
Rapid photo-oxidation reactions of imidazole derivatives accelerated by planar quinoid oxidation-state structures†
Yue
Yu
ab,
Bohan
Wang
b,
Jianai
Chen
a,
Yujie
Dong
 a,
Wang
Zhan
a,
Shitong
Zhang
c,
Weijun
Li
*a,
Bing
Yang
c,
Cheng
Zhang
a and
Yuguang
Ma
*b
a,
Wang
Zhan
a,
Shitong
Zhang
c,
Weijun
Li
*a,
Bing
Yang
c,
Cheng
Zhang
a and
Yuguang
Ma
*b
aInternational Sci. & Tech. Cooperation Base of Energy Materials and Application, College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, P. R. China. E-mail: liwj@zjut.edu.cn
bInstitute of Polymer Optoelectronic Materials and Devices, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, China. E-mail: ygma@scut.edu.cn
cState Key Lab of Supramolecular Structure and Materials, Jilin University, 2699 Qianjin Avenue, Changchun 130012, P. R. China
First published on 28th January 2023
Abstract
The photo-oxidation reaction of 2,4,5-triphenylimidazole (lophine) and its derivatives has been studied in-depth since the discovery of its chemiluminescence phenomenon. It has been well agreed that the photostability of lophine could be maintained if hydrogen in imidazole is substituted by benzyl or alkyl groups. However, recently, it has been discovered that a lophine derivative (DPA-PIM) substituted with benzene at the N position of the imidazole ring undergoes a rapid photo-oxidation reaction after the introduction of diphenylamine as the donor group into lophine. Based on DPA-PIM, a series of lophine derivatives with different donor groups were designed and synthesized. In situ absorption spectra indicated that lophine derivatives linked with the p-C position of the arylamine group exhibit photo-oxidation activity under UV irradiation. In comparison, when the N position of the corresponding arylamine group is linked to benzene-substituted lophine, the photostability of derivatives can be maintained. ESR and electrochemical measurements indicated that the arylamine group linked in the p-C position would help lophine derivatives to form a rearranged stable planar quinoid oxidation state structure under UV irradiation, which tends to be easily attacked by self-sensitized singlet oxygen. The planar quinoid structure greatly contributes to the rapid reaction rate of this photo-oxidation reaction. Therefore, we tentatively put forward the mechanism for this kind of photo-oxidation reaction with two main intermediate states: the quinoid oxidation-state structure and the 1,2-dioxetane-like intermediate. It is believed that this finding can deepen the knowledge of the photostability of lophine derivatives or imidazole-based materials. Owing to their rapid reaction rate, some of the imidazole derivatives can also serve as high-sensitivity oxygen sensor materials.
Introduction
2,4,5-Triphenylimidazole (lophine),1 as the first observed chemiluminescence molecule in 1877 by Radziszewski,2 was discovered to exhibit light emission under basic conditions upon exposure to oxygen at room temperature. In this process, lophine was oxidized to provide ammonia as the product. Since then, the oxidation reaction of lophine derivatives has been widely researched. In 1962, Hayashi and Maeda proposed a mechanism for the oxidation of lophine: the anion of lophine formed in ethanolic potassium hydroxide undergoes oxidation to yield the free radical 2,4,5-triphenylimidazyl, from which the corresponding peroxide is produced with molecular oxygen and is finally decomposed to yield ammonia with simultaneous light emission.3 However, they could not figure out the molecular structure of the corresponding peroxide of lophine. In 1964, two research groups developed Hayashi's mechanism and reported independently that lophine hydroperoxide is the important intermediate and could be obtained by the irradiation of lophine in the presence of oxygen.4,5 In 1965, White and Harding published a review of lophine and its derivatives based on the previous work, and then proposed the reaction mechanism for the chemiluminescence of lophine: the important intermediate of hydroperoxide anion could be formed by the attack of oxygen on the imidazole anion or by proton abstraction from hydroperoxide oxygen on carbon; and then a nucleophilic attack of hydroperoxide oxygen on carbon yielded diaroylamidine, which could generate light and provide ammonia.6 In this mechanism, hydrogen on nitrogen in imidazole was obviously very important to this reaction. For this purpose, White conducted an in-depth study and found that when hydrogen on nitrogen in imidazole of lophine was substituted by benzyl or alkyl groups; the resulting lophine derivatives could not be oxidized by oxygen. This phenomenon was also proved by Kang in 2002,7 except for one case, in which Wasserman in 1968 used methylene blue as the sensitizer and a 150 watt floodlamp as the light source and found that benzene-substituted lophine (PIM, shown in Scheme 1) could also be oxidized.8 However, the reaction observed by Wasserman required two weeks for full oxidation with a slow reaction rate. These related reported reactions of lophine and its derivatives are summarized in Scheme 1. Therefore, it can be concluded that lophine derivatives substituted with the phenyl group for nitrogen of the imidazole ring, such as PIM, could be seen as relatively inert to undergo the photo-oxidation reaction.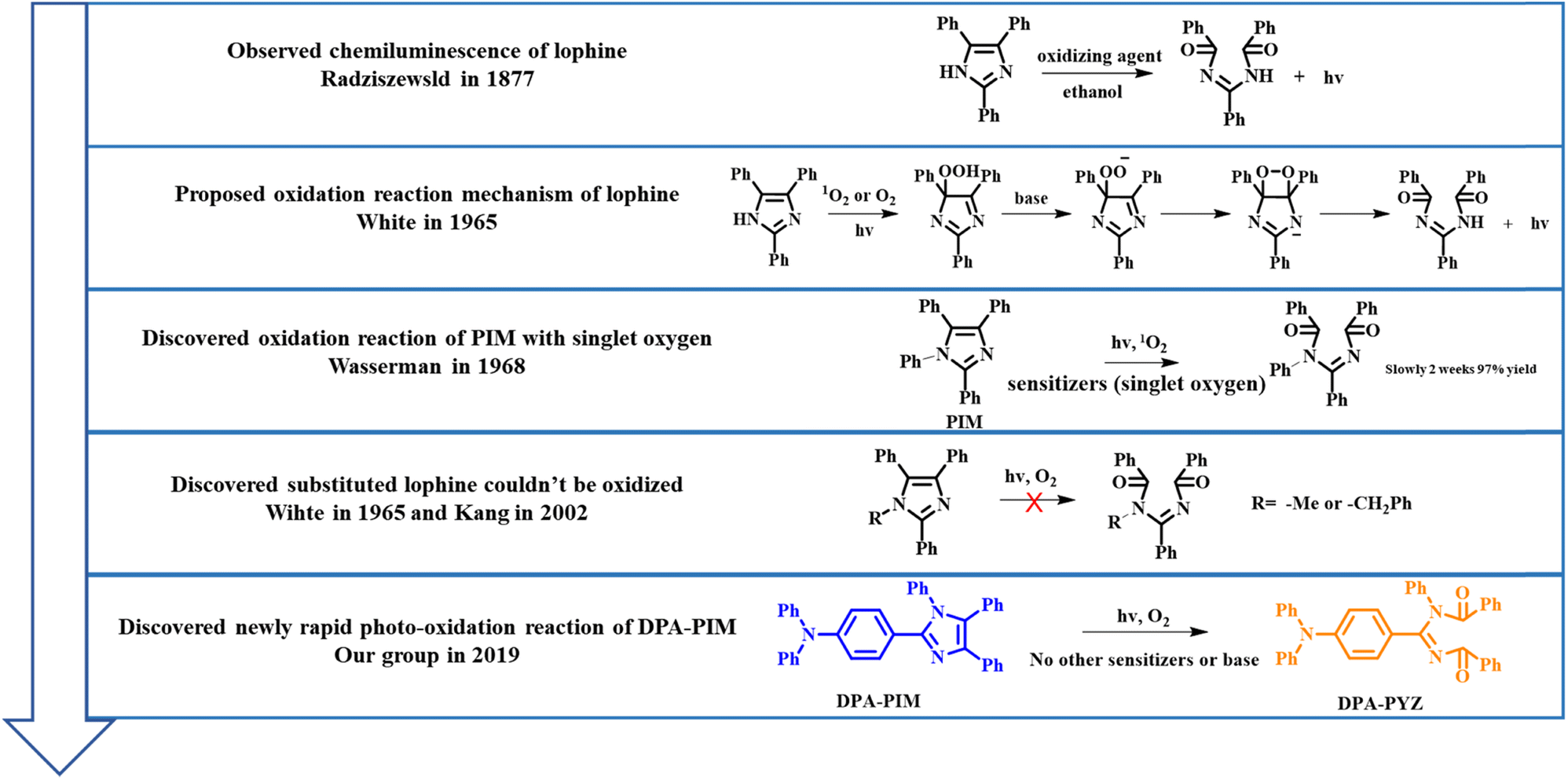 | ||
| Scheme 1 Oxidation reaction of lophine and its derivatives, as reported so far. | ||
Based on this feasible photostability, many PIM derivatives were designed and utilized as very important photo- or electro-luminescent materials for optoelectronic applications.9–15
Recently, our group reported a diphenylamine-linking PIM derivative DPA-PIM (Scheme 1), which can serve as a deep-blue electroluminescent material with a surprisingly narrow emission peak and excellent color purity,9 and found that it also underwent rapid oxidation by oxygen under UV irradiation with a similar amidine product obtained finally.16–19 However, unlike those oxidation reactions of lophine derivatives reported before, this photo-oxidation reaction does not need any base or sensitizers (singlet oxygen), and occurred with a very rapid reaction rate within 90 min for the complete reaction, as indicated by NMR data. This is against our knowledge of the oxidation reaction of lophine derivatives (Scheme 1). The only reasonable explanation is that the introduction of diphenylamine has a great influence on the oxidation reaction of lophine derivatives.
In this work, a series of PIM derivatives were designed and synthesized to deeply investigate this photo-oxidation reaction. It has been found that different linking positions of similar donor groups have a great influence on this kind of photo-oxidation reaction. Through the UV spectra (under potential) and ESR spectra, the planar quinoid oxidation-state structure was confirmed to play a key role in the photo-oxidation reaction of such PIM derivatives. The p-C position linking of the arylamine group can help the molecule to form a rearranged planar quinoid oxidation-state structure, which tends to be easily attacked by self-sensitized singlet oxygen. As for derivatives whose donor linking position is the N atom directly, a large steric hindrance between the PIM and the corresponding conjugated planes is formed to generate a twisted structure, which hinders the formation of quinoid oxidation-state structures. As far as we know, it is the first report of intermediate quinoid oxidation-state structures for the oxidation reaction of lophine derivatives. At last, a new mechanism of this kind of photo-oxidation reaction was put forward based on two main intermediates: the newly discovered quinoid oxidation-state radical and the well-known 1,2-dioxetane-like intermediate. Based on this mechanism, high-sensitivity oxygen sensing materials20–23 can also be designed.
Results and discussion
Photo-oxidation reactions of PIM derivatives
Based on the previous work of our group,16–19 it is known the introduction of diphenylamine is a determinant to promote this kind of photo-oxidation reaction. In order to study this photo-oxidation reaction in-depth, a series of lophine derivatives, compounds 1–27 including PIM (1) and DPA-PIM (19), were designed and synthesized, as shown in Scheme 2. Among these lophine derivatives, PIM is substituted by different electron donor or acceptor groups in R1, R2, and R3 positions to investigate the photo-oxidation reaction. The oxidation reactions of these lophine derivatives were tracked using in situ UV-vis absorption spectra and judged by the absorption signals of lophine derivatives as well as their corresponding oxidation products, with their toluene solution irradiated by a UV light (6 W) for a different time in the air. The molecular structures of lophine derivatives and the photo-oxidation reaction status from the absorption spectra results are shown in Scheme S1 and Table S1† respectively. It is worth mentioning that PIM, which was reported to undergo a mild oxidation reaction when using methylene blue as the sensitizer and a 150 watt floodlamp as the light source,8 could not be oxidized under our experimental condition without any sensitizers. There are no new peaks corresponding to oxidation-reaction products that emerged in the absorption spectra of PIM even after the irradiation time of 60 min (Fig. S1a†). Obviously, lophine derivatives 1–18 with R1 substituted with hydrogen or bromine could not be oxidized no matter what group was linked on R2 and R3, while compounds 19–27 with R1 substituted with diphenylamine can undergo the photo-oxidation reaction. Besides, there is almost no effect observed on the photo-oxidation reaction if different substituent groups are linked on R2 and R3. The corresponding product could be obtained by purification if a photo-oxidation reaction is available. Thus, it can be concluded that the substituted group on the R1 position of PIM derivatives has a significant impact on this photo-oxidation reaction, and arylamine analogs seem to benefit the occurrence of this photo-oxidation reaction.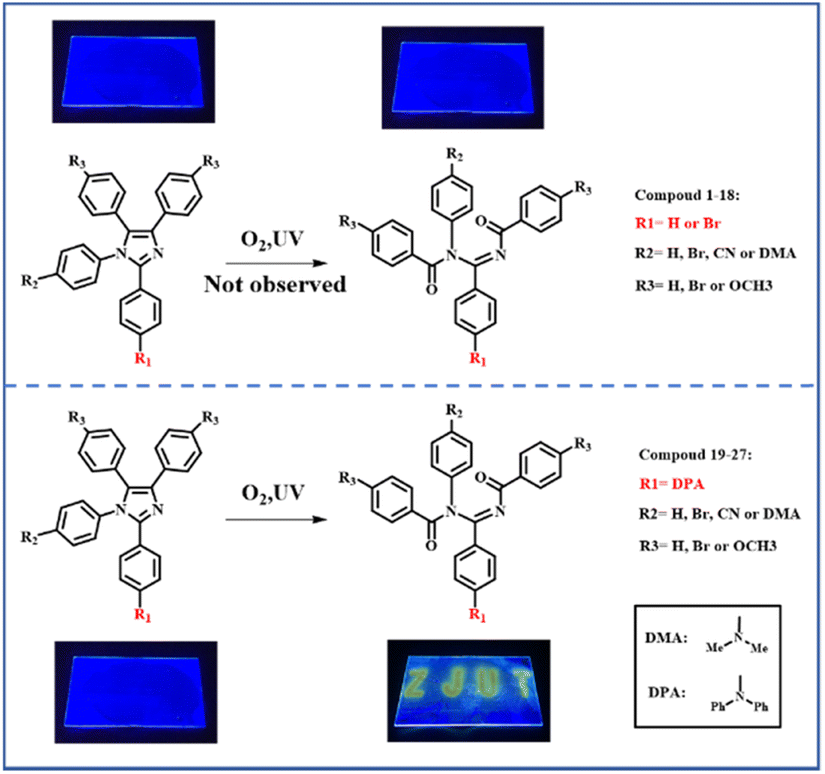 | ||
| Scheme 2 Photo-oxidation reaction of PIM derivatives with donor or acceptor group on different substitution positions. | ||
Another series of lophine derivatives with arylamine structures such as carbazole/dimethylacridan/phenoxazine groups, substituted at the R1 position of PIM, were also synthesized via a one-step cyclization reaction or the Ullmann coupling reaction (P10, Table S1†), and the detailed synthesis routes are shown in Scheme S1.† Their structures and photo-oxidation reaction activities were also studied, and the results are summarized in Table 1. In situ absorption spectra of CZ-PIM, DMAC-PIM, and POZ-PIM indicate that they could not undergo the photo-oxidation reactions, as shown in Fig. S2.† As for DMA-PIM, p-CZ-PIM, p-DMAC-PIM, and p-POZ-PIM, obvious photo-oxidation reactions were observed because the new absorption peaks belonging to reaction products emerged and increased with the prolonged UV irradiation time. Apparently, the three pairs of PIM derivatives (CZ-PIM and p-CZ-PIM, DMAC-PIM and p-DMAC-PIM, and POZ-PIM and p-POZ-PIM) have a similar D–A structure with the same donor group but different substitution positions. Their different photo-oxidation reaction activities indicated that the electron-donating ability of arylamine analogs seemed not to be the sole significant factor for this kind of photo-oxidation, though p-DMAC-PIM and p-POZ-PIM with stronger electron-donating abilities exhibited an obviously higher photo-oxidation reaction activity than that of p-CZ-PIM. The corresponding products were also obtained by purification if the photo-oxidation reaction was available (in the ESI† part). Thus, besides its electron-donating ability, the configuration of arylamine donor groups (i.e. linking type of arylamine group) substituted on R1 of the PIM structure has a more important influence on this photo-oxidation reaction.
