
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering polymorphs in colloidal metal dichalcogenides: precursor-mediated phase control, molecular insights into crystallisation kinetics and promising electrochemical activity†
Nilotpal
Kapuria‡
 ,
Niraj Nitish
Patil‡
,
Abinaya
Sankaran
,
Fathima
Laffir
,
Hugh
Geaney
,
Edmond
Magner
,
Micheal
Scanlon
,
Kevin M.
Ryan
and
Shalini
Singh
*
,
Niraj Nitish
Patil‡
,
Abinaya
Sankaran
,
Fathima
Laffir
,
Hugh
Geaney
,
Edmond
Magner
,
Micheal
Scanlon
,
Kevin M.
Ryan
and
Shalini
Singh
*
Department of Chemical Sciences and Bernal Institute, University of Limerick, Limerick, Ireland. E-mail: Shalini.Singh@ul.ie
First published on 20th April 2023
Abstract
Controlling the crystal phase in layered transition metal dichalcogenides (TMDs) is critical for their diverse, flexible applications. However, due to the thermodynamic stability of 2H over other polymorphs, fine synthesis control over polymorphism in TMDs is challenging, restricting the entire range of characteristics associated with other polymorphs. Herein, we present a solution-based crystal phase engineering approach for layered transition metal disulphide nanosheets by modulating the reactivity of the molecular precursors. By tuning precursor–ligand chemistry, 2H, 1T′ and polytypic MoS2 and WS2 are synthesised. The flexibility to selectively modify the reactivity of S and metal precursors allowed control over the proportion of specific phases in synthesised nanosheets. The formation of 1T′ is facilitated by the highly reactive metal and S source, whereas less reactive sources lead to the formation of thermodynamically stable 2H. The electrocatalytic properties of the synthesised TMDs were examined for the oxygen reduction reaction. The polytypic MoS2 comprising a mix of 2H–1T′ displayed the most positive potential of 0.82 V (vs. RHE). The comprehensive mechanistic interpretation of the chemical transformations provided in this study will be instrumental in designing scalable solution-based pathways for phase engineering in layered transition metal dichalcogenides. Furthermore, this synthesis approach has the potential to be extended to various TMD compositions, enabling exquisite control over polymorphism in TMDs.
 Shalini Singh | Dr Shalini Singh is a lecturer in the Department of Chemical Sciences and the Bernal Institute at the University of Limerick, Ireland. She received her PhD in chemistry from the University of Limerick in 2016 and was then an FWO (Research Foundation, Flanders, Belgium) Postdoctoral Fellow at Ghent University, Belgium until 2019. From 2020, she is leading the ‘Functional Nanomaterial Research Group’ at the University of Limerick. Her research interest is focused on the development of novel colloidal semiconductors and metallic nanocrystals for energy conversion and storage applications. |
Introduction
Transition metal dichalcogenides (TMDs) such as MoS2, WSe2, ZrS2, and TiS2 have emerged as a class of key materials due to their intriguing properties applicable in catalysis, energy storage, electronics, and optoelectronics.1–4 The TMDs typically occur in 2H or 1T/1T′ crystal phases. The 2H phase displays trigonal prismatic coordination, whereas, in 1T or 1T′, the metal atoms display octahedral coordination.5 The metal coordination significantly affects the properties of these polymorphs, where the 2H phase is semiconducting in nature and known for its optoelectronic properties.6 The 1T/1T′ phase outperforms the 2H phase in terms of catalytic activity due to its metallic nature and superior electrical conductivity.7–9From a thermodynamic standpoint, the formation of the 2H phase is more favourable than the formation of the metastable phase like 1T and its derivatives. Most of the synthesis procedures lead to thermodynamically stable 2H phase formation. The octahedral coordination in the 1T phase tends to convert into distorted octahedral coordination resulting in 1T′ or related semi-metallic phases based on the distortion. In this context, the heterogeneities in metal coordination in a single structure will allow effective tuning and switching of physical and electrical parameters by forming polytypic (mixture of two or more different phases sharing interface) transition metal dichalcogenides. Several techniques have been explored recently for synthesising 1T-TMDs, including exfoliation, plasma hot-electron transfer, electron irradiation, mechanical stresses, and hydrothermal reactions.10–15 However, the conversion from the 1T/1T′ to the 2H phase is easily triggered by various factors, including contamination, oxidation, and the development of intermediate phases.
Compared to thermodynamically stable phases, metastable TMD materials require higher formation energy, which is challenging to achieve in conventional synthesis approaches like chemical vapour deposition and vapour transport. Traditional high-temperature solid-state procedures do not give enough control to form metastable compounds. These approaches primarily rely on feeding a continuous excessive heat to the reaction vessel to overcome the kinetic barriers of solid-state diffusion, which normally drives the formation of the most thermodynamically stable products.16 Contrary to this, solution-phase colloidal synthesis approaches allow more kinetic control by attaining an optimum monomer supply rate.5,17–19 In recent years, colloidal chemistry provided a viable way to obtain metastable phases and thermodynamically stable phases of various metal chalcogenides. The variable phases can be obtained by strategically manipulating the precursor and ligand chemistry to access kinetic growth domains.20,21 For instance, the metastable wurtzite-like CuInSe2 phase was synthesised over the thermodynamically favoured chalcopyrite phase by slowing down nucleation, using precursors with higher C–Se bond dissociation energy.22,23 Similarly, several different polymorphs of iron sulfide NCs were synthesised by exploiting the difference in bond dissociation energy of C–S in a series of organosulfur precursors.24 The interaction of the growing crystal with its chemical environment, which can be tuned in solution phase synthesis, was found to be beneficial in altering the free energy of different polymorphs, lowering the energy barrier for the preferential formation of metastable polymorphs and potentially preventing their transformation into thermodynamically stable ones. For example, coordinating ligands played an important role in the formation of metastable orthorhombic AgInSe2 nanocrystals, and ligand-mediated topotactic cation exchange converts orthorhombic Ag2Se to orthorhombic AgInSe2.25 Besides controlling the polymorphism of metal chalcogenide NCs, precursor chemistry has been advantageous in controlling the ratio of different polymorphs. For example, Wang et al. showcased that switching between two different selenium precursors (diphenyldiselenide and selenium powder) and keeping a metal precursor the same, polytypic cubic-wurtzite and monophasic cubic phases of Cu2SnSe3 NCs were synthesised.26,27 Recent advancements in colloidal synthesis have led to synthetic colloidal protocols for TMDs forming specific phases, such as 2H, 1T/1T′, by selecting suitable precursors and ligands.4,28–31 The majority of research relating ligand-precursor coordination to nanocrystal crystal phase has primarily focused on non-layered covalently linked nanocrystallites, and a generic and simple approach for phase engineering in layered van der Waals structures has yet to be established. Further, in terms of catalytic activity, hierarchical heterostructures containing interfacing mixed phases resulted in enhancements in catalytic activity attributed to improved synergistic effects from electrical conductivity and active sites.32 The studies show that at the heterojunction of the 1T′/2H mixed phase heterostructure, the kinetic barrier drops and electron transport enhances significantly, resulting in an increased number of active sites. In addition to this, the interaction of the 2H phase stabilises the metastable 1T′ phase, considerably increasing its catalytic activity.33,34 Thus, the control of polytypism and its relationship with electrocatalytic activity tuning requires thorough understanding.
Herein we demonstrate phase engineering of colloidal TMD nanosheets (MoS2 and WS2) by tuning precursor reactivity in solution. We showcased that altering the reactivity of metal and S sources resulted in the formation of different polymorphs of monophasic 2H (semiconducting), 1T′ (metallic) transition metal disulfide nanosheets and nanosheets with mixed phase composition (2H–1T′ polytypes). The wide tunable reactivity of the chalcogen precursors facilitated the phase modulation of polytypic transition metal disulfides. Altering the reactivity of the S sources affects the supply rate of S atoms impacting the nucleation rate and favouring the formation of certain polymorphs. Further, the solvent played an important role in controlling the polymorphs; the use of a non-coordinating solvent speeds up reaction kinetics, leading to the formation of a kinetically favourable 1T′ product. In this context, we provide in-depth mechanistic insights that enable improved predictive synthesis of particular phases. In addition, we carried out a retrosynthetic approach that predicted chemical intermediates during the nanosheet formation, leading to the specific polymorph formation. Furthermore, we demonstrate the advantage of the polytypic transition metal sulfide nanosheets over the monophasic nanosheets displaying better catalytic performance with higher onset potentials and diffusion-limited current density during the oxygen reduction reaction (ORR).
