to form a metal complex and then the uniform adsorption and dispersion of the metal complex on the carbon black. The metal complex-containing carbon precursor was then pyrolyzed and treated in acid to remove undesirable metal nanoparticles, thus yielding the final FeCu-SAC. Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and extended X-ray absorption fine structure (EXAFS) spectroscopy confirmed that the Fe and Cu atoms were atomically dispersed on the nitrogen-doped carbon. Electron energy loss spectroscopy (EELS) indicated that the Fe and Cu atoms co-existed in a tiny area; therefore, they were close enough to interact with the adjacent Fe and Cu sites. The as-synthesized FeCu-SAC catalyst exhibited excellent ORR performance with high half-wave potential (E1/2) of 0.926 V and good stability in 0.1 M KOH, which was significantly higher than the control Fe-SAC or Cu-SAC samples. The DFT calculations demonstrated that the introduction of Cu sites can modify the electron density on Fe sites, thereby leading to a lower energy barrier for ORR intermediates and higher ORR activity. The excellent ORR activity enabled the constructed Zn–air battery to have a high power density of 201.4 mW cm−2 and a high specific energy of 827.68 W h kg−1.
Experimental
Chemicals
Iron(II) acetate and dimethyl sulfoxide were purchased from J&K Scientific Co., Ltd. Copper(II) acetate monohydrate was purchased from Xilong Scientific Co., Ltd. 1,10-phenanthroline monohydrate was purchased from Sinopharm Chemical Reagent Co., Ltd. Potassium hydroxide was purchased from Shanghai Aladdin Co., Ltd. Ketjen black EC-300J was produced by lion corporation. Cabot Vulcan XC72 and Cabot Black Pearls 2000 were produced by the Cabot Corporation. Commercial Pt/C was produced by E-teK. Deionized water (18.2 MΩ) was used throughout the experiment. All chemicals were analytically pure and used as received without further purification.Synthesis of FeCu-SAC, Fe-SAC and Cu-SAC
FeCu-SAC was synthesized by applying a modified ligand-mediated method.45 Typically, 6.08 mg of iron(II) acetate, 6.98 mg of copper(II) acetate monohydrate and 58.8 mg of 1,10-phenanthroline monohydrate were dissolved in 2 mL of dimethyl sulfoxide under magnetic stirring. Subsequently, 69.6 mg of carbon black (Ketjen black EC-300J) was added into the solution and then heated in an oil bath at 60 °C under magnetic stirring for 4 h. The resulting dispersion was heated at 190 °C for 12 h to evaporate the dimethyl sulfoxide, and a black solid was obtained. Then, the produced black solid was ground with a mortar and subsequently pyrolyzed in a tube furnace at 800 °C under an argon atmosphere for 2 h. After cooling to room temperature, the black powder was soaked in 2 M HCl at 80 °C with continuous magnetic stirring for 12 h. Then, the products were washed with deionized water 3 times and ethanol 2 times under centrifugal, and the FeCu-SAC was obtained. The Fe-SAC was synthesized with a similar method except that only 6.08 mg of iron(II) acetate and 29.4 mg of 1,10-phenanthroline monohydrate were used. The Cu-SAC was synthesized with a similar method except that only 6.98 mg of copper(II) acetate monohydrate and 29.4 mg of 1,10-phenanthroline monohydrate were used. To synthesize FeCu-SAC loaded on various carbon substrates, all experimental protocols were the same as those for FeCu-SAC except that different carbon substrates (Cabot Vulcan XC72 and Cabot Black Pearls 2000) were used.Details of the electrochemical measurements, characterization and simulation methods can be found in the ESI.†
Results and discussion
Characterization of FeCu-SAC, Fe-SAC and Cu-SAC
FeCu-SAC was synthesized using a modified ligand-mediated method.45 For comparison, Fe-SAC and Cu-SAC were also synthesized using the same method (Ketjen black EC-300J was used as the carbon substrate without specified notation). Transmission electron microscopy (TEM) images showed that the FeCu-SAC (Fig. 1a) maintained a similar morphology to the carbon substrate (Fig. S1†), and no metal nanoparticles were observed. The inset selected area electron diffraction (SAED) image further confirmed the absence of metal nanoparticles in FeCu-SAC, whereas the diffraction rings corresponded to carbon, and no diffraction dots were observed. Similar TEM (Fig. S2 and S3†) and SAED results (Fig. S4 and S5†) were also observed in the control Fe-SAC and Cu-SAC samples. Powder X-ray diffraction (XRD) further provided crystal structure information at the macroscale (Fig. S6†). For all three samples, there were only two broad peaks located at about 24.8° and 43.4°, corresponding to the (002) and (100) planes of graphite, and no crystal peaks corresponding to metal species were found. Energy dispersive X-ray (EDX) element maps for FeCu-SAC indicate that the Fe, Cu and N were uniformly dispersed over the carbon substrate (Fig. 1b and c). The individual Fe and Cu were also uniformly distributed over the carbon substrates in Fe-SAC (Fig. S7†) and Cu-SAC (Fig. S8†), respectively. | ||
| Fig. 1 (a) TEM and SAED (insert) images of FeCu-SAC. (b) HAADF-STEM image of FeCu-SAC. (c) EDX elemental maps of FeCu-SAC. | ||
The metal content was analysed by inductively coupled plasma optical emission spectroscopy. The Fe and Cu contents in FeCu-SAC were determined to be 1.29 wt% and 0.25 wt%, respectively. Similar Fe and Cu loadings were also achieved in Fe-SAC (1.19 wt%) and Cu-SAC (0.34 wt%), respectively. Raman spectra (Fig. S9†) exhibited two broad peaks located at about 1359 and 1595 cm−1, which were assigned to disordered sp3 carbon (D band) and graphitic sp2 carbon (G band), respectively. The FeCu-SAC, Fe-SAC and Cu-SAC samples exhibited similar ID/IG ratios (1.06–1.08), indicating similar defect concentrations and graphitization degrees.
HAADF-STEM was used to determine the morphology of the Fe and Cu sites for the FeCu-SAC on an atomic scale, in which the individual bright dots (Fig. 2a) indicated the atomic dispersion of Fe and Cu atoms on the carbon substrate. However, the similar atomic numbers of Fe (26) and Cu (29) made it challenging to distinguish between the Fe and Cu sites in terms of the bright dots in the HAADF-STEM image. The coexistence of Fe and Cu atoms in the FeCu-SAC was further validated by EELS (Fig. 2b), whereas the Fe EELS spectrum at 711 eV and the Cu EELS spectrum at 973 eV were captured with the detecting spot in the size of ∼4 nm2 (red square area in Fig. 2a), indicating very closed distances of Fe and Cu atoms. The Fe and Cu atoms were also atomically dispersed in Fe-SAC and Cu-SAC, respectively (Fig. S10 and S11†). EXAFS further provided average metal coordination information for the Fe and Cu sites in the FeCu-SAC. Both Fe K-edge and Cu K-edge EXAFS R space plots of FeCu-SAC (Fig. 2c and d) displayed only an intense peak at approximately 1.56 Å and 1.5 Å, respectively, similar to the metal–N bond in FePc and CuPc. No metal scattering paths were observed compared to metal foil and metal oxides, indicating the absence of a metal–metal bond. EXAFS fitting for the first shell coordination demonstrated that both Fe and Cu atoms were coordinated with 4 nitrogen atoms, referred to as FeN4 and CuN4 sites, respectively (Table S1†). Furthermore, the EXAFS wavelet transform (WT) results displayed the scattering paths of the samples in both the R and K spaces. The Fe K-edge WT-EXAFS and Cu K-edge WT-EXAFS for FeCu-SAC showed maximum WT intensity at around 3.5 Å−1 and 4.0 Å−1, respectively (Fig. 2e and f), which are distinct from those of metal foil and metal oxide references. For Fe-SAC and Cu-SAC control samples, the dominant presence of Fe–N and Cu–N bonds in the EXAFS results also confirmed the atomic dispersion of the metal sites in the two samples, which agrees with the HAADF-STEM results (Fig. S12 and S13†). Detailed analysis of the X-ray absorption near edge spectroscopy (XANES) results and its first-derivative spectra are presented in Fig. S14 and S15.† In the K-edge XANES spectra for FeCu-SAC and Fe-SAC, the Fe absorption edge was located between Fe foil and Fe2O3, indicating that the valence state of Fe was from 0 to +3 (approximately +2). For FeCu-SAC and Cu-SAC, the Cu adsorption edge was slightly lower than that of CuO, indicating that the valence state of Cu was slightly lower than +2 because it was coordinated with N rather than O. Based on the first-derivative spectra, the Fe absorption edge position in FeCu-SAC was slightly lower than that of Fe-SAC, indicating that the Fe oxidation state in FeCu-SAC was slightly lower than that in Fe-SAC. The Cu absorption edge position in FeCu-SAC was slightly higher than that in Cu-SAC, indicating that the Cu oxidation state in FeCu-SAC was slightly higher than that in Cu-SAC. The oxidation state changes can be attributed to the Cu atoms as electron donators enriching the electron density of the Fe atoms.
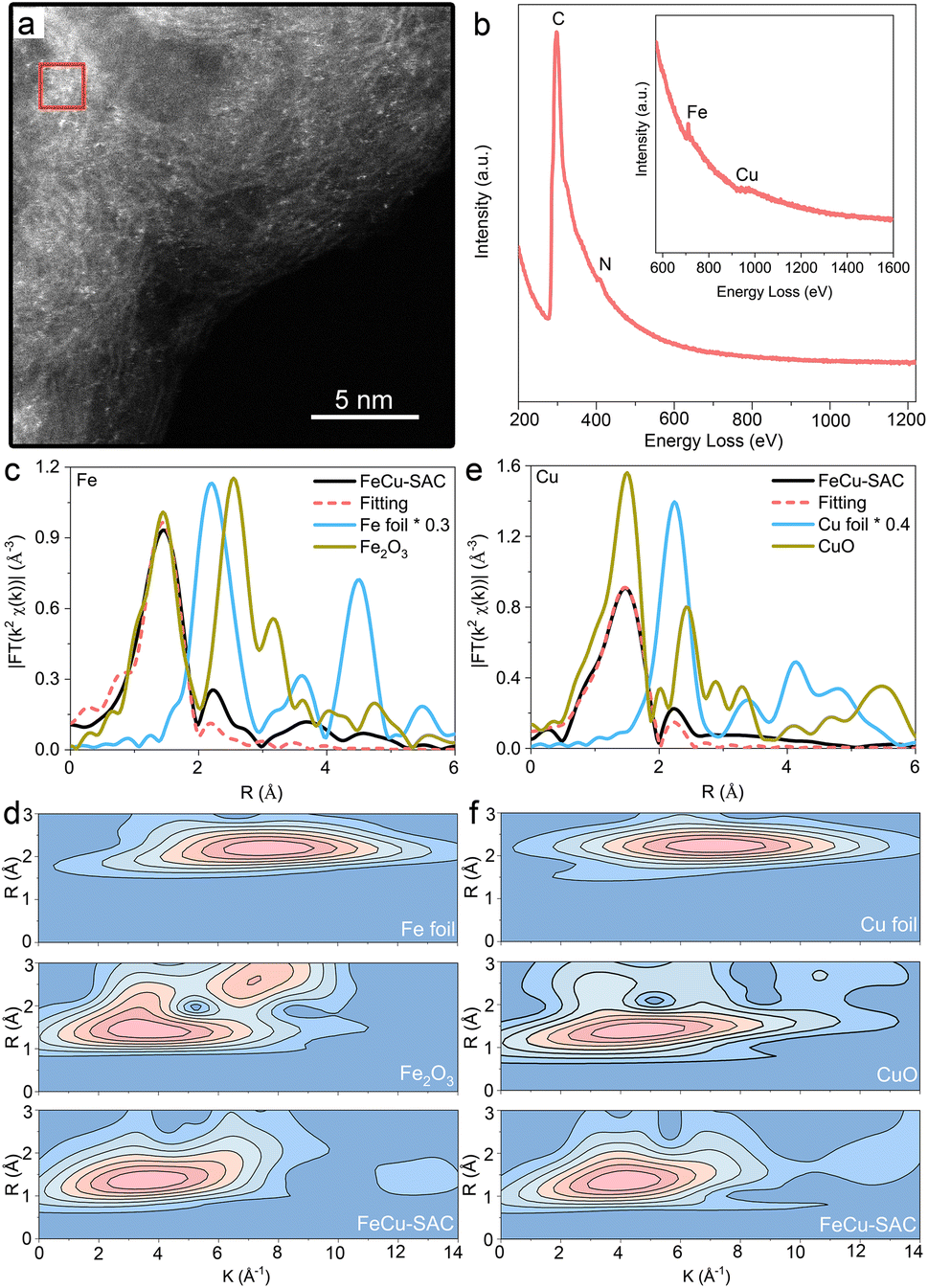 | ||
| Fig. 2 (a) HAADF-STEM image of FeCu-SAC. (b) EELS spectrum for FeCu-SAC (inset: magnified spectrum for Fe and Cu peaks). (c) Fe K-edge EXAFS R space plot and fitting results for FeCu-SAC. (d) Fe K-edge EXAFS WT images of FeCu-SAC and references. (e) Cu K-edge EXAFS R space plot and fitting results of FeCu-SAC. (f) Cu K-edge EXAFS WT images of FeCu-SAC and references. | ||
