
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlow synthesis of hypercrosslinked polymers with additional microporosity that enhances CO2/N2 separation†
Nadhita
Chanchaona
 a,
Liang
Ding
a,
Shiliang
Lin
a,
Sulaiman
Sarwar
a,
Simone
Dimartino
a,
Ashleigh J.
Fletcher
b,
Daniel M.
Dawson
c,
Kristina
Konstas
d,
Matthew R.
Hill
d and
Cher Hon
Lau
*a
a,
Liang
Ding
a,
Shiliang
Lin
a,
Sulaiman
Sarwar
a,
Simone
Dimartino
a,
Ashleigh J.
Fletcher
b,
Daniel M.
Dawson
c,
Kristina
Konstas
d,
Matthew R.
Hill
d and
Cher Hon
Lau
*a
aSchool of Engineering, University of Edinburgh, Robert Stevenson Road, EH9 3FK, UK. E-mail: cherhon.lau@ed.ac.uk
bDepartment of Chemical and Process Engineering, University of Strathclyde, 75 Montrose Street, Glasgow, G1 1XJ, UK
cSchool of Chemistry, EaStCHEM, Centre of Magnetic Resonance, University of St. Andrews, KY16 9ST, UK
dCSIRO Manufacturing Flagship, Private Bag 10, Clayton South, Melbourne, Victoria 3169, Australia
First published on 27th January 2023
Abstract
Hypercrosslinked polymers (HCPs) are typically synthesised over 24 hour batch reactions, limiting productivity rates during scale-up production. Continuous flow synthesis can potentially overcome this limitation. However, the formation of insoluble HCP products, compounded by HCP expansion due to solvent adsorption during synthesis can clog flow reactors. Here, we overcome clogging issues through reactor design and optimisation of synthesis parameters. Using this reactor, we synthesised HCPs via internal, post-, and external crosslinking strategies underpinned by Friedel–Crafts alkylation over various synthesis parameters – residence time, substrate concentration, reagent ratio, and temperature. The space-time-yield (STY) values, a key parameter for productivity rates, of flow synthesis were 32–117 fold higher than those in batch reactions. HCPs produced via internal crosslinking in flow synthesis contained additional microporosity that enhanced CO2/N2 selectivity at 298 K by 850% when compared to HCPs produced in batch reactions. Outcomes from this work could enable high productivity scale-up production of HCPs for post-carbon capture.
Introduction
Hypercrosslinked polymers (HCPs) are excessively crosslinked, amorphous polymers typically synthesised via Friedel–Crafts alkylation (Fig. 1). In this reaction, aromatic protons are replaced with methylene bridges that are linked to an adjacent aromatic molecule. This is facilitated by an electrophilic attack on the aromatic ring by a carbocation. This can be achieved by post-, internal, and external crosslinking.1 Post-crosslinking uses halogenated crosslinkers to link up the chains of polymers such as polystyrene or polystyrene-co-divinylbenzene. Internal crosslinking is akin to a condensation reaction in which halogenated compounds are crosslinked. External crosslinking exploits formaldehyde dimethyl acetal (FDA) as the crosslinker to link up simple aromatic compounds.2 During the crosslinking process, solvent molecules pre-occupy the free spaces between the newly linked up polymer chains. Removing solvent molecules from the product by drying leads to the formation of porous architectures that underpin Brunauer–Emmett–Teller (BET) surface areas as high as 2000 m2 g−1.3 As such, HCPs are widely deployed in the industry as ion-exchange resins4 such as Purolite's Macronet™,5 DowDupont Inc.'s Amberlite™, and Lanxess AG's Lewatit® series, and potentially for gas storage and separations.6,7 The global market value of HCPs in 2020 was USD$1.4 billion and is expected to reach USD$2 billion by 2028.8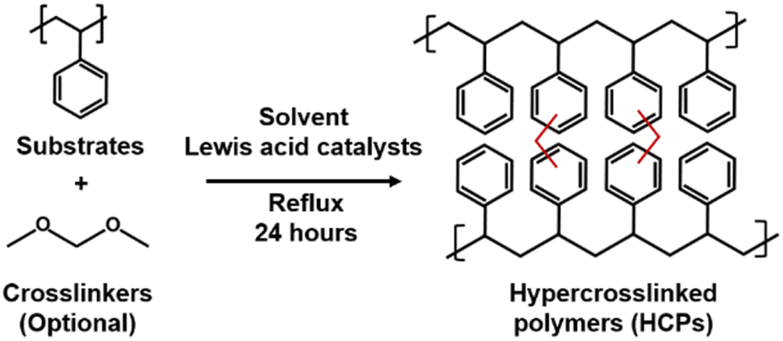 | ||
| Fig. 1 Generic Friedel–Crafts alkylation for HCP synthesis. | ||
Most HCPs are produced via batch reactions over 18–24 hours9 to ensure extensive crosslinking10 and this may be inadequate to produce enough HCPs to meet the growing global demand. In the first hour of batch reactions, more than 80% of the reagents are used up in cross-linking. The pores of the initial polymer will adsorb solvent molecules, creating a gel-like structure. More solvent is then added to disperse the initially-formed polymers, breaking up the gel-like material to ensure complete crosslinking.
Most studies in this field are focused on developing new HCP materials with unprecedented adsorption capacity,11,12 identifying alternative reagents (solvents, monomers) for the sustainable synthesis of HCPs,13 and improving batch productivity rates.14 For example, low HCP productivity rates production can be improved by reducing synthesis time from 24 hours to 5–35 minutes via solvent-free mechanochemical ball milling and liquid-assisted grinding.15 This significant reduction in synthesis duration does not impact HCP quality, with specific surface areas reaching 625–1720 m2 g−1. Another technique suitable for improving the productivity of HCP production is flow synthesis.
The key benefit of replacing batch reactions with flow synthesis is improving productivity rates. To date, flow chemistry had been used for synthesising microporous materials, such as polymers of intrinsic microporosity (PIM),16 metal–organic frameworks (MOFs),17 covalent organic frameworks (COFs),18 and HCPs.10 Flow syntheses of MOFs and COFs enhanced the space-time yield (STY) – a key indicator of productivity rates, comprising product yield, operating time, and reactor volume,17 by 30 fold,19 and 94 fold,18 respectively. These significant STY improvements were attributed to a reduced production time. Meanwhile, Fritsch deployed continuous flow reactions to reduce the PIM synthesis time by 90%.16 The first reported example of flow synthesis of HCPs was reported in 2000 when Vincent and co-workers reported using a tubular reactor to control the particle size of HCPs formed via post-crosslinking during suspension polymerisation.20 However, this work mainly focused on narrowing the particle size distribution. Our group previously deployed flow synthesis to produce HCPs via the external crosslinking method.10 A key limitation of this proof-of-concept was reactor clogging by the newly formed insoluble HCP products that expanded upon solvent adsorption within the reactor.
In this work, we overcome reactor clogging by optimising reactor design and synthesis parameters – residence time, temperature, reagent concentration, and ratio for flow synthesis of HCPs via internal crosslinking of α,α′-dichloro-p-xylene (DCX), post- and external crosslinking of waste Styrofoam in a bespoke coil reactor. To demonstrate the effects of flow synthesis on HCP quality and productivity rates, we compared the specific surface areas, micropore volumes, and STY value of HCPs produced from the flow synthesis with batch reactions. Results from this work support the theory that flow synthesis could be used to scale the production of good-quality HCPs. This is different from existing approaches that rely on HCP functionalisation,21–23 utilisation of bespoke24 or novel25,26 monomers in HCP synthesis, and modified synthesis protocols.15 Our approach establishes a new method to tailor and enhance HCP CO2/N2 selectivity via reactor design and flow synthesis.
Experimental
Materials and equipment
Styrofoam or waste-expanded polystyrene (PS) was obtained from plastic waste collected at The University of Edinburgh. 1,2-Dichloroethane (DCE) and iron(III) chloride (FeCl3) were purchased from Alfa Aesar. Formaldehyde dimethyl acetal (FDA) and α,α′-dichloro-p-xylene (DCX) were purchased from Sigma-Aldrich. Methanol, chloroform, and acetone were purchased from Thermo Fisher Scientific. All chemicals were used as is.Polytetrafluoroethylene (PTFE) tubes with dimensions of 1/8 in × 0.063 in × 50 ft were purchased from Supelco Inc., while PTFE tube fittings (Y-junction connectors, female union connectors, ferrules, and nuts) were purchased from Nanjing Runze Fluid Control Equipment Co., Ltd. Polypropylene syringes were purchased from MB Fibreglass, while 21G needles were supplied from Becton, Dickinson, and Company. A tube holder was fabricated from polylactic acid (PLA) filaments using a Prusa i3 MK3S 3D printer. The design was based on two different shapes with 7 individual parts, a central ring, and holder wings, which can be hinged on to each other. A peristaltic dosing pump (SEKO Kronos 50) was used to pump the monomer solution as well as cleaning solvents after the operation. A syringe pump (Cole-Parmer® SP210iwz) was deployed to inject the catalyst solution into the reactor.
HCP synthesis in batch reactors
HCPs synthesised in batch reactors were used as control materials here. All batch syntheses were performed in a 250 mL three-neck round-bottom glass flask connected to a reflux condenser. HCPs were synthesised via:(i) Internal crosslinking – 2.884 g of DCX and 2.672 g of FeCl3 were dissolved in 100 mL DCE. The mixture was heated to 373 K (ref. 24) and continuously stirred with a magnetic stirrer for 24 hours.
(ii) Post-crosslinking – 0.385 g of PS, 0.385 g of FeCl3, and 0.385 g of DCX were first dissolved in 100 mL of DCE. The mixture was heated to 353 K (ref. 27) via an oil bath and continuously stirred with a magnetic stirrer for 24 hours.
(iii) External crosslinking – 0.762 g of PS, 3.810 g of FeCl3, and 3.810 g of FDA were dissolved in 100 mL of DCE. The mixture was heated to 353 K (ref. 28) via an oil bath and continuously stirred with a magnetic stirrer for 24 hours.
HCP synthesis in flow reactors
Fig. 2 and S1† show the experimental set-up of the flow reactor used in this work for HCP synthesis via post-, internal, and external crosslinking. The details of the synthesis via the flow system are described as follows: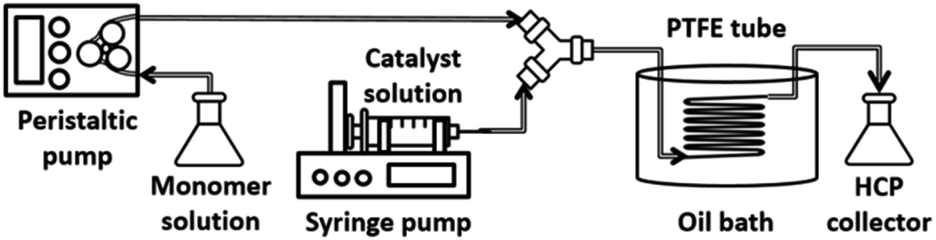 | ||
| Fig. 2 Schematic showing the equipment used in our flow reactor. | ||
(i) Internal crosslinking – the substrate solution was prepared by dissolving 1.154 g of DCX in 20 mL of DCE. A peristaltic pump was used to feed the substrate solution into the system. The catalyst solution was prepared by mixing 1.069 g of FeCl3 in 20 mL of DCE and then loaded into a syringe and pumped into the system by a syringe pump. All reactants were mixed in a Y-junction connector prior to the entry into the reactor at the total volumetric flow rate of 5.40 mL min−1. The reactor was submerged in a heated oil bath setting at 343 K. The slurry product was collected at the end of the reactor.
(ii) Post-crosslinking – the substrate solution was prepared by dissolving 0.154 g of PS in 20 mL of DCE. The catalyst solution was prepared by mixing 1.524 g of FeCl3 and 1.524 g of DCX in 20 mL of DCE and then loaded into a syringe. The substrate solution was fed into the system via a peristaltic pump, while the catalyst solution was fed via a syringe pump. Both solutions were fed into the reactor at the total volumetric flow rate of 2.94 mL min−1 and mixed at a Y-junction. The reactor was submerged in a pre-heated oil bath at 343 K. The slurry product was collected at the end of the reactor.
(iii) External crosslinking – the substrate solution was prepared by dissolving 0.305 g of PS in 20 mL of DCE. The catalyst solution was prepared by mixing 1.524 g of FeCl3 and 1.524 g of FDA in 20 mL of DCE and loaded into a syringe. A peristaltic pump was used for feeding substrate solution into the reactor. A syringe pump was used for feeding the catalyst solution into the system. The total volumetric flow rate was 1.94 mL min−1. Both solutions were mixed at the Y-junction before entering the reactor, which was pre-heated inside the oil bath at 343 K. The slurry product was collected at the end of the reactor.
The reactor volume was approximately 30 mL (1.58 mm inner diameter, 15.21 m length). Our reactor comprised a 50 mm pitch helical coil with multiple curvature radii (34, 39, 44, 49, and 54 mm) designed to minimise the spatial footprint.
Since the reactor volume (V) was fixed, a change in the residence time (τ) depended on the total volumetric feed flow rate (F), as shown in the following equation:
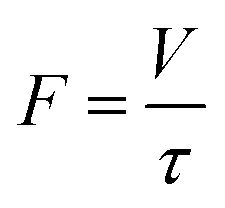
The operating time of flow synthesis (top) was calculated using the following equation where Vfeed was denoted as the volume of the feed solution:
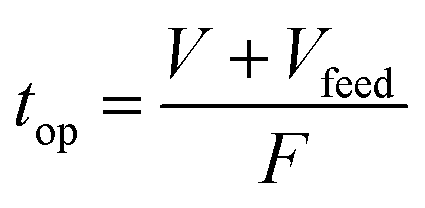
Upon completion of each synthesis process, either in batch or flow, products were washed with chloroform and methanol until the washings became clear. Then, the samples were the soaked overnight in methanol and rewashed in the same way with methanol and acetone. Finally, samples were dried in an oven at 333 K for 8 hours and collected for further characterisation.
Space-time-yield (STY) of HCP synthesis
HCP yield was calculated by dividing the weight of the dried powder produced by the weight of monomers or substrates. The amount of external crosslinkers was not included in this calculation as the crosslinking could be formed from other halogenated compounds used in synthesis, i.e., the chloro group in DCX (substrate) or DCE (solvent).27 This could lead to a yield value of more than 100%.29 Hence, HCP yield was calculated using the following equation:30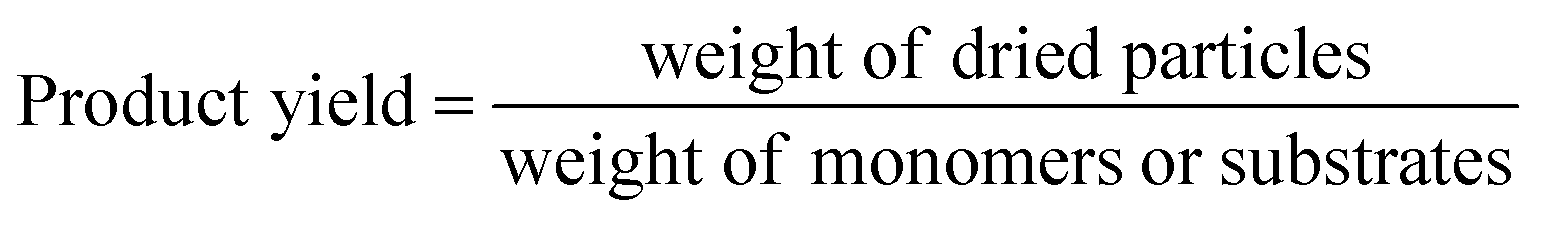
STY is the term used in flow chemistry to determine how much product, i.e., yield is produced in a reactor volume within a time period. As batch reactions here took place over 24 hours, the operating period for the calculation of the flow system was then fixed to per day unit. The STY calculations for batch and flow systems17,31 were shown in the following equations:
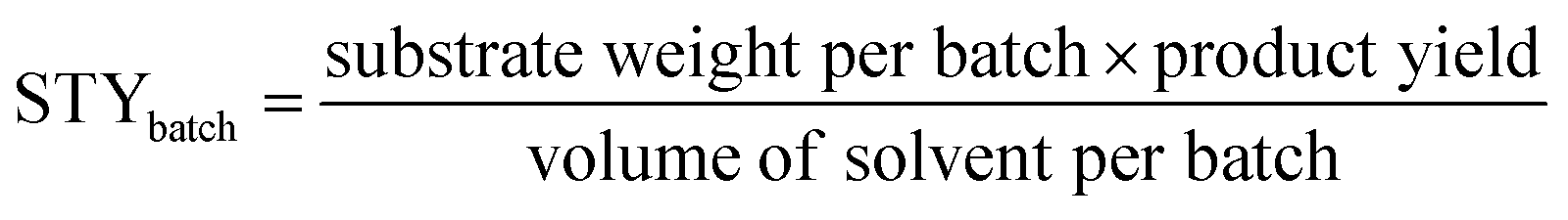
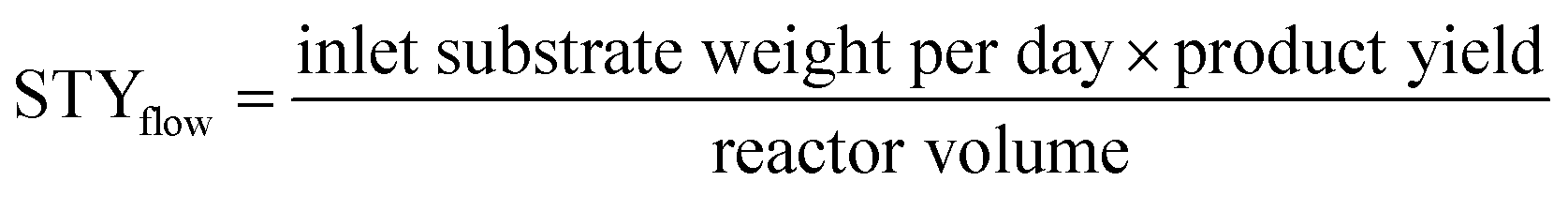
Characterisation
The pore size distributions of HCPs characterize in this work were characterized with N2 gas adsorption at 77 K using various gas analysers at The University of Edinburgh, Strathclyde University, and CSIRO, Australia. These isotherms were used to determine the specific surface area and for porosity analysis using the BET theory and density functional theory (DFT) method, respectively, using the AsiQwin software. Rouquerol's four criteria were applied for BET surface area correction for microporous materials (Fig. S4†).32,33 Samples were degassed in a vacuum at 373 K for 12 hours before characterisation.Fourier transform infrared spectroscopy (FTIR) was used to investigate the chemical structure of the reagents and HCPs synthesised in this work. This was achieved with an ATR-FTIR (Nicolet iS10 with Smart iTX Diamond accessory, Thermo Fisher Scientific, Waltham, MA) with 64 scans per sample and a resolution of 0.48 cm−1. The chemical structures of the synthesised HCPs were validated with solid-state 13C cross polarisation (CP) magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectroscopy using a Bruker Avance III spectrometer with a 9.4 T superconducting magnet.
N2 and CO2 adsorption studies at 298 K were performed and the adsorption isotherms were used to calculate CO2/N2 selectivity (SCO2/N2) based on the ideal adsorption solution theory (IAST). The standard conditions34 of 298 K and 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85 molar ratio of the inlet gas mixture (yCO2 = 0.15, yN2 = 0.85) were chosen for this calculation using following equations:
:85 molar ratio of the inlet gas mixture (yCO2 = 0.15, yN2 = 0.85) were chosen for this calculation using following equations:
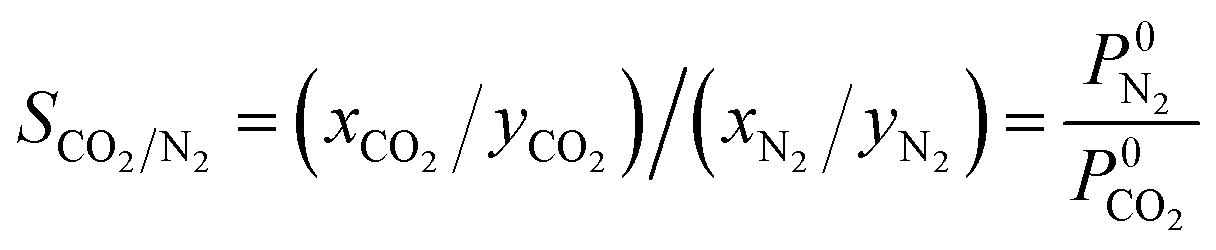
| Ptyi = P0ixi |
| xCO2 + xN2 = 1 |
| yCO2 + yN2 = 1 |
xi and yi denote the mole fraction of gas i (where i = CO2, N2) in the adsorbed phase and gas phase, respectively. P0i is the equilibrium pressure for pure i assuming that spreading pressure (π) was constant during isothermal adsorption. The single-site Langmuir–Freundlich model (SSLF) was selected to associate the adsorbed amount of gas (Qi) with the function of pressure (P), as shown in the following equation:
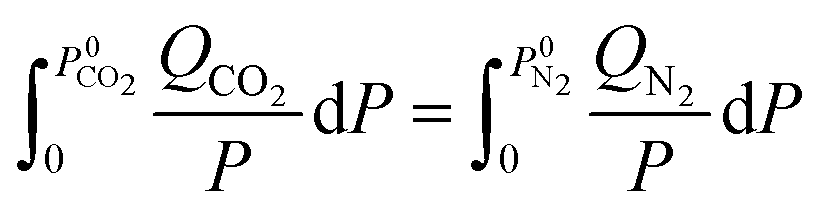
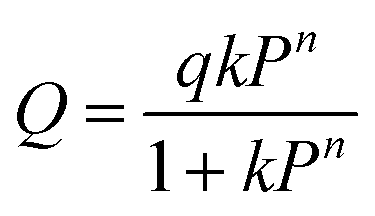
The parameters from the SSLF model equation, q, k, and n, represented the maximum-adsorbed amount of pure gas at the adsorption site, the affinity parameter, and the solid heterogeneity respectively. These parameters were obtained from the regression analysis of the experimental adsorption isotherm data.
Transmission electron spectroscopy (TEM) was deployed to characterise HCP morphology. The samples were dispersed in DCE and deposited onto carbon-coated copper grids (Agar Scientific). Excess solvent was blotted away and the grid was air-dried. Samples were imaged in an FEI F20 microscope operating at 200 kV. The images were taken under low-dose conditions using a Gatan Rio CCD camera. The micrographs (Fig. S8) are shown in ESI.†
Results and discussions
The productivity of the flow processes
Here, we show that HCP synthesis duration could be reduced, from 24 hours to 5–15 minutes in a flow reactor via internal-, post-, and external crosslinking strategies (Table 1). This was attributed to the enhanced heat and mass transfer rates associated with the higher specific area (the ratio of the surface area to volume)35 of the flow reactor deployed in this work. The specific area of the flow reactor deployed in this work was 5087 m2 m−3, 68 fold higher than the 75 m2 m−3 of the batch reactor used here (Table S4†). Reactors with a high surface area to volume ratio35 typically possess the advantages of rapid heat and mass transfers that enhance the reaction kinetics.36| Crosslinking approach | Substrate–crosslinker type | Substrate concentration [% w/v] | Substrate:FeCl3:crosslinker |
Temperature [K] | Volumetric flow rate [mL min−1] | Reaction timea [min] | HCP yield [%] | Sampleb |
|---|---|---|---|---|---|---|---|---|
| a Residence time is an equivalent term for reaction time in the flow system. b The samples were named X-Y-HCP in which X denotes hypercrosslinking approach (I for internal crosslinking, P for post crosslinking, E for external crosslinking) and Y denotes synthesis system (B for batch reactions, F for flow synthesis). | ||||||||
| Internal crosslinking | DCX–N.A. | 2.884 | 1:1:0 (mol) |
373 | N.A. | 1440 | 58.50 | I-B-HCP |
| 2.884 | 1:1:0 (mol) |
343 | 5.40 | 5 | 7.24 | I-F-HCP | ||
| Post-crosslinking | PS–DCX | 0.385 | 1:1:1 (wt) |
353 | N.A. | 1440 | 155.38 | P-B-HCP |
| 0.385 | 1:1:1 (wt) |
343 | 2.94 | 10 | 126.72 | P-F-HCP | ||
| External crosslinking | PS–FDA | 0.762 | 1:5:5 (wt) |
353 | N.A. | 1440 | 156.63 | E-B-HCP |
| 0.762 | 1:5:5 (wt) |
343 | 1.94 | 15 | 114.30 | E-F-HCP | ||
In our work, heat transfer was mainly impacted by this significant difference in reactor-specific surface area. The larger specific surface area of the flow reactor enhanced heat transfer, i.e., the energy flow from the oil bath to the reactor walls that were in close contact with the reactants inside the reactor. At the same time, the small volume of the flow reactor offered short diffusion pathways (better mass transfer) to enable faster mixing.37 The combination of these factors was favourable for polymerisation/crosslinking kinetics. The effects of the thermal conductivity of the reactor material and heat source temperature were negligible here. The difference in thermal conductivities between the glass batch reactor (1 W m−1 K−1)38 and polytetrafluoroethylene (PTFE) flow reactor (0.25 W m−1 K−1)39 was 4 times, while the difference in heat source temperatures used in both batch and flow syntheses was only 10 K.
In terms of mass transfer, a higher degree of mixing is favourable for the reaction as reagent particles have more access to the reactor surface where the highest heat energy is provided.17 The mixing efficiency could be correlated to the dimensionless Reynolds number (Re) which describes fluid flow patterns – laminar, transient, and turbulence. Turbulent flow is typically associated with good mixing as an external driving force is applied to induce particle diffusion, rather than relying solely on molecular diffusion.17 In a stirred tank reactor, the turbulent region is where Re > 10000,40 while turbulent fluid flow in a helical coil reactor is where Re > 40.41
Using an equation for a propeller turbine mixer tank,42 we estimated that the Re number in our batch reactor reached a value of 6524 (see ESI†). This indicated transient fluid flow (the transition region between laminar and turbulent flow pattern, 100 < Re < 10000) in our batch reactor, i.e., moderate mixing efficiency. Meanwhile, the Re number in our coil flow reactor (Table S5†) during the synthesis of E-F-HCP (15 minutes residence time) was estimated to be 65. This indicated turbulent mixing in our flow reactor. Clearly, the coiled reactor used in this work provided better mixing rates, enhancing polymerisation/crosslinking kinetics.20
The advantage of short reaction times in flow synthesis resulted in an increase in STY (Fig. 3). With flow synthesis, the STY value improved by 32 fold for internal crosslinking, 117 fold for post-crosslinking, and 68 fold for external crosslinking. This addressed the issue of low HCP productivity rates due to time constraints required in batch reactions that we have now overcome with flow synthesis.
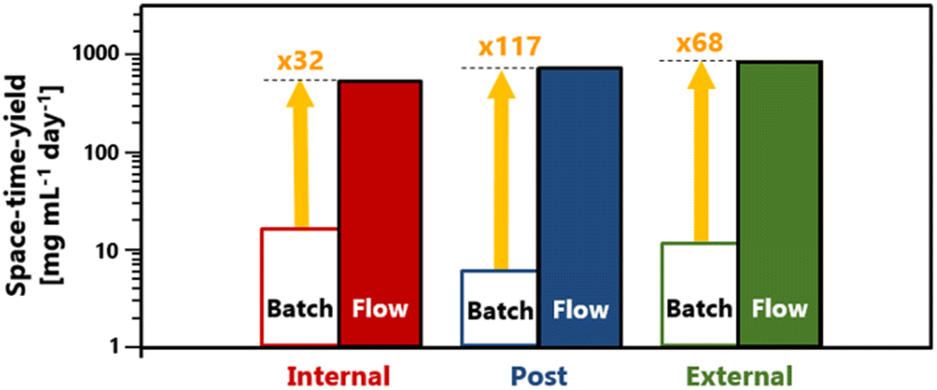 | ||
| Fig. 3 STY of HCP from batch and flow production. | ||
Chemical structures of flow-produced HCPs
The chemical structures of these HCPs were characterised and validated with FTIR and 13C-NMR analyses. HCPs synthesised in batch reactors were used as control materials. The nomenclature for HCPs synthesised in this work is summarised in Table 1, alongside the type of reagents and operating conditions.Comparing the FTIR spectra of HCPs synthesised by internal crosslinking in batch (I-B-HCP) and continuous flow (I-F-HCP) reactions, we observed that in both samples, the infrared peak intensities correlated to C–Cl vibrations27,43,44 at 668 and 1260 cm−1 and C–H bending44 at 854 cm−1 were reduced when compared to the unreacted DCX precursor (Fig. 4a). This was ascribed to chloromethyl substitution at the para position, suggesting that the chloro group was eliminated during Friedel–Crafts alkylation. The intensities of peaks correlated to the skeletal benzene rings, such as aromatic C–H stretching43 at 2971 and 2866 cm−1 and aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching43,45 at 1421, 1445, and 1512 cm−1 were also reduced. This suggested the success of DCX crosslinking.45 At the same time, the peak corresponding to C–H stretching45 at 2923 cm−1 from HCP broadened when compared to that of DCX. This was due to the overlapping original C–H stretching peak with the newly formed methylene bridge (–CH2CH2–) crosslinks.45 This type of crosslinked bridge was formed from DCE molecules that functioned as both a solvent and a crosslinker reagent.46
C stretching43,45 at 1421, 1445, and 1512 cm−1 were also reduced. This suggested the success of DCX crosslinking.45 At the same time, the peak corresponding to C–H stretching45 at 2923 cm−1 from HCP broadened when compared to that of DCX. This was due to the overlapping original C–H stretching peak with the newly formed methylene bridge (–CH2CH2–) crosslinks.45 This type of crosslinked bridge was formed from DCE molecules that functioned as both a solvent and a crosslinker reagent.46
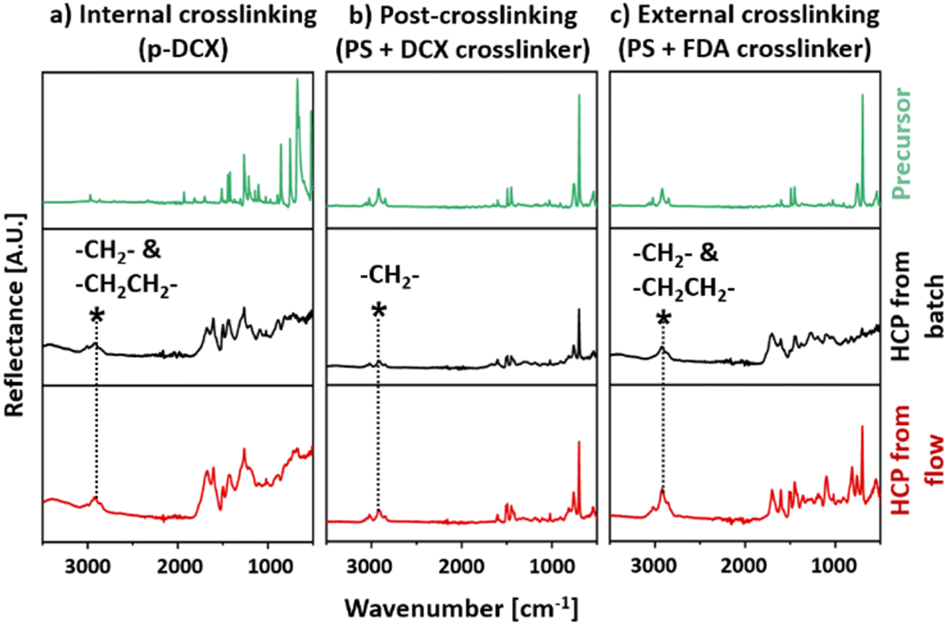 | ||
| Fig. 4 FTIR spectra of the HCPs and their precursors: (a) internal crosslinking (p-DCX), (b) post-crosslinking (PS + DCX crosslinker), (c) external crosslinking (PS + FDA crosslinker). | ||
The FTIR spectra of HCPs synthesised via post-crosslinking, P-B-HCP, and P-F-HCP (Fig. 4b), resembled that of PS. We observed peaks that corresponded to aromatic C–H stretching at 3087, 3061, 3027 cm−1, C–H bending at 756, 696 cm−1, aromatic CC stretching at 1603, 1493, 1453 cm−1, C–H bending at 854 cm−1 and backbone C–H stretching at 2921 and 2849 cm−1.28 However, these peaks were less intense than those of PS. These indicated that the aromatic benzene rings of PS were modified during post-crosslinking. These trends were also presented in the FTIR spectra of HCPs synthesised via external crosslinking, E-B-HCP, and E-F-HCP (Fig. 4c). The broadened peak corresponding to C–H stretching45 centred at 2921 cm−1 similar to the spectra of internally crosslinked HCPs was observed in these spectra. This indicated that –CH2CH2– crosslinks were also formed in the externally crosslinked HCP. The presence of newly formed crosslinking methylene bridges in these HCPs was also validated with 13C-NMR.
The 13C-NMR spectra of I-B-HCP and I-F-HCP (Fig. 5a) contained major peaks centred at 138, 35, and 16 ppm. These peaks were correlated to the substituted aromatic carbon,47 the crosslinked bridges from via FDA28,47 and DCE,27 respectively. There were also unreacted DCX24,48 in these samples as we also observed peaks centred at 43 ppm (carbon in –CH2Cl) and 128 ppm (non-substituted aromatic ring). The spectra of P-B-HCP and P-F-HCP (Fig. 5b) also contained a peak centred at 146 ppm correlated to the quaternary non-substituted aromatic carbon48,49 from the parent PS molecule. These spectra also contained peaks at 136 and 128 ppm, which corresponded to substituted aromatic carbon and non-substituted aromatic rings from both PS and DCX, respectively. The broad peak centred at 40 ppm in these spectra was a mixture of signals attributed to aliphatic methylene carbon, methine backbone carbon, and methylene crosslink bridges.48 We did not observe any peaks corresponding to chloromethyl carbon in these samples. The 13C-NMR spectra of E-B-HCP and E-F-HCP (Fig. 5c) were like those of HCPs produced from post-crosslinking.
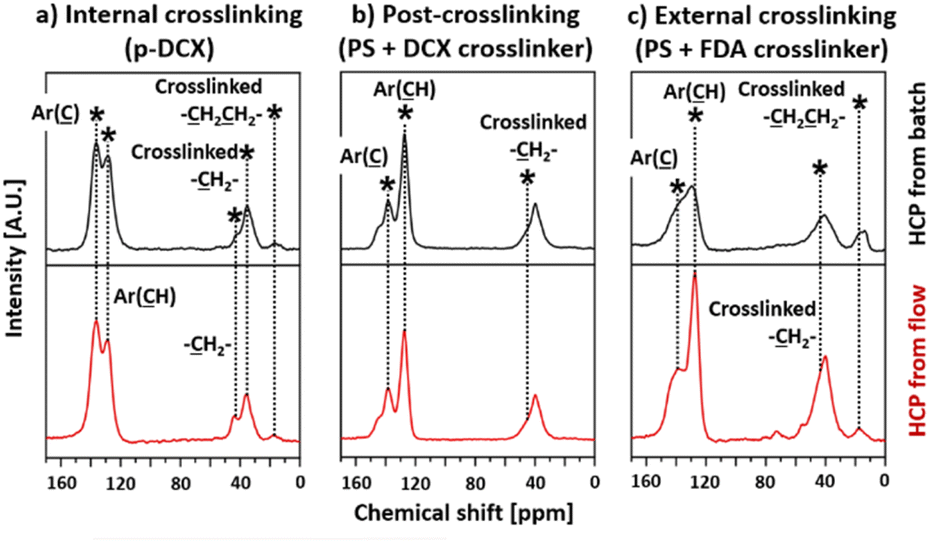 | ||
| Fig. 5 13C-NMR spectra of the HCPs: (a) internal crosslinking (p-DCX), (b) post-crosslinking (PS + DCX crosslinker), (c) external crosslinking (PS + FDA crosslinker). | ||
HCP microporosity
Porosity in HCPs is formed as a function of the crosslinking degree.50 As the degree of crosslinking increases, the BET surface area increases, resulting from the creation of micropores (<2 nm).46,50 However, as the micropores are excessively developed within the HCPs, this then results in the decline of BET surface area when a certain degree of crosslinking is achieved.1 Here we observed that the BET surface areas of the flow-produced HCPs were in the range between 800–1160 m2 g−1 similar to reported batch-produced HCP counterparts from literature.24,27,28 Regardless of crosslinking strategies, the BET surface areas were lower than those of HCPs synthesised in batch reactors by 1.5–9.6%, while the total pore volume of flow-produced HCPs was on average 21% lower (Table 2). However, the enhancement of micropore volume in the flow-produced HCPs synthesised via internal and external crosslinking leads to the formation of narrower pores between 0–10 nm (Fig. 6). These results indicate that flow synthesis created more microporosity in such HCPs. This difference in micropore volume between HCP synthesised from batch and flow reactions could be attributed to different heat-mass transfer rates of reactor types, leading to different crosslinking rates and intensities. For batch synthesis, we used a 250 mL three-neck round-bottom glass flask as a reactor. Meanwhile, for flow synthesis, we used a 30 mL PTFE helical coil tube as a reactor. The smaller volume of the coiled tube induced faster mixing of reactants,36 and enhanced mass transfer35 when compared to the glass flask. The helical coil also increased the vortex flow pattern inside the tube.41 Moreover, the contact area for transferring heat energy from the oil bath to the reaction mixture is larger in the flow reactor. The increment in microporosity in flow-produced HCP could potentially benefit applications, such as gas separation and storage.| Samples | BET surface area [m2 g−1] | DFT total pore volume [cm3 g−1] | DFT micropore volume [cm3 g−1] |
|---|---|---|---|
| a DFT micropore volume data were obtained from the cumulative pore volume of the pore size up to 2 nm. | |||
| I-B-HCP | 964 | 1.263 | 0.042 |
| I-F-HCP | 950 | 1.014 | 0.281 |
| (−1.5%) | (−19.7%) | (+569%) | |
| P-B-HCP | 903 | 0.788 | 0.203 |
| P-F-HCP | 816 | 0.650 | 0.195 |
| (−9.6%) | (−17.3%) | (−3.9%) | |
| E-B-HCP | 1162 | 1.219 | 0.184 |
| E-F-HCP | 1092 | 0.917 | 0.293 |
| (−6.0%) | (−24.8%) | (+59.2%) | |
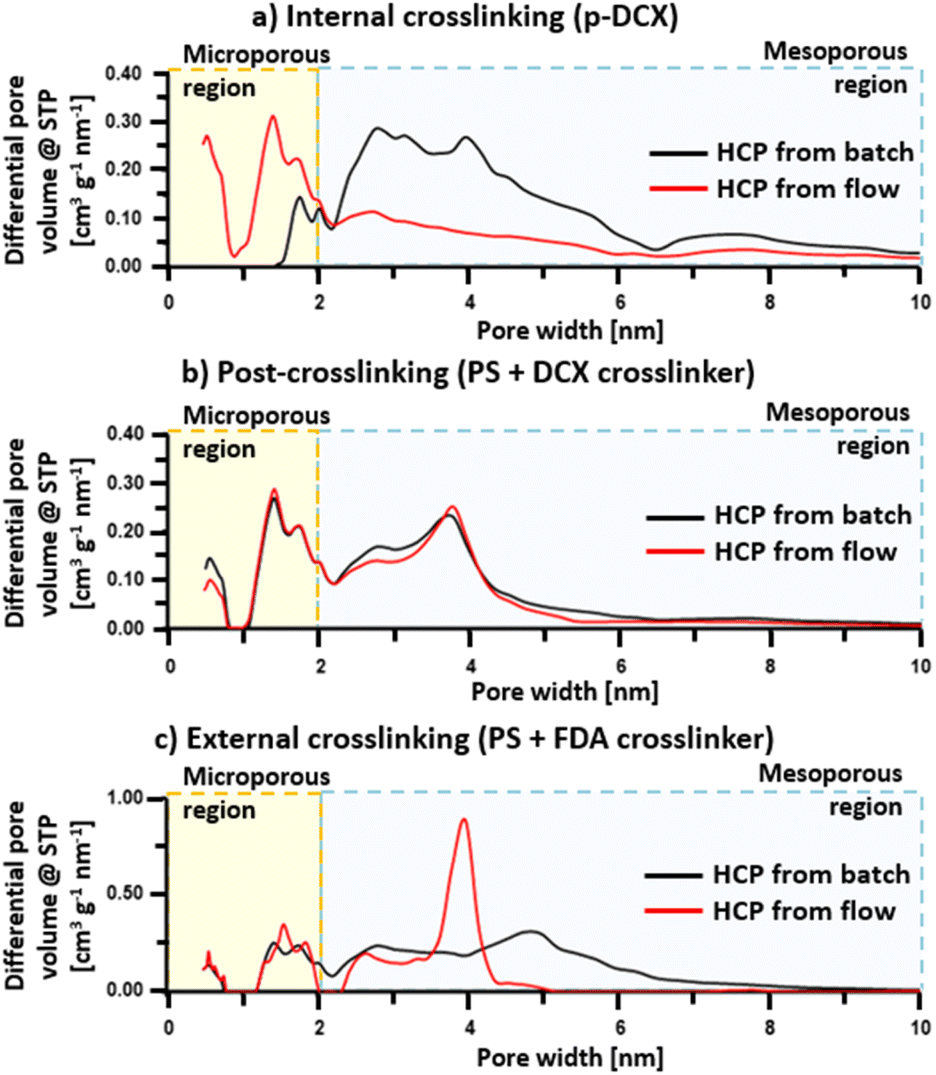 | ||
| Fig. 6 Pore size distribution of flow-produced HCPs and their batch equivalents. | ||
The existence of micropores was confirmed through the steep slope at lower relative pressure, where P/P0 < 0.05 (Fig. S5†). Based on IUPAC classification,51 the isotherms of internally crosslinked HCPs were a mixture of type I and IV. Moreover, the presence of macropores (>50 nm) in the internally crosslinked HCPs could be observed in the isotherm at P/P0 > 0.9. It should be noted that this was the only HCP type in this study that formed macropores. The isotherms of HCPs synthesised by post-crosslinking and external crosslinking were classified as type IV, where ink bottle-like pore shapes were more prevalent. The hysteresis loops of these HCPs indicated that there were significant amounts of small mesopores (2–10 nm) within these HCPs, which correlated to the broad peaks in the pore size distributions (Fig. 6).
The microporosity of HCPs studied here could be tailored by varying synthesis parameters: residence time, feed solution composition and concentration, and temperature. Here we observed that at a fixed substrate concentration, reagent ratios, and temperatures, an increase in residence time from 10 to 15 minutes typically enhanced the micropore volume of externally crosslinked HCPs by 595%. However, a further increase of residence time to 20 minutes reduced micropore volume by 65.5% (Table S3†). Similar trends were observed in batch reactions over 12 and 24 hour periods.1 Prolonged reaction times could lead to excessive crosslinking, forming pores that are smaller than the kinetic diameter of N2, the gas used to probe porosity content during gas adsorption experiments. Another reason for an optimised micropore volume in HCPs produced via 15 minutes of residence time in flow reactors could be the mixing intensity. Our calculation (Table S5†) showed that with a residence time of 15 minutes, the Reynolds number of our reactor reached a value of 65. This was closest to the optimal Reynolds number of 40 in helical reactors.41 Here it is important to highlight that the optimisation of HCP porosity by tailoring residence time did not drastically affect STY, i.e., productivity rate. The highest STY value of 965 mg mL−1 day−1 was achieved with a residence time of 10 minutes. The increase of residence time to 15 minutes reduced this STY value by 28.3%, reaching 692 mg mL−1 day−1.
The enhancements in both micropore volume and STY were observed (Table S3†) when the weight ratio of PS by FeCl3 by FDA increased from 1:1:1 to 1:5:5 while fixing the other three synthesis parameters. No solid was formed at the weight ratio of 1:1:1. This indicates that crosslinking did not occur. Although a similar test in the batch system showed that the crosslinking using a 1:1:1 ratio of PS to FeCl3 to FDA is achievable,28 shorter residence time and lower temperature in flow reactors might be insufficient to induce crosslinking when there is a low ratio of catalyst and crosslinker to the substrate. HCP products were obtained when the weight ratio was increased to 1:3:3, with a STY of 981 mg mL−1 day−1. However, the micropore volume of 0.006543 cm3 g−1 was the lowest among all the externally crosslinked HCPs. The increase in micropore volume and the STY when increasing the ratio from 1:3:3 to 1:5:5 was 1493% and 47.5%, respectively. Similar trends showing the improvement of the specific surface area, as a result of pore generation, and product yield were also observed in batch HCP synthesis.24,28 The higher amount of the catalyst and crosslinker in the system indicated a higher possibility of the substrate getting involved in the reaction. Hence, the suitable weight ratio of substrate to the catalyst to crosslinker for flow synthesis in our system was 1:5:5.
Increasing the reaction temperature while fixing the residence time, substrate concentration, and chemical ratio did not enhance STY (average of 830 mg mL−1 day−1), however, micropore volumes were enhanced. The increase in the temperature from 333 to 343 K increased the micropore volume by 84.8% from 0.059 to 0.109 cm3 g−1. This indicated that elevating temperature by a 10 K difference in our system affected the crosslinking degree in HCPs.
The impact of PS concentration on micropore volume was subtle compared with the other three synthetic parameters. The amount of micropore volume was increased by just 37.5% from 0.4% to 0.6% w/v of PS. Increasing the substrate concentration from 0.6 to 0.8% w/v and then to 1.0% w/v slightly increased and decreased micropore volume by 5.7% and 9.5%, respectively. This trend was also observed in batch synthesis,24,46 which suggested that the fabrication of microporous polymers, i.e., HCP is not sensitive to the monomer concentration as much as macroporous polymers.24 On the other hand, the substrate concentration caused the largest influence on STY among all four synthetic parameters, as the correlation between PS concentration and STY was directly proportional. Increasing the PS concentration from 0.4% to 1.0% w/v enhanced STY by 3 fold from 457 to 1447 mg mL−1 day−1.
With these optimal synthesis parameters, we were able to enhance the STY of HCP synthesis using flow reactions, as shown earlier in Fig. 3. This inferred that we were able to not only enhance the microporosity of the internally crosslinked HCPs but also improve the STY of such HCPs.
CO2/N2 separation
Due to higher total pore volumes in batch-produced HCPs (Table 2), the CO2 and N2 uptakes (Fig. 7) of such HCPs, regardless of crosslinking strategies, were higher than those synthesised under a continuous flow. Amongst all the HCPs studied here, the total pore volume of HCPs synthesised via internal crosslinking in batch reactions were the highest, reaching 1.26 cm3 g−1. As such, the CO2 and N2 uptakes of HCPs synthesised via internal crosslinking reached 0.99 and 0.27 mmol g−1, respectively. With a total pore volume of 1.22 cm3 g−1, the CO2 and N2 uptakes of HCPs synthesised in batch reactions via external crosslinking reached 0.8 and 0.1 mmol g−1, respectively. These were 19% and 63% lower than those of I-B-HCP. With the lowest total pore volume of 0.79 cm3 g−1, HCPs synthesised through post-crosslinking in batch reactions only uptake 0.55 and 0.05 mmol g−1 of CO2 and N2, respectively. Although continuous flow synthesis reduced the total pore volume of HCPs by 17–25% (Table 2), leading to lower CO2 and N2 uptakes, this approach enhanced the micropore volumes in internally and externally crosslinked HCPs by 569% and 59%, respectively. The 569% increase in the micropore volume in internally crosslinked HCPs underpinned a CO2/N2 selectivity (at 298 K and 1000 mbar) of 71.5, 850% higher than that of batch-produced internally crosslinked HCPs, which was the lowest amongst all samples studied here, at 7.5 (Fig. 7). This significant difference in CO2/N2 selectivity between the flow and the batch produced internally crosslinked HCPs could be attributed to the amount and type of pores present. With 25% more total pore volume, more CO2 and N2 could be adsorbed in I-B-HCPs. However, with only 3.2% of micropores, the ability to separate CO2 from N2 in I-B-HCPs is diminished. Due to the similar kinetic diameters of N2 (3.64 Å) and CO2 (3.30 Å),52 materials with small pore sizes and narrow pore size distributions are required for effective CO2/N2 separations, especially for post-carbon capture53 and ideally, the pore sizes should be between 5 to 7.8 Å.53 In this work, such pores were only observed in HCPs synthesised in flow reactions via internal crosslinking. Hence, the CO2/N2 selectivity (calculated using IAST theory with 0.15 bar CO2 partial pressure, 298 K and 1 atm) of these HCPs were the highest amongst all HCPs studied here. This outcome demonstrated that HCP CO2/N2 selectivity could be improved through optimisation of the reaction parameters via reactor design, i.e., from batch to flow reactor type. This is unique from existing strategies such as HCP functionalisation,21–23 the use of bespoke monomers25,26 and tailoring synthesis conditions (heat, duration, etc.).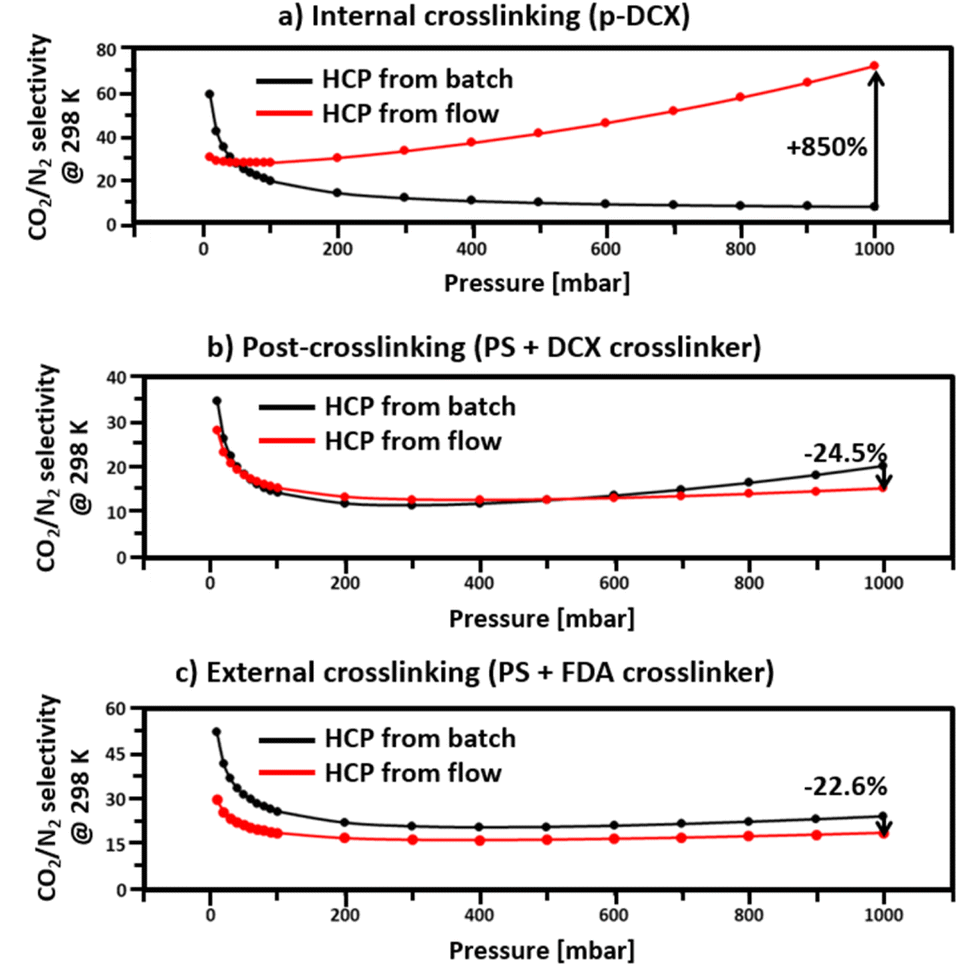 | ||
| Fig. 7 CO2/N2 selectivity at 298 K, 1 atm, 15:85 of CO2:N2 inlet gas mixture. | ||
Conclusions
This study highlights two key benefits of the implementation of continuous flow synthesis in HCP production. First, we observed higher microporosity in HCPs synthesised in flow reactors. Regardless of their lower total pore volumes and BET surface areas, high volumes of micropores within flow-produced HCPs enhanced CO2/N2, reaching a value of 71.5. We also verified that the porosity can be tailored through the alteration of flow synthetic parameters. When comparing our optimised internally crosslinked HCPs with their batch counterpart, the HCP from flow synthesis showed an increase in micropore volume by 569% resulting in an 850% increase in CO2/N2 separation. Second, the optimal residence time was reduced to 5 minutes with flow synthesis, instead of 24 hours as in a batch operation. This enhanced the productivity of STY by 32 fold, which resulted in a HCP production rate of 563 mg cm−3 day−1. We also demonstrated the versatility of flow reactions for HCP synthesis via post-crosslinking and external crosslinking approaches. The findings from this work show the potential to scale up HCP synthesis with flow reactors to meet the global demand for such materials, and the potential for carbon capture. The pilot scale of this process should be the next step, focusing on the optimisation of operating parameters.Author contributions
The study was proposed by C. H. L. The conceptualised idea was discussed between N. C. and C. H. L. The synthesis process was conducted by N. C. The contribution of N2 gas analysis characterisation was shared between L. D. and A. J. F. FTIR characterisation was conducted by S. L. and S. S. 13C-NMR characterisation was done by D. M. D. CO2 and N2 adsorption tests for CO2/N2 separation were performed by K. K., and M. R. H. All the results were analysed by N. C. The paper was prepared by N. C. with supervision from C. H. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Metal and Materials Technology Center, Thailand. The authors express their appreciation towards Mr Fergus Dingwall for assistance in material characterisation.Notes and references
- H. Liu, S. Li, H. Yang, S. Liu, L. Chen, Z. Tang, R. Fu and D. Wu, Adv. Mater., 2017, 29, 1700723 CrossRef PubMed.
- Y. Luo, B. Li, W. Wang, K. Wu and B. Tan, Adv. Mater., 2012, 24, 5703–5707 CrossRef CAS PubMed.
- J. Huang and S. R. Turner, Polym. Rev., 2018, 58, 1–41 CrossRef CAS.
- V. A. Davankov and M. P. Tsyurupa, React. Polym., 1990, 13, 27–42 CrossRef CAS.
- M. P. Tsyurupa and V. A. Davankov, React. Funct. Polym., 2006, 66, 768–779 CrossRef CAS.
- C. H. Lau, X. Mulet, K. Konstas, C. M. Doherty, M.-A. Sani, F. Separovic, M. R. Hill and C. D. Wood, Angew. Chem., Int. Ed., 2016, 55, 1998–2001 CrossRef CAS PubMed.
- H. Ramezanipour Penchah, H. Ghanadzadeh Gilani and A. Ghaemi, J. Chem. Eng. Data, 2020, 65, 4905–4913 CrossRef CAS.
- Materials and Chemicals - Ion Exchange Resin Market, Ion Exchange Resin Market Study, By Type (Cationic Resins, Anionic Resins, Other), By End Use Industry (Power, Chemical and Petrochemical, Water and Wastewater Treatment, Food and Beverages, Pharmaceuticals, Electrical and Electronics, Metal and Mining), Forecasts To 2028, 2021, available at: https://www.reportsanddata.com/report-detail/ion-exchange-resin-market, accessed: 9th July 2021 Search PubMed.
- L. Tan and B. Tan, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC.
- C. H. Lau, T.-d. Lu, S.-P. Sun, X. Chen, M. Carta and D. M. Dawson, Chem. Commun., 2019, 55, 8571–8574 RSC.
- Y. Liu, X. Chen, X. Jia, X. Fan, B. Zhang, A. Zhang and Q. Zhang, Ind. Eng. Chem. Res., 2018, 57, 17259–17265 CrossRef CAS.
- T. Ratvijitvech, J. Polym. Environ., 2020, 28, 2211–2218 CrossRef CAS.
- F. Björnerbäck and N. Hedin, ChemSusChem, 2019, 12, 839–847 CrossRef PubMed.
- J.-S. M. Lee, T. Kurihara and S. Horike, Chem. Mater., 2020, 32, 7694–7702 CrossRef CAS.
- S. Grätz, S. Zink, H. Kraffczyk, M. Rose and L. Borchardt, Beilstein J. Org. Chem., 2019, 15, 1154–1161 CrossRef PubMed.
- N. B. McKeown, Sci. China: Chem., 2017, 60, 1023–1032 CrossRef CAS.
- M. Rubio-Martinez, T. D. Hadley, M. P. Batten, K. Constanti-Carey, T. Barton, D. Marley, A. Mönch, K.-S. Lim and M. R. Hill, ChemSusChem, 2016, 9, 938–941 CrossRef CAS PubMed.
- Y. Peng, W. K. Wong, Z. Hu, Y. Cheng, D. Yuan, S. A. Khan and D. Zhao, Chem. Mater., 2016, 28, 5095–5101 CrossRef CAS.
- M. Rubio-Martinez, M. P. Batten, A. Polyzos, K.-C. Carey, J. I. Mardel, K.-S. Lim and M. R. Hill, Sci. Rep., 2014, 4, 5443 CrossRef CAS PubMed.
- P. J. Dowding, J. W. Goodwin and B. Vincent, Colloid Polym. Sci., 2000, 278, 346–351 CrossRef CAS.
- S. Krishnan and C. V. Suneesh, J. Solid State Chem., 2021, 299, 122152 CrossRef CAS.
- Y. Liu, X. Jia, J. Liu, X. Fan, B. Zhang, A. Zhang and Q. Zhang, Appl. Organomet. Chem., 2019, 33, e5025 Search PubMed.
- Y. Qiao, Z. Zhan, Y. Yang, M. Liu, Q. Huang, B. Tan, X. Ke and C. Wu, Mater. Today Commun., 2021, 27, 102338 CrossRef CAS.
- C. D. Wood, B. Tan, A. Trewin, H. Niu, D. Bradshaw, M. J. Rosseinsky, Y. Z. Khimyak, N. L. Campbell, R. Kirk, E. Stöckel and A. I. Cooper, Chem. Mater., 2007, 19, 2034–2048 CrossRef CAS.
- Q. B. Meng and J. Weber, ChemSusChem, 2014, 7, 3312–3318 CrossRef CAS PubMed.
- M. Saleh, H. M. Lee, K. C. Kemp and K. S. Kim, ACS Appl. Mater. Interfaces, 2014, 6, 7325–7333 CrossRef CAS PubMed.
- Z. Fu, I. M. A. Mohamed, J. Li and C. Liu, J. Taiwan Inst. Chem. Eng., 2019, 97, 381–388 CrossRef CAS.
- X. Dong, A. Akram, B. Comesaña-Gándara, X. Dong, Q. Ge, K. Wang, S.-P. Sun, B. Jin and C. H. Lau, ACS Appl. Polym. Mater., 2020, 2, 2586–2593 CrossRef CAS.
- L. Tan, B. Li, X. Yang, W. Wang and B. Tan, Polymer, 2015, 70, 336–342 CrossRef CAS.
- V. Rajangam, A. Abidov, M. Peng, C. Babu, M. Palanichamy, W. Cha and H. Jang, Fibers Polym., 2015, 16, 1458–1467 CrossRef.
- J. Wegner, S. Ceylan and A. Kirschning, Chem. Commun., 2011, 47, 4583–4592 RSC.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, Stud. Surf. Sci. Catal., 2007, 160, 49–56 CrossRef CAS.
- K. Shi, E. E. Santiso and K. E. Gubbins, in Porous Materials: Theory and its Application for Environmental Remediation, ed. J. C. Moreno-Piraján, L. Giraldo-Gutierrez and F. Gómez-Granados, Springer International Publishing, Cham, 2021, pp. 315–340, DOI:10.1007/978-3-030-65991-2_12.
- A. Hassanpouryouzband, J. Yang, B. Tohidi, E. Chuvilin, V. Istomin, B. Bukhanov and A. Cheremisin, Environ. Sci. Technol., 2018, 52, 4324–4330 CrossRef CAS PubMed.
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed. Engl., 2011, 50, 7502–7519 CrossRef CAS PubMed.
- F. E. Valera, M. Quaranta, A. Moran, J. Blacker, A. Armstrong, J. T. Cabral and D. G. Blackmond, Angew. Chem., Int. Ed., 2010, 49, 2478–2485 CrossRef CAS PubMed.
- D. Wilms, J. Klos and H. Frey, Macromol. Chem. Phys., 2008, 209, 343–356 CrossRef CAS.
- Y. Cengel, Heat and Mass Transfer: Fundamentals and Applications, McGraw-Hill Higher Education, 2014 Search PubMed.
- D. M. Price and M. Jarratt, Thermochim. Acta, 2002, 392, 231–236 CrossRef.
- G. Tchobanoglous, F. L. Burton, H. D. Stensel and F. Burton, Wastewater Engineering: Treatment and Reuse, McGraw-Hill Education, 2003 Search PubMed.
- M. Mansour, P. Khot, D. Thévenin, K. D. P. Nigam and K. Zähringer, Chem. Eng. Sci., 2020, 214, 114522 CrossRef CAS.
- J. J. Derksen, M. S. Doelman and H. E. A. Van den Akker, Exp. Fluids, 1999, 27, 522–532 CrossRef.
- Z. Wei, Q. Chen and H. Liu, New J. Chem., 2021, 45, 11607–11617 RSC.
- Y. Liu, X. Fan, X. Jia, B. Zhang, H. Zhang, A. Zhang and Q. Zhang, J. Mater. Sci., 2016, 51, 8579–8592 CrossRef CAS.
- L. Sun, K. Chai, L. Zhou, D. Liao and H. Ji, Int. J. Biol. Macromol., 2021, 175, 396–405 CrossRef CAS PubMed.
- Z. Fu, J. Jia, J. Li and C. Liu, Chem. Eng. J., 2017, 323, 557–564 CrossRef CAS.
- Z.-A. Qiao, S.-H. Chai, K. Nelson, Z. Bi, J. Chen, S. M. Mahurin, X. Zhu and S. Dai, Nat. Commun., 2014, 5, 3705 CrossRef PubMed.
- R. Joseph, W. T. Ford, S. Zhang, M. P. Tsyurupa, A. V. Pastukhov and V. A. Davankov, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 695–701 CrossRef CAS.
- C. Dalla Valle, M. Zecca, F. Rastrelli, C. Tubaro and P. Centomo, Polymers, 2020, 12, 600 CrossRef CAS PubMed.
- Q.-Q. Liu, L. Wang, A.-G. Xiao, H.-J. Yu and Q.-H. Tan, Eur. Polym. J., 2008, 44, 2516–2522 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- E. S. Kentish, A. C. Scholes and W. G. Stevens, Recent Pat. Chem. Eng., 2008, 1, 52–66 CrossRef.
- L. Zou, Y. Sun, S. Che, X. Yang, X. Wang, M. Bosch, Q. Wang, H. Li, M. Smith, S. Yuan, Z. Perry and H.-C. Zhou, Adv. Mater., 2017, 29, 1700229 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta09253k |
| This journal is © The Royal Society of Chemistry 2023 |