Experimental section
Reagents
Molybdenum hexacarbonyl [Mo(CO)6, technical grade, lot# BCBB6657], molybdenum pentachloride (MoCl5, ≥95%, lot# MKCJ9980), tungsten hexacarbonyl [W(CO)6, 97%, lot# MKCD5432], tungsten hexachloride (WCl6, ≥99.9%, lot# MKCK4583), tert-butyl disulphide (TBDS, 97%, lot# 05019EJ), thiourea (≥99%, lot# BCBG6245V), 1-dodecanethiol (1-DDT, ≥98%, lot# STBK4241), oleyl amine (OLA, 70%, lot# STBK3552), squalane (96%, lot# BCCD3542), oleic acid (OA, 90%, lot# MKCH4532), phenyl disulphide (99%, STBC7722V), and tri-octylphosphine (TOP, 97%, MKCP6805) were purchased from Sigma Aldrich. Toluene, ethanol, and isopropanol (IPA) were purchased from Lennox, Ireland. Tetradecyl phosphonic acid (TDPA, 97%, AK1803131) was purchased from Plasmachem. KOH pellets (≥85%, lot# L0080) were purchased from Honeywell. All reagents were used as received without further purification.Synthesis
Transition metal disulphide nanosheets were prepared using a colloidal hot injection setup. Based on the desired product, using reaction condition Rx (where x = 1–6 in Table 1), the Mo-precursor source was prepared by dissolving 0.25 mmol of Mo(Co)6 or MoCl5 in the desired amount of solvent and 10 mol% of tetradecyl phosphonic acid with subsequent heating to 130 °C on a hotplate kept in an Ar atmosphere in a glovebox. Similarly, under reaction condition Rx′ (where x′ = 1–6 in Table 1), the metal precursor sources used were W(Co)6 or WCl6. 1 mmol of a sulphur source was dissolved in the appropriate solvent and prepared as per conditions R (Mo) and R′ (W) as described in Table 1. Both the cationic and anionic precursor sources were prepared and mixed in an oxygen and moisture-free atmosphere at 130 °C. The appropriate volume of solvent was placed in a 3-neck round-bottomed flask and connected to a Schlenk line setup. Table 1 lists the details of the reaction parameters. The precursor solution was evacuated at 120 °C for 20 min and heated to 320 °C under Ar; the mixed solution was injected at a rate of ∼0.7 ml min−1. The reaction mixture was kept at 320 °C for 2 h. The NCs were washed in a mixture of 10 ml toluene and 10 ml ethanol at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 min. After the first wash, the NCs were dispersed in toluene and washed with IPA.
000 rpm for 5 min. After the first wash, the NCs were dispersed in toluene and washed with IPA.
| Reaction no. | Nanocrystal | Solvent (volume) | Metal source | S-source | S-source preparation temperature in °C |
|---|---|---|---|---|---|
| R1 | MoS2 | OLA (7 ml) | 1 eq. Mo(CO)6 in 2 ml OA and 10 mol% TDPA | 4 eq. S-powder in 5 ml OLA | 100 |
| R1′ | WS2 | OLA (7 ml) | 1 eq. W(CO)6 in 2 ml OA and 10 mol% TDPA | 4 eq. S-powder in 5 ml OLA | 100 |
| R2 | MoS2 | OA (7 ml) | 1 eq. Mo(CO)6 in 5 ml OA and 10 mol% TDPA | 4 eq. S-powder in 2 ml TOP | RT |
| R2′ | WS2 | OA (7 ml) | 1 eq. W(CO)6 in 5 ml OA and 10 mol% TDPA | 4 eq. S-powder in 2 ml TOP | RT |
| R3 | MoS2 | OLA (7 ml) | 1 eq. Mo(CO)6 in 2 ml OA and 10 mol% TDPA | 4 eq. tert-butyl disulphide in 5 ml OLA | RT |
| R3′ | WS2 | OLA (7 ml) | 1 eq. W(CO)6 in 2 ml OA and 10 mol% TDPA | 4 eq. tert-butyl disulphide in 5 ml OLA | RT |
| R4 | MoS2 | OLA (7 ml) | 1 eq. Mo(CO)6 in 2 ml OA and 10 mol% TDPA | 4 eq. DDT in 5 ml OLA | RT |
| R4′ | WS2 | OLA (7 ml) | 1 eq. W(CO)6 in 2 ml OA and 10 mol% TDPA | 4 eq. DDT in 5 ml OLA | RT |
| R5 | MoS2 | OLA (7 ml) | 1 eq. Mo(CO)6 in 2 ml OA and 10 mol% TDPA | 4 eq. thiourea in 5 ml OLA | 200 |
| R5′ | WS2 | OLA (7 ml) | 1 eq. W(CO)6 in 2 ml OA and 10 mol% TDPA | 4 eq. thiourea in 5 ml OLA | 200 |
| R6 | MoS2 | OA (7 ml) | 1 eq. MoCl5 in 5 ml OA and 10 mol% TDPA | 4 eq. S-powder in 2 ml squalane | 140 |
| R6′ | WS2 | OA (7 ml) | 1 eq. WCl6 in 5 ml OA and 10 mol% TDPA | 4 eq. S-powder in 2 ml squalane | 140 |
Material characterisation
For transmission electron microscopy (TEM) analysis, the NCs were dispersed in toluene and drop cast on continuous carbon-coated 200 mesh nickel grids (source). Low-resolution and high-resolution TEM (HRTEM) and dark-field scanning transmission electron microscopy (DFSTEM) were conducted on a 200 kV JEOL JEM-2100F field emission microscope, equipped with a Gatan Ultra Scan CCD camera and an EDAX Genesis energy dispersive X-ray spectroscopy (EDS) detector. Interplanar distances and particle orientation were determined from FFT analysis of the selected area in Gatan GMS3 software.Raman spectroscopy was performed using a Horiba Labraman 300 spectrometer system equipped with a 532 nm laser.
UV-Vis-NIR spectroscopy was done on a Cary 5000 spectrophotometer in cuvettes of 1 cm path length using toluene as solvent.
Electrochemical measurement
Cyclic voltammetry (CV) and linear sweep voltammetry (LSV) experiments for all the synthesized NCs were performed in a standard three-electrode system using catalyst-coated glassy carbon as the working electrode, a platinum wire as a counter electrode, and Ag/AgCl (3 M NaCl) as the reference electrode in 0.1 M KOH solution. The studies were performed using the rotating disk electrode (RDE) setup, and the PGSTAT204 potentiostat was from Metrohm (Netherlands) and used in conjunction with NOVA software version 2.1.2. Approximately 3.55 mg of the NC catalyst was homogeneously dispersed in 300 μl of ethanol, 100 μl of IPA, and 25 μl of 5 wt% Nafion (binder) by 60 min ultrasonication forming casting ink. 4 μl of this prepared ink is drop cast upon the pre-polished glassy carbon electrode to form a 0.5 mg cm−2 active catalyst layer. Cyclic voltammetry studies were performed in both O2 and N2 saturated 0.1 M KOH solution at a scan rate of 50 mV s−1 over the potential range of −1 to 0.2 V (vs. Ag/AgCl (3 M NaCl)). The recorded potentials are reported vs. the reversible hydrogen electrode (RHE) by converting them using the following Nernst equations, where EoAg/AgCl (3 M NaCl) = 0.1976 V vs. the standard hydrogen electrode (SHE) at 25 °C:| ERHE = EAg/AgCl + 0.059pH + EoAg/AgCl(3 M NaCl) | (1) |
At different rotation rates from 400 to 2400 rpm, linear sweep voltammograms were recorded using O2 saturated 0.1 M KOH solution at a scan rate of 5 mV s−1 over the potential range of 0.2 to −1 V (vs. Ag/AgCl (3 M NaCl)) to understand the diffusion kinetics of the catalyst layer.35 The Koutechy–Levich (K–L) plots were derived from the LSV curves using the following equations:
| 1/j = 1/jl + 1/jk = 1/Bω1/2 + 1/jk | (2) |
| jk = nFkC0 |
| B = 0.62nFC0D−2/30v−1/6 |
485 C mol−1), k is the electron transfer rate constant, C0 = 1.2 × 10−6 mol cm−3 is the concentration of O2 molecules in 0.1 M KOH, D0 = 1.90 × 10−5 cm2 s−1 is the diffusion coefficient of O2 in pure water at 25 °C and 1.0 atm O2 pressure, and v = 0.01 cm2 s−1 is the kinematic viscosity of 0.1 M KOH.36 For the stability test, the polytypic NCs were cycled for a longer duration under the same conditions mentioned above. LSVs were recorded at 1600 rpm under similar conditions as above after 2500 CV cycles for MoS2 polytypic NCs and 1000 cycles for WS2 polytypic NCs.
Results and discussion
Synthesis of TMD nanosheets
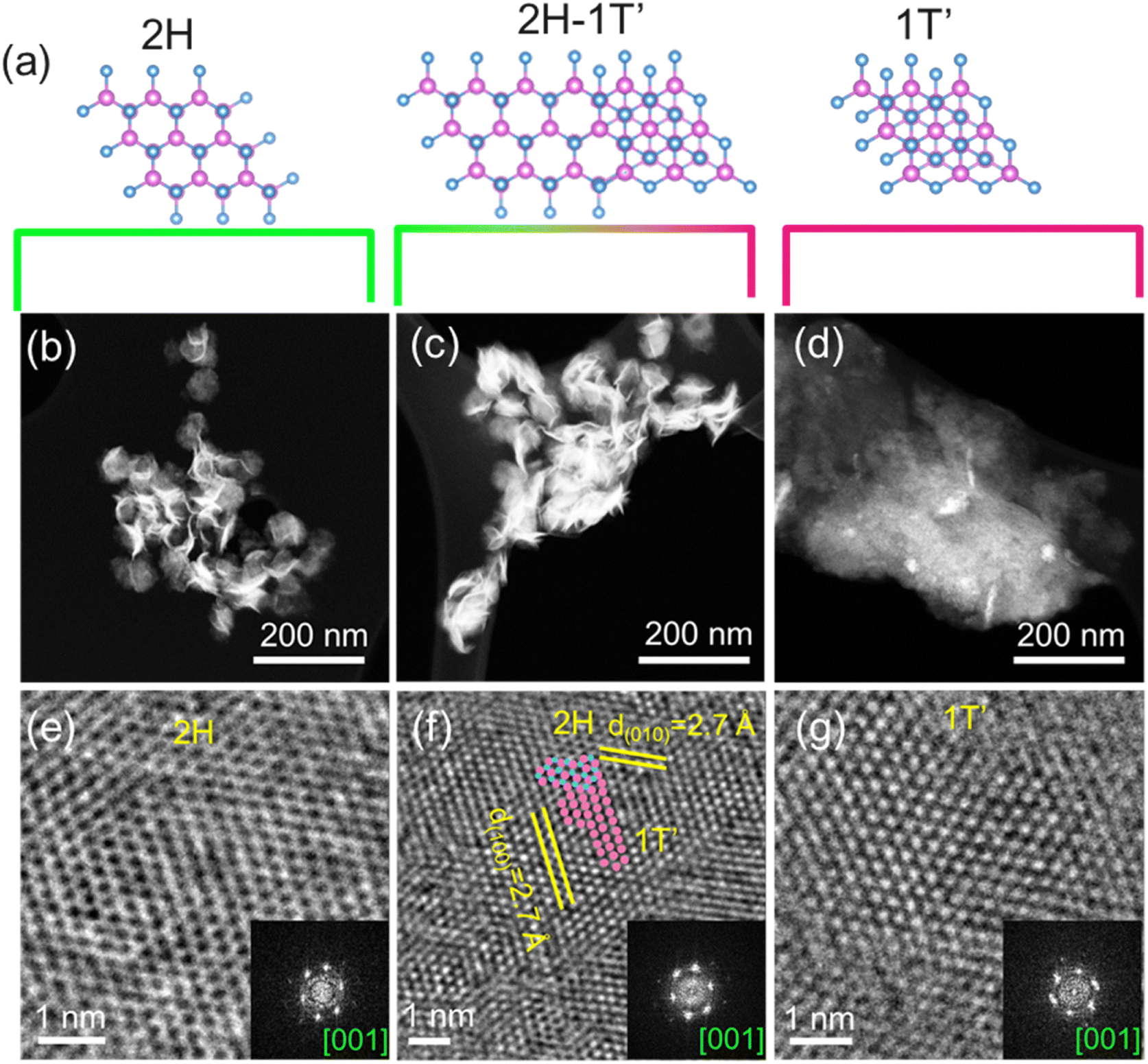
Using a controlled injection (∼0.7 ml min−1) of metal source dissolved in oleic acid (OA) and added with S-source mixed in a range of hot co-ordinating and non-coordinating solvents resulted in the formation of MS2 NCs with different percentages of 2H and 1T′ phases. Based on the reactivity of different metals and S-sources, a series of reactions were carried out as listed in Table 1 to control the percentage of a particular crystal phase in polytypic MS2 NCs (see the Experimental section for details). Annular dark-field scanning electron micrographs (ADF-STEM) (Fig. 1b–d and 2b–d, ESI S1†) of the MoS2 and WS2 nanocrystals (NCs) display nanosheet-like morphologies where multiple layers are bundled together. The contrast in symmetry of the trigonal coordinated 2H phase and octahedral coordinated 1T′ phase of the TMD is identified by the high-resolution TEM (HRTEM) of the NCs (Fig. 1a, e–g, and k–m). | ||
| Fig. 1 (a) Schematic showing the atomic arrangement of 2H, 2H–1T′ polytype, and 1T′ TMDs projected from the c-axis, the blue atoms correspond to S and pink atoms correspond to Mo or W; low magnification STEM micrograph of MoS2 NCs prepared under conditions (b) R1, (c) R3, (d) R6 and the corresponding HRTEM images for the (e) 2H phase, (f) 2H–1T′ phase and (g) 1T′ phases of MoS2 prepared under conditions R1, R3 and R6 respectively. | ||
 | ||
| Fig. 2 (a) Schematic showing the atomic arrangement of 2H, 2H–1T′ polytype, and 1T′ TMDs projected from the c-axis low magnification STEM micrograph of WS2 NCs prepared under conditions (b) R1′, (c) R3′, (d) R6′ and the corresponding HRTEM images for the (e) 2H phase dominated WS2 where pink patches denote the 1T′ phase, (f) 2H–1T′ phase and (g) 1T′ dominated phases of WS2 where green patches denote the 2H phase prepared under conditions R1′, R3′, and R6′, respectively. | ||
For MoS2, the HRTEM of the NCs synthesized using S–OLA in condition R1 showcases a honeycomb-like atomic arrangement with a d-spacing of 2.7 Å corresponding to the (100) and (010) planes which are characteristic features for 2H phase (Fig. 1e). Suspension of S powder in non-coordinating solvent squalane in synthesis R6 resulted in 1T′ phase formation with octahedral Mo–S coordination where the nearest Mo–Mo distance is 3.2 Å as calculated from HRTEM (Fig. 1g). The alternating Mo–Mo distance of 2.7 Å along with the typical Mo–Mo distance of 3.2 Å as shown in the ESI† (Fig. S2a†) explains the distortion present in the Mo–S octahedral coordination in the 1T′ structure. Using tert-butyl disulfide (TBDS) in condition R3, the presence of both 2H and 1T′ phases is observed in the MoS2 nanosheets as shown in Fig. 1f. Similarly, for WS2, conditions R1′, R3′, and R6′ resulted in 2H dominated, a mixed 2H–1T′ and 1T′ dominated phases respectively (Fig. 2e–g). Furthermore, in UV-Vis-NIR absorption spectra (Fig. S3†), the 2H MoS2 and 2H-dominated WS2 exhibit three excitonic peaks. The intensity of these peaks is reduced in the polytypic 2H–1T′ phases of MoS2 and WS2, whereas the absorbance appears to be featureless in the spectra of 1T′ phases of MoS2 and WS2.
Polytypism
The effect of S-precursor reactivity on polytypism of the 2H–1T′ phase was studied using Raman spectra and XPS analysis of the as-synthesised nanosheets. The presence of the peaks at 378 (E12g) and 405 (A1g) cm−1 (Fig. 3a) for MoS2 confirms the formation of the 2H phase when S–OLA is used with Mo(CO)6. Similar reaction conditions for WS2 display 2H rich phase with Raman peaks 350 (E12g) and 415 (A1g) (Fig. 3b). The diminished intensity of these two peaks with the appearance of peaks at 185–195 and 225 cm−1 corresponding to the phonon modes and a peak at 350 cm−1 (J3) indicates the formation of the 1T′-phase MoS2 when S–squalane is used with MoCl5.37,38 Additionally, a less intense peak at ∼300 cm−1 (E1g) suggests the occurrence of octahedral Mo–S coordination.30 Similarly, for WS2 nanocrystals, the appearance of 180 cm−1 peaks resulting from phonon mode and 267 cm−1 (J2) peak and suppression of (E12g) and A1g peaks indicate the occurrence of the 1T′ phase (Fig. 3b).30 Gradual increase of reactivity of S-source induced polytypism in nanosheets with increased 1T′ phase formation when TOP–S, TBDS, and 1-DDT are used as the S-source (Fig. 3c). For MoS2, the peaks at 228.5 (3d5/2) and 231.6 eV (3d3/2) correspond to the 1T′ phase whereas the characteristic peaks of 2H MoS2 phase appear at 229.2 (Mo 3d5/2) and 232.3 eV (Mo 3d3/2) (Fig. 4a). For 1T′ WS2, the characteristic W4f peaks correspond to 31.5 (W4f7/2) and 33.7 eV (W4f5/2) with a binding energy of 0.9 eV lower than the 2H phase (Fig. 4b). Upon deconvolution of the XPS spectra, the 1T′ phase percentage with respect to the 2H phase is determined (Fig. 4c and d). When highly reactive S–squalane and WCl6 were employed, the synthesis resulted in 100% of 1T′ MoS2 and 83% of 1T′ WS2 phase formation (R6 and R6′). The estimated 1T′ phase % in the NCs synthesized using TBDS and W(CO)6 is 36 and 35%, respectively, for MoS2 and WS2 (R3 & R3′). When 1-DDT is used as the S source with metal carbonyls, the 1T′ phase % further increases beyond 50% of the phase composition (R4 & R4′).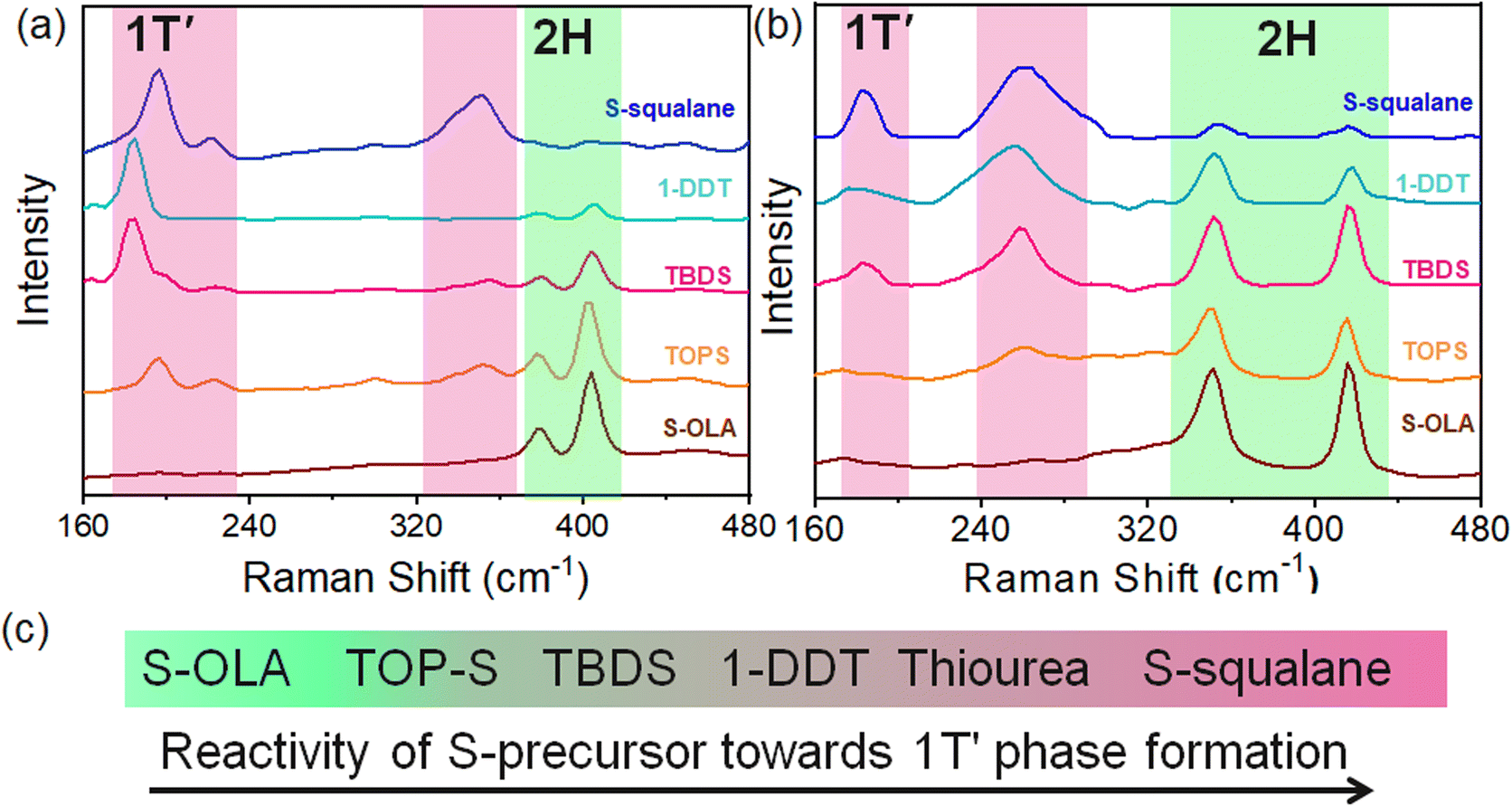 | ||
| Fig. 3 Raman spectra of (a) MoS2 and (b) WS2 NCs prepared by manipulation of precursor chemistry of S and the metal source; (c) reactivity trend of S-sources follow S–OLA (R1, R1′) < TOP–S (R2, R2′) < TBDS (R3, R3′) < 1-DDT (R4, R4′) < S–squalane (R6, R6′) for 1T′ phase formation. | ||
 | ||
| Fig. 4 XPS analysis of as prepared (a) MoS2 NCs, (b) WS2 NCs synthesized using different S-sources and metal sources; phase percentage analysis of (c) MoS2 and (d) WS2 from deconvolution of the XPS spectra. | ||
Mechanistic insights
We have investigated the role of precursor–ligand chemistry in modulating the TMD nanosheets' crystal phases. The metastable phases of MS2 nanocrystals are achieved by establishing kinetic growth conditions. The monomer supply rate generally decides the reaction kinetics. We utilized the reactivity differences of the commercially available S-sources to avail active sulphur species to control the metastable phase formation. The most commonly used S-source, S powder occurs as polysulphide (Sn). Upon the reaction with OLA, it undergoes S–S scission to form radical anions and alkanethioamide as per eqn (3).39 To understand the growth condition, S–OLA solution was heated to 320 °C under reaction condition R1 and analysed with 13C NMR. The presence of the peak at ∼169 ppm from C of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S centre and the peak at ∼61 ppm from the α-C of RCH2CSNH2 support the formation of alkanethioamide (Fig. 5a). Furthermore, in the 1H NMR, the α-proton peak diminishes after alkanethioamide formation (Fig. S7†). This rate-limiting step is responsible for a slow rate of sulphide species formation during MS2 synthesis.
S centre and the peak at ∼61 ppm from the α-C of RCH2CSNH2 support the formation of alkanethioamide (Fig. 5a). Furthermore, in the 1H NMR, the α-proton peak diminishes after alkanethioamide formation (Fig. S7†). This rate-limiting step is responsible for a slow rate of sulphide species formation during MS2 synthesis. | (3) |
 | ||
| Fig. 5 13C NMR of (a) S–oleylamine solution and (b) S–squalane suspension heated under reaction conditions. CDCl3 used as a reference displays a peak at ∼77.2 ppm. | ||
Here, the direct use of alkanethioamide or thioamide group will expedite the monomer supply rate. To increase the lability of the –CS in thioamides, an electron-donating group such –NH2 (e.g., thiourea) will further facilitate the reaction kinetics as illustrated in eqn (4). As a result, more of the 1T′ phase forms when thiourea is used as the S-source as observed in the XPS spectra of the TMDs (Fig. S6†).
 | (4) |
The reactivity of the C–S bond can be further varied using organyl disulfide sources. Reactivity in organyl disulphides varies depending on the bond dissociation energy of C–S. The tert-butyl disulphide (TBDS), which displays C–S bond dissociation energies (BDE) of ∼58 kcal mol−1, undergoes a slow process to produce S22− species preceding a sluggish sulphide species formation reaction.40 When S powder is used with TOP, the strong interaction of TOP with S (TOPS) slows the free sulphide formation.41 However, adding excess oleic acid facilitates the S2− formation from TOP–S as per eqn (5), making it more reactive than S–OLA but less reactive than tert-butyl disulphide. Similar to TBDS, 1-DDT shows C–S BDE between ∼55 and 60 kcal mol−1. However, the presence of OLA forms a Lewis acid–base adduct (R1CH2C–Sδ−–Hδ+⋯NH2R) with 1-DDT, increasing the reactivity of the thiol compared to TBDS.
 | (5) |
In contrast to the previous systems, squalane as a non-coordinating solvent does not show any interaction with the sulphur source; hence it tends to supply a high amount of Sn2−. A possible side reaction based on the millimolar concentration of sulfur radicals is forming alkyl disulphide or alkanethiols when reacting with alkane, as shown in eqn (6).
 | (6) |
The presence of the alkyl thiol was confirmed from the 13C NMR peak around ∼36 ppm (Fig. 5b). The formed alkyl thiol will also stabilize the metastable form. Thus, S-sources' reactivity follows the following trend: S–squalane > thiourea > tert-butyl disulfide > TOP–S > S–OLA.
Furthermore, a comprehensive picture of the influence of precursor–ligand coordination on the crystallinity of the nanosheets can only be built when the metal sources are considered. Among the utilised metal sources in this study, metal carbonyls are comparatively less reactive than metal chlorides due to the ability to exert readily available metal cations than the metal carbonyl, where a π M → CO back bonding exists.42 Thus, employing metal chlorides as a precursor favours a greater monomer supply rate forming 1T′ dominated polytypes as confirmed from the Raman spectra of the nanosheets formed using metal chlorides (Fig. S4 and S5†). Considering the reaction outcome and reactivity trend of precursors, we propose that MS2 nucleation initiates with metastable 1T′ phase formation. Hence, manipulation of precursor chemistry which would allow a faster rate of free sulphide species (Sn2−) and free metal cation (Mn+) formation will ultimately arrest the distorted octahedral M–S coordination of the kinetically metastable phase – 1T′. For example, 1T′ phase formation dominates when S–squalane is used with reactive MClx. In contrast, when S–squalane or thiourea (Fig. S6†) is replaced with a less reactive S–OLA or TBDS, a slow monomer supply rate provides ample time for reorganisation of octahedral M–S co-ordination to form 2H phase dominated TMDs. Hence, the reaction trend of R1 < R2 < R3 < R4 < R5 < R6 is observed towards a faster nucleation rate, resulting in the formation of 1T′ dominated phases.
Furthermore, to check our hypothesis, the aliquots from reactions of MoS2 performed with four different S-sources with variable reactivity (S–OLA < TBDS < 1-DDT < S–squalane) were collected at different growth times of 1, 5, 10, 30, 60 min at 320 °C and were screened with UV-Vis-NIR absorption spectroscopy. All the aliquots collected at 1 min showed the presence of 1T′ phase irrespective of the S-sources used (Fig. 6). However, for low reactive S-sources such as S–OLA, the formation of the 2H-phase started just after 5 min of growth time. TBDS shows higher reactivity than S–OLA; thus, the formation of the 2H phase occurs after 10 min. Further increased reactivity realised using 1-DDT exhibited 2H phase formation only after 30 min of growth time. However, the reaction system using highly reactive S–squalane and MoCl5 did not exhibit any occurrence of the 2H phase even after 60 min of growth time.
 | ||
| Fig. 6 UV-Vis-NIR spectra of MoS2 reaction aliquots withdrawn at 1, 5, 10, 30, and 60 min of growth time at 320 °C using (a) S–squalane and MoCl5, (b) 1-DDT–oleylamine and Mo(CO)6, (c) TBDS–oleylamine and Mo(CO)6, and (d) Mo(CO)6, and S–OLA suspension. | ||
Ostwald's step rule for nucleation suggests that a metastable phase forms first before it's conversion into a thermodynamically stable phase.43 Thus, in the mentioned systems, the 1T′ phase forms first as the metastable phase, as shown in Scheme 1. The slower reactivity S-source provided enough time for the distorted M–S octahedral coordination to re-organise into stable trigonal prismatic M–S coordination as in the 2H phase. However, higher reactivity of the S-source arrests the distorted octahedral M–S co-ordination via continuous supply of S-monomer, preventing re-organisation into trigonal prismatic coordination.
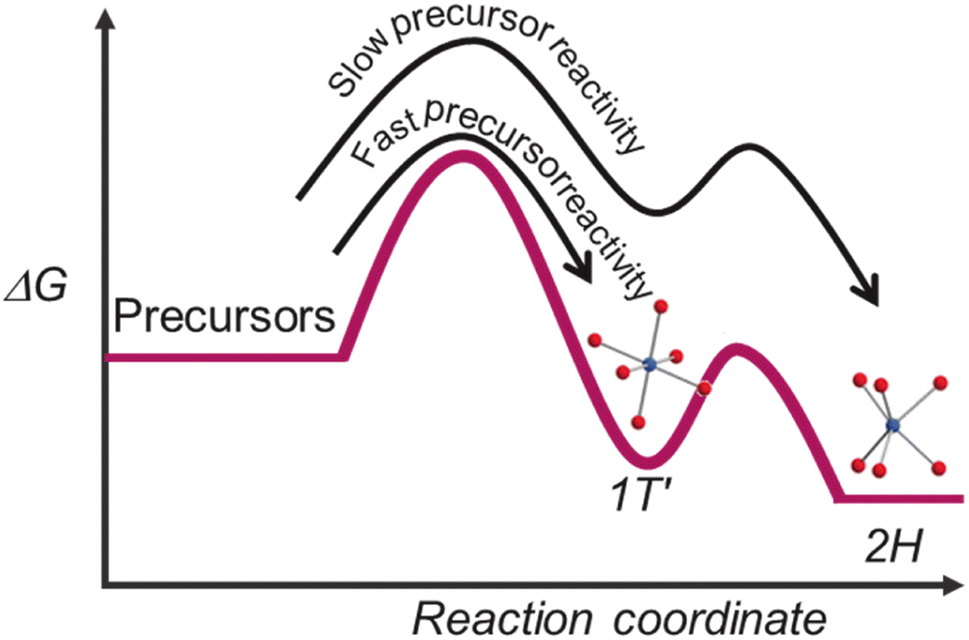 | ||
| Scheme 1 Representation of transition metal disulphide formation based on precursor reactivity trends. The blue spheres denote metal, and the red spheres represent sulphur. | ||
Electrochemical performance
We study the ORR activity of synthesized 2H, 1T′, and polytypic 2H–1T′ MS2 NCs in alkaline media using CV and LSV techniques. The LSV curves of MoS2 polymorphs recorded in an O2 saturated environment at 1600 rpm show that polytypic 2H–1T′ MoS2 has superior ORR kinetics, with an onset potential, E0 = 0.82 V vs. RHE compared to 2H MoS2 (E0 = 0.76 V vs. RHE) and 1T′ MoS2 (E0 = 0.79 V vs. RHE), see Fig. 7a. In addition, the polytypic 2H–1T′ MoS2 exhibits a ∼35 mV positive shift in half-wave potential and a higher diffusion-limited current density (jL = −3.5 mA cm−2) compared to 1T′ MoS2 (jL = −1.8 mA cm−2). | ||
| Fig. 7 ORR activity of MoS2 and WS2 samples. Rotating disc voltammograms (at 1600 rpm) of (a) 2H-, 1T′- and polytypic 2H–1T′ MoS2 NCs, and (b) 2H-, 1T′- and polytypic 2H–1T′ WS2 NCs in O2 saturated 0.1 M KOH at 1600 rpm; cyclic voltammograms of (c) polytypic 2H–1T′ MoS2 and (d) 2H–1T′ WS2 in O2 and N2 saturated 0.1 M KOH. | ||
A similar trend is observed for WS2 nanosheet samples where the onset potential for polytypic 2H–1T′, 1T′, and 2H WS2 is 0.79, 0.77, and 0.74 V vs. RHE, respectively (Fig. 7b). The polytypic 2H–1T′ WS2 exhibits a ∼41 mV positive shift in half-wave potential and a higher diffusion-limited current density (jL = −2.2 mA cm−2) compared to 1T′ MoS2 (jL = −1.5 mA cm−2). Though 2H MS2 is a more stable structure, the active sites are only located at the edges leading to poor electrochemical activity. In contrast, the 1T′ phase exhibits metallic behaviour where both the basal sites and edges are active towards molecular intermediate binding during electrocatalysis.44 As a result, 1T′ nanosheets display better catalytic activity but suffer from poor stability. Phase engineering of the MS2 to form polytypic NCs results in enhanced electrocatalytic activity with onset values comparable to most reports (Table S2†).45,46 In our study, we see improved reduction kinetics for the polytypes as the integration of the 1T′ phase is beneficial to improve the number of active sites and electronic conductivity and the 2H phase provides stability to the 1T′ phase.47 Additionally, the interface between 2H–1T′ can show chalcogen vacancies and accumulated charges inducing enhanced charge transfer beneficial for improved ORR kinetics, thus, performing better than the monophases.48–50 This was further corroborated by decreased Tafel slope values of the polytypes compared to 1T′ monophases (Fig. S9†). Additionally, the MoS2 polytype shows improved stability while maintaining its polytypic nature during the long-term cycling of ORR, as shown in Fig. S10 and S11a.† However, in the case of WS2, though the mixed phase is maintained (Fig. S11b†), a slight negative shift in onset potential and half-wave potential is observed (Fig. S10b†) because of structural deterioration as confirmed in post-mortem TEM analysis (Fig. S12†).
The corresponding CVs of the best performing polytypic 2H–1T′ MoS2 (Fig. 7c) and 2H–1T′ WS2 (Fig. 7d) recorded in an O2 or N2 saturated 0.1 M KOH electrolyte are shown. The diffusion peaks observed in an O2-saturated environment with onset potential values of 0.82 vs. RHE (2H–1T′ MoS2) and 0.79 V vs. RHE (2H–1T′ WS2) represent the catalytic reduction of O2. The absence of a reduction peak in the CV scans in a N2 saturated environment confirms the oxygen reduction activity of the NCs.
Linear sweep voltammograms of MS2 NCs recorded in an O2 environment at different rotational speeds from 400 to 2400 rpm are shown in Fig. S8a and c.† The current densities are independent of the rotation speed between 0.9 and 0.85 V vs. RHE, which indicates kinetic current density (jk). The diffusion current density increases with the rotation speed between 0.6 and 0.45 V vs. RHE. This increase in the current density at increased rotating speed is due to the reduced capacitance between the electrode surface and reactant, a high O2 diffusion rate, and mass transfer. The corresponding K–L plot was drawn from the LSV curves to understand the order of the reaction. MS2 (MoS2 and WS2). The number of electrons (n) transferred per O2 molecule during the ORR increases from 2.67 to 3.94 for MoS2 (Fig. S8b†) and 2.37 to 4.30 for WS2 (Fig. S8d†) in the potential range of 0.6 to 0.2 V. Thus, it corroborates the 2nd order reaction kinetics involving a 2-electron process as per the following pathways:51
| O2 + H2O + 2e− → OH− + HO2− | (7) |
| H2O + HO2− + 2e− → 3OH− | (8) |
The synthesized MS2 MoS2 and WS2 NCs exhibit the sluggish 2-electron peroxide pathway rather than a faster 4-electron pathway like Pt–C, as reported in several other studies.52 Yet, the phase engineering has led to the enhanced degree of activation owing to interfacing 2H–1T' phases present in the polytypic nanosheets.53 The controlled reaction to form polytypic MoS2 nanosheets showed an increase in the number of electrons transferred (Fig. S8b and d†), leading to better reduction kinetics than the WS2 nanosheets.
Conclusions
In summary, alteration of precursor reactivity is demonstrated as an effective phase control tool for layered transition metal disulphide NC synthesis. Our system exploits the difference in reactivity of a series of commercially available S-sources which displayed a reactivity trend of S–OLA < TOP–S < tert-butyl sulphide < 1-DDT < S–squalane towards controlled 1T′ phase formation. Similarly, reactive metal sources such as metal chlorides displayed a higher tendency to form 1T′ phases than metal carbonyls. The significance of polytypism is realised by applying these NCs as electrode materials for the oxygen reduction reaction where the polytypes showed superior performance compared to the monophasic counterparts. The polytypic MoS2 displayed a higher onset potential of 0.82 V (vs. RHE) with a current diffusion limited density of −3.5 mA cm−2 and polytypic WS2 displayed an onset potential of 0.79 V (vs. RHE) with a current diffusion limited density of −2.2 mA cm−2. The present study provides important insights into chalcogen precursor reactivity regulation to create a suitable library of polytypic TMS NCs to significantly enhance the physiochemical properties without foreign metal doping.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NK acknowledges funding from the Irish Research Council (IRC) under Grant Number IRCLA/2017/285. NNP acknowledges the funding from the Faculty of Science and Engineering, University of Limerick. K. M. R. acknowledges Science Foundation Ireland (SFI) under the Principal Investigator Program under contract no. 16/IA/4629 and under grant no. SFI 16/M-ERA/3419. S. S. acknowledges the funding and support from the Department of Chemical Sciences, University of Limerick.References
- C. L. Tan, X. H. Cao, X. J. Wu, Q. Y. He, J. Yang, X. Zhang, J. Z. Chen, W. Zhao, S. K. Han, G. H. Nam, M. Sindoro and H. Zhang, Recent Advances in Ultrathin Two-Dimensional Nanomaterials, Chem. Rev., 2017, 117, 6225, DOI:10.1021/acs.chemrev.6b00558
.
- C. L. Tan and H. Zhang, Two-dimensional transition metal dichalcogenide nanosheet-based composites, Chem. Soc. Rev., 2015, 44, 2713, 10.1039/c4cs00182f
- P. S. Zhou, G. Collins, Z. Hens, K. M. Ryan, H. Geaney and S. Singh, Colloidal WSe2 nanocrystals as anodes for lithium-ion batteries, Nanoscale, 2020, 12, 22307 RSC
- P. S. Zhou, P. Schiettecatte, M. Vandichel, A. Rousaki, P. Vandenabeele, Z. Hens and S. Singh, Synthesis of Colloidal WSe2 Nanocrystals: Polymorphism Control by Precursor-Ligand Chemistry, Cryst. Growth Des., 2021, 21, 1451, DOI:10.1021/acs.cgd.0c01036
- A. Coogan and Y. K. Gun'ko, Solution-based "bottom-up" synthesis of group VI transition metal dichalcogenides and their applications, Mater. Adv., 2021, 2, 146, 10.1039/d0ma00697a
- K. F. Mak and J. Shan, Photonics and optoelectronics of 2D semiconductor transition metal dichalcogenides, Nat. Photonics, 2016, 10, 216, DOI:10.1038/Nphoton.2015.282
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. S. Li and S. Jin, Enhanced Hydrogen Evolution Catalysis from Chemically Exfoliated Metallic MoS2 Nanosheets, J. Am. Chem. Soc., 2013, 135, 10274, DOI:10.1021/ja404523s
- D. Voiry, M. Salehi, R. Silva, T. Fujita, M. W. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Conducting MoS2 Nanosheets as Catalysts for Hydrogen Evolution Reaction, Nano Lett., 2013, 13, 6222, DOI:10.1021/nl403661s
- G. Q. Li, D. Zhang, Q. Qiao, Y. F. Yu, D. Peterson, A. Zafar, R. Kumar, S. Curtarolo, F. Hunte, S. Shannon, Y. M. Zhu, W. T. Yang and L. Y. Cao, All The Catalytic Active Sites of MoS2 for Hydrogen Evolution, J. Am. Chem. Soc., 2016, 138, 16632, DOI:10.1021/jacs.6b05940
- C. Lee, Q. Y. Li, W. Kalb, X. Z. Liu, H. Berger, R. W. Carpick and J. Hone, Frictional Characteristics of Atomically Thin Sheets, Science, 2010, 328, 76, DOI:10.1126/science.1184167
- Y. M. Kang, S. Najmaei, Z. Liu, Y. J. Bao, Y. M. Wang, X. Zhu, N. J. Halas, P. Nordlander, P. M. Ajayan, J. Lou and Z. Y. Fang, Plasmonic Hot Electron Induced Structural Phase Transition in a MoS2 Monolayer, Adv. Mater., 2014, 26, 6467, DOI:10.1002/adma.201401802
- Y. C. Lin, D. O. Dumcencon, Y. S. Huang and K. Suenaga, Atomic mechanism of the semiconducting-to-metallic phase transition in single-layered MoS2, Nat. Nanotechnol., 2014, 9, 391, DOI:10.1038/Nnano.2014.64
- F. Dybala, M. P. Polak, J. Kopaczek, P. Scharoch, K. Wu, S. Tongay and R. Kudrawiec, Pressure coefficients for direct optical transitions in MoS2, MoSe2, WS2, and WSe2 crystals and semiconductor to metal transitions, Sci. Rep., 2016, 6, 26663, DOI:10.1038/srep26663
- A. P. Nayak, S. Bhattacharyya, J. Zhu, J. Liu, X. Wu, T. Pandey, C. Q. Jin, A. K. Singh, D. Akinwande and J. F. Lin, Pressure-induced semiconducting to metallic transition in multilayered molybdenum disulphide, Nat. Commun., 2014, 5, 3731, DOI:10.1038/ncomms4731
- X. M. Geng, Y. L. Zhang, Y. Han, J. X. Li, L. Yang, M. Benamara, L. Chen and H. L. Zhu, Two-Dimensional Water-Coupled Metallic MoS2 with Nanochannels for Ultrafast Supercapacitors, Nano Lett., 2017, 17, 1825, DOI:10.1021/acs.nanolett.6b05134
-
J. Reedijk and K. R. Poeppelmeier, Comprehensive Inorganic Chemistry II: from Elements to Applications, Elsevier, 2nd edn, 2013, p. 7544 Search PubMed
- J. Polte, Fundamental growth principles of colloidal metal nanoparticles - a new perspective, Crystengcomm, 2015, 17, 6809, 10.1039/c5ce01014d
- N. Kapuria, M. Conroy, V. A. Lebedev, T. E. Adegoke, Y. Zhang, I. S. Amiinu, U. Bangert, A. Cabot, S. Singh and K. M. Ryan, Subsuming the Metal Seed to Transform Binary Metal Chalcogenide Nanocrystals into Multinary Compositions, ACS Nano, 2022, 16, 8917 CrossRef CAS PubMed
- N. Kapuria, S. Imtiaz, A. Sankaran, H. Geaney, T. Kennedy, S. Singh and K. M. Ryan, Multipod Bi(Cu2-xS)n Nanocrystals formed by Dynamic Cation–Ligand Complexation and Their Use as Anodes for Potassium-Ion Batteries, Nano Lett., 2022, 22, 10120, DOI:10.1021/acs.nanolett.2c03933
- N. Kapuria, N. N. Patil, K. M. Ryan and S. Singh, Two-dimensional copper based colloidal nanocrystals: synthesis and applications, Nanoscale, 2022, 14, 2885, 10.1039/d1nr06990j
- N. Kapuria, U. V. Ghorpade, M. Zubair, M. Mishra, S. Singh and K. M. Ryan, Metal chalcogenide semiconductor nanocrystals synthesized from ion-conducting seeds and their applications, J. Mater. Chem. C, 2020, 8, 13868, 10.1039/d0tc02895a
- B. A. Tappan, G. Barim, J. C. Kwok and R. L. Brutchey, Utilizing Diselenide Precursors toward Rationally Controlled Synthesis of Metastable CuInSe2 Nanocrystals, Chem. Mater., 2018, 30, 5704, DOI:10.1021/acs.chemmater.8b02205
- E. A. Hernandez-Pagan, E. H. Robinson, A. D. La Croix and J. E. Macdonald, Direct Synthesis of Novel Cu2-xSe Wurtzite Phase, Chem. Mater., 2019, 31, 4619, DOI:10.1021/acs.chemmater.9b02019
- J. M. Rhodes, C. A. Jones, L. B. Thal and J. E. Macdonald, Phase-Controlled Colloidal Syntheses of Iron Sulfide Nanocrystals via Sulfur Precursor Reactivity and Direct Pyrite Precipitation, Chem. Mater., 2017, 29, 8521, DOI:10.1021/acs.chemmater.7b03550
- B. A. Tappan, M. K. Horton and R. L. Brutchey, Ligand-Mediated Phase Control in Colloidal AgInSe2 Nanocrystals, Chem. Mater., 2020, 32, 2935, DOI:10.1021/acs.chemmater.9b05163
- J. J. Wang, A. Singh, P. Liu, S. Singh, C. Coughlan, Y. N. Guo and K. M. Ryan, Colloidal Synthesis of Cu2SnSe3 Tetrapod Nanocrystals, J. Am. Chem. Soc., 2013, 135, 7835, DOI:10.1021/ja403083p
- S. Singh and K. M. Ryan, Occurrence of Polytypism in Compound Colloidal Metal Chalcogenide Nanocrystals, Opportunities, and Challenges, J. Phys. Chem. Lett., 2015, 6, 3141, DOI:10.1021/acs.jpclett.5b01311
- J. Q. Geisenhoff, A. K. Tamura and A. M. Schimpf, Manipulation of Precursor Reactivity for the Facile Synthesis of Heterostructured and Hollow Metal Selenide Nanocrystals, Chem. Mater., 2020, 32, 2304, DOI:10.1021/acs.chemmater.9b04305
- M. S. Sokolikova, P. C. Sherrell, P. Palczynski, V. L. Bemmer and C. Mattevi, Direct solution-phase synthesis of 1T ' WSe2 nanosheets, Nat. Commun., 2019, 10, 712, DOI:10.1038/s41467-019-08594-3
- Z. Liu, K. Nie, X. Qu, X. Li, B. Li, Y. Yuan, S. Chong, P. Liu, Y. Li, Z. Yin and W. Huang, General Bottom-Up Colloidal Synthesis of Nano-Monolayer Transition-Metal Dichalcogenides with High 1T′-Phase Purity, J. Am. Chem. Soc., 2022, 144, 4863, DOI:10.1021/jacs.1c12379
- J. N. Yang, Q. C. Xu, Y. T. Zheng, Z. M. Tian, Y. Y. Shi, C. X. Ma, G. Y. Liu, B. Peng, Z. Wang and W. J. Zheng, Phase Engineering of Metastable Transition Metal Dichalcogenides via Ionic Liquid Assisted Synthesis, ACS Nano, 2022, 16, 15215, DOI:10.1021/acsnano.2c06549
- L. Wu, Q. Wang, T. T. Zhuang, G. Z. Zhang, Y. Li, H. H. Li, F. J. Fan and S. H. Yu, A library of polytypic copper-based quaternary sulfide nanocrystals enables efficient solar-to-hydrogen conversion, Nat. Commun., 2022, 13, 5414, DOI:10.1038/s41467-022-33065-7
- S. Das, G. Swain and K. Parida, One step towards the 1T/2H-MoS2 mixed phase: a journey from synthesis to application, Mater. Chem. Front., 2021, 5, 2143, 10.1039/d0qm00802h
- P. Prabhu, V. Jose and J. M. Lee, Design Strategies for Development of TMD-Based Heterostructures in Electrochemical Energy Systems, Matter, 2020, 2, 526, DOI:10.1016/j.matt.2020.01.001
- R. F. Zhou, Y. Zheng, M. Jaroniec and S. Z. Qiao, Determination of the Electron Transfer Number for the Oxygen Reduction Reaction: From Theory to Experiment, ACS Catal., 2016, 6, 4720, DOI:10.1021/acscatal.6b01581
- S. Abinaya, P. Moni, V. Parthiban, A. K. Sahu and M. Wilhelm, Metal Silicide Nanosphere Decorated Carbon-Rich Polymer-Derived Ceramics: Bifunctional Electrocatalysts towards Oxygen and their Application in Anion Exchange Membrane Fuel Cells, Chemelectrochem, 2019, 6, 3268, DOI:10.1002/celc.201900475
- B. Shang, X. Q. Cui, L. Jiao, K. Qi, Y. W. Wang, J. C. Fan, Y. Y. Yue, H. Y. Wang, Q. L. Bao, X. F. Fan, S. T. Wei, W. Song, Z. L. Cheng, S. J. Guo and W. T. Zheng, Lattice -Mismatch-Induced Ultrastable 1T-Phase MoS2-Pd/Au for Plasmon-Enhanced Hydrogen Evolution, Nano Lett., 2019, 19, 2758, DOI:10.1021/acs.nanolett.8b04104
- Y. Y. Liu, J. J. Wu, K. P. Hackenberg, J. Zhang, Y. M. Wang, Y. C. Yang, K. Keyshar, J. Gu, T. Ogitsu, R. Vajtai, J. Lou, P. M. Ajayan, B. C. Wood and B. I. Yakobson, Self-optimizing, highly surface-active layered metal dichalcogenide catalysts for hydrogen evolution, Nat. Energy, 2017, 2, 17127, DOI:10.1038/nenergy.2017.127
- J. W. Thomson, K. Nagashima, P. M. Macdonald and G. A. Ozin, From Sulfur-Amine Solutions to Metal Sulfide Nanocrystals: Peering into the Oleylamine-Sulfur Black Box, J. Am. Chem. Soc., 2011, 133, 5036, DOI:10.1021/ja1109997
- R. L. Brutchey, Diorganyl Dichalcogenides as Useful Synthons for Colloidal Semiconductor Nanocrystals, Acc. Chem. Res., 2015, 48, 2918, DOI:10.1021/acs.accounts.5b00362
- B. C. Steimle, R. W. Lord and R. E. Schaak, Phosphine-Induced Phase Transition in Copper Sulfide Nanoparticles Prior to Initiation of a Cation Exchange Reaction, J. Am. Chem. Soc., 2020, 142, 13345, DOI:10.1021/jacs.0c06602
- G. Frenking, I. Fernandez, N. Holzmann, S. Pan, I. Krossing and M. F. Zhou, Metal-CO Bonding in Mononuclear Transition Metal Carbonyl Complexes, JACS Au, 2021, 1, 623, DOI:10.1021/jacsau.1c00106
- A. L. Washington, M. E. Foley, S. Cheong, L. Quffa, C. J. Breshike, J. Watt, R. D. Tilley and G. F. Strouse, Ostwald's Rule of Stages and Its Role in CdSe Quantum Dot Crystallization, J. Am. Chem. Soc., 2012, 134, 17046, DOI:10.1021/ja302964e
- G. P. Gao, Y. Jiao, F. X. Ma, Y. L. Jiao, E. Waclawik and A. J. Du, Charge Mediated Semiconducting-to-Metallic Phase Transition in Molybdenum Disulfide Monolayer and Hydrogen Evolution Reaction in New 1T' Phase, J. Phys. Chem. C, 2015, 119, 13124, DOI:10.1021/acs.jpcc.5b04658
- S. Jayabal, G. Saranya, J. Wu, Y. Q. Liu, D. S. Geng and X. B. Meng, Understanding the high-electrocatalytic performance of two-dimensional MoS2 nanosheets and their composite materials, J. Mater. Chem. A, 2017, 5, 24540, 10.1039/c7ta08327k
- Y. Cao, Roadmap and Direction toward High-Performance MoS2 Hydrogen Evolution Catalysts, ACS Nano, 2021, 15, 11014, DOI:10.1021/acsnano.1c01879
- J. Q. Zhu, Z. C. Wang, H. J. Dai, Q. Q. Wang, R. Yang, H. Yu, M. Z. Liao, J. Zhang, W. Chen, Z. Wei, N. Li, L. J. Du, D. X. Shi, W. L. Wang, L. X. Zhang, Y. Jiang and G. Y. Zhang, Boundary activated hydrogen evolution reaction on monolayer MoS2, Nat. Commun., 2019, 10, 1348, DOI:10.1038/s41467-019-09269-9
- Y. Lee, N. Ling, D. Kim, M. Zhao, Y. A. Eshete, E. Kim, S. Cho and H. Yang, Heterophase Boundary for Active Hydrogen Evolution in MoTe2, Adv. Funct. Mater., 2022, 32, 2105675, DOI:10.1002/adfm.202105675
- X. Y. Lu, Y. Yang, Y. Yin, Z. L. Wang, L. Sutrisno, C. L. Yan and O. G. Schmidt, Atomic Heterointerface Boosts the Catalytic Activity toward Oxygen Reduction/Evolution Reaction, Adv. Energy Mater., 2021, 11, 2102235, DOI:10.1002/aenm.202102235
- J. N. Song, Y. Chen, H. J. Huang, J. J. Wang, S. C. Huang, Y. F. Liao, A. E. Fetohi, F. Hu, H. Y. Chen, L. L. Li, X. P. Han, K. M. El-Khatib and S. J. Peng, Heterointerface Engineering of Hierarchically Assembling Layered Double Hydroxides on Cobalt Selenide as Efficient Trifunctional Electrocatalysts for Water Splitting and Zinc-Air Battery, Adv. Sci., 2022, 9, 2104522, DOI:10.1002/advs.202104522
- K. Huang, K. Bi, J. C. Xu, C. Liang, S. Lin, W. J. Wang, T. Z. Yang, Y. X. Du, R. Zhang, H. J. Yang, D. Y. Fan, Y. G. Wang and M. Lei, Novel graphite-carbon encased tungsten carbide nanocomposites by solid-state reaction and their ORR electrocatalytic performance in alkaline medium, Electrochim. Acta, 2015, 174, 172, DOI:10.1016/j.electacta.2015.05.152
- G. Solomon, M. G. Kohan, M. Vagin, F. Rigoni, R. Mazzaro, M. M. Natile, S. J. You, V. Morandi, I. Concina and A. Vomiero, Decorating vertically aligned MoS2 nanoflakes with silver nanoparticles for inducing a bifunctional electrocatalyst towards oxygen evolution and oxygen reduction reaction, Nano Energy, 2021, 81, 105664, DOI:10.1016/j.nanoen.2020.105664
- J. X. Mao, P. Liu, C. C. Du, D. X. Liang, J. Y. Yan and W. B. Song, Tailoring 2D MoS2 heterointerfaces for promising oxygen reduction reaction electrocatalysis, J. Mater. Chem. A, 2019, 7, 8785, 10.1039/c9ta01321k
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta09892j |
| ‡ Nilotpal Kapuria and Niraj Nitish Patil have contributed equally and share joint first author |
| This journal is © The Royal Society of Chemistry 2023 |