
A covalent organic framework constructed from a donor–acceptor–donor motif monomer for photocatalytic hydrogen evolution from water†
Guang-Bo
Wang‡
a,
Hai-Peng
Xu‡
a,
Ke-Hui
Xie
a,
Jing-Lan
Kan
a,
Jianzhong
Fan
 b,
Yan-Jing
Wang
a,
Yan
Geng
*a and
Yu-Bin
Dong
*a
b,
Yan-Jing
Wang
a,
Yan
Geng
*a and
Yu-Bin
Dong
*a
aCollege of Chemistry, Chemical Engineering and Materials Science, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, Key Laboratory of Molecular and Nano Probes, Ministry of Education, Shandong Normal University, Jinan 250014, P. R. China. E-mail: gengyan@sdnu.edu.cn; yubindong@sdnu.edu.cn
bSchool of Physics and Electronics, Shandong Normal University, Jinan 250014, P. R. China
First published on 6th January 2023
Abstract
Covalent organic frameworks (COFs), featuring semiconductor-like behavior, have recently garnered widespread interest for applications in photocatalysis by virtue of their well-defined and tailorable porous structures, high surface areas and excellent chemical stability. A facile strategy for designing COFs exerting efficient charge transfer and separation as well as suppressing charge carrier recombination is the precise integration of electron-donating and electron-withdrawing moieties to form long-range ordered donor–acceptor (D–A) type structures. In this work, we rationally designed and synthesized a novel imine-linked COF (DABT-Py-COF) by the condensation of a newly designed D–A–D type monomer of 4,4′,4′′,4′′′-(benzo[c][1,2,5]thiadiazole-4,7-diylbis(9,9-dimethyl-9,10-dihydroacridine-10,2,7-triyl))tetrabenzaldehyde and 1,3,6,8-tetrakis(4-aminophenyl)pyrene under solvothermal conditions. Remarkably, the obtained DABT-Py-COF exhibited outstanding and steady hydrogen production with a maximum hydrogen evolution rate (HER) of 5458 μmol g−1 h−1 under visible-light irradiation (AM 1.5). This work has paved the way for the rational design and preparation of more efficient D–A type COFs for photocatalysis.
Introduction
Since Honda and Fujishima pioneered water photolysis to generate hydrogen on an n-type TiO2 electrode in 1972,1 visible-light driven photocatalytic hydrogen evolution over certain photocatalysts has been considered as one of the most promising strategies for clean hydrogen energy production and enormous efforts have been devoted to develop more efficient photocatalysts ranging from inorganic semiconductors to conjugated microporous polymers for hydrogen production during the past few decades.2–6 Nonetheless, the weak light harvesting, fast charge recombination and the limited structural tunability of most of these photocatalysts have significantly inhibited their photocatalytic performances.Covalent organic frameworks (COFs), as a burgeoning family of advanced crystalline porous materials that allow the atomically precise assembly of different building units into extended two-dimensional (2D) or three-dimensional (3D) frameworks with periodically ordered skeletons,7 have received increasing interest and become a rapidly growing research field for a wide range of applications in recent years.8–13 In particular, COFs have recently exhibited promising potential application in hydrogen production by virtue of their multiple merits including π–π stacking structures, excellent visible-light response ability, tunable band gaps and remarkable physiochemical stability, which are rather challenging to be simultaneously realized within previously reported inorganic and organic amorphous photocatalysts.14,15 In 2014, Lotsch and co-workers pioneered the employment of a hydrazone-linked COF as the photocatalyst loaded with a Pt co-catalyst in the presence of a certain sacrificial electron donor (SED) for photocatalytic H2 evolution under visible-light irradiation;16 subsequently, a variety of COFs with different linkages or topologies were designed and reported for efficient hydrogen evolution17–22 and the hydrogen evolution rate (HER) could even reach