To investigate the chemical composition and elemental valence states, X-ray photoelectron spectroscopy (XPS) was performed. The C 1s spectrum of FeCu-SAC exhibited an intense peak at about 284.8 eV, and it can be deconvoluted into two peaks, corresponding to C–C or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–N species (Fig. S16a†). The N 1s spectrum of FeCu-SAC can be deconvoluted into four peaks, corresponding to pyridinic-N, pyrrolic-N, graphitic-N, and oxidized-N (Fig. S16b†). The Fe 2p spectrum showed a Fe 2p3/2 peak at 711.07 eV corresponding to Fe3+ (Fig. S16c†). The Cu 2p spectrum exhibited a Cu 2p3/2 peak at 933.06 eV corresponding to Cu2+ (Fig. S16d†). The metal valence state was slightly higher than the XANES results, which may result from the surface oxidization. For XPS spectra of Fe-SAC (Fig. S17†) and Cu-SAC (Fig. S18†), the C 1s spectra showed similar results with FeCu-SAC. However, for the N 1s spectra, the concentration of different N species was different (Table S2†).
C and C–N species (Fig. S16a†). The N 1s spectrum of FeCu-SAC can be deconvoluted into four peaks, corresponding to pyridinic-N, pyrrolic-N, graphitic-N, and oxidized-N (Fig. S16b†). The Fe 2p spectrum showed a Fe 2p3/2 peak at 711.07 eV corresponding to Fe3+ (Fig. S16c†). The Cu 2p spectrum exhibited a Cu 2p3/2 peak at 933.06 eV corresponding to Cu2+ (Fig. S16d†). The metal valence state was slightly higher than the XANES results, which may result from the surface oxidization. For XPS spectra of Fe-SAC (Fig. S17†) and Cu-SAC (Fig. S18†), the C 1s spectra showed similar results with FeCu-SAC. However, for the N 1s spectra, the concentration of different N species was different (Table S2†).
ORR performance
The above characterization results revealed that the FeCu-SAC contained FeN4 and CuN4 sites that were expected to have a synergistic effect for promoting ORR activity. This motivated us to investigate the ORR performance comprehensively. First, cyclic voltammetry (CV) curves were measured in O2 and N2 saturated 0.1 M KOH. As shown in Fig. S19,† the CV curve of FeCu-SAC exhibited a reduction peak at 0.870 V (all potentials were converted to versus reversible hydrogen electrode (RHE) in this study) in O2 saturated electrolyte, which disappeared in N2 saturated electrolyte, indicating that this peak originated from ORR. The reduction peaks in the CV curves of Fe-SAC (Fig. S20†) and Cu-SAC (Fig. S21†) appeared at lower potentials, 0.867 and 0.749 V, respectively.Furthermore, the ORR activity was evaluated using linear sweep voltammograms (LSV). Fig. 3a depicts the LSV curves for FeCu-SAC, Fe-SAC and Cu-SAC. The FeCu-SAC showed excellent ORR performance with a high E1/2 of 0.926 V, which was significantly higher than that of commercial Pt/C (0.884 V), Fe-SAC (0.899 V) and Cu-SAC (0.810 V). Moreover, the FeCu-SAC exhibited good ORR performance in 0.5 M H2SO4 with a half wave potential of 0.779 V (Fig. S22†). The higher activity of FeCu-SAC might be attributed to the synergistic effect between the Fe and Cu atoms. Fig. 3b shows the E1/2 and kinetic current density (Jk) at 0.9 V calculated from the LSV curves in Fig. 3a. The FeCu-SAC exhibited the highest Jk of 16.33 mA cm−2, which was 2.9 and 161.3 times higher than those of Fe-SAC and Cu-SAC, respectively, indicating the outstanding intrinsic ORR activity of FeCu-SAC. Moreover, the FeCu-SAC showed a low Tafel slope of 53.6 mV dec−1, which was slightly lower than those of Fe-SAC (61.7 mV dec−1) and Cu-SAC (54.2 mV dec−1) (Fig. 3c). This demonstrates the fast kinetics of FeCu-SAC for ORR. To probe the ORR path (i.e. 2e- or 4e-transfer process), a rotating ring-disk electrode (RRDE) measurement was performed. FeCu-SAC showed the lowest H2O2 yield (lower than 2.4%) and the highest electron transfer number (3.93), indicating the efficient four-electron transfer ORR process of FeCu-SAC (Fig. 3d). Moreover, the electron transfer number was also evaluated by employing Koutecký–Levich plots. As shown in Fig. S23–S25,† the FeCu-SAC exhibited the highest electron transfer number of 3.80, and the Fe-SAC and Cu-SAC exhibited an electron transfer number of 3.72 and 2.96, respectively, which agreed with the RRDE results. In addition, the FeCu-SAC exhibited excellent methanol tolerance during the ORR process (Fig. 3e), whereas the current density of the FeCu-SAC exhibited negligible decay after the injection of 2 mL of methanol. However, commercial Pt/C showed 36% current decay after the methanol injection for 5000 s. Stability was also an important parameter used to evaluate the ORR performance of the catalyst. The stability of the catalysts was evaluated using the amperometric i–t method (Fig. 3f). FeCu-SAC exhibited good stability with only 15% current decay over 24 h, which was much lower than those of Fe-SAC (39%) and commercial Pt/C (49%). These results indicate that the addition of Cu atoms to FeCu-SAC can simultaneously improve the ORR activity and stability,16 and the as-synthesized FeCu-SAC is one of the best dual metal SACs for ORR compared with reported studies (Table S3†).28–30,40,44–57
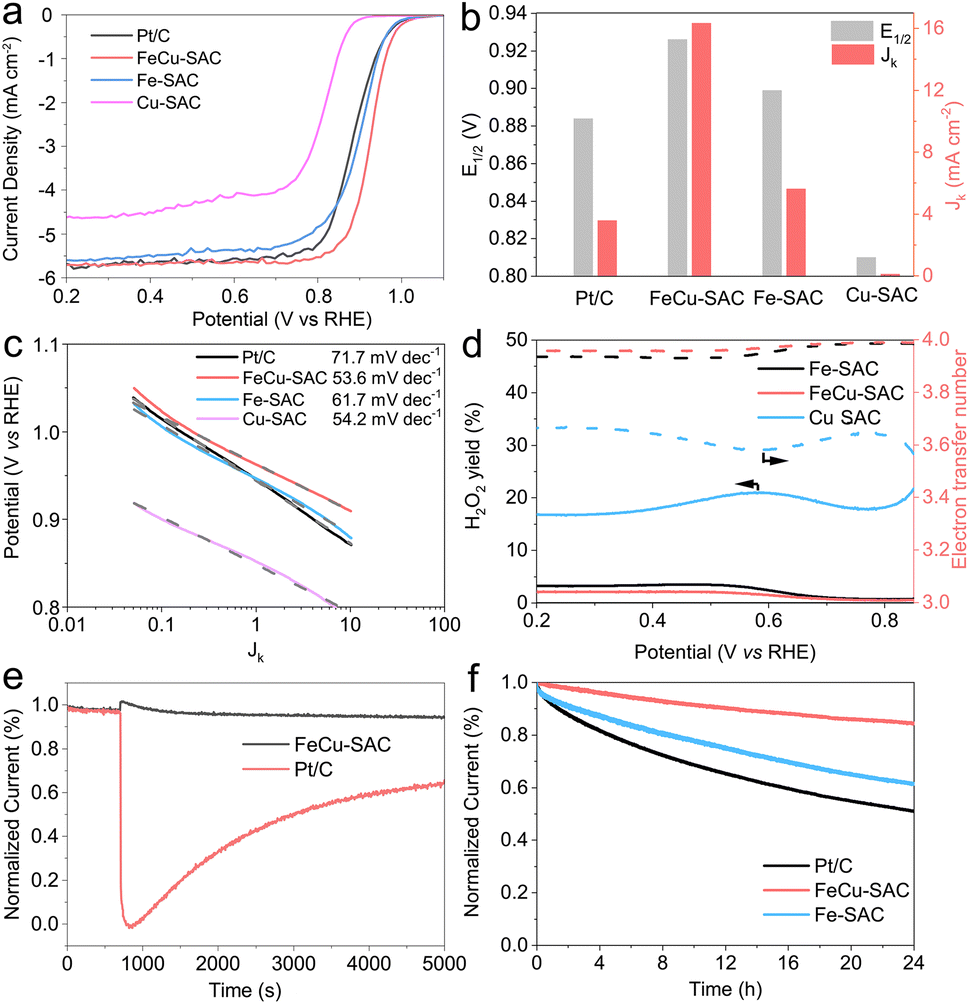 | ||
| Fig. 3 ORR performance of FeCu-SAC, Fe-SAC, Cu-SAC and commercial Pt/C. (a) LSV curves. (b) Half-wave potential and kinetic current density at 0.9 V. (c) Tafel slope of FeCu-SAC, Fe-SAC, Cu-SAC and commercial Pt/C. (d) H2O2 yield and calculated electron transfer tested by RRDE. (e) Methanol tolerance of FeCu-SAC and commercial Pt/C. (f) Amperometric i–t curves of FeCu-SAC, Fe-SAC and commercial Pt/C. | ||
Dual metal synergistic effect and DFT simulation
To understand the synergistic effect between Fe and Cu atoms, a physically mixed Fe-SAC and Cu-SAC sample was also measured. Compared to the FeCu-SAC, the lower E1/2 of the physically mixed sample (0.882 V, Fig. S26†) implied negligible interactions between Fe and Cu atoms, which was due to the distinct neighboring distance between Fe and Cu atoms. In contrast, the neighboring distance between Fe and Cu atoms was very close in FeCu-SAC owing to the synergistic interaction between Fe and Cu atoms. To further illustrate the synergistic effect of Fe and Cu atoms on the ORR activity of FeCu-SAC, DFT calculations were performed. The structural model for FeCu-SAC was based on the fact that one FeN4 site and one CuN4 site sit adjacent to the graphene matrix (Fig. 4a), while only the FeN4 site or CuN4 site was present in the Fe-SAC (Fig. 4b) or Cu-SAC (Fig. S27b†) samples. The possible intermediate active sites were carefully examined using the ab initio calculation.58,59 The optimized structure revolution processes for Pt, Fe-SAC, Cu-SAC and FeCu-SAC during ORR are presented in Fig. S27.† Based on the optimized ORR intermediates, the Fe sites in FeCu-SAC served as the active sites for ORR, while the Fe or Cu sites functioned in Fe-SAC or Cu-SAC, respectively. The electron density difference analysis (Fig. 4c and d) indicated that the Fe sites in FeCu-SAC had a higher electron density than those in Fe-SAC. This implies that the neighboring Cu sites with a high electron density (d10) can serve as an electron donator enriching the electron density of Fe sites. Further, the free-energy evolution for ORR of Pt, Fe-SAC, Cu-SAC and FeCu-SAC are displayed in Fig. 4e. It showed that the FeCu-SAC had different rate determining steps than that of Fe-SAC and Cu-SAC. The FeCu-SAC encountered the same rate determining step as that of Pt metal, i.e. the O* to OH* step, while another two steps were the ORR barriers for Fe-SAC and Cu-SAC, i.e. the OH* to OH− step and O2 to OOH* step. The energy barrier of the rate determining step for FeCu-SAC was 0.47 eV, even lower than that of Pt (0.62 eV), whereas there were much higher energy barriers for Fe-SAC (0.55 eV) and Cu-SAC (1.19 eV). These results indicate that the introduction of CuN4 sites in FeCu-SAC endowed the FeN4 site with an enriched electron density. Therefore, the modified Fe sites displayed optimized adsorption/desorption energy on the ORR intermediates. The calculation of the density of states (Fig. 4f and S28†) further provided evidence that the occupied density of states across the Femi level of FeCu-SAC was beneficial to the electron interaction and conduction of FeCu-SAC during the ORR process.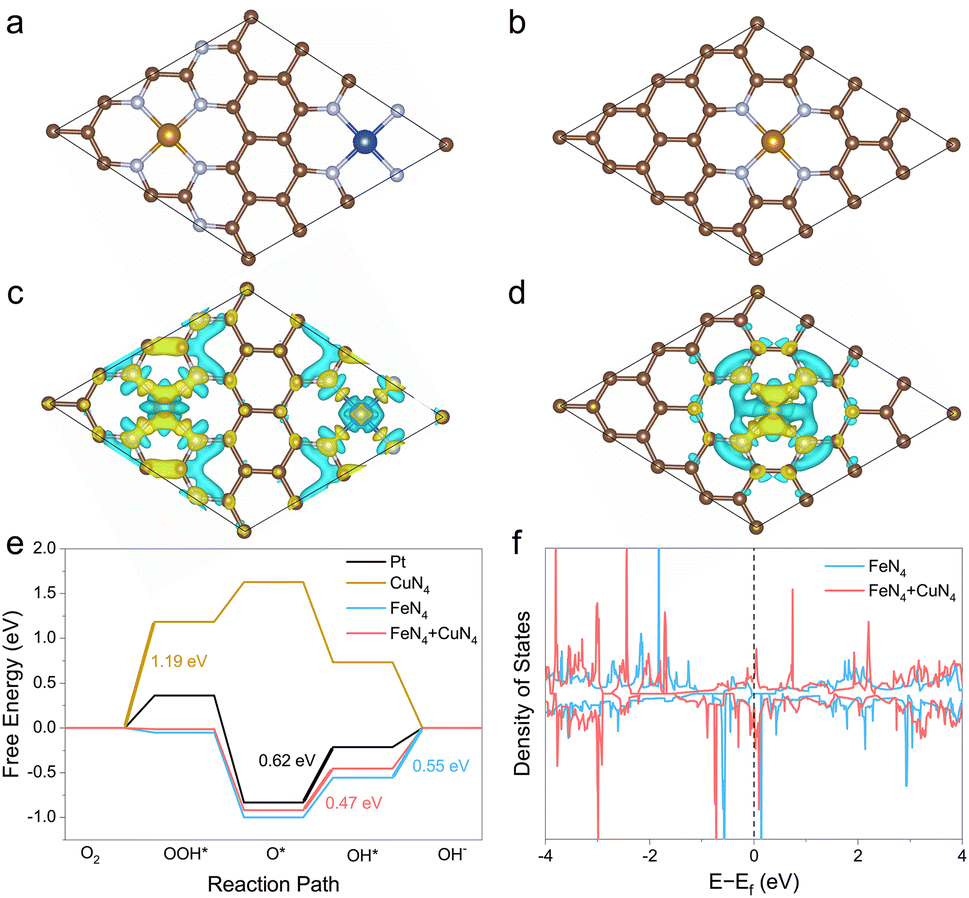 | ||
| Fig. 4 (a) Optimized structure of FeCu-SAC. (b) Optimized structure of Fe-SAC. (C: brown, N: gray, Fe: khaki, and Cu: blue). (c) Electron density difference analysis of FeCu-SAC. (d) Electron density difference analysis of Fe-SAC. (e) Calculated free-energy evolution diagram for ORR of Pt, Fe-SAC, Cu-SAC and FeCu-SAC at 1.23 V. (f) Density of states for FeN4 + CuN4 and FeN4. | ||
The influence of carbon substrates
The use of commercial carbon black with excellent conductivity can avoid the abundant usage of expensive organic precursors to achieve the large-scale, robust and low-cost preparation of SAC. However, there is limited research on which commercial carbon black is more suitable for synthesizing high-efficiency SAC. To investigate the influence of carbon substrates, FeCu-SAC was also synthesized with other typical commercial carbon blacks, such as Cabot Vulcan XC72 and Cabot Black Pearls 2000. Accordingly, the prepared SACs were named FeCu-SAC-XC 72 and FeCu-SAC-BP 2000, respectively (our standard FeCu-SAC catalyst would be FeCu-SAC-300J using this notation). FeCu-SAC-300J exhibited the highest ORR performance with a high E1/2 of 0.926 V (Fig. 5a), which was significantly higher than those of FeCu-SAC-XC 72 (0.839 V) and FeCu-SAC-BP 2000 (0.868 V). To illustrate why Ketjen black EC-300J was more suitable for ORR, N2 adsorption/desorption was performed to detect the surface area and pore structure of these catalysts. As shown in Fig. S29,† all these catalysts exhibited type-IV isotherms, in which both FeCu-SAC-BP2000 (375.85 m2 g−1) and FeCu-SAC-300J (337.88 m2 g−1) exhibited significantly higher surface areas than that of FeCu-SAC-XC 72 (92.79 m2 g−1). This explains why FeCu-SAC-XC 72 showed much lower ORR activity than the other two control samples. Further, although the FeCu-SAC-BP2000 has a slightly higher surface area than that of FeCu-SAC-300J, the FeCu-SAC-BP 2000 mainly contained micropores, which mainly induce an internal surface but cannot be utilized for ORR (Fig. 5b).60–62 Instead, the hierarchical pore structure of FeCu-SAC-300J enabled the accommodation of single-atom sites in the micropores, while the meso/macropores enabled the efficient mass transfer of oxygen intermediates.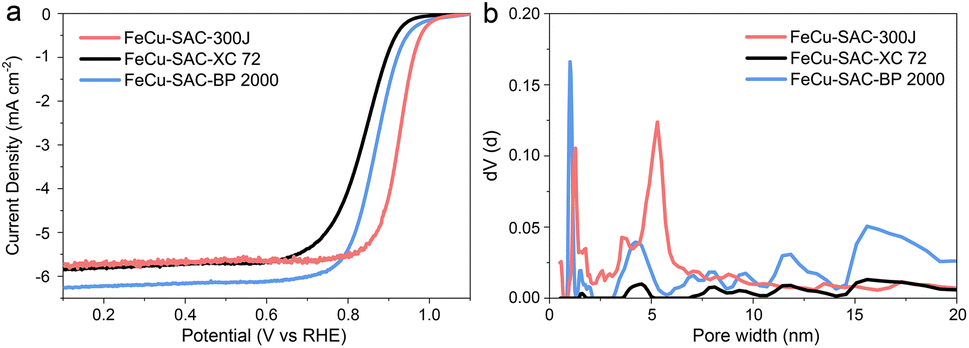 | ||
| Fig. 5 (a) LSV curves of FeCu-SACs with various carbon substrates. (b) Pore size distribution of FeCu-SACs with various carbon substrates. | ||
Zn–air battery
The Zn–air battery was considered as a promising secondary battery owing to its low cost and high capacity.63–65 Inspired by the efficient ORR performance, FeCu-SAC was used to assemble an aqueous primary Zn–air battery to evaluate its performance in practical energy devices. As shown in Fig. S30,† the Zn–air battery constructed using FeCu-SAC exhibited the highest power density of 201.4 mW cm−2 at 358.2 mA cm−2, which was significantly higher than that of the benchmark commercial Pt/C (128.2 mW cm−2 at 197.9 mA cm−2). Moreover, the Zn–air battery constructed by FeCu-SAC also exhibited superior specific energy of 827.68 W h kg−1 at 20 mA cm−2 to the Zn–air battery constructed by commercial Pt/C (817.79 W h kg−1 at 20 mA cm−2, Fig. S31†).Conclusions
In summary, Fe, Cu dual metal SAC was successfully synthesized using a facile and low-cost ligand-mediated method. Owing to Cu atoms dispersed nearby around Fe atoms, the synergistic effect between Fe and Cu resulted in improved ORR activity and stability for FeCu-SAC. A high half-wave potential of 0.926 V was achieved in the alkaline media. DFT calculations confirmed that the addition of Cu atoms can decrease the energy barrier for ORR and therefore facilitate ORR activity. Furthermore, the influence of commercial carbon black supports was also investigated, and Ketjen EC-300J supported Fe, Cu dual metal SAC exhibited superior ORR performance owing to its high surface area and suitable pore structure. The excellent ORR activity and durability enabled the constructed Zn–air battery to have a high power density of 201.4 mW cm−2 and a high specific energy of 827.68 W h kg−1. This study offers a paradigm for using Cu as a promoter to boost the ORR activity of Fe-SAC, and we expect that this study will inspire researchers to design low-cost and highly efficient ORR catalysts.Author contributions
Tierui Zhang and Lu Shang conceived the idea and designed the experiments. Tierui Zhang and Shouguo Wang supervised the project. Hongzhou Yang carried out the experiments, characterizations. He Huang helped to carry out the DFT simulations. Qing Wang help to analyse EXAFS results and write the manuscript. Hongzhou Yang, Lu Shang, Tierui Zhang and Shouguo Wang wrote the manuscript. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the National Key Research and Development Program of China (2019YFB2005800), the National Natural Science Foundation of China (52130103, 51825205, 21871279 and 51971026), the Beijing Natural Science Foundation (2222080), and the Youth Innovation Promotion Association of the CAS (Y2021011). The XAFS experiments were conducted at the 1W1B beamline of Beijing Synchrotron Radiation Facility (BSRF).Notes and references
- Y. He, S. Liu, C. Priest, Q. Shi and G. Wu, Chem. Soc. Rev., 2020, 49, 3484–3524 RSC.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- S. Sultan, J. Tiwari, J. Jang, A. Harzandi, F. Salehnia, S. Yoo and K. Kim, Adv. Energy Mater., 2018, 8, 1801002 CrossRef.
- J. Tiwari, W. Lee, S. Sultan, M. Yousuf, A. Harzandi, V. Vij and K. Kim, ACS Nano, 2017, 11, 7729–7735 CrossRef CAS PubMed.
- D. Zhao, Z. Zhuang, X. Cao, C. Zhang, Q. Peng, C. Chen and Y. Li, Chem. Soc. Rev., 2020, 49, 2215–2264 RSC.
- Y. Wang, N. Zhao, B. Fang, H. Li, X. Bi and H. Wang, Chem. Rev., 2015, 115, 3433–3467 CrossRef CAS PubMed.
- Z. Yang, H. Yang, L. Shang and T. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202113278 CAS.
- J. Tiwari, K. Nath, S. Kumar, R. Tiwari, K. Kemp, N. Le, D. Youn, J. Lee and K. Kim, Nat. Commun., 2013, 4, 2221 CrossRef PubMed.
- J. Tiwari, K. Kemp, K. Nath, R. Tiwari, H. Nam and K. Kim, ACS Nano, 2013, 7, 9223–9231 CrossRef CAS PubMed.
- H. Yang, R. Shi, L. Shang and T. Zhang, Small Struct., 2021, 2, 2100007 CrossRef CAS.
- D. Wang, P. Yang, L. Liu, W. Wang and Z. Chen, Mater. Today Energy, 2022, 26, 101017 CrossRef CAS.
- Y. Zhu, C. Shang, Z. Wang, J. Zhang, M. Yang, H. Cheng and Z. Lu, Rare Met., 2021, 40, 90–95 CrossRef.
- M. Lefevre, E. Proietti, F. Jaouen and J. Dodelet, Science, 2009, 324, 71–74 CrossRef CAS PubMed.
- L. Wu, R. Zhao, G. Du, H. Wang, M. Hou, W. Zhang, P. Sun and T. Chen, Green Energy Environ., 2022 DOI:10.1016/j.gee.2022.03.014.
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W. Cheong, R. Shen, X. Wen, L. Zheng, A. Rykov, S. Cai, H. Tang, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 5422 CrossRef CAS PubMed.
- L. Jiao, J. Zhu, Y. Zhang, W. Yang, S. Zhou, A. Li, C. Xie, X. Zheng, W. Zhou, S. Yu and H. Jiang, J. Am. Chem. Soc., 2021, 143, 19417–19424 CrossRef CAS PubMed.
- G. Wu, K. More, C. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- W. Zhang, Y. Chao, W. Zhang, J. Zhou, F. Lv, K. Wang, F. Lin, H. Luo, J. Li, M. Tong, E. Wang and S. Guo, Adv. Mater., 2021, 33, 2102576 CrossRef CAS PubMed.
- Y. Yan, H. Cheng, Z. Qu, R. Yu, F. Liu, Q. Ma, S. Zhao, H. Hu, Y. Cheng, C. Yang, Z. Li, X. Wang, S. Hao, Y. Chen and M. Liu, J. Mater. Chem. A, 2021, 9, 19489–19507 RSC.
- R. Li and D. Wang, Adv. Energy Mater., 2022, 12, 2103564 CrossRef CAS.
- Q. An, J. Jiang, W. Cheng, H. Su, Y. Jiang and Q. Liu, Small Methods, 2022, 6, 202200408 Search PubMed.
- C. Xu, L. Chen, Y. Wen, S. Qin, H. Li, Z. Hou, Z. Huang, H. Zhou and Y. Kuang, Mater. Today Energy, 2021, 21, 100721 CrossRef CAS.
- M. Ha, D. Kim, M. Umer, V. Gladkikh, C. Myung and K. Kim, Energy Environ. Sci., 2021, 14, 3455–3468 RSC.
- W. Li, J. Yang and D. Wang, Angew. Chem., Int. Ed., 2022, 61, e202213318 CAS.
- X. Zheng, B. Li, Q. Wang, D. Wang and Y. Li, Nano Res., 2022, 15, 7806–7839 CrossRef CAS.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS PubMed.
- J. Chen, H. Li, C. Fan, Q. Meng, Y. Tang, X. Qiu, G. Fu and T. Ma, Adv. Mater., 2020, 32, e2003134 CrossRef PubMed.
- Z. Chen, X. Liao, C. Sun, K. Zhao, D. Ye, J. Li, G. Wu, J. Fang, H. Zhao and J. Zhang, Appl. Catal., B, 2021, 288, 120021 CrossRef CAS.
- T. Cui, Y. Wang, T. Ye, J. Wu, Z. Chen, J. Li, Y. Lei, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202115219 CAS.
- D. Liu, B. Wang, H. Li, S. Huang, M. Liu, J. Wang, Q. Wang, J. Zhang and Y. Zhao, Nano Energy, 2019, 58, 277–283 CrossRef CAS.
- Y. Qu, Z. Li, W. Chen, Y. Lin, T. Yuan, Z. Yang, C. Zhao, J. Wang, C. Zhao, X. Wang, F. Zhou, Z. Zhuang, Y. Wu and Y. Li, Nat. Catal., 2018, 1, 781–786 CrossRef CAS.
- L. Cui, L. Cui, Z. Li, J. Zhang, H. Wang, S. Lu and Y. Xiang, J. Mater. Chem. A, 2019, 7, 16690–16695 RSC.
- J. Yang, W. Liu, M. Xu, X. Liu, H. Qi, L. Zhang, X. Yang, S. Niu, D. Zhou, Y. Liu, Y. Su, J. Li, Z. Tian, W. Zhou, A. Wang and T. Zhang, J. Am. Chem. Soc., 2021, 143, 14530–14539 CrossRef CAS PubMed.
- F. Li, G. Han, H. Noh, S. Kim, Y. Lu, H. Jeong, Z. Fu and J. Baek, Energy Environ. Sci., 2018, 11, 2263–2269 RSC.
- C. Huang, L. Zheng, W. Feng, A. Guo, X. Gao, Z. Long and X. Qiu, ACS Sustainable Chem. Eng., 2020, 8, 14030–14038 CrossRef CAS.
- D. Wang, C. Ao, X. Liu, S. Fang, Y. Lin, W. Liu, W. Zhang, X. Zheng, L. Zhang and T. Yao, ACS Appl. Energy Mater., 2019, 2, 6497–6504 CrossRef CAS.
- J. Xu, A. Elangovan, J. Li and B. Liu, J. Phys. Chem. C, 2021, 125, 2334–2344 CrossRef CAS.
- H. Wu, J. Yan, X. Xu, Q. Yuan, J. Wang, J. Cui and A. Lin, Chem. Eng. J., 2022, 428, 132611 CrossRef CAS.
- C. Du, Y. Gao, H. Chen, P. Li, S. Zhu, J. Wang, Q. He and W. Chen, J. Mater. Chem. A, 2020, 8, 16994–17001 RSC.
- X. Wei, S. Wei, S. Cao, Y. Hu, S. Zhou, S. Liu, Z. Wang and X. Lu, Appl. Surf. Sci., 2021, 564, 150423 CrossRef CAS.
- C. Li, D. Zhao, H. Long and M. Li, Rare Met., 2021, 40, 2657–2689 CrossRef CAS.
- J. Zhang, C. Asokan, G. Zakem, P. Christopher and J. Medlin, Green Energy Environ., 2022, 7, 1263–1269 CrossRef CAS.
- Z. Wang, H. Jin, T. Meng, K. Liao, W. Meng, J. Yang, D. He, Y. Xiong and S. Mu, Adv. Funct. Mater., 2018, 28, 1802596 CrossRef.
- Z. Xiao, P. Sun, Z. Qiao, K. Qiao, H. Xu, S. Wang and D. Cao, Chem. Eng. J., 2022, 446, 137112 CrossRef CAS.
- H. Yang, L. Shang, Q. Zhang, R. Shi, G. Waterhouse, L. Gu and T. Zhang, Nat. Commun., 2019, 10, 4585 CrossRef PubMed.
- T. He, Y. Chen, Q. Liu, B. Lu, X. Song, H. Liu, M. Liu, Y. Liu, Y. Zhang, X. Ouyang and S. Chen, Angew. Chem., Int. Ed., 2022, 61, e202201007 CAS.
- J. Li, J. Chen, H. Wan, J. Xiao, Y. Tang, M. Liu and H. Wang, Appl. Catal., B, 2019, 242, 209–217 CrossRef CAS.
- Y. He, X. Yang, Y. Li, L. Liu, S. Guo, C. Shu, F. Liu, Y. Liu, Q. Tan and G. Wu, ACS Catal., 2022, 12, 1216–1227 CrossRef CAS.
- K. Wang, J. Liu, Z. Tang, L. Li, Z. Wang, M. Zubair, F. Ciucci, L. Thomsen, J. Wright and N. Bedford, J. Mater. Chem. A, 2021, 9, 13044–13055 RSC.
- Z. Wang, X. Jin, C. Zhu, Y. Liu, H. Tan, R. Ku, Y. Zhang, L. Zhou, Z. Liu, S. Hwang and H. Fan, Adv. Mater., 2021, 33, e2104718 CrossRef PubMed.
- G. Zhang, Y. Jia, C. Zhang, X. Xiong, K. Sun, R. Chen, W. Chen, Y. Kuang, L. Zheng, H. Tang, W. Liu, J. Liu, X. Sun, W. Lin and H. Dai, Energy Environ. Sci., 2019, 12, 1317–1325 RSC.
- X. Zhou, J. Gao, Y. Hu, Z. Jin, K. Hu, K. Reddy, Q. Yuan, X. Lin and H. Qiu, Nano Lett., 2022, 22, 3392–3399 CrossRef CAS PubMed.
- D. Yu, Y. Ma, F. Hu, C. Lin, L. Li, H. Chen, X. Han and S. Peng, Adv. Energy Mater., 2021, 11, 2101242 CrossRef CAS.
- Z. Chen, X. Su, J. Ding, N. Yang, W. Zuo, Q. He, Z. Wei, Q. Zhang, J. Huang and Y. Zhai, Appl. Catal., B, 2022, 308, 121206 CrossRef CAS.
- A. Han, X. Wang, K. Tang, Z. Zhang, C. Ye, K. Kong, H. Hu, L. Zheng, P. Jiang, C. Zhao, Q. Zhang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 19262–19271 CrossRef CAS PubMed.
- Q. Miao, S. Yang, Q. Xu, M. Liu, P. Wu, G. Liu, C. Yu, Z. Jiang, Y. Sun and G. Zeng, Small Struct., 2022, 3, 2100225 CrossRef CAS.
- M. Tong, F. Sun, Y. Xie, Y. Wang, Y. Yang, C. Tian, L. Wang and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 14005–14012 CrossRef CAS PubMed.
- V. Vij, S. Sultan, A. Harzandi, A. Meena, J. Tiwari, W. Lee, T. Yoon and K. Kim, ACS Catal., 2017, 7, 7196–7225 CrossRef CAS.
- H. Huang, H. Wu, C. Chi, B. Huang and T. Zhang, J. Mater. Chem. A, 2019, 7, 8897–8904 RSC.
- X. Wan, X. Liu, Y. Li, R. Yu, L. Zheng, W. Yan, H. Wang, M. Xu and J. Shui, Nat. Catal., 2019, 2, 259–268 CrossRef CAS.
- Q. Wang, Y. Yang, F. Sun, G. Chen, J. Wang, L. Peng, W. Chen, L. Shang, J. Zhao, D. Sun-Waterhouse, T. Zhang and G. Waterhouse, Adv. Energy Mater., 2021, 11, 2100219 CrossRef CAS.
- X. Xie, L. Peng, H. Yang, G. Waterhouse, L. Shang and T. Zhang, Adv. Mater., 2021, 33, 2101038 CrossRef CAS PubMed.
- J. Fu, Z. Cano, M. Park, A. Yu, M. Fowler and Z. Chen, Adv. Mater., 2017, 29, 1604685 CrossRef PubMed.
- M. Dong, X. Liu, L. Jiang, Z. Zhu, Y. Shu, S. Chen, Y. Dou, P. Liu, H. Yin and H. Zhao, Green Energy Environ., 2020, 5, 499–505 CrossRef.
- R. Anand, B. Ram, M. Umer, M. Zafari, S. Umer, G. Lee and K. Kim, J. Mater. Chem. A, 2022, 10, 22500–22511 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta09922e |
| ‡ Hongzhou Yang and He Huang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |