
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdjusting the band gap of CsPbBr3−yXy (X = Cl, I) for optimal interfacial charge transfer and enhanced photocatalytic hydrogen generation†
Marija
Knezevic
 a,
Vien-Duong
Quach
a,
Isabelle
Lampre
a,
Marie
Erard
a,
Pascal
Pernot
a,
David
Berardan
b,
Christophe
Colbeau-Justin
a and
Mohamed Nawfal
Ghazzal
*a
a,
Vien-Duong
Quach
a,
Isabelle
Lampre
a,
Marie
Erard
a,
Pascal
Pernot
a,
David
Berardan
b,
Christophe
Colbeau-Justin
a and
Mohamed Nawfal
Ghazzal
*a
aInstitut de Chimie Physique, UMR 8000 CNRS, Université Paris-Saclay, F-91405 Orsay, France. E-mail: mohamed-nawfal.ghazzal@universite-paris-saclay.fr
bInstitut de Chimie Moléculaire et des Matériaux d'Orsay, UMR 8182 CNRS, Université Paris-Saclay, F-91405 Orsay, France
First published on 27th February 2023
Abstract
Metal halide perovskites (MHPs, CsPbX3: X = Cl, Br, I) demonstrate high photogenerated charge-carrier production and mobility, which makes them promising candidates for photocatalysis. In this work, we investigated how adjusting the band gap energy of MHPs at room temperature by anion exchange (CsPbBr3−yXy: X = Cl, Br, I) leads to optimal interfacial electron transfer from CsPbBr3−yXy to TiO2 by means of transient absorption spectroscopy (TAS), time-resolved photoluminescence (TRPL), and time-resolved microwave conductivity (TRMC). We found that the formation of indirect excitons at the MHPs/TiO2 interface results in slower charge-carrier relaxation, which is essential for photocatalysis. The substitution of bromide with chloride reduces the trapping states (healing effect), which favors charge-carrier relaxation to the ground state and leads to higher charge recombination and lower photocatalytic activity. The iodine, on its side, acts as a hole trapper proposing an optimal band gap facilitating fast charge injection in the TiO2. The charge-carrier injection from one material to another suppresses recombination, leading to impressive H2 generation.
Introduction
The recent energy crisis and constant energy demand growth have been prompting the need for new sustainable energy sources. Metal-halide perovskites (MHPs, CsPbX3: X = Cl, Br, I), known for their extraordinary light-harvesting capacity and charge-carrier dynamics1 (i.e., high photogenerated charge-carrier production and mobility), could play an important role in resolving the current energy-matter. MHPs have been extensively investigated in the field of optoelectronic devices and solar cells, reaching a record of energy conversion efficiency of up to 32.5% for perovskite-silicon tandem solar cells, and 25.7% for single-junction perovskite solar cells.2 Due to their impressive photophysical properties, MHPs are a valid candidate for photocatalysis. However, their inherent instability upon exposure to moisture, polar solvents, and UV irradiation, enhances their interactions with CO2, O2, and H2O, resulting in anodic corrosion, i.e. PbO formation, decelerating their immediate application in photocatalysis.3Therefore, significant attention has been paid to MHPs encapsulation with non-toxic and stable materials. MHPs encapsulation has been beneficial for CO2 photocatalytic reduction when coupled to graphdiyne (GDY),4 graphene oxide,5 titanium dioxide,6 boron imidazolate frameworks (BIFs)7 zeolite,8 poly(3-hexylthiophene-2,5-diyl) (P3HT) polymer,9 fullerene,10 MXene nanosheets,11 and porous g-C3N4 (PCN),12 while hydrogen production was improved by encapsulation in polymers such as polyaniline (PANI), and metal–organic-frameworks derived Co3O4/N-doped C core/shell composite.13,14 MHPs are also capable of singlet oxygen generation through energy transfer during methyl orange photocatalytic degradation when coated with stable and inert SiO2.15 Furthermore, coupling with metallic co-catalysts such as Ni(tpy)16 and Re(CO)3Br(dcbpy) (dcbpy = 4,4′-dicarboxy-2,2′-bipyridine)17 complex molecules improves photocatalytic efficiency of CO2 reduction, e.g., Ni(tpy) can act as the electron sink, preventing electron–hole recombination in CsPbBr3 nanocrystals (NCs). In general, heterojunctions improve the efficiency of the photogenerated charge carriers (i.e., electron–hole separation) and the CO2 capture. It is worth noting that one must consider the oxidation of carbon-based materials to CO2 and CO during the photocatalytic reaction since it could influence the yield of CO.18 Moreover, it is possible to maximize MHPs efficiency by using non-polar solvents e.g., mixed metal halide perovskite CsPb(Br0.5/Cl0.5)3 demonstrated the effective reduction of CO2 from ethyl acetate saturated solution.19
The growing interest of the photocatalytic community for MHPs has led to the development of different strategies to promote the photoelectronic and photocatalytic performance of MHPs, including selective control of MHP synthetic procedures, offering great versatility in morphology control, and electronic band gap engineering. Such an approach considers the replacement of long-chain oleic acid with either short-chain glycine or methyl acetate, directly ameliorating photocatalytic activity in CO2 reduction, due to the large available surface-active area.20,21 The latter could also be improved through multifaceted morphology control, eventually resulting in enhanced photocatalytic efficiency.22 In general, the MHPs band gap modification has been typically introduced by anion substitution reaction and metallic doping e.g. Fe(II), Co(II), Mn(II), and Zn(II).23–26 Nevertheless, the relationship between adjusting the band gap of MHP, interfacial charge lifetime, and photocatalytic activity has been scarcely investigated.
In this work, we adjusted interfacial charge transfer and lifetime by constructing tunable band gap energy of a heterojunction between MHP and TiO2. The band gap of MHP was controlled by post-anion substitution at room temperature. Then, we successfully encapsulated MHPs by sol–gel coating, constructing stable MHPs covered by TiO2 overlayer. The lifetime of photogenerated charges was followed at a variable time scale (from fs to μs) using transient absorption spectroscopy (TAS, fs), time-resolved photoluminescence (TRPL, ns), and time-resolved microwave conductivity (TRMC, μs). The optimal band gap configuration exhibits highly efficient charge injection and demonstrates stable photocatalytic hydrogen production in an aqueous solution, compared to solely MHPs. We found that chloride substitution enhances photoluminescence in the Br/Cl mixed halide perovskites, whereas iodide is the best candidate to promote photocatalytic H2 generation.
Experimental section
Synthetic procedures
Sol–gel synthesis of CsPbBr3 perovskites covered by TiO2 overlayer nanostructure (CsPbBr3@TiO2)
Titanium isopropoxide (TTiP, 20 μL) was dissolved in 1 mL of hexane and added dropwise in 10 mL colloidal solution of CsPbBr3 in hexane (1 mg mL−1) under vigorous stirring. The solution was aged for 3 h under stirring at ambient temperature. Then, the obtained nanoparticles were recovered by centrifugation and dried overnight at 80 °C. The final MHP@TiO2 nanomaterials were calcinated at 300 °C under air for 2 h. The same procedure was employed for titanium diisopropoxide bis(acetylacetonate) (TAA), titanium(IV) butoxide (TBOT), and titanium(IV) chloride (TiCl4).Characterization
Diffusion reflectance spectra (DRS) were recorded using UV-vis-NIR Cary 5000 spectrophotometer (Agilent Technologies), equipped with an integrating sphere for diffuse and total reflection measurements. The maximum reflectance was set to 100% using BaSO4 as a reference in the 200 to 1100 nm wavelength range.The photoluminescence (PL) emission spectra of colloidal MHPs in hexane were recorded using Fluorolog 3 HORIBA fluorimeter. All samples were excited at 400 nm (5 nm slit), and emission was monitored in the 420 to 700 nm range (1 nm slit).
X-ray diffraction (XRD) measurements were carried out by room temperature powder X-ray diffraction (P-XRD). Patterns were recorded by a Panalytical X'Pert diffractometer with a Ge (111) incident monochromator (Cu Kα radiation) and an X'cellerator detector.
Transmission electron microscopy (TEM, JEOL JEM 2100Plus, operating at 200 kV) was used to study the morphology of samples. The samples were ground, dispersed in hexane, and then drop-casted on carbon-coated copper grids. The grids were dried at ambient atmosphere before the measurements. The size of synthesized nanostructures was determined by ImageJ software.
Scanning transmission electron microscopy (STEM) experiments were performed on a Titan3 G2 80-300 microscope, operating at 200 kV equipped with a high angle annular dark field (HAADF) detector and an extra-high-brightness field emission gun (XFEG) for energy-dispersive X-ray spectroscopy (EDS) mapping.
Femtosecond transient-absorption spectroscopy TAS spectra of CsPbBr3−yXy and CsPbBr3−yXy/TiO2 thin films were recorded with a homemade pump–probe set-up based on a commercial amplified titanium sapphire laser (Amplitude Laser) that delivered pulses (780 nm, 110 fs) at a repetition rate of 1 kHz. The pump beam (390 nm) was generated using 90% of the fundamental beam by second harmonic generation in a 2 mm thick BBO crystal. The 10% left of the fundamental beam was used to generate a white light continuum (WLC) (400–800 nm) in a rotating fused silica plate. A broadband beam splitter was then used to divide the WLC into probe and reference beams. The probe and reference beams were transported and focused after attenuation on the entrance slit of a polychromator equipped with a charge-coupled device (CCD) camera (Princeton Instruments). The resolution of the system was about 200 fs. The 390 nm pump pulses were focused on the samples to a diameter of 780 ± 50 μm, and their energy was adjusted to vary the fluence (209 and 418 μJ cm−2). The time-resolved spectra were recorded from 430 to 650 nm and corrected for the group velocity dispersion. The spectro-kinetics data were then analyzed by global target analysis with sequential models with increasing lifetimes, using SK-Ana software.30–32
Time-resolved laser scanning time-correlated single photon counting (TCSPC) microscopy was performed with a homemade setup based on a TE2000 Nikon microscope equipped with a 60× 1.2 NA water immersion objective. The pulsed excitation source was a LDH 440 nm pulsed diode (80 ps FWHM, 20 MHz of repetition rate, PicoQuant). The emitted luminescence was selected with the appropriate filter (480 +/− 30 nm or 535 +/− 20 nm) and detected by a MCP-PMT (Hamamatsu), which is connected to the PicoHarp 300 TCSPC module (PicoQuant). Lifetime measurements were analyzed by the Pico-quant SymPhoTime64 software (v5.3.2).33
Time-resolved microwave conductivity (TRMC) signals were recorded using a nanosecond laser system (EKSPLA) integrating a Nd:YAG laser and an optical parametric oscillator (OPO) which delivers 8 ns pulses at a tunable wavelength from 225 to 2000 nm and a repetition rate of 10 Hz.34,35 All TRMC measurements were performed at the excitation wavelength of 360 and 440 nm, with a laser energy of 1.3 mJ and 2.6 mJ, respectively. The incident microwave was generated using a 30 GHz Gunn diode.
Photoelectrochemical (PEC) measurements were carried out in a quartz photoelectrochemical cell with a three-electrode setup, involving a Pt disk counter electrode, a non-aqueous reference electrode Ag|(AgNO3 0.01 M, (Bu4N)(PF6) 0.1 M in CH3CN), and a working electrode. The working electrodes were prepared by depositing MHPs previously dispersed in hexane on fluorine-doped tin oxide-coated glass (FTO); the amount of deposited MHPs and the surface area of the FTO slides remained unchanged for all samples. Amperometric transient photocurrent (TPC) was recorded at a bias voltage of 0.7 V (vs. ref), under 20 seconds solar light illumination delivered by solar light stimulator AM 1.5G, using (Bu4N)(PF6) 0.1 M in a mixture of CH3OH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH3CN (1:3 v/v) as a supporting electrolyte. The electrolyte was replaced by (Bu4N)(PF6) 0.1 M in dichloromethane (DCM) for cyclic voltammetry (CV) measurements. The formal potential of the non-aqueous reference electrode Ag|Ag+ was calibrated by adding 2 mM ferrocene/ferrocenium (Fc/Fc+) as an internal standard at the end of measurements. All measurements were performed on PGSTAT101 Metrohm Autolab potentiostat.
:CH3CN (1:3 v/v) as a supporting electrolyte. The electrolyte was replaced by (Bu4N)(PF6) 0.1 M in dichloromethane (DCM) for cyclic voltammetry (CV) measurements. The formal potential of the non-aqueous reference electrode Ag|Ag+ was calibrated by adding 2 mM ferrocene/ferrocenium (Fc/Fc+) as an internal standard at the end of measurements. All measurements were performed on PGSTAT101 Metrohm Autolab potentiostat.
Photocatalytic evolution of H2. In order to assess the photocatalytic activity of MHP@TiO2 nanostructures, in a quartz reactor, 3 mg of the photocatalyst was dispersed in 3 mL of the aqueous solution of 25 vol% glycerol and degassed under a continuous flow of Ar gas (>99%, Air liquid) to remove dissolved oxygen. The samples were irradiated using an Oriel 300 W Xenon lamp with an infrared water filter for 3 h under stirring, and the gas sample was analyzed every 30 min by gas chromatography (GC) (Chemlys gas chromatographer).
Results and discussion
Synthesis and physicochemical characterization of CsPbBr3−yXy and CsPbBr3−yXy@TiO2 nanopowders
Cesium-lead bromide (CsPbBr3) nanoparticles are synthesized by a hot injection colloidal synthesis at 200 °C.27,28 The post-anion exchange reactions are performed by adjusting the amount of lead halide salts (PbX2) in CsPbBr3 colloidal solution at room temperature, yielding CsPbBr1.65Cl1.35, CsPbBr1.95Cl1.05, CsPbBr1.95I1.05, and CsPbBr1.65I1.35, where Br/X represents their molar ratio in crude solution (Fig. 1a). The post-anion substitution, which differs from the usually reported one-pot hot injection synthesis process,36 took up to several minutes (under stirring) to reach an equilibrium, and allow efficient and non-reversible anion insertion into the crystalline structure. The anion exchange enables a fine band gap tuning all over the visible range, as demonstrated by the red and blue shift observed in the UV-visible spectra (Fig. 1b). The PL spectra of such exchanged nanocrystalline materials are adequately shifted with respect to the optical absorption spectra (Fig. 1c). We estimated the optical band gap energy to vary from 2.95 eV (CsPbCl3) to 2.06 eV (CsPbBr1.65I1.35) using Kubelka–Munk modified function (Fig. 1c and Table S1†). Luminescent properties of colloidal MHPs in hexane were explored under UV light excitation (360 nm), demonstrating the emission from blue (CsPbBr1.65Cl1.35), green (CsPbBr3) to yellow (CsPbBr1.65I1.35) (Fig. 1c, inset). The prevalence of iodide dramatically reduces the PL intensity of colloidal NCs, which almost completely vanished when samples were precipitated and dried (not shown), demonstrating the crucial role of the ligand/solvent environment for defect passivation.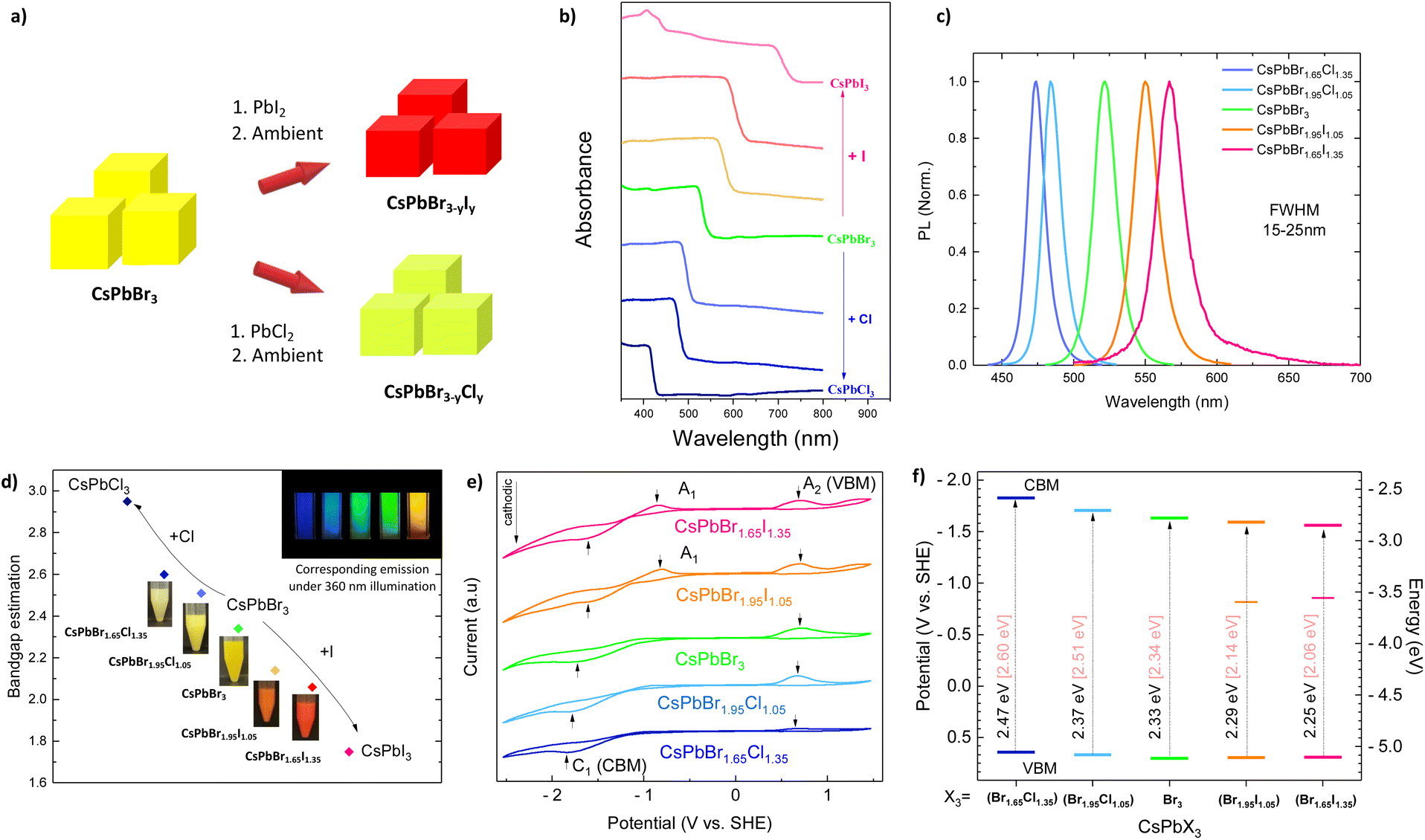 | ||
| Fig. 1 (a) Schematic representation of CsPbBr3−yXy and CsPbBr3−yXy (X = Cl, I) synthesis; (b) absorption spectra of solid CsPbBr3−yXy MHPs, (c) PL spectra of colloidal CsPbBr3−yXy MHPs, and (d) Kubelka–Munk optical band gap estimation of CsPbBr3−yXy MHPs, inset in (d) photographs of CsPbBr1.65Cl1.35, CsPbBr1.35Cl1.05, CsPbBr3, CsPbBr1.35I1.05, and CsPbBr1.65I1.35 colloidal solution in hexane. (e) Cyclic voltammograms of CsPbBr3−yXy recorded at 50 mV s−1 in a solution of 100 mM (Bu4N)(PF6) in DCM showing oxidation (A2) and reduction (C1) peaks, A1 peaks were marked out for iodide perovskites. (f) Energy band edge diagram of MHPs deduced from (e). Electrochemical band gap values (Eqpg, black) were compared to optical ones (Eoptg, pale pink). | ||
Introducing chloride and iodide in the structure leads to a shift in the valence band maximum (VBM) and conduction band minimum (CBM), although halide contribution is more significant in the valence band, consisting of either 3p, 4p, or 5p orbitals of chloride, bromide, and iodide, respectively.37 We determined VBM and CBM of MHPs by cyclic voltammetry. The charge transfer between CsPbBr3 and FTO conductive glass at working electrodes was reflected by anodic (A2) and cathodic (C1) peaks, respectively. The potential gap between anodic and cathodic peaks reveals the so-called quasi-particle band gap (Eqpg) and its value should be relatively close to the optical band gap (Eoptg) extracted from the Kubelka–Munk function.38 The irreversible voltammogram of CsPbBr3 was observed as a negative-sweeping potential scan from 0.5 (V vs. SHE) up to −1.63 (C1) (V vs. SHE), with the continuous current increase due to further reduction of NCs. Upon the reverse of the potential scan, oxidation of MHPs occurred and produced an anodic wave; we recorded an oxidation peak (A2) at 0.70 (V vs. SHE). The irreversibility could be ascribed to the partial degradation of MHPs after charge transfer (Fig. S1a†).38,39Fig. 1e depicts cyclic voltammograms of the mix-halide MHPs, including CsPb(Br1.65Cl1.35), CsPb(Br1.95Cl1.05), CsPb(Br1.95I1.05), CsPb(Br1.65I1.35) in comparison with CsPbBr3. In addition to anodic (A2) and cathodic (C1) peaks, we observed an additional peak A1 in the case of iodide-substituted perovskites. This can be attributed to trap-to-band transition.40 The potential gap between the peaks was similar to the optical band gap estimation (Fig. 1f).
The hot-injection synthesis yields orthorhombic CsPbBr3 NCs with an average size of 18 nm (Fig. 2a and S2†). A room-temperature anion substitution provides well-crystallized metal-halide perovskites, where both Br/Cl and Br/I halogen mixed perovskites keep the same orthorhombic crystal structure as pristine CsPbBr3 (Fig. 2b). The lattice parameters, space group, and crystal symmetry of CsPbBr3−yXy (X = Cl, I) are given in Table S2.† The evolution of the average unit cell spacing was compatible with anion substitution, either iodide or chloride, in which the partial substitution exhibits intermediate cell parameters between those of the parent particles. The synthesis of pure CsPbI3 results in the quick transformation from dark to a yellow orthorhombic non-perovskite phase, while CsPbCl3 crystallizes in a tetragonal phase (Fig. S3†). The anion substitution rate was estimated by STEM-EDS mapping (Fig. 2c–e and S4†), revealing easier chloride penetration e.g., CsPbBr1.95X1.05 substitution yields CsPbBr2.2Cl0.6, and CsPb1.2Br2I0.16, with an average particle size of 25 and 22 nm, respectively, similar to the size of CsPbBr3 NCs (Fig. 2c–e). An easier penetration of chloride is attributed to the smaller atomic size of chloride compared to iodide, allowing facile diffusion in CsPbBr3 nanocubes. It is worth mentioning that during the STEM and TEM analysis, the long beam exposure induces the degradation of MHPs and leads to the formation of lead nanoparticles (dots at the surface).41
 | ||
| Fig. 2 (a) XRD patterns of CsPbBr3−yXy (X = Cl, I); (b) STEM image of CsPbBr3; STEM-EDS mapping of (c) CsPbBr3, (d) CsPbBr1.95I1.05, and (e) CsPbBr1.95Cl1.05. | ||
Considering MHPs stability issues and interfacial charge separation, we coated MHPs with a thin TiO2 overlayer (Fig. 3a). We elaborated MHPs coating with different TiO2 precursors (e.g., TAA, TTiP, TBOT, and TiCl4). We found that TTiP and TBOT are homogeneously coated on the MHP surface (Fig. 3 and S5†), whereas TAA altered the cubic particle morphology. We could not observe a distinctive overlayer when TiCl4 was used as a precursor. Moreover, TiCl4 easily triggers Cl substitution, which could be observed as a blue-shifted absorption spectral line (Fig. S6†). We performed all the experiments with TTiP, due to its optimal hydrolysis reaction and better NCs dispersion. The sol–gel process enables obtaining perovskite nanoparticles covered by a thin TiOx layer. However, we have to avoid MHPs dispersion in the solution by ultrasonication before the coating procedure. Indeed, ultrasonication should be avoided since the perovskites are easily dissociated in solution (destroyed). Therefore, the obtained material is an agglomerate-like perovskite fully covered by a TiOx layer (labeled CsPbBr3@TiOx) (Fig. 3b, c and S7†).
 | ||
| Fig. 3 (a) Schematic representation of the sol–gel synthesis of CsPbBr3 covered by TiOx (TTiP) overlayer (CsPbBr3@TiOx), (b and c) TEM images of CsPbBr3@TiOx, (d) TEM and (e) EDS mapping images of CsPbBr3@TiO2 upon calcination at 300 °C. The scale bar corresponds to 300 nm. | ||
As synthesized CsPbBr3@TiOx NCs were further calcined at 300 °C to obtain a crystalline TiO2 overlayer (CsPbBr3@TiO2) (Fig. 3d). STEM-EDS mapping showed a homogenous distribution of the Ti and O signal all over the Cs, Pb, and Br, indicating the successful deposition of TiO2 layer, which is in agreement with the TEM images, and EDS mapping (Fig. 3e and S8†). The obtained CsPbBr3@TiO2 nanostructures retained their optical properties and orthorhombic crystal structure upon thermal treatment, as evidenced by XRD (Fig. S9†). The optical properties of CsPbBr3 and CsPbBr3@TiO2 exhibit similar behavior as shown in Fig. S10.† The composite nanostructure shows a shift of its absorption edge to 533 nm, with an enlarged (shoulder-like) absorption probably due to carbon residues.42 The optical band gap of CsPbBr3 and CsPbBr3@TiO2 is estimated using the Kubelka–Munk function to be similar to 2.34 eV and 2.35 eV, respectively. The TiO2 porous layer allows anion diffusion resulting in a band gap tuning (Fig. 3a and S11†). The UV-vis spectra show gradual redshift and blueshift when Br is substituted by I and Cl, respectively (Fig. S11†). We have performed gradual anion exchange substitution with an increasing amount of the adequate precursor (PbI2 and PbCl2), and we have observed uniform red and blue shifts (Fig. S11†). At higher concentrations of PbI2, no significant change in the absorption spectrum was observed after CsPbBr1.44I1.56@TiO2, indicating a maximum ion substitution is reached. The emission spectra of CsPbBr3−yXy@TiO2 (X = Cl, I) demonstrate homogenous blue- and redshifts (Fig. S12†). However, photoluminescence intensity is significantly quenched compared to the non-coated MHPs, having been completely diminished after CsPbBr1.95I1.05@TiO2. STEM-EDS mapping of anion exchanged CsPbBr1.95Cl1.05@TiO2 and CsPbBr1.95I1.05@TiO2 shows a homogeneous distribution of bromide, iodide, and chloride, and well-covered MHPs with TiO2 overlayer (Fig. S13–S16†). It is worth noting that upon calcination, we observe a growth of the TiO2 layer due to crystallization, resulting in a thicker overlayer than the amorphous TiOx. As prepared MHPs@TiO2 were investigated for photocatalytic activity.
Femtosecond transient absorption spectra of CsPbBr3−yXy and CsPbBr3−yXy/TiO2 (X = Cl, I) thin films
 | ||
| Fig. 4 (a, c and e) TA spectra, and (b, d and f) kinetics traces of CsPbBr3, CsPbBr1.95Cl1.05, and CsPbBr1.95I1.05 excited at 390 nm, with a fluence of 209 μJ cm−2. | ||
We investigated the complex temporal evolution of the spectra at different fluences (209 and 418 μJ cm−2) by a global target analysis, using a sequential kinetic model with four components (SAS) (eqn (1)).30
 | (1) |
SAS0 is the instantaneous rising signal, which decays roughly within the pulse duration (t0 = 1/k0 ≈ 300 fs) (Tables 1 and S3†). Since k0 cannot be estimated with a high degree of certainty, we will focus on constants k1 and k2. SAS1 appears (almost instantaneously) with the rate constant k0 and decays with k1. SAS2 appears with k1 and disappears with k2 leading to SAS3, which decays with k3 (t3 = 1/k3 ≫ 100 ps, i.e., out of the scan range). The species-associated spectra (SAS) and the temporal evolution obtained from the global analysis are presented in Fig. S17.† The three distinctive lifetimes are attributed to the intraband cooling (t0), state filling of the higher excitonic levels (t1), and Auger recombination (t2). At higher excitation fluence (418 μJ cm−2) pristine CsPbBr3 shows a slower relaxation (t1 = 3.3 ps), than at the lower excitation fluence (209 μJ cm−2) (t1 = 0.77 ps) (Tables 1 and S3†). This behavior corresponds to the charge-carrier relaxation mediated through the hot-phonon bottleneck.47 It is worth noting that such slow charge-carrier relaxation could stem from polaron formation, which could also screen and reduce charge-carrier mobility, resulting in slower charge-carrier cooling dynamics.44 Furthermore, Auger recombination lifetime, t2, was estimated to be 52 ps and 27 ps at 209 and 418 μJ cm−2 excitation fluence, respectively, being in accordance with the previous reports.48
| Samples | t 0 (ps) | t 1 (ps) | t 2 (ps) | Samples | t 0 (ps) | t 1 (ps) | t 2 (ps) |
|---|---|---|---|---|---|---|---|
| CsPbBr1.65Cl1.35 | 0.26 ± 0.14 | 2.30 ± 0.21 | 25.6 ± 0.80 | CsPbBr1.65Cl1.35/TiO2 | 0.53 ± 0.04 | 5.0 ± 0.40 | 29.4 ± 1.6 |
| CsPbBr1.95Cl1.05 | 0.42 ± 0.13 | 3.45 ± 0.21 | 47.6 ± 1.90 | CsPbBr1.95Cl1.05/TiO2 | 0.50 ± 0.04 | 4.5 ± 0.30 | 26.3 ± 1.1 |
| CsPbBr3 | 0.29 ± 0.26 | 0.77 ± 0.05 | 52.6 ± 3.15 | CsPbBr3/TiO2 | 0.20 ± 0.02 | 2.1 ± 0.08 | 33.3 ± 1.3 |
| CsPbBr1.95I1.05 | <0.1 | 1.0 ± 0.02 | 26.3 ± 2.01 | CsPbBr1.95I1.05/TiO2 | 0.40 ± 0.01 | 3.45 ± 0.10 | 26.3 ± 0.5 |
| CsPbBr1.65I1.35 | 0.20 ± 0.02 | 0.67 ± 0.05 | 21.7 ± 1.58 | CsPbBr1.65I1.35/TiO2 | 0.48 ± 0.01 | 3.7 ± 0.15 | 22.7 ± 0.5 |
Our findings contrast with previous reports that demonstrate a negative effect of chloride due to the incorporation of both shallow trap states and deep ones in the conduction band.49,50 We credit this discrepancy to the room temperature substitution method and the lower incorporated amount of chloride. Substitution with iodide significantly altered the intensity of TA spectra (Fig. 4e and S20†). The samples CsPbBr1.65I1.35 and CsPbBr1.95I1.05 were excited with the energy of 1.54 Eoptg and 1.48 Eoptg, respectively. In general, hole dynamics dominate iodide-based MHPs Fig. 4.51,52
One can observe redshift at early delay times, which is a signature of hole trapping by iodide when excited above the band gap.52 Moreover, the PB intensity, i.e., the state-filling effect, is significantly reduced due to hole trapping. In mixed CsPbBr3−yIy, hole transfer from bromide to iodide-rich states is energetically favored, resulting in redshifted bleach (Fig. 4e and S20a†). It was possible to monitor kinetics only at 560 nm (Fig. 4f), due to the very low intensity of PB band. At higher excitation fluence (418 μJ cm−2) iodide-based perovskite showed a slower state-filling effect and enhanced Auger recombination. (Fig. S18c and d†).
From our results, we can conclude that chloride substitution reduces the density of trapping states (healing effect), especially in CsPbBr1.95Cl1.05, which is beneficial for charge-carrier relaxation to the ground state, whereas iodide act as a hole-trapping center, subjecting electrons to Auger recombination.
Charge-carrier dynamics in CsPbBr3−yXy and CsPbBr3−yXy@TiO2 nanopowders: time-resolved photoluminescence and time-resolved microwave conductivity
To further study charge-carrier dynamics, we assessed the PL lifetime of mixed halide solid samples hybridized or not with TiO2 (Fig. 5a and d). We observed that the used room temperature anion substitution improves PL lifetime once a small amount of Br is substituted with Cl, while a large amount of Cl induces a decrease (Fig. 5a). The average PL lifetime is estimated to be ≈ 2.42 ns for CsPbBr3, increasing to ≈ 3.28 ns in CsPbBr1.95Cl1.05 and decreasing to 1.53 ns for CsPbBr1.65Cl1.35 (Table S4†). The PL lifetimes were estimated using a multiexponential function, and it is worth noting that t4 is of the same order of magnitude as the instrument response function. Interestingly, chloride prevents hole trapping in lead vacancies due to its high electronegativity by repulsing holes to the top of the valence band.54 The healing effect of chloride contributes to slower thermalization due to the accumulation of the positive charges at the top valence band, in agreement with TAS measurements. In the case of iodide substitution, PL is quenched up to ≈ 0.39 ns in CsPbBr1.95I1.05, pointing out that iodide acts as a hole scavenger that translates as a non-radiative process.54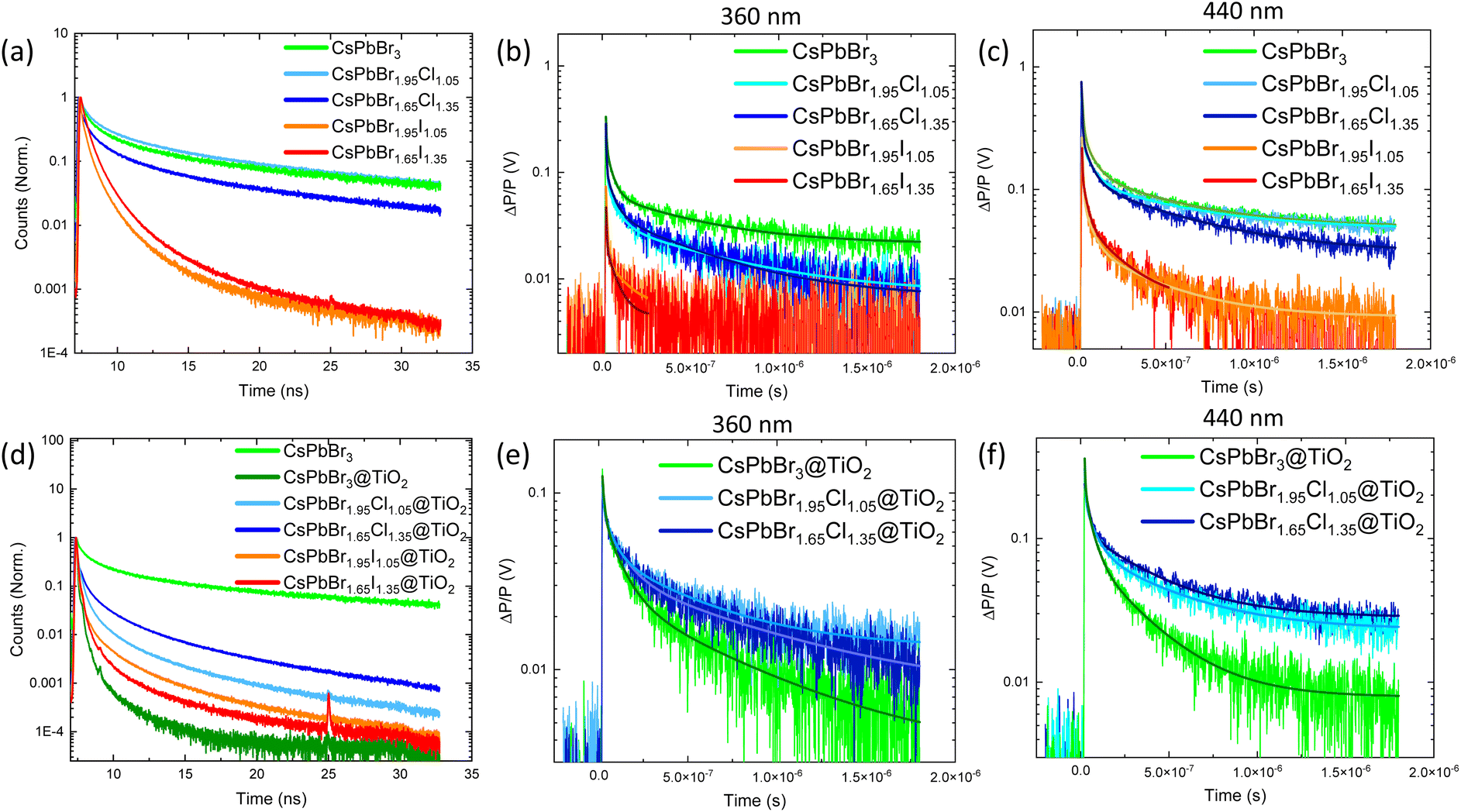 | ||
| Fig. 5 TRPL lifetime decay of (a) CsPbBr3−yXy and (d) CsPbBr3−yXy @TiO2 (X = Cl, I) nanostructures; TRMC signals under (b and e) UV (360 nm) and (c and f) visible (440) pulsed illumination and of CsPbBr3−yXy and CsPbBr3−yXy @TiO2 (X = Cl, I) nanostructures, respectively. | ||
We further investigated charge-carrier dynamics by means of TRMC, which enables the evaluation of the charge-carrier dynamics of free electrons and holes.55 TRMC signals obtained under UV and visible excitation (Fig. 5b and c) showed long charge-carrier lifetimes of bromide and chloride-based MHPs, exceeding μs timescale, whereas iodide-based MHPs charge-carrier lifetime does not exceed nanoscale, being in accordance with PL measurements. We complemented this study with transient photocurrent (TPC) response (to simulate natural photoactivation condition) of MHPs upon anion substitution (Fig. S24†). We observed that iodide-substituted perovskite experienced higher photocurrent response, indicating superior charge-carrier separation. We attribute this phenomenon to the holes trapped by iodide, resulting in suppressed recombination of electron–hole pairs. The photocurrent response of iodide-based perovskites exhibits a slow rise demonstrating low charge carrier mobility. This could be explained by the fast trapping of charges that is likely to screen an inner electric field and thus hinder charge transport. In contrast, CsPbBr3 and chloride-based perovskites demonstrate relatively steady photocurrent responses due to shallow trapping in which electrons are captured and then slowly re-emitted.
Coating MHPs with a thin TiO2 overlayer leads to a drastic decrease in the PL lifetime, with the average PL lifetime being below the resolution of the system (Fig. 5d), indicating electron transfer from MHP to TiO2. TRMC signals also confirmed the electron accumulation in TiO2 at a longer time scale (Fig. 5e and f). In the MHPs@TiO2 nanostructures, charge-carrier dynamics is quite similar, however, the number of created charge-carrier is lower (ΔP/P); we attribute this behavior to the electron screening upon the electron transfer. Interestingly, iodide-based perovskites covered by TiO2 overlayer provide only a sharp signal (not shown), and charge-carrier lifetime decay was not observed, which is in agreement with PL lifetime analysis indicating rapid quenching at the picosecond scale. A schematic representation of the charge-carrier transfer mechanism is presented in Fig. S25† under UV (a) and visible (b) light excitation.
Photocatalytic generation of H2 by CsPbBr3−yXy@TiO2
The photocatalytic activity of the MHP@TiO2 nanostructures was evaluated for H2 generation in glycerol/water solution (1:3 v/v) under xenon lamp illumination for 3 h, and the results are shown in Fig. 6. The photocatalytic reaction of glycerol and H2O under UV-vis illumination in the presence of a photocatalyst generates hydrogen (eqn (2) and (3)):56 | (2) |
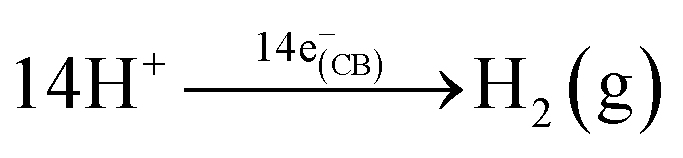 | (3) |
 | ||
| Fig. 6 (a) Photocatalytic activity and (b) H2 evolution rate of the MHP@TiO2 nanostructures. | ||
MHPs alone showed fast deactivation (low stability) in aqueous solution and under light excitation compared to the composite nanostructure, observed as rapid dissolution once introduced in the glycerol/water mixture. For instance, MHPs@TiO2 nanostructure demonstrated a stable appearance up to 3 h of photocatalytic reaction (Fig. S26†), while uncoated MHPs are dissolved after 10 min in the solution. The photocatalytic activity of pristine CsPbBr3@TiO2 demonstrated low H2 production (Fig. 6a and b). The chloride substitution resulted in an overall decrease in the hydrogen production rate (Fig. 6b). As previously mentioned, chloride contributes to the healing defect in the valence band (VB), enhancing hole migration toward the valence band. Consequently, chloride enhances charge-carrier recombination, thus such a low H2 production is expected. CsPbBr1.95I1.05@TiO2 exhibit impressive hydrogen production, reaching up to 250 μmol g−1 after 3 h irradiation (Fig. 6b). Since iodide acts as a hole trapper, it inhibits charge carrier recombination, allowing more electrons in the conduction band (CB) to be available for proton reduction. Moreover, the fast electron transfer to TiO2 and hole injection from TiO2 to VB of perovskites contributes to the enhancement of interfacial charge separation, which is beneficial for the H2 generation. However, larger iodide amount in CsPbBr1.65I1.35@TiO2 does not lead to a further enhancement of H2 evolution, probably due to increased hole trapping centers that cannot be compensated with hole injection from TiO2 (Fig. 6b). The optimal photocatalytic H2 evolution reaction is obtained with CsPbBr1.95I1.05@TiO2, showing an optimal interfacial charge-carrier separation.
Conclusions
In this work, we demonstrated room temperature anion substitution in CsPbBr3 nanocrystals, that yields stable highly emissive mixed halide Cl/Br perovskites and low emissive orthorhombic mixed halide I/Br perovskites. A room temperature method offers a possibility of defect healing through the addition of small amounts of chloride, directly influencing the enhancement of photoluminescence lifetime. However, the addition of iodide results in the formation of hole-trapping centers that prevent radiative charge-carrier recombination. We further investigated the electron transfer from metal-halide perovskite to TiO2. Ultrafast charge carrier dynamics (TAS) proved slower charge carrier relaxation due to the formation of indirect excitons between MHPs and the TiO2 layer, which is significantly pronounced in CsPbBr1.95I1.05/TiO2 and CsPbBr1.65I1.35/TiO2 thin films. CsPbBr1.95I1.05@TiO2 demonstrated the highest photocatalytic hydrogen production, owing to electron transfer to TiO2 and hole injection from TiO2 to CsPbBr1.95I1.05, essential for water splitting. These results represent a step forward in hydrogen generation from aqueous solutions.Author contributions
M. K. performed the experiments, curated the data, and wrote the first draft of the ms. V.-D. Q. carried out and discussed the electrochemical measurements and characterizations. I. L. discussed the TAS results. D. B. performed and discussed the XRD results. P. P. helped curating the TAS data. M. E. performed the TRPL analysis and discussed the results. C. C.-J. discussed the work. M. N. G. conceived and supervised the work. The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MK acknowledges the French “Ministère de l'enseignement supérieur et de la recherche” (MESR) for the PhD Grant. MNG thanks the public grant overseen by the French National Research Agency (ANR), through the IngenCat project (ANR-20-CE43-0014), and NEXTCCUS project as part of the ERANET-ACT3 call program for the financial support. The authors thank François Brisset for his valuable help in performing STEM and TEM analysis.References
- R. A. Scheidt, E. Kerns and P. V. Kamat, J. Phys. Chem. Lett., 2018, 9, 5962–5969 CrossRef CAS PubMed
.
- https://www.nrel.gov/pv/cell-efficiency.html .
- J. Li, L. Wang, X. Yuan, B. Bo, H. Li, J. Zhao and X. Gao, Mater. Res. Bull., 2018, 102, 86–91 CrossRef CAS
- K. Su, G. X. Dong, W. Zhang, Z. L. Liu, M. Zhang and T. B. Lu, ACS Appl. Mater. Interfaces, 2020, 12, 50464–50471 CrossRef CAS PubMed
- Y. F. Xu, M. Z. Yang, B. X. Chen, X. D. Wang, H. Y. Chen, D. Bin Kuang and C. Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS PubMed
- Y. F. Xu, X. D. Wang, J. F. Liao, B. X. Chen, H. Y. Chen and D. Bin Kuang, Adv. Mater. Interfaces, 2018, 5, 1801015 CrossRef
- Z. Y. Chen, Q. L. Hong, H. X. Zhang and J. Zhang, ACS Appl. Energy Mater., 2022, 5, 1175–1182 CrossRef CAS
- Z. C. Kong, J. F. Liao, Y. J. Dong, Y. F. Xu, H. Y. Chen, D. Bin Kuang and C. Y. Su, ACS Energy Lett., 2018, 3, 2656–2662 CrossRef CAS
- L. Li, Z. Zhang, C. Ding and J Xu, Chem. Eng. J., 2021, 419, 129543 CrossRef CAS
- Z. Zhang, M. Shu, Y. Jiang and J. Xu, Chem. Eng. J., 2021, 414, 128889 CrossRef CAS
- A. Pan, X. Ma, S. Huang, Y. Wu, M. Jia, Y. Shi, Y. Liu, P. Wangyang, L. He and Y. Liu, J. Phys. Chem. Lett., 2019, 10, 6590–6597 CrossRef CAS PubMed
- M. Ou, W. Tu, S. Yin, W. Xing, S. Wu, H. Wang, S. Wan, Q. Zhong and R. Xu, Angew. Chem., Int. Ed., 2018, 57, 13570–13574 CrossRef CAS PubMed
- W. Song, Y. Wang, C. Wang, B. Wang, J. Feng, W. Luo, C. Wu, Y. Yao and Z. Zou, ChemCatChem, 2021, 13, 1711–1716 CrossRef CAS
- R. Tang, S. Zhou, H. Li, R. Chen, L. Zhang and L. Yin, Appl. Catal., B, 2020, 265, 118583 CrossRef CAS
- K. Gu, Y. Wang, J. Shen, J. Zhu, Y. Zhu and C. Li, ChemSusChem, 2020, 13, 682–687 CrossRef CAS PubMed
- Z. Chen, Y. Hu, J. Wang, Q. Shen, Y. Zhang, C. Ding, Y. Bai, G. Jiang, Z. Li and N. Gaponik, Chem. Mater., 2020, 32, 1517–1525 CrossRef CAS
- Z. C. Kong, H. H. Zhang, J. F. Liao, Y. J. Dong, Y. Jiang, H. Y. Chen and D. Bin Kuang, Sol. RRL, 2020, 4, 1900365 CrossRef CAS
-
L. Zhou, Theory Model. Dispersed Multiph. Turbul. React. Flows, 2018, pp. 15–70 Search PubMed
- S.-H. Guo, J. Zhou, X. Zhao, C.-Y. Sun, S.-Q. You, X.-L. Wang and Z.-M. Su, J.
Catal., 2019, 369, 201–208 CrossRef CAS
- Y. Xu, W. Zhang, K. Su, Y. X. Feng, Y. F. Mu, M. Zhang and T. B. Lu, Chem.–Eur. J., 2021, 27, 2305–2309 CrossRef CAS PubMed
- Y. Li, Q. Shu, Q. Du, Y. Dai, S. Zhao, J. Zhang, L. Li and K. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 451–460 CrossRef CAS PubMed
- S. Shyamal, S. K. Dutta, T. Das, S. Sen, S. Chakraborty and N. Pradhan, J. Phys. Chem. Lett., 2020, 11, 3608–3614 CrossRef CAS PubMed
- S. Shyamal, S. K. Dutta and N. Pradhan, J. Phys. Chem. Lett., 2019, 10, 7965–7969 CrossRef CAS PubMed
- Y. W. Liu, S. H. Guo, S. Q. You, C. Y. Sun, X. L. Wang, L. Zhao and Z. M. Su, Nanotechnology, 2020, 31, 215605 CrossRef CAS PubMed
- Y. F. Mu, W. Zhang, X. X. Guo, G. X. Dong, M. Zhang and T. B. Lu, ChemSusChem, 2019, 12, 4769–4774 CrossRef CAS PubMed
- L. Ding, C. Shen, Y. Zhao, Y. Chen, L. Yuan, H. Yang, X. Liang, W. Xiang and L. Li, Mol. Catal., 2020, 483, 1–7 Search PubMed
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed
- C. Lu, M. W. Wright, X. Ma, H. Li, D. S. Itanze, J. A. Carter, C. A. Hewitt, G. L. Donati, D. L. Carroll, P. M. Lundin and S. M. Geyer, Chem. Mater., 2019, 31, 62–67 CrossRef CAS
- N. M. Ghazzal, N. Chaoui, E. Aubry, A. Koch and D. Robert, J. Photochem. Photobiol., A, 2010, 215, 11–16 CrossRef CAS
-
P. Pernot, SK-ana, 2018, DOI:10.5281/zenodo.1064370
- C. Ruckebusch, M. Sliwa, P. Pernot, A. de Juan and R. Tauler, J. Photochem. Photobiol., C, 2012, 13, 1–27 CrossRef CAS
- I. H. M. Van Stokkum, D. S. Larsen and R. Van Grondelle, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS PubMed
- M. Erard, A. Fredj, H. Pasquier, D. B. Beltolngar, Y. Bousmah, V. Derrien, P. Vincent and F. Merola, Mol. BioSyst., 2013, 9, 258–267 RSC
- G. D. Gesesse, C. Li, E. Paineau, Y. Habibi, H. Remita, C. Colbeau-Justin and M. N. Ghazzal, Chem. Mater., 2019, 31, 4851–4863 CrossRef CAS
- C. Wang, J. Li, E. Paineau, A. Laachachi, C. Colbeau-Justin, H. Remita and M. N. Ghazzal, J. Mater. Chem. A, 2020, 8, 10779–10786 RSC
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS PubMed
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef CAS PubMed
- S. N. Inamdar, P. P. Ingole and S. K. Haram, ChemPhysChem, 2008, 9, 2574–2579 CrossRef CAS PubMed
- S. K. Haram, B. M. Quinn and A. J. Bard, J. Am. Chem. Soc., 2001, 123, 8860–8861 CrossRef CAS PubMed
- F. Liu, C. Ding, Y. Zhang, T. S. Ripolles, T. Kamisaka, T. Toyoda, S. Hayase, T. Minemoto, K. Yoshino, S. Dai, M. Yanagida, H. Noguchi and Q. Shen, J. Am. Chem. Soc., 2017, 139, 16708–16719 CrossRef CAS PubMed
- Z. Dang, J. Shamsi, F. Palazon, M. Imran, Q. A. Akkerman, S. Park, G. Bertoni, M. Prato, R. Brescia and L. Manna, ACS Nano, 2017, 11, 2124–2132 CrossRef CAS PubMed
- Z. J. Li, E. Hofman, J. Li, A. H. Davis, C. H. Tung, L. Z. Wu and W. Zheng, Adv. Funct. Mater., 2018, 28, 1704288 CrossRef
- A. Mondal, J. Aneesh, V. Kumar Ravi, R. Sharma, W. J. Mir, M. C. Beard, A. Nag and K. V Adarsh, Phys. Rev. B, 2018, 98, 115418 CrossRef CAS
- G. Kaur and H. N. Ghosh, J. Phys. Chem. Lett., 2020, 11, 8765–8776 CrossRef CAS PubMed
- J. Butkus, P. Vashishtha, K. Chen, J. K. Gallaher, S. K. K. Prasad, D. Z. Metin, G. Laufersky, N. Gaston, J. E. Halpert and J. M. Hodgkiss, Chem. Mater., 2017, 29, 3644–3652 CrossRef CAS
- J. Aneesh, A. Swarnkar, V. Kumar Ravi, R. Sharma, A. Nag and K. V Adarsh, J. Phys. Chem. C, 2017, 121, 4734–4739 CrossRef CAS
- A. Mondal, J. Aneesh, V. Kumar Ravi, R. Sharma, W. J. Mir, M. C. Beard, A. Nag and K. V. Adarsh, Phys. Rev. B, 2018, 98, 115418 CrossRef CAS
- A. Mondal, J. Aneesh, V. Kumar Ravi, R. Sharma, W. J. Mir, M. C. Beard, A. Nag and K. V. Adarsh, Phys. Rev. B, 2018, 98, 115418 CrossRef CAS
- N. Soetan, A. Puretzky, K. Reid, A. Boulesbaa, H. F. Zarick, A. Hunt, O. Rose, S. Rosenthal, D. B. Geohegan and R. Bardhan, ACS Photonics, 2018, 5, 3575–3583 CrossRef CAS
- S. Mandal, S. Ghosh, S. Mukherjee, C. K. De, D. Roy, T. Samanta and P. K. Mandal, Nanoscale, 2021, 13, 3654–3661 RSC
- H. Chung, S. Il Jung, H. J. Kim, W. Cha, E. Sim, D. Kim, W.-K. Koh and J. Kim, Angew. Chem., Int. Ed., 2017, 56, 4160–4164 CrossRef CAS PubMed
- N. Mondal and A. Samanta, Nanoscale, 2017, 9, 1878–1885 RSC
- Z. Gevorkian, V. Gasparian and Y. Lozovik, Appl. Phys. Lett., 2016, 108, 51109 CrossRef
- G. Nan, X. Zhang, M. Abdi-Jalebi, Z. Andaji-Garmaroudi, S. D. Stranks, G. Lu and D. Beljonne, Adv. Energy Mater., 2018, 8, 1–9 Search PubMed
- E. M. Hutter and T. J. Savenije, ACS Energy Lett., 2018, 3, 2068–2069 CrossRef CAS PubMed
- V. Kumaravel, M. D. Imam, A. Badreldin, R. K. Chava, J. Y. Do, M. Kang and A. Abdel-Wahab, Catal, 2019, 9, 276 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta09920a |
| This journal is © The Royal Society of Chemistry 2023 |