|
|
||||
|---|---|---|---|---|
| Compound | R | Linking type | Reaction conditiona | Whether reactb |
| a The wavelength of the irradiation source is determined by the absorption spectra of analogs. b Determined by the change in the absorption spectra of analog compounds under UV irradiation in the air for 60 min. | ||||
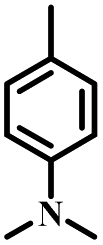
| DMA-PIM |
|
N | O2, UV (365 nm) | Yes |
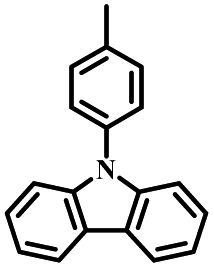
| CZ-PIM |
|
N | O2, UV (330 nm, 365 nm) | Not observed |
| p-CZ-PIM |
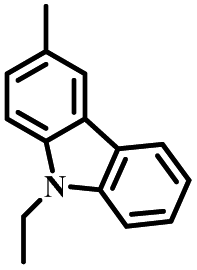
|
p-C | O2, UV (365 nm) | Yes |
| DMAC-PIM |
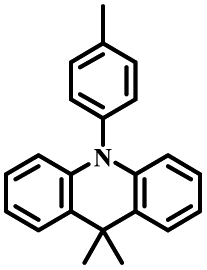
|
N | O2, UV (365 nm) | Not observed |
| p-DMAC-PIM |
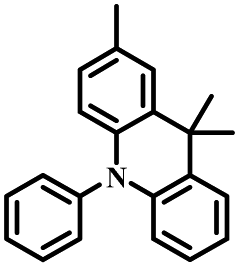
|
p-C | O2, UV (365 nm) | Yes |
| POZ-PIM |
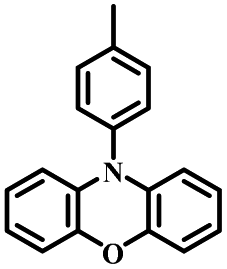
|
N | O2, UV (365 nm) | Not observed |
| p-POZ-PIM |
|
p-C | O2, UV (365 nm) | Yes |
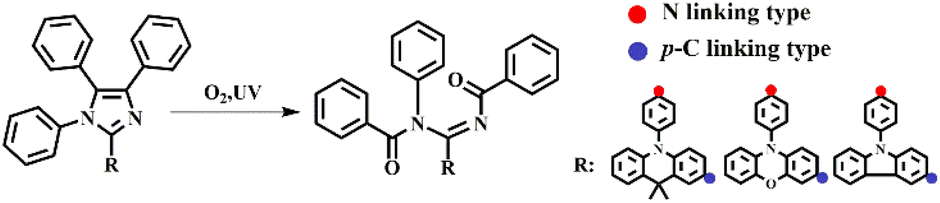
Therefore, it can be concluded that this photo-oxidation reaction occurs in PIM derivatives only when the R1 position of PIM is substituted with arylamine groups whose linking type plays a key role. Moreover, this photo-oxidation reaction needs the indispensable participation of UV irradiation and oxygen, but without any other sensitizers (singlet oxygen) or basic condition. It is worth mentioning that this photo-oxidation reaction occurs with a very rapid reaction rate, which can be reflected by the complete conversion of reactants within 90 minutes in solutions as well as the impossibility to obtain their intrinsic PL spectra in the air (a new redshifted PL peak emerged around 550 nm corresponding to that of the photo-oxidation reaction product). These characters make us believe that the photo-oxidation reaction observed in the cases of PIM derivatives is of a new type, which is different from the previous similar oxidation reaction of lophine derivatives reported in the literature, though the same terminal product of ammonia is obtained.2–8 Therefore, we carried out further studies of the photo-oxidation reaction to try to reveal the reaction mechanism.
Mechanism of the photo-oxidation reaction
Referring to the photo-oxidation reaction, the reactive oxygen species such as singlet oxygen (1O2) and superoxide anion radicals (−˙O2) should be the key factors to promote the occurrence of the reaction. Therefore, as shown in Fig. 1, electron spin resonance (ESR) was adopted first to study this photo-oxidation in the cases of DPA-PIM, p-DMAC-PIM, p-CZ-PIM, and p-POZ-PIM while using unreacted PIM, DMAC-PIM, CZ-PIM, and POZ-PIM respectively as reference. During the test, 2,2,6,6-tetramethylpiperidinooxy (TEMPO) and 5,5-dimethyl-1-pyrroline N-oxide (DMPO) were adopted as spin trapping agents for singlet oxygen24,25 and superoxide anion radicals26,27 respectively. The featured quartet signals of superoxide anion radicals (Fig. 1a–d) and the triplet 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 peak of singlet oxygen (Fig. 1e–h) demonstrate that both superoxide anion radicals and singlet oxygen have been detected in all the eight PIM derivatives. But they differ from each other in signal intensity, which could reflect the quantity of singlet oxygen or superoxide anion radicals based on the controlled identical measurement condition. From the ESR data, it is obvious that in the cases of DPA-PIM, p-DMAC-PIM, p-CZ-PIM, and p-POZ-PIM capable of undergoing the photo-oxidation reaction, there exist large amounts of superoxide anion radicals in them, while for singlet oxygen, the situation seems to be the inverse. Large amounts of singlet oxygen are detected in the cases of PIM, DMAC-PIM, CZ-PIM, and POZ-PIM that are unable to observe the photo-oxidation reaction, and the detected singlet oxygen amounts in DPA-PIM, p-DMAC-PIM, p-CZ-PIM, and p-POZ-PIM are relatively less in quantity. However, in our previous report,16 singlet oxygen had been verified to be the determinate oxidant for this photo-oxidation reaction rather than superoxide anion radicals, because the addition of a singlet oxygen quencher28 would terminate the oxidation reaction effectively and a similar phenomenon was not observed by adding a trapping agent of superoxide anion radical. A reasonable explanation for the less quantity of detected singlet oxygen in cases of DPA-PIM, p-DMAC-PIM, p-CZ-PIM, and p-POZ-PIM is that most of the singlet oxygen has been consumed during the photo-oxidation reaction. Consequently, a large confusion is that in the cases of PIM, DMAC-PIM, CZ-PIM, and POZ-PIM, large amounts of singlet oxygen have been detected, but the photo-oxidation reaction has not been observed. This makes us confirm that the observed photo-oxidation reaction induced by the singlet oxygen in our cases does not occur at the ground-state molecule of imidazole derivatives, but at some other intermediate state of imidazole molecules.
:1:1 peak of singlet oxygen (Fig. 1e–h) demonstrate that both superoxide anion radicals and singlet oxygen have been detected in all the eight PIM derivatives. But they differ from each other in signal intensity, which could reflect the quantity of singlet oxygen or superoxide anion radicals based on the controlled identical measurement condition. From the ESR data, it is obvious that in the cases of DPA-PIM, p-DMAC-PIM, p-CZ-PIM, and p-POZ-PIM capable of undergoing the photo-oxidation reaction, there exist large amounts of superoxide anion radicals in them, while for singlet oxygen, the situation seems to be the inverse. Large amounts of singlet oxygen are detected in the cases of PIM, DMAC-PIM, CZ-PIM, and POZ-PIM that are unable to observe the photo-oxidation reaction, and the detected singlet oxygen amounts in DPA-PIM, p-DMAC-PIM, p-CZ-PIM, and p-POZ-PIM are relatively less in quantity. However, in our previous report,16 singlet oxygen had been verified to be the determinate oxidant for this photo-oxidation reaction rather than superoxide anion radicals, because the addition of a singlet oxygen quencher28 would terminate the oxidation reaction effectively and a similar phenomenon was not observed by adding a trapping agent of superoxide anion radical. A reasonable explanation for the less quantity of detected singlet oxygen in cases of DPA-PIM, p-DMAC-PIM, p-CZ-PIM, and p-POZ-PIM is that most of the singlet oxygen has been consumed during the photo-oxidation reaction. Consequently, a large confusion is that in the cases of PIM, DMAC-PIM, CZ-PIM, and POZ-PIM, large amounts of singlet oxygen have been detected, but the photo-oxidation reaction has not been observed. This makes us confirm that the observed photo-oxidation reaction induced by the singlet oxygen in our cases does not occur at the ground-state molecule of imidazole derivatives, but at some other intermediate state of imidazole molecules.
 | ||
| Fig. 1 ESR spectra of 100 μl 0.5 mg ml−1 (a) PIM and DPA-PIM, (b) DMAC-PIM and p-DMAC-PIM, (c) CZ-PIM and p-CZ-PIM and (d) POZ-PIM and p-POZ-PIM solutions in toluene with 10 ppm TEMPO and (e) PIM and DPA-PIM, (f) DMAC-PIM and p-DMAC-PIM, (g) CZ-PIM and p-CZ-PIM and (h) POZ-PIM and p-POZ-PIM solutions in toluene with 10 ppm DMPO after 365 nm UV irradiation for 10 min. | ||
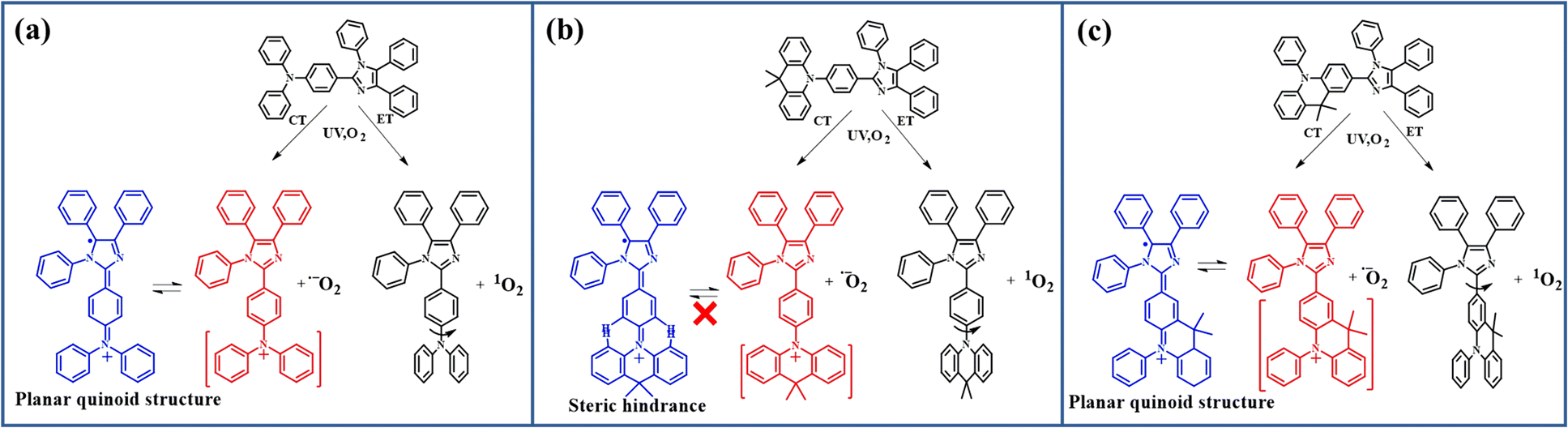
Commonly, singlet oxygen is generated by an energy transfer (ET) process from excited-state molecules to ground-state triplet oxygen,29 while the superoxide anion radical is usually generated by a charge transfer (CT) process from the excited-state molecule to ground-state triplet oxygen. The introduction of the donor group into PIM derivatives will facilitate the CT process from the excited-state molecule to ground-state triplet oxygen owing to its ease to lose an electron from the donor group. Besides, it can help generate a donor–acceptor structure, leading to an intra-molecular CT transition, which further promotes the intersystem crossing from singlet to triplet excited states and benefits the generation of more singlet oxygen. As we learned, the generation of one superoxide anion radical should accompany the production of an oxidation-state PIM molecule at the same time.30 There are large amounts of superoxide anion radicals detected in the above-mentioned PIM derivatives that are capable of undergoing the photo-oxidation reaction. It inspired us to believe that there were also many oxidation-state PIM derivatives generated in them. Provided that the observed photo-oxidation reaction in our cases does not occur between the singlet oxygen and their ground-state molecule, it would be only attributed to one occurring between the singlet oxygen and oxidation-state structures of PIM derivatives.
However, there is another fact that the different linking types of arylamine donor groups also influence the photo-oxidation reaction. This makes us come to a further thought that this rapid photo-oxidation reaction requires a special oxidation-state structure, in which the imidazole ring tends to be easily attacked by singlet oxygen. For CZ-PIM, DMAC-PIM, and POZ-PIM, the N atom in the donor group is directly connected to the acceptor group PIM, leading to a relatively stable twisted donor–acceptor structure in their molecular configurations due to the strong steric hindrance. While for the p-CZ-PIM, p-DMAC-PIM, and p-POZ-PIM, the carbon atom in the para-position of the donor group is directly linked with the imidazole ring, resulting in a smaller twist angle or close to planar D–A structure in their molecular configurations. The largest difference between the above-mentioned six molecules should be the molecular configurations deriving from the twist angle difference between the acceptor group imidazole ring and donor group arylamine. This makes us focus on the effect of twist angle on the relative properties of these molecules in the following studies.
Actually, as shown in Fig. 2, there are two possible reaction routes for PIM derivatives with O2 under UV irradiation. One is that compounds get to the excited state by UV irradiation and then transfer the energy to ground-state triplet oxygen with singlet oxygen generated by the ET process. The other one is that oxygen gains electrons from the excited-state compounds by CT to produce superoxide anion radicals, and the compounds form the corresponding oxidation-state radical after losing an electron. However, the different linking types for the PIM derivatives will produce different oxidation-state structures. As shown in Fig. 2b, S3a and S3c,† the spatially adjacent hydrogen atoms in the cases of twisted donor–acceptor molecules DMAC-PIM, CZ-PIM, and POZ-PIM will produce a large steric hindrance between the PIM and the conjugated planes of corresponding donor groups 9,9-dimethyl-9,10-dihydroacridine, carbazole and phenoxazine respectively. This linking type makes it difficult to form a free radical at the imidazole ring. By contrast, as for p-CZ-PIM, p-DMC-PIM, and p-POZ-PIM as well as DPA-PIM, there are no big steric hindrances between the donor group and imidazole ring, as shown in Fig. 2c, S3b and S3d.† Once the CT process from their molecules to oxygen occurs with a cation radical formed at the donor part in the oxidation-state molecules, the relatively flexible arylamine groups could assist them to easily undergo the structural rearrangement and form a planar quinoid oxidation-state structure, in which a free radical of carbon could be generated at the imidazole ring, and then the singlet oxygen can easily attack this free radical to promote the photo-oxidation reaction. This assumption can also explain the large amounts of superoxide anion radicals present in DPA-PIM, p-DMAC-PIM, p-CZ-PIM, and p-POZ-PIM (Fig. 1): the formation of planar quinoid oxidation-state structure is in favor of the CT process.
 | ||
| Fig. 2 Possible conformation change of (a) DPA-PIM (b) DMAC-PIM and (c) p-DMAC-PIM under UV irradiation in air. | ||
In order to prove the quinoid oxidation-state structure formed when losing electrons in DPA-PIM and p-C linking D–A PIM derivatives, their oxidation-state properties were studied by the spectroelectrochemistry method (the UV-vis absorption spectra under different applied electrochemistry potentials). First, their electrochemical properties were studied. As shown in Fig. S4a,†PIM shows an oxidation process with an onset potential of 1.23 V. Compared to PIM, the lower oxidation onset potentials were observed for derivatives at 1.18 V, 0.94 V, 0.89 V, 0.86 V, 0.75 V, 0.68 V and 0.66 V for CZ-PIM, p-CZ-PIM, DPA-PIM, DMAC-PIM, p-DMAC-PIM, POZ-PIM, and p-POZ-PIM, respectively. The changing rules of these redox potentials are in accordance with their corresponding donors, presenting a decreasing trend with the increase in electron-donating ability. From Fig. S4b–d,† it could be known the p-C linking derivatives having a similar oxidation onset potentials to their N-linking analogs. The electrochemistry results indicate the first redox of these D–A molecules should happen mainly at the donor part rather than the PIM part.
When a positive potential is applied to reach their oxidation state, the PIM derivatives with different molecular structures would exhibit different UV absorption spectra owing to the oxidation-state structural difference. As shown in Fig. 3, the initial oxidation potential of PIM was 1.23 V, but there was no change in the UV-vis absorption spectra for its film until the voltage of 1.50 V, indicating that it is difficult to form an observable oxidation state. As for DPA-PIM, when a voltage of 1.20 V was applied, there were two new peaks ranging from 400 to 650 nm in blue gridlines and 650 to 1000 nm in red gridlines respectively, which emerged in the absorption spectra. These two new absorption peaks should be attributed to different structural changes of the diphenylamine group under applied potentials according to the literature.31–33
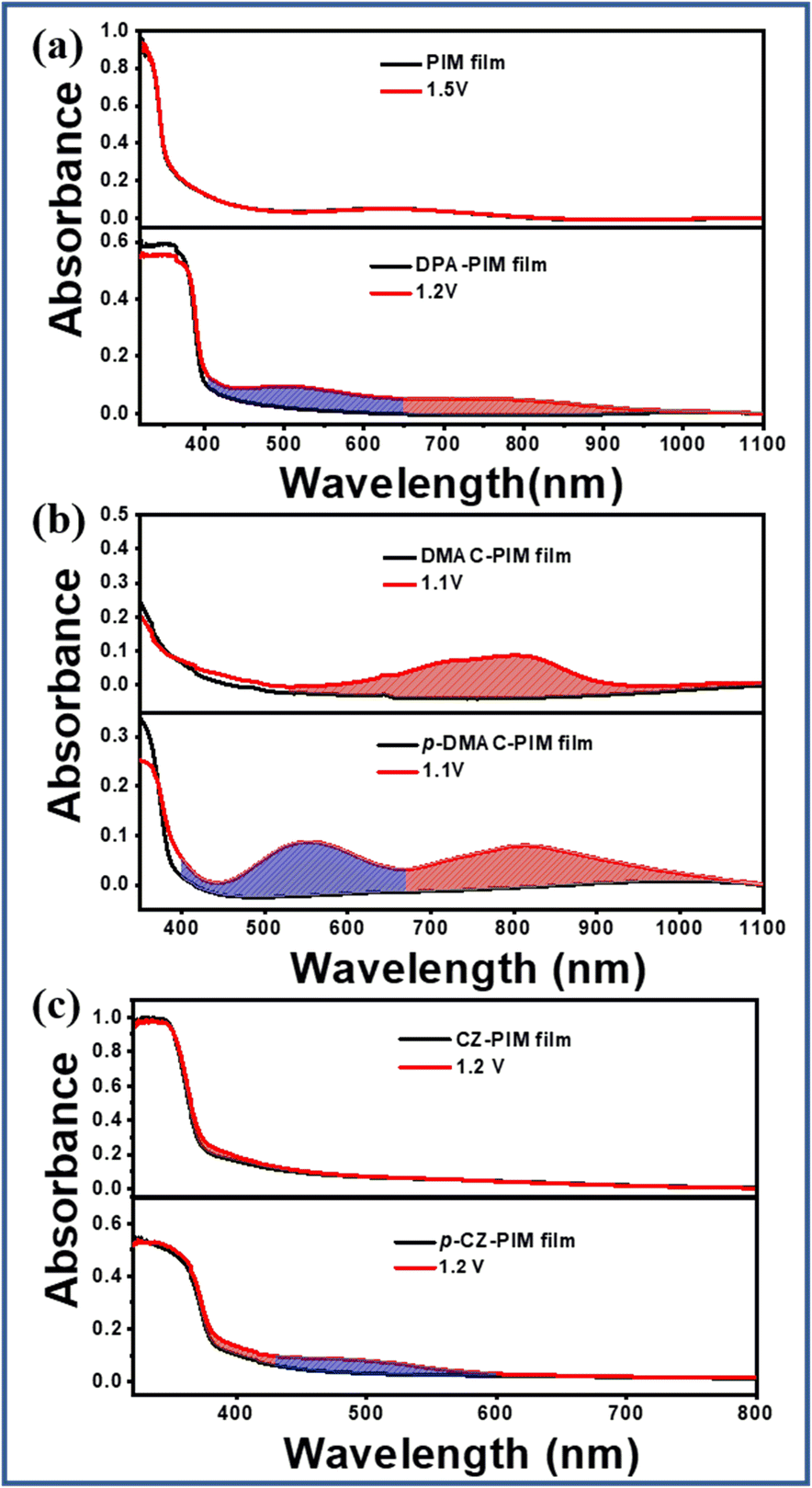 | ||
| Fig. 3 Absorption spectra of (a) PIM and DPA-PIM (b) DMAC-PIM and p-DMAC-PIM (c) CZ-PIM and p-CZ-PIM film before and after applying voltage (p-POZ-PIM was not stable under applied potentials and its related data was not given). | ||
In the cases of DMAC-PIM vs. p-DMAC-PIM, and CZ-PIM vs. p-CZ-PIM films, as shown in Fig. 3b and c, their UV absorption spectra under different potentials are also different. Except for the similar absorption peak in red gridlines around 800 nm for DMAC-PIM and p-DMAC-PIM, and 400 nm for CZ-PIM and p-CZ-PIM, there were new absorption bands ranging from 400 to 660 nm for p-DMAC-PIM and 420 to 600 nm for p-CZ-PIM (blue gridlines) that could not be observed for DMAC-PIM and CZ-PIM respectively. The newly emerged special absorption bands (blue gridlines) in p-C linking-type PIM derivatives should be attributed to their planar quinoid oxidation-state structure. Thus, it can be speculated that, in the case of DPA-PIM, the shorter-wavelength absorption band (blue gridlines, Fig. 3a) should also be attributed to the planar quinoid oxidation-state structure. In addition, such quinoid structures in a film state can be maintained for a long time even after removing voltage, as shown in Fig. S5.† This long lifetime of the quinoid structure is in favor of the attack of singlet oxygen. The above-mentioned results indicated that the p-C linking type of the donor group can help PIM derivatives to form the planar quinoid oxidation-state structure in comparison to the N-linking type, which is helpful to the occurrence of photo-oxidation reactions.
From the experimental results, the photo-oxidation reaction mechanism of PIM derivatives was proposed by taking DPA-PIM as the example, as shown in Scheme 3. Under UV irradiation, DPA-PIM donates an electron to O2 by a CT process, which produces the superoxide anion radical and generates the planar oxidation state radical (I), with a carbon radical formed in the imidazole ring and an aminium structure of a positive charge in the arylamine part. At the same time, singlet oxygen can also be generated by the ET process from excited-state DPA-PIM. The singlet oxygen quickly attacks the carbon radical to produce the peroxide species (II), which further attacks the adjacent carbon atom of the vinyl bond to form the imine hydroperoxide intermediate (III). Then, the intermediate (III) undergoes bond breaking (or decomposition) and recombines with another electron (or radical elimination) to eventually convert into the terminal benzoylimino-benzamide compound DPA-PYZ. In this process, the planar quinoid oxidation-state structure is supposed to be the dominant and most important stage, which contributes much to the rapid reaction rate.
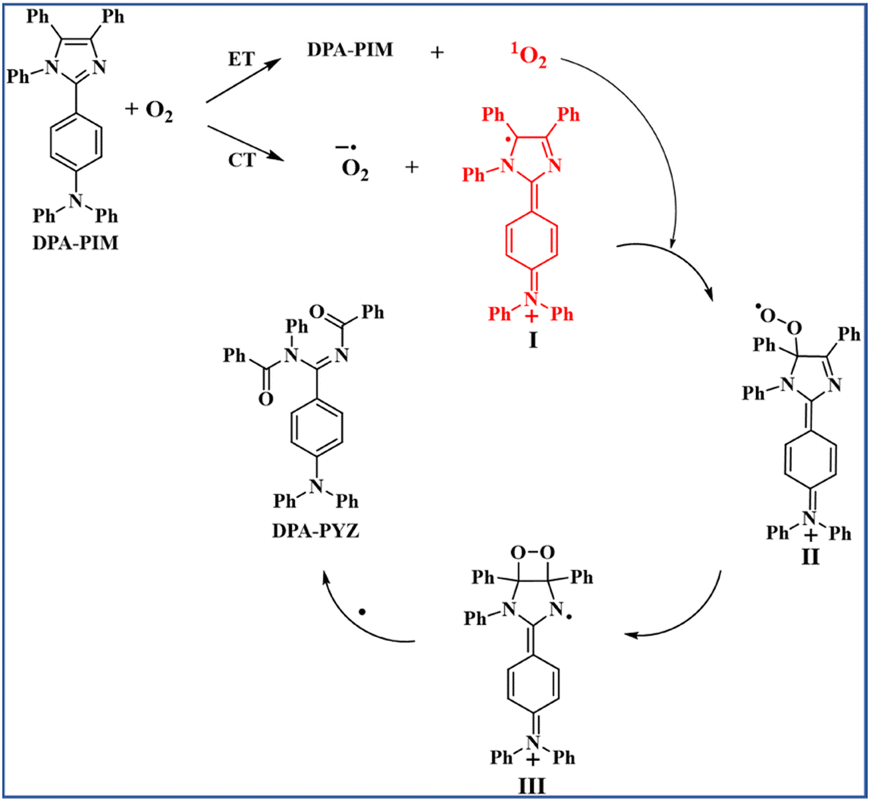 | ||
| Scheme 3 Proposed mechanism for the p-C linking PIM derivative photo-oxidation reaction (with DPA-PIM as an example). | ||
The imine hydroperoxide intermediate (III) is another key stage in this photo-oxidation reaction. The similar decomposition procedure of the intermediate imine hydroperoxides is well known in the mechanism of chemiluminescence of lophine derivatives proposed by Emil H. White etc. in 1965.6 It is worth noting that all the currently observed experimental facts are in good accordance with the mechanism we proposed here for this photo-oxidation reaction. In addition, this mechanism applied equally to the PIM derivatives whose linking way of arylamine donor group is p-C type.
Oxygen sensor
Based on the rapid reaction rate, these PIM derivatives with photo-oxidation activity can be used as fluorescent probes for oxygen detectors. Compared to DPA-PIM, p-DMAC-PIM had higher oxygen sensitivity for its lower oxidation onset potentials and more planar structure tending to form the quinoid oxidation-state structure. As shown in Fig. 4a and b, the p-DMAC-PIM film displayed totally different PL spectra in different O2 concentrations. In addition, the lowest detectable oxygen volume ratio was about 0.2%, which is among the best results and has great potential in applications of trace oxygen detection compared to most of the oxygen sensors whose sensitivities ranged from 0.1% to 5%.20 Moreover, the p-DMAC-PIM film can be used as the fluorescent probe material to detect oxygen in an aqueous solution. As shown in Fig. 4d, e and g, the p-DMAC-PIM film in H2O exhibited a rapid fluorescence change from dark blue to green under UV irradiation (6 W, 365 nm) within 1 minute. In addition, as shown in Fig. 4c and f, the ratios of fluorescence intensities of the oxidation-reaction product and p-DMAC-PIM increased more and more slowly due to the concentration of reactants (oxygen and lophine derivatives respectively) reducing. Similar phenomena can also be observed in the other p-C linking derivatives, as shown in Fig. S6–S9.† Owing to these advantages, these materials show great potential in applications such as in vivo reactive oxygen detection,34 fire alarms,35 and water quality monitoring areas.36 We believe that according to the photo-oxidation mechanism we proposed, by structure modification (donor group in special), on the one hand, we can increase the oxygen sensitivity of this kind of material further, and on the other hand, we can adjust the fluorescence color before and after photo-oxidation to match more application situations.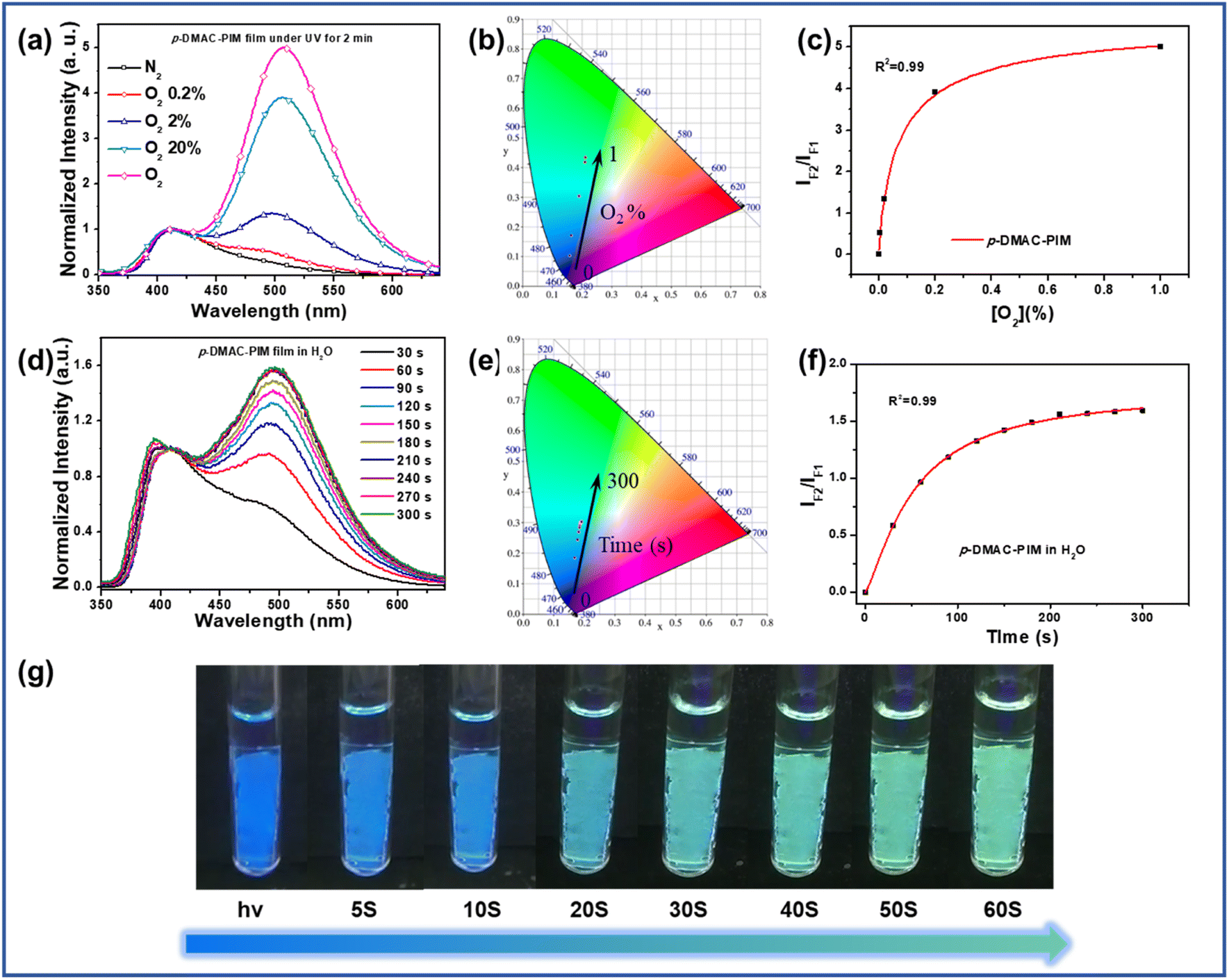 | ||
| Fig. 4 (a) Normalized PL spectra, (b) CIE colorimetry, (c) plot of IF2/IF1 against [O2] of the p-DMAC-PIM film in the gas mixture of N2 and O2 (different volume ratios). (d) Normalized PL spectra, (e) CIE colorimetry, (f) plot of IF2/IF1 against [O2], and (g) photograph of the p-DMAC-PIM film in H2O after UV irradiation (365 nm, 6 W) for different time periods. | ||
Conclusions
In summary, a series of PIM derivatives have been synthesized to study a new kind of photo-oxidation reaction. In situ UV-vis absorption spectra indicated that the substituted group of R1 position of PIM derivatives has a significant impact on this photo-oxidation reaction. Moreover, for the R1 position, lophine derivatives with the p-C linking type of arylamine groups such as p-CZ-PIM, p-DMAC-PIM and p-POZ-PIM can undergo the photo-oxidation reaction under UV irradiation. In comparison, when the N-linking type of the corresponding arylamine group is adopted, the photostability of derivatives CZ-PIM, DMAC-PIM and POZ-PIM can be maintained. ESR and electrochemical measurements indicate that the arylamine group in the p-C linking type will help lophine derivatives to form a rearranged stable planar quinoid oxidation-state structure under UV irradiation, which tends to be easily attacked by self-sensitized singlet oxygen. The quinoid oxidation-state structure contributes much to the rapid reaction rate of this photo-oxidation reaction. Thus, the mechanism for this kind of photo-oxidation reaction was tentatively put forward based on two main intermediates: the newly discovered planar quinoid oxidation-state structure and the well-known 1,2-dioxetane-like intermediate. It is believed that this finding can deepen the knowledge of lophine derivatives and highly contribute to the photoelectronic investigations of imidazole-based materials. Furthermore, via the accurate structural design of specific lophine derivatives according to the proposed mechanism, high-sensitivity oxygen sensor materials with multicolors have been obtained, which show great potential in the application of in vivo reactive oxygen detection areas.Experiment
Spin trapping-ESR tests
First, 0.5 mg ml−1 toluene solution of the sample was dispersed ultrasonically for 30 min, and 100 μl of the obtained solution was mixed with 10 ppm 2,2,6,6-tetramethylpiperidinooxy (TEMPO). After illumination for 10 min (365 nm), the mixture was characterized using a Bruker A300 ESR spectrometer. The superoxide anion radical O2˙− trapping-ESR tests were also performed as described above, except for the use of 5,5-dimethyl-1-pyproline N-oxide (DMPO) as the spin-trapping agent. The feature of the signal about singlet oxygen was a triplet 1:1:1 peak with a nearly equal intensity. The description of the featured triplet signals of singlet oxygen could be found in many other works reported in the literature.24,25 As for the superoxide anion radical, the feature of the signal is a quartet peak, whose relative peak intensity differs from that of hydroxyl radicals (the quartet 1:2:2:1 peak) and may vary little in the relative intensity of peaks in different testing environments. The description of featured quartet signals of superoxide anion radicals could also be found in many studies reported in the literature.26,27
Electrochemical testing of PIM derivatives
The UV-vis absorption spectra under different applied electrochemistry potentials were recorded on a CHI760D electrochemical workstation in a three-electrode system. PIM derivative spin-coating films on ITO were prepared as the working electrode. A 0.1 M solution of lithium perchlorate in dry H2O (for oxidation) avoiding dissolving film was used as the supporting electrolyte, a titanium (99.99%) plate was used as the counter electrode, and the voltage was kept constant during testing. The oxidation potentials were analysed on a CHI760D electrochemical workstation. The measurements used a glassy carbon working electrode and a Pt wire counter electrode at a scanning rate of 50 mV s−1 against an Ag/Ag+ (0.01 M of AgNO3 in acetonitrile) reference electrode in a nitrogen-saturated anhydrous acetonitrile and dichloromethane (DCM) solution of 0.1 mol l−1 Bu4NPF6 as the electrolyte.Oxygen detector
The ratio of N2 to O2 was controlled by the speed of gas flow respectively, which was adjusted using a flowmeter. N2 and O2 were thoroughly mixed until stable in a big container before measurement. Then, the speed of gas flow for N2 and O2 was controlled to be 500/1, 100/2, and 5/1 ml min−1 respectively to get the ratio of about 0.2%, 2%, and 20%. The film of the sample was prepared by spin coating 5 mg ml−1 dichloromethane solution, the spin speed was 1000 rpm, and the spin time was 60 seconds. PL spectra measurement for oxygen detection was carried out using a HORIBA Fluorolog-3 instrument.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Our works are grateful for support from the National Natural Science Foundation of China (52103232, 51603185), the National Key R&D Program of China (2020YFA0714604) and the Zhejiang Provincial Natural Science Foundation of China (LY19E030006 and LQ19E030016). This work was also supported by the Open Fund of the State Key Laboratory of Luminescent Materials and Devices (South China University of Technology).Notes and references
- W. Miao, Chem. Rev., 2008, 108, 2506 CrossRef CAS PubMed.
- B. R. Radziszewski, Ber. Dtsch. Chem. Ges., 1877, 10, 70–75 CrossRef.
- T. Hayashi and K. Maeda, Bull. Chem. Soc. Jpn., 1962, 35, 2057 CrossRef CAS.
- E. H. White and M. J. C. Harding, J. Am. Chem. Soc., 1964, 86, 5686 CrossRef CAS.
- J. Sonnenberg and D. M. White, J. Am. Chem. Soc., 1964, 86, 5685 CrossRef CAS.
- E. H. White and M. J. C. Harding, Photochem. Photobiol., 1965, 4, 1129 CrossRef CAS.
- P. Kang and C. S. Foote, J. Am. Chem. Soc., 2002, 124, 9629 CrossRef CAS PubMed.
- H. H. Wasserman, K. Stiller and M. B. Floyd, Tetrahedron Lett., 1968, 9, 3277 CrossRef.
- W. Li, L. Yao, H. Liu, Z. Wang, S. Zhang, R. Xiao, H. Zhang, P. Lu, B. Yang and Y. Ma, J. Mater. Chem. C, 2014, 2, 4733 RSC.
- S. Karmakar, S. Mardanya, S. Das and S. Baitalik, J. Phys. Chem. C, 2015, 119, 6793 CrossRef CAS.
- Y. Kwon, S. H. Han, S. Yu, J. Y. Lee and K. M. Lee, J. Mater. Chem. C, 2018, 6, 4565 RSC.
- K. Udagawa, H. Sasabe, F. Igarashi and J. Kido, Adv. Opt. Mater., 2016, 4, 86 CrossRef CAS.
- K. Udagawa, H. Sasabe, C. Cai and J. Kido, Adv. Mater., 2014, 26, 5062 CrossRef CAS PubMed.
- S. Xiao, Y. Gao, R. Wang, H. Liu, W. Li, C. Zhou, S. Xue, S. Zhang, B. Yang and Y. Ma, Chem. Eng. J., 2022, 440, 135911 CrossRef CAS.
- C. Du, T. Lu, Z. Cheng, Y. Chang, H. Liu, J. Wang, L. Wan, Y. Lv and P. Lu, J. Mater. Chem. C, 2022, 10, 14186 RSC.
- Y. Yu, R. Zhao, C. Zhou, X. Sun, S. Wang, Y. Gao, W. Li, P. Lu, B. Yang and C. Zhang, Chem. Commun., 2019, 55, 977 RSC.
- Y. Yu, J. Chen, S. Tan, C. Zhou, W. Li, Y. Dong and C. Zhang, J. Mater. Chem. C, 2021, 9, 888 RSC.
- J. Liu, J. Chen, Y. Dong, Y. Yu, S. Zhang, J. Wang, Q. Song, W. Li and C. Zhang, Mater. Chem. Front., 2020, 4, 1411 RSC.
- J. Chen, X. Guo, Y. Yu, C. Liu, W. Li, S. Zhang, J. Liu, Y. Dong, C. Zhang and W. Wong, J. Lumin., 2022, 244, 118690 CrossRef CAS.
- X. Wang and O. S. Wolfbeis, Chem. Soc. Rev., 2014, 43, 3666 RSC.
- R. Ruiz-González, R. Bresolí-Obach, Ò. Gulías, M. Agut, H. Savoie, R. W. Boyle, S. Nonell and F. Giuntini, Angew. Chem., Int. Ed., 2017, 56, 2885 CrossRef PubMed.
- P. Lehner, C. Staudinger, S. M. Borisov and I. Klimant, Nat. Commun., 2014, 5, 4460 CrossRef CAS PubMed.
- H. Liu, G. Pan, Z. Yang, Y. Wen, X. Zhang, S. Zhang, W. Li and B. Yang, Adv. Opt. Mater., 2022, 10, 2102814 CrossRef CAS.
- J. Ge, M. Lan, B. Zhou, W. Liu, L. Guo, H. Wang, Q. Jia, G. Niu, X. Huang, H. Zhou, X. Meng, P. Wang, C. Lee, W. Zhang and X. Han, Nat. Commun., 2014, 5, 4596 CrossRef CAS PubMed.
- H. Wang, X. Yang, W. Shao, S. Chen, J. Xie, X. Zhang, J. Wang and Y. Xie, J. Am. Chem. Soc., 2015, 137, 11376 CrossRef CAS PubMed.
- C. Xia, R. Fernandes, F. H. Cho, N. Sudhakar, B. Buonacorsi, S. Walker, M. Xu, J. Baugh and L. F. Nazar, J. Am. Chem. Soc., 2016, 138, 11219 CrossRef CAS PubMed.
- Y. Xu, Y. Chen and W. F. Fu, Appl. Catal., B, 2018, 236, 176 CrossRef CAS.
- E. Baciocchi, T. D. Giacco, F. Elisei, M. F. Gerini, M. Guerra, A. Lapi and P. Liberali, J. Am. Chem. Soc., 2003, 125, 16444 CrossRef CAS PubMed.
- A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994 CrossRef CAS PubMed.
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029 CrossRef CAS PubMed.
- G. S. Liou, S. H. Hsiao and T. H. Su, J. Mater. Chem., 2005, 15, 1812 RSC.
- H. M. Wang, S. H. Hsiao, G. S. Liou and C. H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4775 CrossRef CAS.
- J. H. Wu and G. S. Liou, Adv. Funct. Mater., 2014, 24, 6422 CrossRef CAS.
- K. Kundu, S. F. Knight, N. Willett, S. Lee, W. R. Taylor and N. Murthy, Angew. Chem., Int. Ed., 2009, 48, 299 CrossRef CAS PubMed.
- M. Zhang, M. Wang, M. Zhang, C. Yang, Y. Li, Y. Zhang, J. Hu and G. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 47456 CrossRef CAS PubMed.
- W. T. Wallace, D. B. Gazda, T. F. Limero, J. M. Minton, A. V. Macatangay, P. Dwivedi and F. M. Fernández, Anal. Chem., 2015, 87, 5981 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta09587d |
| This journal is © The Royal Society of Chemistry 2023 |