up to 100 mmol g−1 h−1 under visible-light irradiation (λ ≥ 420 nm).17 Among all the reported COF materials, the combination of electron-rich and electron-deficient moieties within a COF backbone to form a periodically ordered donor (D)–acceptor (A) structure has proven to be an effective approach to achieve more efficient charge transport and separation, leading to enhanced photocatalytic performance.18,23,24 For example, we have designed and reported a benzodifuran-based D–A type COF material (BDF-TAPT-COF) by the condensation of electron-donating 4,4′-(benzo[1,2-b:4,5-b′]difuran-4,8-diyl)dibenzaldehyde and electron-withdrawing tris-(4-aminophenyl)triazine, which exhibited an excellent and steady HER of 1390 μmol g−1 h−1 and an outstanding apparent quantum yield of 7.8% at 420 nm.18 The advantages of the introduction of the donor–acceptor–donor type monomer mainly include the enhanced visible-light absorption ability, higher charge transfer and separation efficiency together with a lower charge recombination rate. In this regard, the polycondensation of a D–A–D type of monomer consisting of two electron-donating groups and one electron-withdrawing group within one building unit with another building unit to construct D–A type COFs is expected to facilitate stronger intramolecular charge transfer (ICT) and therefore realizing enhanced charge separation and transport,25,26 while it is still rarely explored thus far.27 In this context, we herein report a novel D–A type imine-linked COF material, termed DABT-Py-COF, by condensation of a newly designed D–A–D type monomer of 4,4′,4′′,4′′′-(benzo[c][1,2,5]thiadiazole-4,7-diylbis(9,9-dimethyl-9,10-dihydroacridine-10,2,7-triyl))tetrabenzaldehyde (DABT-4CHO), and 1,3,6,8-tetrakis(4-aminophenyl)pyrene (PyTA) under conventional solvothermal conditions. The obtained DABT-Py-COF features high crystallinity, permanent porosity, broad light absorption range and excellent chemical stability. More importantly, the DABT-Py-COF exhibited remarkable sunlight-driven hydrogen evolution with a maximum HER of 5458 μmol g−1 h−1 under visible-light irradiation (AM 1.5) and it can be reused for at least 5 consecutive cycles over 20 h with negligible loss of its photoactivity and stability.
Experimental section
Materials
All the solvents were purchased from commercial suppliers and used as received without further purification. 4,7-Bis(9,9-dimethylacridin-10(9H)-l)benzo[c][1,2,5]thiadiazole (DABT) was synthesized according to the previously reported literature.28Synthesis of 4,7-bis(2,7-dibromo-9,9-dimethylacridin-10(9H)-yl)benzo[c][1,2,5]thiadiazole (DABT-4Br)
The N-bromosuccinimide (NBS) (3.916 g, 22 mmol) solution (CHCl3, 30 mL) was added dropwise into the DABT (2.746 g, 5 mmol) solution (CHCl3, 30 mL) at 0 °C. Afterwards, the reaction mixture was stirred for half an hour at 0 °C, before being stirred at room temperature for 12 hours. The crude product was obtained, after the solvent was removed under vacuum, then purified by column chromatography on silica gel (CH2Cl2/petroleum ether = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) to afford the product as reddish-brown powder (4.12 g, 95% yield). 1H NMR (400 MHz, chloroform-d) δ 7.95 (s, 2H), 7.62 (d, J = 2.2 Hz, 4H), 7.11 (dd, J = 8.7, 2.2 Hz, 4H), 6.17 (d, J = 8.7 Hz, 4H), 1.78 (s, 12H). 13C NMR (101 MHz, chloroform-d) δ = 154.43, 138.90, 133.21, 133.15, 132.66, 129.59, 128.50, 115.43, 114.47, 36.58, 30.60. HRMS (ESI) m/z calcd. for C36H27Br4N4S: 866.8649; found: 866.8691 [M + H]+.
:2) to afford the product as reddish-brown powder (4.12 g, 95% yield). 1H NMR (400 MHz, chloroform-d) δ 7.95 (s, 2H), 7.62 (d, J = 2.2 Hz, 4H), 7.11 (dd, J = 8.7, 2.2 Hz, 4H), 6.17 (d, J = 8.7 Hz, 4H), 1.78 (s, 12H). 13C NMR (101 MHz, chloroform-d) δ = 154.43, 138.90, 133.21, 133.15, 132.66, 129.59, 128.50, 115.43, 114.47, 36.58, 30.60. HRMS (ESI) m/z calcd. for C36H27Br4N4S: 866.8649; found: 866.8691 [M + H]+.
Synthesis of 4,4′,4′′,4′′′-(benzo[c][1,2,5]thiadiazole-4,7-diylbis(9,9-dimethyl-9,10-dihydroacridine-10,2,7-triyl))tetrabenzaldehyde (DABT-4CHO)
DABT-4Br (4.3 g, 5 mmol), 4-formylphenylboronic acid (4.5 g, 30 mmol), anhydrous Na2CO3 (5.0 g, 47.18 mmol), and Pd(PPh3)4 (0.5 g, 0.05 mmol) were dissolved in the mixed solvent of toluene/ethanol/water (4:2:1, v/v/v, 70 mL). Under a N2 atmosphere, the reaction mixture was stirred at 80 °C for 24 hours. After the reaction mixture was cooled to room temperature, 50 mL of water was added. The layers were separated. The aqueous phase was extracted with CH2Cl2 (3 × 25 mL), and the combined organic phases were dried over magnesium sulfate, filtered, and then the solvent was removed. The crude product was purified by column chromatography on silica gel (CH2Cl2/petroleum ether = 1:4) to afford DABT-4CHO as a dark red solid (2.66 g, 55%). 1H NMR (400 MHz, DMSO-d6) δ = 10.05 (s, 4H), 8.31 (s, 2H), 8.08–7.92 (m, 20H), 7.50 (dd, J = 8.6, 2.1 Hz, 4H), 6.57 (d, J = 8.6 Hz, 4H), 1.96 (s, 12H). 13C NMR (101 MHz, chloroform-d) δ 191.80, 154.64, 146.78, 140.07, 134.76, 133.23, 131.41, 130.39, 126.95, 125.87, 124.91, 114.47, 36.66, 31.42. HRMS (ESI) calcd. for C64H47N4O4S 967.3318; found: 967.3326 [M + H]+.
Synthesis of DABT-Py-COF
A Pyrex tube was charged with DABT-4CHO (38.64 mg, 0.04 mmol), 1,3,6,8-tetrakis(4-aminophenyl)pyrene (PyTA) (22.68 mg, 0.04 mmol), 1.0 mL mesitylene and 1.0 mL 1,4-dioxane, and then the mixture was sonicated for 20 min, followed by slow addition of 0.2 mL of 6 M aqueous acetic acid. Afterwards, the tube was flash frozen at 77 K and degassed by three freeze–pump–thaw cycles, sealed under vacuum and heated at 120 °C for 5 days. After the constant temperature process was completed, the tube was cooled to room temperature. It was opened and the resulting precipitate was filtered off, thoroughly washed with THF, methanol and acetone, and subjected to Soxhlet extractions with tetrahydrofuran and dichloromethane for 12 h, respectively. Finally, the resulting yellowish orange powder was dried under vacuum at 80 °C overnight to give DABT-Py-COF (56 mg, 92%).Photocatalytic H2 evolution experiments
For a typical H2 evolution experiment, a Pyrex tube was charged with activated DABT-Py-COF (5 mg), 0.1 M ascorbic acid water solution (10 mL) and hexachloroplatinic acid solution (4 μL, 3 wt% aqueous solution) as a platinum precursor. After deoxygenation with argon for 30 min, CH4 gas is injected into the tube as an internal standard to quantify the amount of photogenerated H2 gas. The system was connected to the device and irradiated with a 300 W Xenon lamp (Beijing Perfectlight) equipped with a cut-off filter of AM 1.5 under constant agitation and fan cooling for real time quantitative hydrogen detection. The generated H2 was quantitatively measured by the drainage gas-collecting method using a gas-tight syringe and analyzed using a gas chromatograph (Fuli, 9790 II (PLF-01)) equipped with a thermal conductivity detector (TCD) and a 5 Å molecular sieve column with argon as the carrier gas. For every series of H2 evolution experiments, two independent measurements were repeated for reproducibility. After the photocatalysis experiment, DABT-Py-COF was recovered by washing with water and acetone and then dried under vacuum at 120 °C for multiple runs.Results and discussion
The donor–acceptor–donor motif monomer DABT-4CHO was designed and synthesized (Scheme 1a and Fig. S1–S5, ESI†), and was further applied to construct DABT-Py-COF by polycondensation with PyTA in the solvent mixture of mesitylene/1,4-dioxane/6 M acetic acid solution (5/5/1, by vol.) at 120 °C for 5 days, affording yellowish orange crystalline powder in 92% yield (Scheme 1b).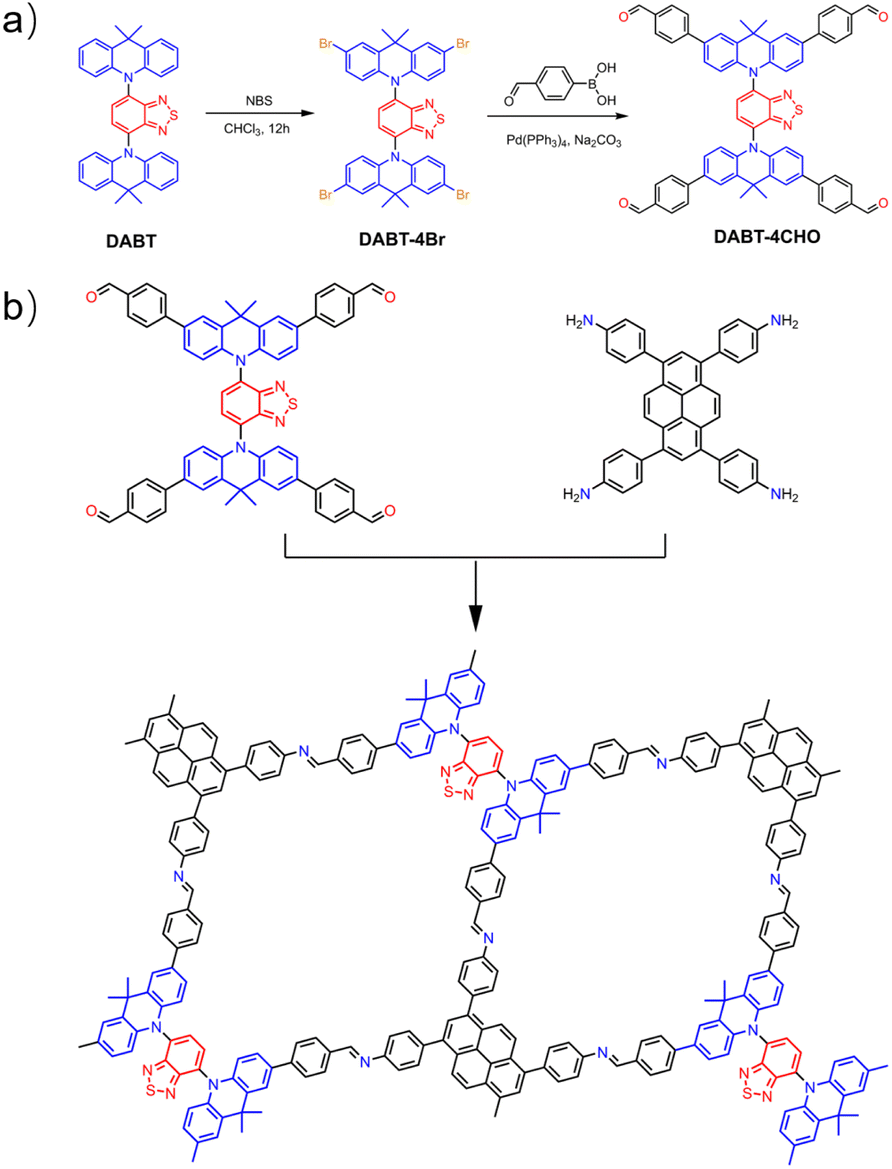 | ||
| Scheme 1 (a) Synthetic route of DABT-4CHO. (b) Schematic diagram for the synthesis of DABT-Py-COF. | ||
The high crystallinity and structure of DABT-Py-COF were confirmed by powder X-ray diffraction (PXRD) together with corresponding structural simulations and Pawley refinements using Materials Studio. As shown in Fig. S6,† no diffraction peaks related to the starting materials were detected. The diffraction peaks at 2.64°, 4.54° and 5.27° could be assigned to the (100), (010) and (200) facets, respectively (Fig. 1a). The experimental PXRD pattern of DABT-Py-COF matched well with the simulated slipped AA stacking model (Fig. 1b and S7, ESI†) and the Pawley refinement showed negligible differences between the simulated and experimental PXRD patterns (Rwp = 5.05% and RP = 3.95%). And the two-dimensional layers in the crystalline state were nonplanar with a large dihedral angle of 54.4° between benzothiadiazole and acridine moieties, which could be applied to explain the tendency for the slipped AA stacking model in DABT-Py-COF (Fig. S8, ESI†). DABT-Py-COF was assigned to the space group of P1 with the unit cell parameters of a = 33.4252 Å, b = 28.0981 Å, and c = 7.0436 Å; α = 44.9959°, β = 90.0502°, and γ = 90.0440° (Table S1, ESI†).
 | ||
| Fig. 1 (a) PXRD pattern of DABT-Py-COF (red) with corresponding Pawley refinement (black) indicating negligible differences (blue) in the experimental data. (b) Top and side views of the space-filling models of DABT-Py-COF in slipped AA stacking mode. | ||
As indicated by the FT-IR spectra (Fig. S9, ESI†), the characteristic peak at 1624 cm−1 clearly confirmed the formation of the imine linkages,29 while the weak peak at 1695 cm−1 is assigned to the free aldehyde groups, indicating the existence of bonding defects.30 As for the solid-state 13C CP-MAS NMR spectrum of DABT-Py-COF (Fig. S10, ESI†), the chemical shifts at approximately 23 and 35 ppm are related to the acridine carbons, while the typical signal at 152 ppm corresponds to the formed imine-linkages31 and other signals from 112 to 150 ppm can be attributed to the aromatic carbons of DABT-Py-COF.32,33 In addition, the X-ray photoelectron spectroscopy (XPS) data further disclosed the structural information of DABT-Py-COF. For example, in the N 1s spectrum, the peaks at 399.0 ± 0.1 and 400.1 ± 0.1 eV are attributed to CN and C–N, respectively. The S 2p spectrum shows a doublet characteristic (at 165.7 ± 0.1 and 166.8 ± 0.1 eV) for the sulfur of the benzothiadiazole groups (Fig. S11, ESI†).19 These results confirmedly verified the successful condensation of the building units. The scanning electron microscopy (SEM) image shows that DABT-Py-COF possesses a sheet-like morphology up to micrometres (Fig. S12, ESI†) and the layered structure of the framework could be obviously observed from its corresponding high-resolution transmission electron microscopy (TEM) image (Fig. S13, ESI†).
The permanent porosity of DABT-Py-COF was investigated by N2 adsorption and desorption measurements performed at 77 K (Fig. 2a). The Brunauer–Emmett–Teller (BET) surface area was estimated to be 549 m2 g−1 with a total pore volume of 0.50 cm3 g−1 at P/P0 = 0.99. The pore size distribution of DABT-Py-COF was centered at 1.18 and 2.16 nm (Fig. 2a, inset) by the nonlocal density functional theory (NLDFT), which is consistent with its theoretical value. To evaluate the chemical stability of the obtained DABT-Py-COF, the DABT-Py-COF samples (10 mg) were respectively immersed in various solvents, including ascorbic acid/water, DMSO, DMF, 3 M HCl and 3 M NaOH, for 1 week. Remarkably, the crystallinity and chemical structures of DABT-Py-COF were well preserved as confirmed by using corresponding PXRD patterns and FT-IR spectra (Fig. 2b and S14, ESI†). Thermogravimetric analysis (TGA) indicated that DABT-Py-COF can be stable up to 260 °C under nitrogen (Fig. S15, ESI†).
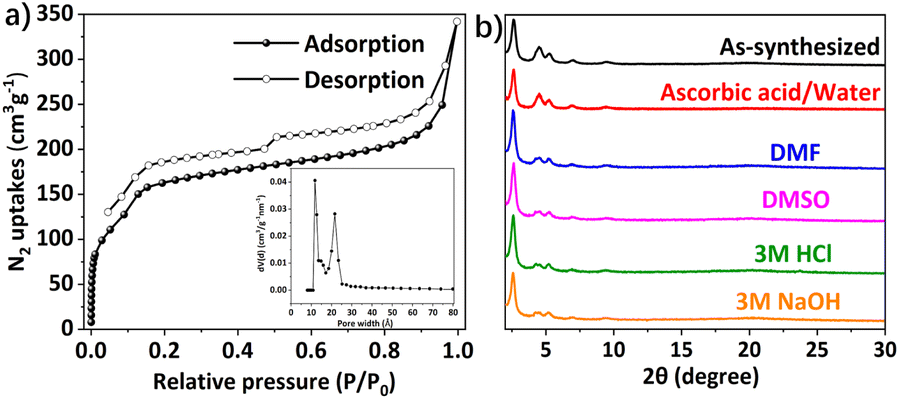 | ||
| Fig. 2 (a) N2 adsorption (solid symbols) and desorption (empty symbols) isotherm of DABT-Py-COF measured at 77 K. Inset, the pore size distribution of DABT-Py-COF derived from the nonlocal density functional theory (NLDFT). (b) Chemical stability test of DABT-Py-COF by immersing in different solvents for 1 week. | ||
The solid-state UV-Vis diffuse reflectance spectrum of DABT-Py-COF exhibits an absorbance tail extending up to 700 nm (Fig. 3a), indicating its wide light harvesting range and the optical band gap (Eg) was evaluated to be 2.00 eV based on the absorption band using the Tauc-plot34 (Fig. 3a, inset). The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) levels of DABT-Py-COF were respectively calculated to be −6.22 and −4.22 eV by cyclic voltammetry (CV) measurements (Fig. S16 and S17, ESI†). Furthermore, the HOMO and LUMO of the segments of DABT-Py-COF were calculated and visualized by employing density functional theory (DFT) computations. As shown in Fig. 3b, the HOMO and LUMO of the segments are mainly located at the acridine and benzothiadiazole moieties, respectively, indicating sufficient ICT within the framework.
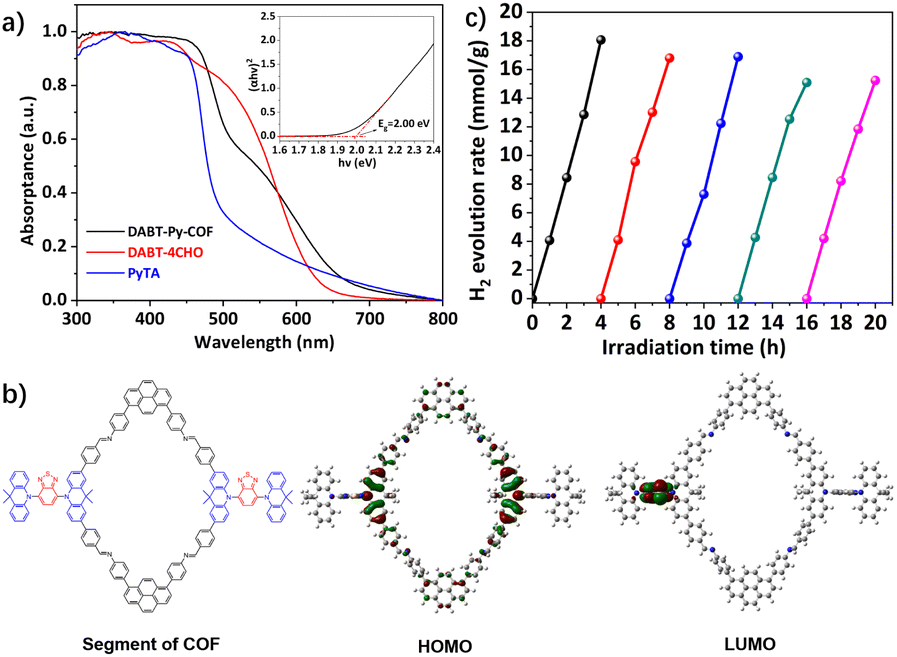 | ||
| Fig. 3 (a) UV-Vis diffuse reflectance spectra (DRS) of the monomers and DABT-Py-COF measured in the solid state. Inset: Tauc-plot for the determination of the optical band gap. (b) Optimized molecular geometries of frontier molecular orbitals for the segments of DABT-Py-COF. (c) Photostability test of DABT-Py-COF for cycling runs over 20 h under visible-light irradiation (AM 1.5). | ||
Subsequently, the photocatalytic hydrogen evolution performance of DABT-Py-COF was investigated in an aqueous solution with in situ photogenerated Pt nanoparticles as the co-catalyst and ascorbic acid or TEOA as the sacrificial electron donor (SED) under visible-light irradiation (AM 1.5), as illustrated in Fig. S18.† The control experiments indicated no activity in the absence of light or the photocatalyst, precluding any possible reaction that probably leads to the hydrogen production and only a little amount of hydrogen (299 μmol g−1 h−1) was observed in the absence of the Pt co-catalyst (Fig. S19, ESI†). Furthermore, the photodeposited Pt content and electron donor type also affect the hydrogen production activity. For example, when TEOA was used as the SED, the HER of DABT-Py-COF was only 130 μmol g−1 h−1 and it can reach up to 4726 μmol g−1 h−1 with ascorbic acid as the SED (Fig. S19, ESI†). Moreover, the hydrogen evolution activity of DABT-Py-COF first increased and then decreased with the increase of the photodeposited Pt content (Fig. S20, ESI†). Thus, in a typical photocatalytic reaction, 5 mg of activated DABT-Py-COF was dispersed in 10 mL solution of H2O containing ascorbic acid (0.1 M) and hexachloroplatinic acid (4 μL, 3 wt% aqueous solution) as a platinum precursor. Under the optimized reaction conditions, a remarkable increase of the produced hydrogen amount with the reaction time was observed and a maximum hydrogen evolution rate (HER) of 5458 μmol g−1 h−1 was observed, higher than most of previously reported CMPs and COF materials35,36 (Table S2, ESI†). The photostability of the photocatalyst was further confirmed by multiple H2 evolution tests. Notably, DABT-Py-COF exhibited a steady hydrogen evolution rate under the optimized conditions without significant decay through 5 consecutive cycles of reaction over 20 h (Fig. 3c). Analysis of DABT-Py-COF after photocatalytic reactions by PXRD, FT-IR, SEM and TEM/EDS mapping revealed negligible compositional or morphological changes of the photocatalyst compared with those of the pristine material (Fig. S21–S24, ESI†), and thereby attested to the excellent stability and reusability of DABT-Py-COF. Next, the size of the photodeposited Pt nanoparticles was investigated via TEM and the in situ formed Pt nanoparticles exhibited a similar morphology with an average size of 2.35 nm (Fig. 4a and b).
 | ||
| Fig. 4 (a) TEM image of DABT-Py-COF after 5 cycles of photocatalytic reactions and (b) the corresponding diameter distribution of the Pt nanoparticles loaded on the DABT-Py-COF material. (c) Photocurrent responses (i–t) of the DABT-Py-COF photocatalyst in the presence or absence of the Pt co-catalyst. (d) Time-resolved photoluminescence spectrum of DABT-Py-COF (excitation at 710 nm). | ||
To further understand the photocatalytic performance of DABT-Py-COF, electrochemical impedance spectroscopy (EIS), photocurrent density (i–t) and time-resolved photoluminescence (TRPL) spectroscopy were performed. First of all, electrochemical impedance spectroscopy was used to evaluate the electrical conductivity of the photocatalysts. As shown in Fig. S25,† the radius of the Nyquist curve of DABT-Py-COF remarkably decreased in the presence of the Pt co-catalyst, suggesting the reduced resistance and enhanced charge migration ability. The significant charge transfer efficiency was further confirmed by the photocurrent response of the photocatalyst (Fig. 4c). The photocurrent response of DABT-Py-COF with Pt was significantly enhanced when the light was switched on in comparison with that of pristine DABT-Py-COF, indicating the low charge recombination rates and high charge transfer efficiency. Furthermore, time-resolved photoluminescence (TRPL) spectroscopy indicated that the average lifetime of DABT-Py-COF was 8.20 ns, beneficial for efficient charge separation and transportation (Fig. 4d).
Conclusions
In summary, we have rationally designed and synthesized a novel imine-linked D–A type DABT-Py-COF, by the condensation of a newly designed D–A–D type monomer of 4,4′,4′′,4′′′-(benzo[c][1,2,5]thiadiazole-4,7-diylbis(9,9-dimethyl-9,10-dihydroacridine-10,2,7-triyl))tetrabenzaldehyde and 1,3,6,8-tetrakis(4-aminophenyl)pyrene under solvothermal conditions. The obtained DABT-Py-COF exhibited high crystallinity, permanent porosity, wide light absorption range and outstanding chemical stability. More importantly, DABT-Py-COF exhibited remarkable and steady hydrogen production with a maximum hydrogen evolution rate of 5458 μmol g−1 h−1 under visible-light irradiation (AM 1.5), surpassing most of the previously reported COF materials. The present work is expected to be beneficial for the development and facile design of more efficient COF-based photocatalysts for potential sunlight-driven hydrogen evolution applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (No. 22171169, 22001153 and 21971153), the Shandong Provincial Natural Science Foundation (No. ZR2020QB036), the Postdoctoral Science Foundation of China (No. 2020M672118), the Taishan Scholars Climbing Program of Shandong Province, the Taishan Youth Scholars Project of Shandong Province, and the Major Basic Research Projects of Shandong Natural Science Foundation (No. ZR2020ZD32).Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- G. Zhang, Z.-A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- C. Dai and B. Liu, Energy Environ. Sci., 2020, 13, 24–52 RSC.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef PubMed.
- G.-B. Wang, K.-H. Xie, H.-P. Xu, Y.-J. Wang, F. Zhao, Y. Geng and Y.-B. Dong, Coord. Chem. Rev., 2022, 472, 214774 CrossRef CAS.
- Q. Guan, L.-L. Zhou and Y.-B. Dong, Chem. Soc. Rev., 2022, 51, 6307–6416 RSC.
- G.-B. Wang, S. Li, C. X. Yan, F. C. Zhu, Q. Q. Lin, K. H. Xie, Y. Geng and Y. B. Dong, J. Mater. Chem. A, 2020, 8, 6957–6983 RSC.
- Q. Guan, G.-B. Wang, L.-L. Zhou, W.-Y. Li and Y.-B. Dong, Nanoscale Adv., 2020, 2, 3656–3733 RSC.
- L. Guo, L. Yang, M. Li, L. Kuang, Y. Song and L. Wang, Coord. Chem. Rev., 2021, 440, 213957 CrossRef CAS.
- T. Sun, J. Xie, W. Guo, D.-S. Li and Q. Zhang, Adv. Energy Mater., 2020, 10, 1904199 CrossRef CAS.
- T. He, K. Geng and D. Jiang, ACS Mater. Lett., 2019, 1, 203–208 CrossRef CAS.
- T. Banerjee, K. Gottschling, G. Savasci, C. Ochsenfeld and B. V. Lotsch, ACS Energy Lett., 2018, 3, 400–409 CrossRef CAS PubMed.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- S. Ma, T. Deng, Z. Li, Z. Zhang, J. Jia, G. Wu, H. Xia, S. W. Yang and X. Liu, Angew. Chem., Int. Ed., 2022, 61, e202208919 CAS.
- G.-B. Wang, F.-C. Zhu, Q.-Q. Lin, J.-L. Kan, K.-H. Xie, S. Li, Y. Geng and Y.-B. Dong, Chem. Commun., 2021, 57, 4464–4467 RSC.
- G.-B. Wang, S. Li, C. X. Yan, Q. Q. Lin, F. C. Zhu, Y. Geng and Y.-B. Dong, Chem. Commun., 2020, 56, 12612–12615 RSC.
- R. Chen, Y. Wang, Y. Ma, A. Mal, X.-Y. Gao, L. Gao, L. Qiao, X.-B. Li, L.-Z. Wu and C. Wang, Nat. Commun., 2021, 12, 1354 CrossRef CAS PubMed.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- K. Wang, Z. Jia, Y. Bai, X. Wang, S. E. Hodgkiss, L. Chen, S. Y. Chong, X. Wang, H. Yang, Y. Xu, F. Feng, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2020, 142, 11131–11138 CrossRef CAS PubMed.
- W. Li, X. Huang, T. Zeng, Y. A. Liu, W. Hu, H. Yang, Y. B. Zhang and K. Wen, Angew. Chem., Int. Ed., 2021, 60, 1869–1874 CrossRef CAS PubMed.
- J. Yang, S. Ghosh, J. Roeser, A. Acharjya, C. Penschke, Y. Tsutsui, J. Rabeah, T. Wang, S. Y. Djoko Tameu, M.-Y. Ye, J. Grüneberg, S. Li, C. Li, R. Schomäcker, R. Van De Krol, S. Seki, P. Saalfrank and A. Thomas, Nat. Commun., 2022, 13, 6317 CrossRef CAS PubMed.
- C. Liu, Z. Fang, J. Sun, M. Shang, K. Zheng, W. Yang and Z. Ge, Nano Energy, 2022, 93, 106800 CrossRef CAS.
- G. Gogoi, L. Bhattacharya, S. Rahman, N. S. Sarma, S. Sahu, B. K. Rajbongshi and S. Sharma, Mater. Today Commun., 2020, 25, 101364 CrossRef CAS.
- J. Y. Zeng, X. S. Wang, B. R. Xie, M. J. Li and X. Z. Zhang, Angew. Chem., Int. Ed., 2019, 59, 10087–10094 CrossRef PubMed.
- F. Ni, Z. Wu, Z. Zhu, T. Chen, K. Wu, C. Zhong, K. An, D. Wei, D. Ma and C. Yang, J. Mater. Chem. C, 2017, 5, 1363–1368 RSC.
- Z.-B. Zhou, X.-H. Han, Q.-Y. Qi, S.-X. Gan, D.-L. Ma and X. Zhao, J. Am. Chem. Soc., 2022, 144, 1138–1143 CrossRef CAS PubMed.
- Q. Guan, D.-D. Fu, Y.-A. Li, X.-M. Kong, Z.-Y. Wei, W.-Y. Li, S.-J. Zhang and Y.-B. Dong, iScience, 2019, 14, 180–198 CrossRef CAS PubMed.
- W. Chen, L. Wang, D. Mo, F. He, Z. Wen, X. Wu, H. Xu and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 16902–16909 CrossRef CAS PubMed.
- F. Yang, C. C. Li, C. C. Xu, J. L. Kan, B. Tian, H. Y. Qu, Y. Guo, Y. Geng and Y. B. Dong, Chem. Commun., 2022, 58, 1530–1533 RSC.
- J. C. Wang, X. Kan, J. Y. Shang, H. Qiao and Y. B. Dong, J. Am. Chem. Soc., 2020, 142, 16915–16920 CrossRef CAS PubMed.
- J. Liu, T. Yang, Z.-P. Wang, P.-L. Wang, J. Feng, S.-Y. Ding and W. Wang, J. Am. Chem. Soc., 2020, 142, 20956–20961 CrossRef CAS PubMed.
- S. Liu, M. Wang, Y. He, Q. Cheng, T. Qian and C. Yan, Coord. Chem. Rev., 2023, 475, 214882 CrossRef CAS.
- Z. Liang, R. Shen, Y. H. Ng, Y. Fu, T. Ma, P. Zhang, Y. Li and X. Li, Chem. Catal., 2022, 2, 2157–2228 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: General methods, and supplementary figures and tables. See DOI: https://doi.org/10.1039/d2ta09625k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |