
Manganese-based oxide electrocatalysts for the oxygen evolution reaction: a review
Peng
Wang
ab,
Shiqi
Zhang
ab,
Zhaobo
Wang
ab,
Yuhan
Mo
ab,
Xiaoyang
Luo
ab,
Fan
Yang
c,
Meili
Lv
c,
Zhaoxiang
Li
d and
Xuanwen
Liu
 *a
*a
aSchool of Materials Science and Engineering, Northeastern University, Shenyang 110819, PR China. E-mail: lxw@neuq.edu.cn
bKey Laboratory of Dielectric and Electrolyte Functional Material Hebei Province, School of Resources and Materials, Northeastern University at Qinhuangdao, Qinhuangdao 066004, PR China
cQinhuangdao Solid Waste Management Centre, Qinhuangdao 066004, PR China
dQinhuangdao Capital Starlight Environmental Technology Co., Ltd, Qinhuangdao 066004, PR China
First published on 17th February 2023
Abstract
The oxygen evolution reaction (OER), as an essential process in water decomposition and air batteries, has received increasing attention in the context of clean energy production and efficient energy storage. With their abundant composition and morphology, manganese-based oxides (MnOx) offer great possibilities for the exploration and design of OER catalysts. In this paper, three classes of MnOx materials, including MnO2, Mn2O3, and Mn3O4, are systematically reviewed and their development and applications in OER systems are comprehensively presented. Subsequently, the effects of Jahn–Teller distortion and the question of the active site and stability of MnOx in the OER are discussed, and the presence of Mn3+, which is considered essential for OER activity, and strategies for improving performance are proposed. This paper focuses on the impact of crystal structure, catalytic mechanisms, and design strategies on MnOx.
1. Introduction
The energy crisis caused by the exploitation and over-consumption of fossil fuels and the increasingly severe climate change have emerged as two prominent issues constraining human development.1–3 Hydrogen can replace fossil fuels as the main energy source, and improving hydrogen production by electrolysis of water is one of the most promising sustainable methods. Nevertheless, the development of hydrogen evolution in water electrolysis is limited by the anodic oxygen evolution reaction (OER) as a consequence of its moderate multi-electron transfer process. The most commonly used electrocatalysts for the OER are noble metals and their oxides, such as Ir, Ru, IrO2, and RuO2, but their extreme cost and scarcity prevent their large-scale application. Therefore, the development of oxygen-evolving electrocatalysts with excellent catalytic properties, elevated stability, and low cost is essential.4–7 Thus far, transition metal matrices (Fe, Co, etc.) and transition metal oxides, nitrides, carbides, hydroxides, and sulfides have been proven to have elevated catalytic activity in the field of water electrolysis.8–11Manganese-based oxides (MnOx) are attractive materials in various fields due to their low price, natural abundance, and low toxicity. With its wide range of oxidation states, MnOx can be broadly classified as MnO2 (oxidation state of +4), Mn3O4 (mixed state), Mn2O3 (oxidation state of +3), and MnO (oxidation state of +2).12 Take the most common MnO2, for example, there are six crystal structures of MnO2, which means that MnO2 can be prepared by adjusting its preparation method to obtain catalysts with different surface areas and controllable morphology, and it is found that the conversion between different crystal structures of MnO2 will improve its catalytic performance.13 However, there are still insufficient studies on MnOx in the OER direction. The catalytic activity of MnOx is extremely different from that of iron, cobalt, and nickel catalysts, whose limited active sites and low conductivity limit their development. Therefore, it is necessary to explore its catalytic properties in-depth and implement modification strategies such as defect engineering or heteroatom incorporation.14
Based on the current research progress, the conventional adsorption mechanism of the OER in an alkaline environment is first discussed in this paper. Second, the latest research progress of MnOx (MnO2, Mn3O4, and Mn2O3) as OER catalysts is reviewed. Of these MnOx materials, MnO2 is currently the most well studied, followed by Mn3O4 and Mn2O3. After that, the Jahn–Teller distortion, active sites, and stability issues in MnOx catalysts and suggested ways to improve OER activity, including the introduction of vacancies and the formation of heterogeneous interfaces are discussed. We believe that this review can serve as a reference for the development of high-quality manganese oxide catalysts.
2. The OER mechanism
The reaction mechanism of the OER is thought to be through two possible pathways: the adsorption evolution mechanism (AEM) and lattice oxygen mechanism (LOM).15 The AEM is a more general mechanism for OER catalysts (Fig. 1a), and the LOM normally occurs in extremely covalent oxides.4,16,17 In this section, we focus on the proportionality of AEM to different reaction intermediates. The AEM pathway is a four-coordinated proton-electron transfer reaction centered on metal ions, as shown in eqn (1)–(4).18 MOH, MO, and MOOH represent intermediates adsorbed on active sites. The hydroxy anion (OH−) is first adsorbed on the surface of the metal site (M) through the single electron oxidation process, forming adsorbed MOH at the M site. Then, MOH intermediates are oxidized to form MO substances. The subsequent O–O bond formation step is usually considered the rate-determining step for most catalysts in this step where MO reacts with another OH− to form MOOH. Finally, MOOH is oxidized by a single electron transfer process, releasing O2 and restoring the original M active site.19| OH− + M → MOH + e− (ΔG1) | (1) |
| MOH + OH− → MO + H2O + e− (ΔG2) | (2) |
| MO + OH− → MOOH + e− (ΔG3) | (3) |
| MOOH + OH− → O2 + M + H2O + e− (ΔG4) | (4) |
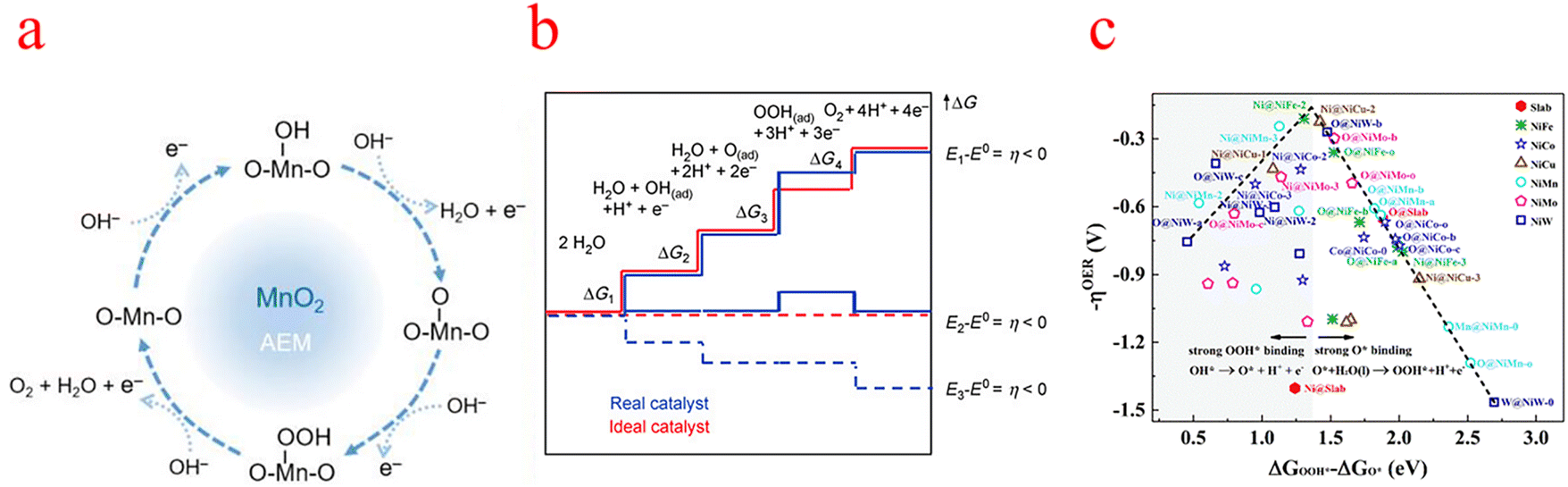 | ||
| Fig. 1 (a) Schematic diagram of the OER mechanism indicated (using MnO2 as an example). (b) Gibbs free energy and reaction coordinate diagram of active intermediates (horizontal line) of the OER. The blue line and red line represent the real (typical) catalyst and the ideal catalyst respectively. (c) Overpotential (−ηOER) Gibbs free binding energy difference with different reaction sites OOH* and O*. (a) Reproduced with permission from ref. 17. Copyright 2023, American Chemical Society. (b) Reproduced with permission from ref. 25. Copyright 2010, John Wiley and Sons. (c) Reproduced with permission from ref. 30. Copyright 2019, Royal Society of Chemistry. | ||
The Gibbs free energy variation of the above four-electron transfer process can be calculated using the following eqn (5)–(8).20,21
| ΔG1 = GMOH − GM − (GOH− − Ge−) | (5) |
| ΔG2 = GMO + GH2O(l) − GMOH − (GOH− − Ge−) | (6) |
| ΔG3 = GMOOH − GMO − (GOH− − Ge−) | (7) |
| ΔG4 = GM + GO2(g) + GH2O(l) − GMOOH − (GOH− − Ge−) | (8) |
Each elementary step has the specific free energy of the corresponding intermediate. The step with the maximum free energy difference (G = Max [ΔG1, ΔG2, ΔG3, ΔG4]) is identified as the rate-determining step and the theoretical overpotential of the overall reaction (η = (G/e) −1.23 V).22 Specifically, catalytic activity is estimated from the magnitude of the potential determining step GOER of the OER.23 That is to say, all adsorption intermediates of MOH, MO, and MOOH should balance the fracture of the O–M bond and the formation of the O–O bond to ensure that the bonding strength of O–M is neither too strong nor too weak.24 An ideal catalyst in thermodynamics requires that all four steps have the same order of reaction-free energy as shown in the figure (Fig. 1b).25 Under this condition, all reaction-free energies are zero at the equilibrium potential (1.23 V), if ignoring the desorption step. Given the constant adsorption energy difference between MOH and MOOH (ΔGMOOH = ΔGMOH + 3.2 ± 0.2 eV), OER overpotential can be determined from MO adsorption energy, which means that the second or third process is the decisive step. According to Sabatier's principle, the ideal catalyst requires that the adsorption strength of key intermediates is neither too strong nor too weak.26–28 As such, ηOER as (ΔGMO − ΔGMOH) leads to a universal volcanic shape relationship (Fig. 1c); an ideal OER electrocatalyst with optimum activity requires an intermediate binding strength and an ΔGMO − ΔGMOH value of 1.6 eV (corresponding to the peak of the volcano).29–31
Although the electrocatalytic decomposition of water has been studied since the last century and the basic mechanism of the anodic OER has been revealed, there are still different debates on the mechanism of water decomposition.32,33 Understanding the underlying mechanisms can only help us to find the source of active sites for different materials and to bring the catalyst performance close to the theoretical thermodynamic limit, which contributes to the understanding of the necessary reactions of MnOx in the OER process. In the following subsections, we focus on MnOx electrocatalysts with different crystal structures and their impact on electrochemical properties.
3. Application of manganese-based oxides in the OER
The oxygen-evolving complex (OEC) of photosystem II uses catalytic Mn4CaOn clusters to generate molecular oxygen from water. It is inspired by this MnOx catalyst that is receiving more and more attention.34,35 Preliminary studies have shown that manganese oxides enhance water oxidation due to their elevated thermodynamic stability, suitability of specific surface areas, and oxidation enthalpy.36,37 Moreover, with the introduction of additional characterization techniques and the development of electrochemistry, more stable and efficient materials have been designed and synthesized to improve the catalytic performance of MnOx. Recent advances in MnOx as an OER electrode material are discussed in detail in this section.3.1 MnO2
MnO2 and its composites have been widely used in supercapacitor and battery applications due to their low cost, environmentally friendly nature, high operating voltage, and theoretical capacity. Notably, MnO2 exists in a variety of crystalline polycrystalline forms, such as α-MnO2, β-MnO2, γ-MnO2, and δ-MnO2 (Fig. 2), and the crystal structure is diversified and controllable.38–40 The MnO2 crystal consists of [MnO6] octahedral units with oxygen atoms at the top of the six corners of the octahedron and manganese atoms in the center. These octahedral units are connected by common corners or edges, resulting in the formation of various tunneling and layered structures. MnO2 crystals (α-MnO2, β-MnO2, γ-MnO2, and δ-MnO2) can be classified into three categories: a one-dimensional tunneling structure, a two-dimensional layered structure (δ-MnO2), and a three-dimensional network structure formed by the common sides of [MnO6] octahedra λ-MnO2.41,42 As a consequence of the multivalent state of Mn and a large number of crystalline phases, MnO2 invariably contains impurities from multiple polycrystalline phases, a complex structure–property relation.43 Different crystalline phases significantly affect the catalytic properties of MnO2 and, in particular, the crystal structure of MnO2 plays a key role in determining the catalytic properties. The OER activity is reported to follow the order α-MnO2 > β-MnO2 > δ-MnO2.44,45 What follows is an elaboration of the physical and chemical properties of MnO2 electrocatalysts with different crystalline phases in terms of catalytic activity, electron/ion conductivity, and electrochemical properties.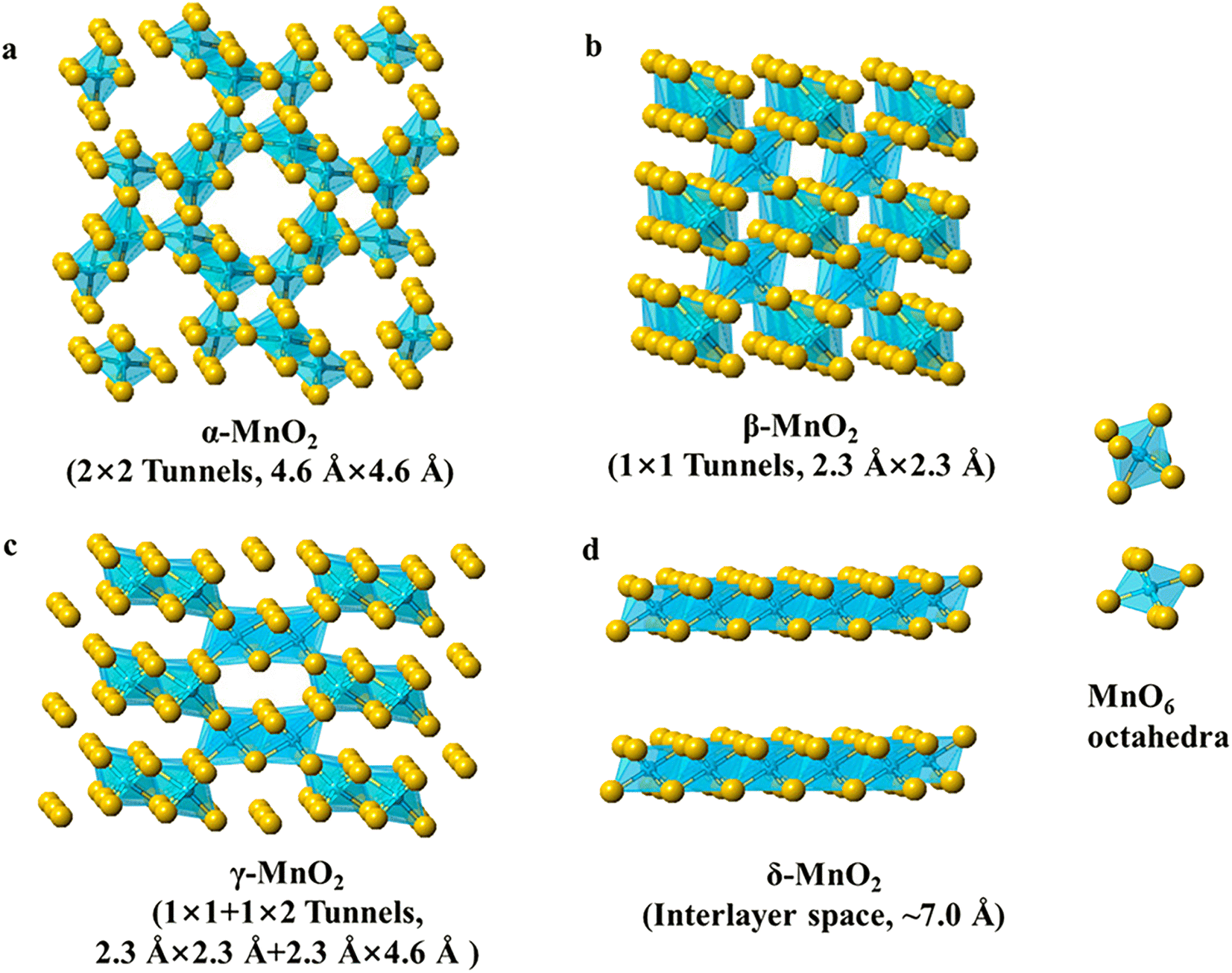 | ||
| Fig. 2 Polyhedral representation of several manganese oxide crystals (α-MnO2, β-MnO2, γ-MnO2, and δ-MnO2). Reproduced with permission from ref. 40. Copyright 2019, American Chemical Society. | ||
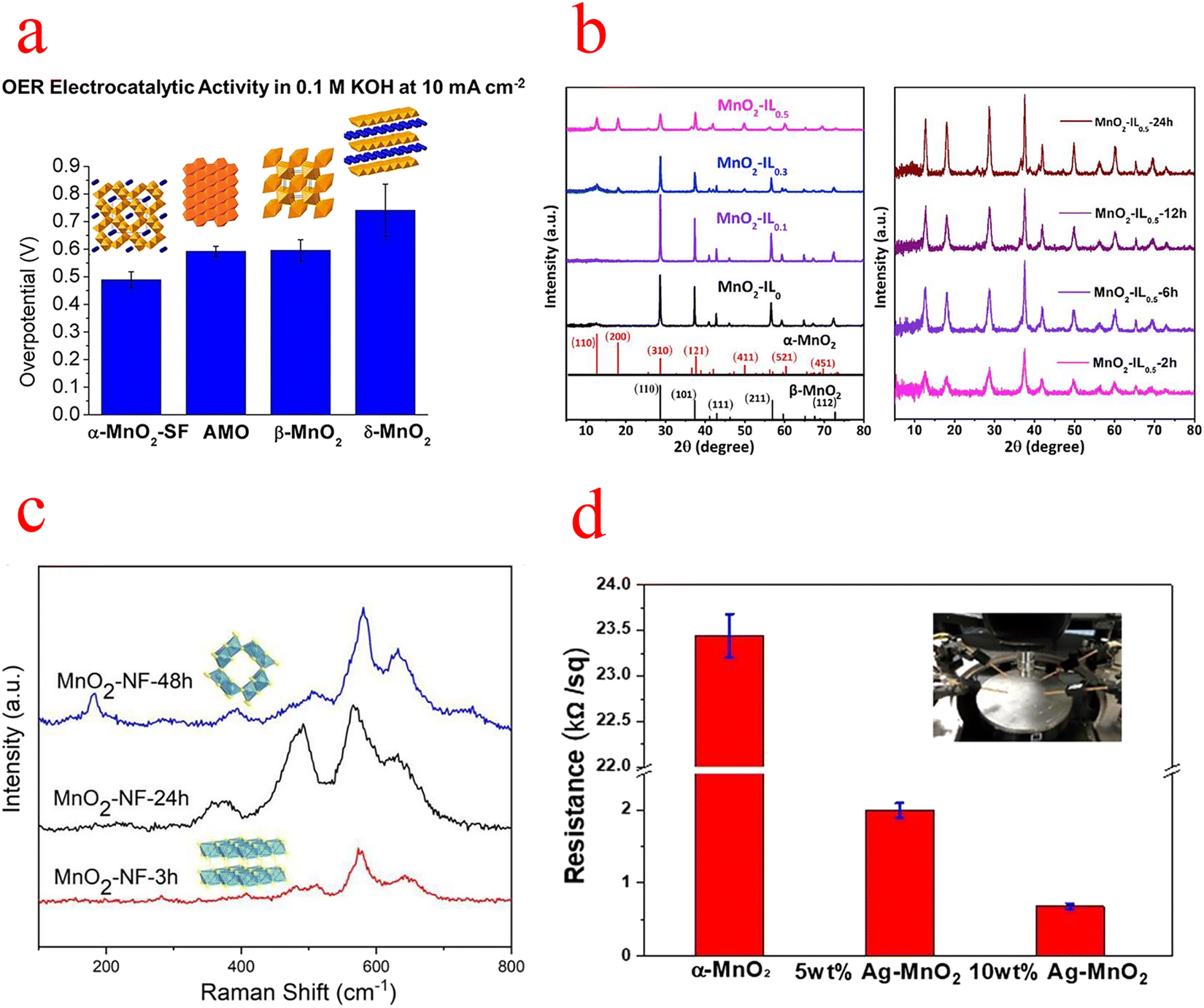 | ||
| Fig. 3 (a) Comparison of the overpotential of MnO2 at 10 mA cm−2. (b) XRD patterns of MnO2 with different IL additions and different reaction times. (c) Raman spectra of MnO2–NF at different hydrothermal times. (d) Resistivity measurements of several types of MnO2. (a) Reproduced with permission from ref. 45. Copyright 2014, American Chemical Society. (b) Reproduced with permission from ref. 49. Copyright 2019, Elsevier. (c) Reproduced with permission from ref. 50. Copyright 2019, Elsevier. (d) Reproduced with permission from ref. 51. Copyright 2019, Elsevier. | ||
Gu used ionic liquids (ILs) as a structural inducer to transform the crystalline structure and morphology of the reaction product from β-MnO2 nanorods to α-MnO2 nanowires with a high specific surface area (Fig. 3b), and X-ray absorption near edge structure (EXANES) and X-ray photoelectron spectroscopy (XPS) analyses show that the transformed α-MnO2 has a high Mn III content and an abundance of oxygen vacancies, which is beneficial for the OER. At 10 mA cm−2, the overpotential is 394 mV and the Tafel slope is low at 49 mV ·dec−1. Also, Gu's research has shown that the crystal structure has a much more decisive and critical influence on the catalytic OER than the surface area.49 Subsequently, Zhou's group similarly used the structural transformation of MnO2 to induce its phase transition from δ to α by merely adjusting the hydrothermal time (Fig. 3c). The transformed α-MnO2 has nanowire and nanosheet morphologies that expand the specific surface area and thus expose more catalytically active sites; the higher content of Mn III ions and oxygen vacancies detected by XPS and electron paramagnetic resonance (EPR) will contribute to better catalytic performance; and density functional theory (DFT) calculations show that α-MnO2 has a more suitable energy band gap (which leads to better electron conductivity and transfer).50 Ni et al. reported that single-atom Ag-doped MnO2 nanowires can catalyze both the OER and oxygen reduction reaction (ORR), due to the strong Ag–O bond weakening the surrounding Mn–O bond, leading to significant crystal distortion, which likely favorably modulates the binding energy of the reaction intermediates, resulting in higher activity. Moreover, the synergistic effect of Ag and MnO2 should enhance the electrocatalytic activity, and the metallic and transition metal oxides modify the electronic structure of the metal and induce a shift of the d-band center. XPS studies confirm that Ag doping produces a large number of surface oxygen vacancies, which open the octahedral walls of MnO2 and facilitate ion diffusion. And the resistivity of α-MnO2 was also measured with a four-point probe, which decreased by more than 20 times after Ag doping (Fig. 3d).51
In order to improve the catalytic activity of β-MnO2, Gu introduced Ru3+ precursor salt in the hydrothermal reaction of β-MnO2. The crystal splitting phenomenon caused by doping drastically affected the OER and ORR activity of β-MnO2. After doping, the obtained nanorods exhibit a fragmented structure as shown in Fig. 4a, and the Fourier-transform extended X-ray absorption fine structure (FT-EXAFS) analysis indicates that the Ru-doped sites are mainly interstitial sites within the tunneling structure of β-MnO2, with some exposure on the surface during crystal splitting, which is the main reason for the elevated OER activity. To explore the active sites for its reaction, DFT confirmed that crystal splitting exposes the atomically dispersed Ru–O species on the MnO2 surface as OER active sites, while the induced lattice strain and Mn3+ state, as well as the abundant oxygen vacancies, can further enhance the ORR activity of the catalyst, which eventually leads to an OER/ORR potential difference of only 0.63 V.55 Yao et al. controlled the concentration of KCl and synthesized β-MnO2 crystals with a bipyramidal structure with tunable morphology using a simple hydrothermal method (Fig. 4b). In situ studies revealed that the K+ cation influences the morphology of β-MnO2 crystals by affecting the formation of the α-K0.5Mn4O8 intermediate phase and the subsequent phase transition process. The elevated concentration of K+ cations reduced the prism length of β-MnO2 crystals by affecting the formation of α-K0.5Mn4O8 nanowires and the tunneling transition process. They speculated that if a suitable surfactant is available to limit the directional attachment of K0.5Mn4O8 nanowires while maintaining the K+ cations, it is possible to significantly reduce the size of the β-MnO2 structure and improve its electrochemical properties. Their work undoubtedly brings current ideas for the design of such materials for applications in supercapacitors, fuel cells, and catalysts.56
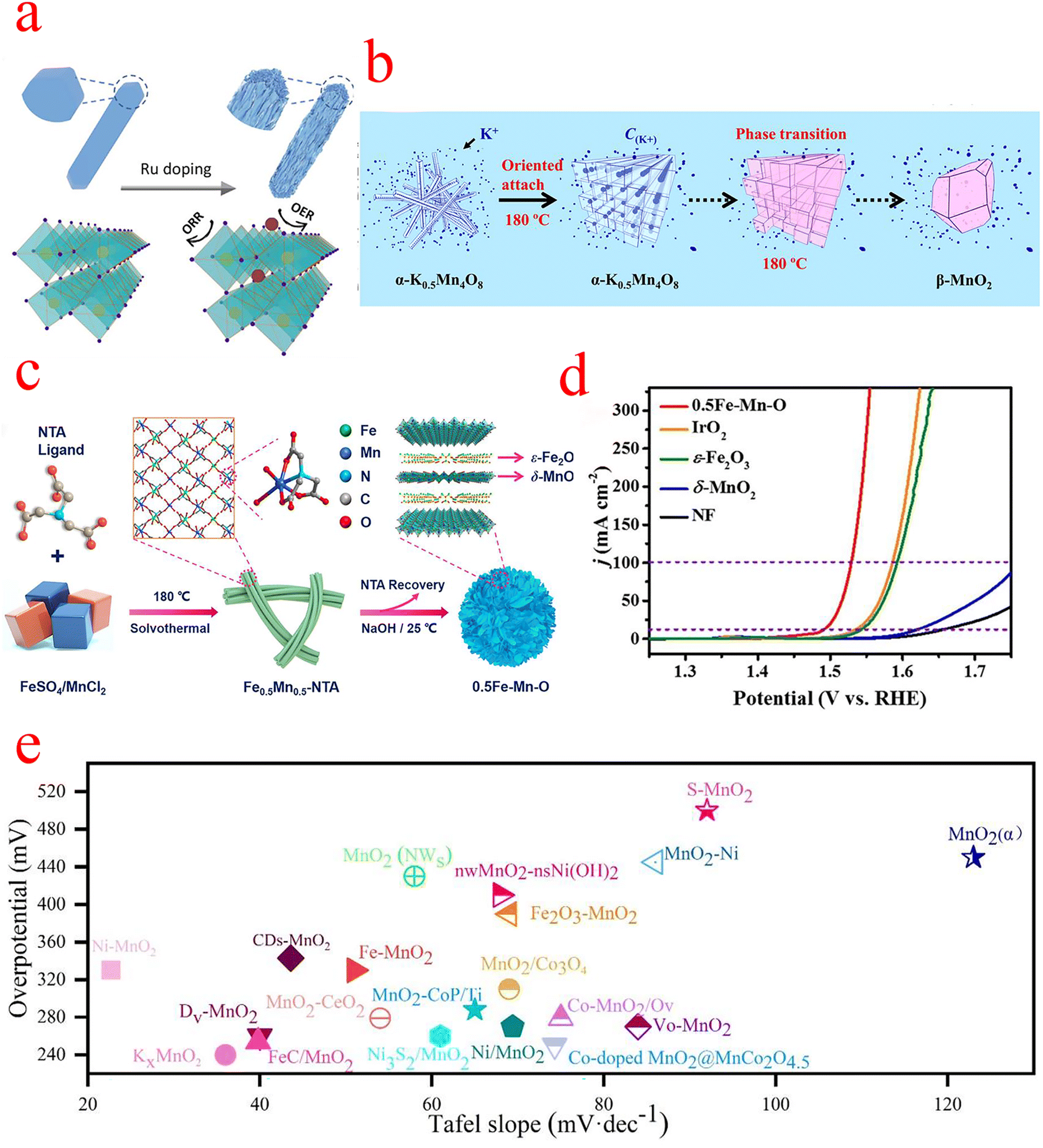 | ||
| Fig. 4 (a) Schematic diagram of Ru doping-induced crystal splitting of β-MnO2 and its OER and ORR sites. (b) The schematic diagram illustrates the directional attachment and phase transformation process. During the phase transition, K+ stable cations are expelled from the crystal structure. (c) Schematic diagram of the synthesis of 0.5Fe–Mn–O nanosheets. (d) Linear sweep voltammetry (LSV) in 1 M KOH aqueous solution. (e) Overpotential and Tafel slope of MnO2 compounded OER electrocatalysts. (a) Reproduced with permission from ref. 55. Copyright 2021, American Chemical Society. (b) Reproduced with permission from ref. 56. Copyright 2018, Elsevier. (c and d) Reproduced with permission from ref. 59. Copyright 2022, Elsevier. | ||
The OER activity of δ-MnO2 primarily depends on the chemical composition, crystal structure, and morphology.58 Cheng et al. prepared ultra-thin amorphous δ-MnO2/ε-Fe2O3 heterojunction nanosheets with oxygen vacancies, denoted as 0.5Fe–Mn–O (Fig. 4c–d), and the catalyst shows high output OER activity, which requires only 299 and 322 mV overpotential to drive current densities of 100 and 300 mA cm−2 and a Tafel slope of only 35.1 mV dec−1. Determination of its indicated chemical state by XPS revealed a higher Mn3+ concentration and proportion of defective oxygen in 0.5Fe–Mn–O compared to δ-MnO2, which was attributed to additional distorted [MnO6] structural units in δ-MnO2 caused by evolved intercalated ε-Fe2O3. The observations further confirm that 0.5Fe–Mn–O consists of δ-MnO2/ε-Fe2O3 heterojunctions with abundant Jahn–Teller active Mn3+, oxygen vacancies, and Fe3+ doping, which facilitates the initiation of interlayer cooperative catalysis and improves OER performance. Further DFT calculations revealed that the oxygen vacancies (Fe–Vo–Mn) immediately adjacent to the Fe and Mn atoms are the optimal OER active sites for the 0.5Fe–Mn–O catalyst, indicating that the oxygen vacancies play an important role in the modified framework for the OER.59 The two-dimensional LDH(+)–Birnessite(−) hybrid catalyst synthesized by Chen et al. exhibited advanced OER catalytic activity and stability with an overpotential of only 407 mV at a current density of 400 mA cm−2 and a Tafel of only 43 mV dec−1, indicating that this hybrid catalyst has good potential for electrolysis of water for hydrogen production. The interlayer NiFe LDH (+) also changes the electronic structure of δ-MnO2 and generates an electric field between NiFe LDH (+) and δ-MnO2(−), which leads to a significant reduction of the overpotential of the OER. DFT calculations confirm the modulation of NiFe LDH during the OER, and the upward shift of Fe-3d orbitals in LDH promotes the electron transfer from δ-MnO2 to LDH, which greatly enhances the performance of the OER.60
Interlayer cations play an essential role in tuning the electronic structure of δ-MnO2 materials. An effective method for increasing the catalytic activity of layered manganite oxides was developed by McKendry et al. through cooperative doping of a two-dimensional lattice and interlayer. The doping of the MnO2 lattice with cobalt allows for more efficient hole transport to the active sites of the interlayer iron oxides; meanwhile, the iron hydrazine intercalation procedure reduces the particle size and increases the concentration of Mn3+ defects. With the addition of intra-layer cobalt and inter-layer iron, a highly efficient OER catalyst was obtained with an overpotential potential drop of 425 mV compared to birnessite.61 Pu synthesized efficient catalysts containing nickel in two-dimensional layered metal oxides (δ-MnO2) by a simple ion-exchange reaction. The intercalation mechanism and oxidation states of nickel and manganese have been explored using the synchrotron X-ray absorption spectroscopy (XAS) technique. It was shown that the intercalation of nickel occurs at the Mn3+ site by reducing Mn3+ to Mn2+ and that the intercalated nickel is the active site for the OER. The synthesis scheme of this work can be broadly applied to provide ideas for the structural development of other layered metal oxides.62
In Fig. 4e, we list the overpotentials and Tafel plots of MnO2 compounded with other materials in recent years, most of which are limited to around 300 mV, and their range of application and catalytic activity is not superior. Although MnO2 has great potential as a non-precious metal oxide catalyst in the field of water electrolysis, it still suffers from problems such as the small number of active sites. Meanwhile, some researchers have designed different MnO2 nanomorphologies to increase the number of active sites on the surface, with nanowires exhibiting the highest OER mass activity due to the highest specific surface area and nanotubes exhibiting the highest specific activity due to the high Mn3+ surface concentration/surface defects.63–65 On the other hand, in addition to the use of monoatomic metals and MnO2 composites to enhance catalytic activity, Xu et al. used MnO2-NWRS/CNTs, a composite of carbon nanotubes and MnO2, to show good catalytic activity as well as conductivity in the ORR and OER.66 We believe that the development of MnO2 in the field of OER electrocatalysts will be accelerated with the preparation of more advanced materials and the design of chemical compositions based on the existing foundation.
3.2 Mn2O3
In the family of MnOx, Mn2O3 has attracted extensive attention in the field of supercapacitors and batteries due to its excellent energy intensity and simple synthesis process. However, there are few types of studies on electrocatalysis.67,68 The crystal structure of Mn2O3 is shown in Fig. 5a, where the Mn3+ cation is coordinated to the O anion in an octahedral, orthogonal crystal system.69 The OER activity of Mn2O3 is attributed to the presence of a Jahn–Teller distorted shared edge octahedron occupied by Mn3+ (d4) centers with significantly elongated Mn–O bonds. Due to the asymmetric occupation of eg orbitals, the bond energy is reduced and the Mn3+–O octahedron is activated to participate in the OER.70 It has been reported that the overpotential value of Mn2O3 is considerably higher than that of other transition metal oxides, and due to the special d-electron structure of Mn2O3, it is very difficult to synthesize pure phases of Mn2O3.71 Zhao monitored the OER process on the Mn2O3 catalyst surface using in situ liquid TEM and observed the development of oxygen nanobubbles around the catalyst in space. In addition, in situ TEM observed that the thickness of the amorphous layer on the Mn2O3 surface is in a variable state, and they conjectured that the high-valence Mn4+ should be reduced to Mn3+ in the generation of molecular O2. This suggests that the amorphous layer on the Mn2O3 surface is the main active region as well as that the valence of Mn is constantly changing, possibly to be more adapted to the different steps of the OER process.72 The poor electronic conductivity and relatively low OER dynamics of Mn2O3 should be improved by combining with an additional state with higher OER activity. Bigiani used the first row of transition metal (Fe, Co, Ni) oxide nanoparticles (NPs) as OER electrocatalysts in alkaline media. Through plasma-assisted fabrication, Fe2O3, Co3O4, and NiO NPs can be efficiently dispersed into Mn2O3 and form tight oxidation interfacial contacts, which can enhance charge carrier transport and facilitate the diffusion of reactants and products.73 The unique hollow RuO2/Mn2O3 composite fiber prepared by Yoon shows excellent OER electrocatalytic performance in alkaline media.74 Another research group, Yang et al. synthesized Ni2P/Mn2O3 ultra-high electrochemically active area catalysts by the electrostatic spinning technique, which required only a low overpotential of 280 mV to provide 10 mA cm−2 current density, superior to many previously reported Mn2O3 catalysts. The large aspect ratio of the one-dimensional nanofibers facilitates electrolyte penetration and oxygen bubble release, and the introduction of Mn modulates the electronic structure of Ni2P and greatly improves the OER performance.75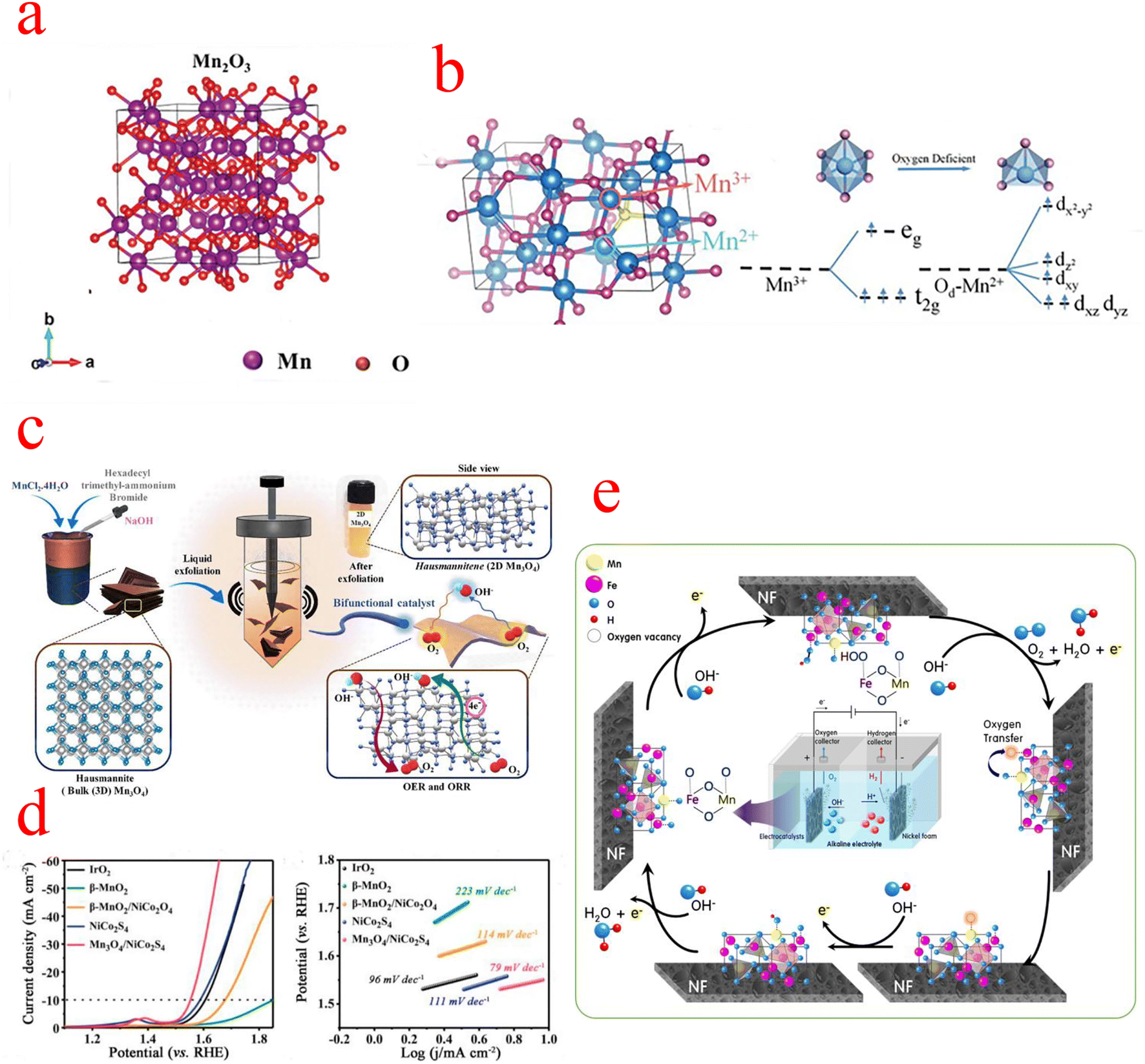 | ||
| Fig. 5 (a) Mn2O3 crystal structure. (b) Crystal structure of Mn3O4, octahedra of [MnO6], and d-orbital splitting conformation. (c) Schematic representation of the liquid phase exfoliation method with Mn3O4 formation. (d) LSV curves and Tafel slope plots. (c) The OER process on the MnFe2O4/NF catalyst. (a) Reproduced with permission from ref. 88. Copyright 2021, John Wiley and Sons. (b) Reproduced with permission from ref. 89. Copyright 2020, John Wiley and Sons. (c) Reproduced with permission from ref. 78. Copyright 2022, Elsevier. (d) Reproduced with permission from ref. 82. Copyright 2020, Elsevier. (e) Reproduced with permission from ref. 87. Copyright 2021, Elsevier. | ||
3.3 Mn3O4
Mn3O4 belongs to the spinel group, where divalent and trivalent manganese ions are distributed in two distinct lattice sites. The oxygen ions are closely arranged in a cubic form, with divalent manganese ions occupying tetrahedral voids and trivalent manganese ions occupying octahedral voids (Fig. 5b).76 The general formula of the spinel type is AB2O4, which consists of a tetrahedral cation A with a divalent charge and an octahedral cation B with a trivalent charge. The spinel structure is nicely conductive due to the excellent electron transfer between the different oxidation states A2+ and B3+.77 Gowda et al. carried out an in-depth study of the origin of the different active sites of the OER and ORR in Mn3O4. They synthesized two-dimensional Mn3O4 by liquid phase exfoliation (LPE), as shown in Fig. 5c, and DFT calculations confirmed the presence of two sites, Mn(III) and Mn(II), on the (112) oriented surface of Mn3O4, which are highly active for the OER and ORR, respectively.78 In the early studies on Mn3O4, electrochemical and spectroscopic techniques were used to characterize their reactivity in the OER.79 For example, MnOx films obtained by electrodeposition have a different composition and polycrystalline morphology; after cyclic activation, the Mn(OH)2 generated by the cathode causes a phase transition of (δ-MnO2) to an intermediate product of (α-Mn3O4) type, and the catalytic activity of MnOx after activation is much higher than that of the original MnOx. It has been shown that different manganite systems in the literature have different OER activity curves, as different manganite compositions and crystals may be introduced by the preparation methods.80Recently, the synergetic chemical coupling effect and interface structure engineering have played a crucial role in the development of Mn3O4 materials. Guo enabled the chemical coupling of active CoO nanoclusters with Mn3O4 octahedra; the strong interaction between the two components promotes the formation of a high-energy interface Mn–O–Co and a highly oxidized state of CoO, and this high-energy structure has a large number of active sites that facilitate rapid charge transfer. The starting potential and catalytic kinetic analysis of the catalysts indicate that the OER activity of CoO/Mn3O4 is mainly derived from CoO, while the coupling of CoO to Mn3O4 further enhances the catalytic performance and provides a higher catalytic activity than the Ru/C OER activity.81 Mn3O4 is the most stable form of MnOx at high temperatures and can be used as a suitable substrate for the construction of composite structures with other active materials. Wang constructed one-dimensional Mn3O4/NiCo2S4 rod-like nanomaterials based on the two-phase composite of metal oxide and sulfide, and its catalytic activity is shown in Fig. 5d. According to the Kirkendall effect, the outward diffusion of Mn4+ and O2− and the sulfidation process of NiCo2O4 proceed almost simultaneously, which leads to the formation of a considerable number of oxygen defects at the interface of Mn3O4 and NiCo2S4 because the diffusion rate of Mn4+ is much faster than the diffusion rate of O2−. The chemical reaction at the interface leads to the formation of vacancies and defects, which can serve as catalytically active sites.82 Huang's work prepared hybrid catalysts of nanoparticles Co3O4/Mn3O4 anchored on (reduced graphene oxide) rGO substrates. Although Co3O4 has excellent ORR/OER activity, it is very unstable at high current densities, and the introduction of Mn3O4 exactly addresses this issue. Meanwhile, the large specific surface area provided by the layered rGO facilitates the contact between the catalyst and the electrolyte, thus improving the utilization of active sites and the reaction kinetics. In addition, DFT calculations showed that after coupling Mn3O4 (211) with Co3O4(111), the ΔGMOH→OH value on the Mn3O4 (211) surface of Co3O4/Mn3O4 was close to −1.23 eV, which is the conceptual value of ΔGMOH→OH for the catalyst at 0.4 V equilibrium potential at pH = 14, yielding that the Co3O4/Mn3O4 Mn3O4 (211) surface is the active surface for the ORR/OER.83 Cho and his team demonstrated a breakthrough synthesis of 4 nm Mn3O4 nanoparticle (NPs) catalysts with an overpotential of only 395 mV at 10 mA cm−2, a performance superior to any other Mn-, Fe-, Co-, and Ni-based electrocatalysts reported so far under neutral conditions. They then further optimized their catalytic performance by Ni doping, and XRD analysis showed Ni doping-induced lattice distortion of the Mn3O4 NPs, which also led to the production of distorted octahedral Mn(III) species, enhancing the number of active sites in the OER. According to electrokinetic studies, the OER catalytic mechanism of pristine and 5% Ni-doped Mn3O4 NPs is similar, namely involving reversible one-electron and one-PCET processes prior to the RDS. This implies that the doping of Ni in Mn3O4 NPs does not affect the OER mechanism under neutral conditions. The enhanced activity of Ni doping may be due to the promotion of this step of O–O bond formation.84–86
Some research groups use low-cost transition metal spinel materials as OER catalysts. Kim synthesized Mn–Fe-based materials with a spinel structure and proposed the OER mechanism on the MnFe2O4/NF electrode during water electrolysis in an alkaline solution (Fig. 5e). The OH− ions decomposed in water combined with the lattice oxygen of Fe2+ and the FeO in MnFe2O4 is oxidized to Fe–OOH (Fe3+). In the next step, Fe–OOH continues to be converted to Fe–OO by OH−, and then O2 is oxidized and desorbed. After O2 is generated, oxygen vacancies are formed in the iron lattice of the anode catalyst. At this point, Mn3+ with strong reducing power moves the oxygen in its lattice through the spinel lattice into the vacancies generated around Fe2+, which is then reduced to Mn2+. After that, the oxygen vacancies formed in the Mn2+ lattice are rapidly filled with OH− ions in water and regenerated into Mn3+ ions. Through this process, the MnFe2O4 spinel lattice is restored to its initial state. In conclusion, the synergistic oxygen transfer process between Fe2+ (or Fe3+) and Mn3+ (or Mn2+) catalytic species in the MnFe2O4/NF of the OER anode contributes to the long-term stability of the catalytic electrode for the OER.87
4. MnOx issues and strategies
MnOx is a potential OER catalyst whose oxides exist in more than 30 different stable polycrystalline forms, and the crystal structure and variable valence states are key to improving its electrostatic properties.44,90,91 However, their electrocatalytic activity is still not sufficient to replace noble metal oxides. To explore efficient MnOx electrocatalysts, it is essential to clarify the dominant factors controlling the electrocatalytic activity of these oxides.92 Above we have comprehensively reviewed the progress of MnO2, Mn3O4, and Mn2O3 materials, many of which have encountered several common problems such as Jahn–Teller distortion, active sites, stability, etc. These critical issues have hindered the understanding of catalytic reaction mechanisms and the development of efficient and stable catalysts.4.1 Jahn−Teller distortion
Jahn–Teller distortion is very common in Mn-based materials, and it is believed that the presence of Mn3+ can induce this phenomenon, thereby explaining the structural degradation of Mn-based electrode materials.93 The high spin electron configuration of the Mn(III) (t2g)3(eg)1 octahedron in the octahedral unit usually experiences Jahn–Teller distortion, resulting in long and flexible Mn–O bonds, for the OER, (eg)1 is considered to be the best electronic configuration for transition metal-based catalysts.94 Compared with manganese(IV), the d4 electron configuration of manganese(III) is more inclined to show the Jahn–Teller effect. In addition, based on the strong Jahn–Teller effect, the transition from Mn4+ to Mn3+ inevitably prolongs the Mn–O bond and causes electron redistribution on the surface.95,96Kang et al. stabilized the Jahn–Teller active Mn3+ species by replacing the Mn4+ ions with the more electronegative Ru4+ ions (Fig. 6a), which led to the stabilization of the Jahn–Teller active Mn3+ species and also provided the electronegative Ru sites. Due to bond competition, the adjacent (Mn–O) bond is weakened by (Ru–O), leading to distorted [MnO6] octahedra with elongated (Mn–O) bond distances, which stabilizes the Mn3+ ion. Although Ru substitution also provides electrocatalytically active Ru sites for α-MnO2, this contribution is smaller than the stabilization of Mn3+ species caused by adjusting the (Mn–O) bond covalency.97 Yang proposed a strategy to thermally induce structural deformation of calcium manganese ore (CaMnO3, denoted as CMO) to improve the oxygen catalytic activity of CMO. When O atoms were partially extracted from CMO (denoted as D-CMO), the structure of [MnO6] octahedra was deformed due to the Jahn–Teller effect (Fig. 6b). They used simulations to model the reaction pathway of the OER (Fig. 6c). The Mn cation on the surface can coordinate with OH− to form an Mn–OH bond, O–Mn–O(OH) is the starting point of the OER process, while the third step with the formation of OOH* (O* + OH− → OOH* + e) is the rate-determining step (RDS), and the RDS shows that the theoretical overpotential of the OER for D-CMO (0.74 V) is lower than that of CMO (0.82 V). When O2 is adsorbed on D-CMO, the average number of electron transfers from Mn to O atoms is more than that of CMO. This means that the Mn–O and strong interactions are shorter, which contributes to the activation of O2 and reduces the Gibbs free energy of the activation step (* + O2 → O2*).98 The above examples demonstrate that the strategy of inducing [MnO6] octahedral deformation using the Jahn–Teller effect can provide a new and effective way to improve the electrocatalytic properties of MnOx.
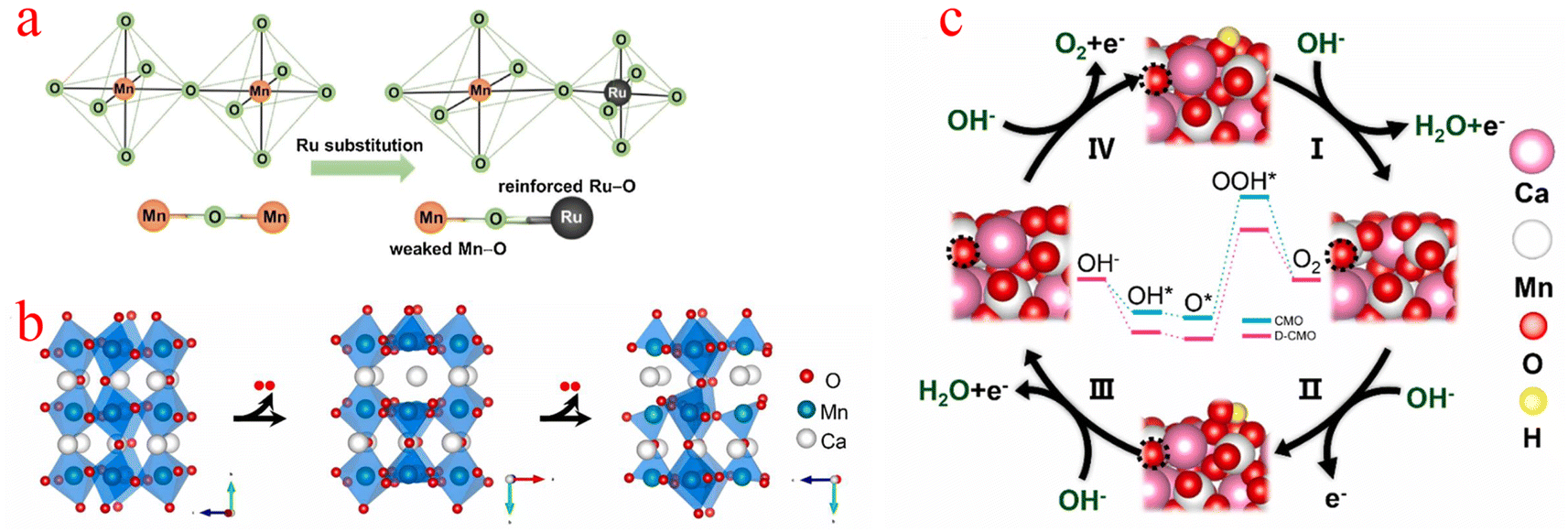 | ||
| Fig. 6 (a) Schematic diagram of the evolution of the adjacent (Mn–O) bond when the (Ru4+–O) bond is incorporated into the α-MnO2 lattice. (b) Schematic diagram of the crystal structure process of O in CaMnO3 being extracted. (c) Reaction pathways and corresponding reaction free energy diagrams for the OER of CMO(121) and D-CMO(121) at 1.23 V potential. (a) Reproduced with permission from ref. 97. Copyright 2018, Elsevier. (b and c) Reproduced with permission from ref. 98. Copyright 2021, Elsevier. | ||
4.2 Active sites
The second is the question of active sites in MnOx. A remarkable correlation between the amount of Mn3+ and the catalytic activity of the OER has been reported. Therefore, strategies to increase the activity of MnOx must take into account the enhancement and stabilization of the Mn3+ species amount. Numerous previous studies have also improved the catalytic performance by introducing Mn(III) in different crystalline phases and found that a large amount of Mn(II) and Mn(III) was oxidized to Mn(IV) in electrocatalytic tests, leading to a decrease in activity.99,100Back in 2012, Takashima's group found that the low catalytic activity of MnO2 under neutral conditions can be attributed to the instability of Mn3+, whose accumulation on the catalytic surface requires Mn2+ oxidation at a potential above 1.4 V, as shown in Fig. 7a. They effectively stabilized the Mn3+ species by the formation of N–Mn bonds through the coordination of amine groups with the Mn sites on the MnO2 electrode surface, and in situ spectroelectrochemical techniques demonstrated that stabilization of surface-associated intermediate Mn3+ species is an effective strategy to reduce the electrolytic water overpotential of MnO2.101 Later, Park, et al. in 2014 chose a pyrophosphate-based Mn compound (Li2MnP2O7) as a model system and observed the effect of the Mn(III) state itself on water oxidation catalysis by adjusting the manganese valence in Li2MnP2O7. Remarkably, the OER catalytic performance increases continuously as the average oxidation state of manganese in Li2−xMnP2O7 increases from 2 to 3, as shown in Fig. 7b.102
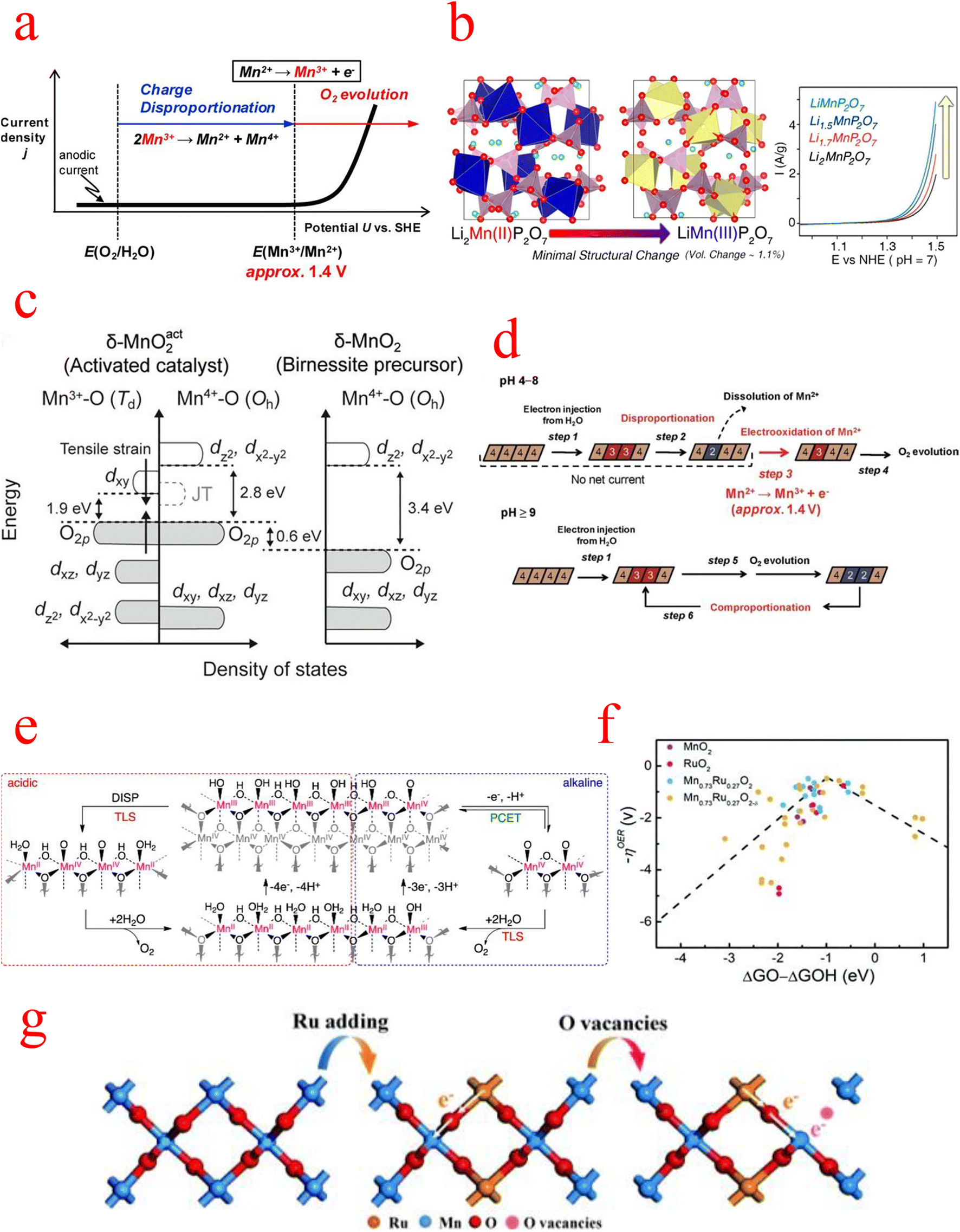 | ||
| Fig. 7 (a) Schematic illustration of a current density (j) vs. potential (U) curve for a pristine δ-MnO2 electrode under neutral pH conditions. (b) Increase in the average oxidation state of Mn from 2 to 3 in Li2−xMnP2O7 and schematic representation of the catalytic performance. (c) Schematic diagram of the band structure of the activated catalyst system derived from the “α-Mn3O4-3e” model. (d) Schematic diagram of the oxidation state of Mn ions in the δ-MnO2 reaction at different pH values. (e) Proposed mechanism for the OER on MnOx under (left) acidic and (right) alkaline pH conditions. (f) Schematic free energy profiles of overpotential results versus the difference between the adsorption energies of O* and OH* intermediates for MnO2, etc. in acid media. (g) A schematic diagram of the charge redistribution process in Mn0.73Ru0.27O2−δ. (a) Reproduced with permission from ref. 101. Copyright 2012, American Chemical Society. (b) Reproduced with permission from ref. 102. Copyright 2014, American Chemical Society. (c) Reproduced with permission from ref. 105. Copyright 2018, Proceedings of the National Academy of Sciences of the United States of America. (d) Reproduced with permission from ref. 107. Copyright 2012, American Chemical Society. (e) Reproduced with permission from ref. 108. Copyright 2014, American Chemical Society. (f and g) Reproduced with permission from ref. 109. Copyright 2022, RSC Publishing. | ||
In recent years, more scholarly studies have pointed out that Mn3+ is the active center of the OER, and its electronic configuration is (t2g)3(eg)1, which plays an important role in the water oxidation reaction because the eg orbital is involved in the σ-bonding to the anion adsorbate, the occupancy of this orbital directly determines the binding energy of the reactants and leads to a volcano-like activity profile.94 Higher Mn3+ content in the manganese group is beneficial for enhancing the adsorption of –OH at the catalyst surface, and higher surface oxidation valence states are also beneficial for improving the OER activity of manganese-based materials.103 In studying the OER activity of β-MnO2 on the (101) and (110) faces, Nakamura's group found that the interlayer charge ratios of Mn2+ and Mn4+ produced two Mn3+ species on the (101) face, resulting in 11 times more Mn3+ coverage on the (101) face than on the (110) face, and thus a higher OER activity on the (101) face.104 The study by Chan et al. explained that the presence of Mn3+ enhances the catalytic effect of the OER. They observed through Raman spectroscopy that the Mn3+ tetrahedron (Td) produces local strain on the oxide lattice. This strain causes the O 2p valence band to be higher than the Mn3+ tetrahedron (Td) and Mn4+ octahedron (Oh) valence bands and leads to the gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) to decrease, while the reduced HOMO–LUMO gap promotes the oxygen evolution reaction. As shown in Fig. 7c, the Mn3+ ions at the Td site are unable to form Mn4+, which causes a rearrangement of the Mn-d and O 2p states and promotes the formation of oxygen vacancies, factors that facilitate the OER to produce oxygen.105 Moreover, in the study of spinel structures, the nature of their OER activity is also closely related to the valence states of Mn. Wei found that Mn in the octahedral sites was identified as the active site of MnCo2O4 and that the ORR/OER activity of MnCo2O4 exhibited a volcanic shape as a function of the Mn valence state in the octahedra, while the apex of the ORR/OER volcanic trend was located at the position of the Mn valence state ≈ +3.106
4.3 Stability of MnOx
Experimental and theoretical work on MnOx catalysts has emphasized the role of Mn(III) as an essential intermediate state for achieving catalytic reactions. Regardless of the initial Mn oxidation state in the catalyst, Mn(III) is generated and participates in the water oxidation process. Therefore, the question of the stability and reconstruction of Mn3+ in MnOx needs to be discussed next.102MnOx can act as an effective electrocatalyst under strongly alkaline conditions but is inefficient under neutral and acidic conditions. In a 2012 review, Takashima et al. investigated the OER process in MnO2 and found fundamentally different mechanisms for the OER in acidic, neutral, and basic conditions. At pH < 9, the larger overpotential is due to the rapid consumption of Mn3+ by the disproportionation reaction, as shown by the following reaction:
| 2Mn3+(aq) + 2H2O → MnO2(s) + Mn2+(aq) + 4H+ |
while at pH ≥ 9, the disproportionation of Mn3+ is effectively inhibited, which leads to a very easy accumulation of Mn3+ in the MnO2 layer, resulting in a large decrease in the overpotential; moreover, in this pH region, the comproportionation reaction between Mn2+ and Mn4+ contributes to the regeneration process of Mn3+ (Fig. 7d). Therefore, the pH of the electrolyte has a significant effect on the productivity and accumulation of Mn3+, which further affects the electrocatalytic performance.107 Subsequently, Huynh et al. performed a kinetic study of the OER mechanism of MnOx over a range of pH values under acidic, neutral, and alkaline conditions. Tafel and reaction order analyses reflect two competing mechanisms: a single-electron single-proton PCET pathway, which dominates under alkaline conditions, and an Mn3+ disproportionation process, which dominates under acidic conditions (Fig. 7e). Another significant point of this study is that it shows that MnOx has a high intrinsic and functional kinetic stability in acidic electrolytes and its ability to remain self-healing for electrolytes with a pH greater than zero, and MnOx can undergo the OER in extremely acidic environments.108 As for the development of MnOx in acidic media, Wang's group has made new progress. They developed Mn–Ru solid-solution oxides with oxygen vacancies (named Mn1−xRuxO2−δ), which is currently the better-performing MnOx catalyst in acidic electrolytes. Among the synthesized catalysts, Mn0.73Ru0.27O2−δ with the optimized electronic structure only requires an overpotential of 208 mV to produce a current density of 10 mA cm−2 in an acidic electrolyte and a Tafel slope of 65.3 mV dec−1, which exceeds RuO2 and most noble metal-based OER electrocatalysts. The volcano diagram and the difference between the overpotential results and the adsorption energies of the O* and OH* intermediates are shown in Fig. 7f. The Mn0.73Ru0.27O2−δ model is at the peak of the volcano diagram, indicating the lowest overpotential value for the OER of all models studied. DFT calculations show that the introduction of Mn can alter the RDS transition from O* → OOH* to OOH* → OO*. In addition, EXANES shows that the valence of the Mn species increases significantly after the OER process, while the valence of the Ru species remains almost unchanged, indicating that the Mn atom is converted into an electron donor and that the Ru species is the true OER active site (Fig. 7g).109
Normally speaking, the (t2g)3(eg)1 orbital and exceptionally stable Mn3+ ions in MnOx are extremely vital to improving their catalytic performance; on the other side, more and more studies have been carried out on the octahedral unit of [MnO6] because the structural changes caused by octahedral distortion can strengthen the orbital interaction between the Mn center and oxygen intermediate, thus considerably promoting the oxygen catalytic kinetics. Therefore, to design high-catalytic activity manganese-based catalysts, it is necessary to consider factors such as the active sites of the electronic orbitals. In the following part of this chapter, we propose some improvements for reference, such as defect engineering (introducing vacancies), interface engineering (heterogeneous interfaces), etc.
4.4 Improvement strategies
| –Mn4+–O2–Mn4+– → –Mn3+–*–Mn3+–+1/2O2 |
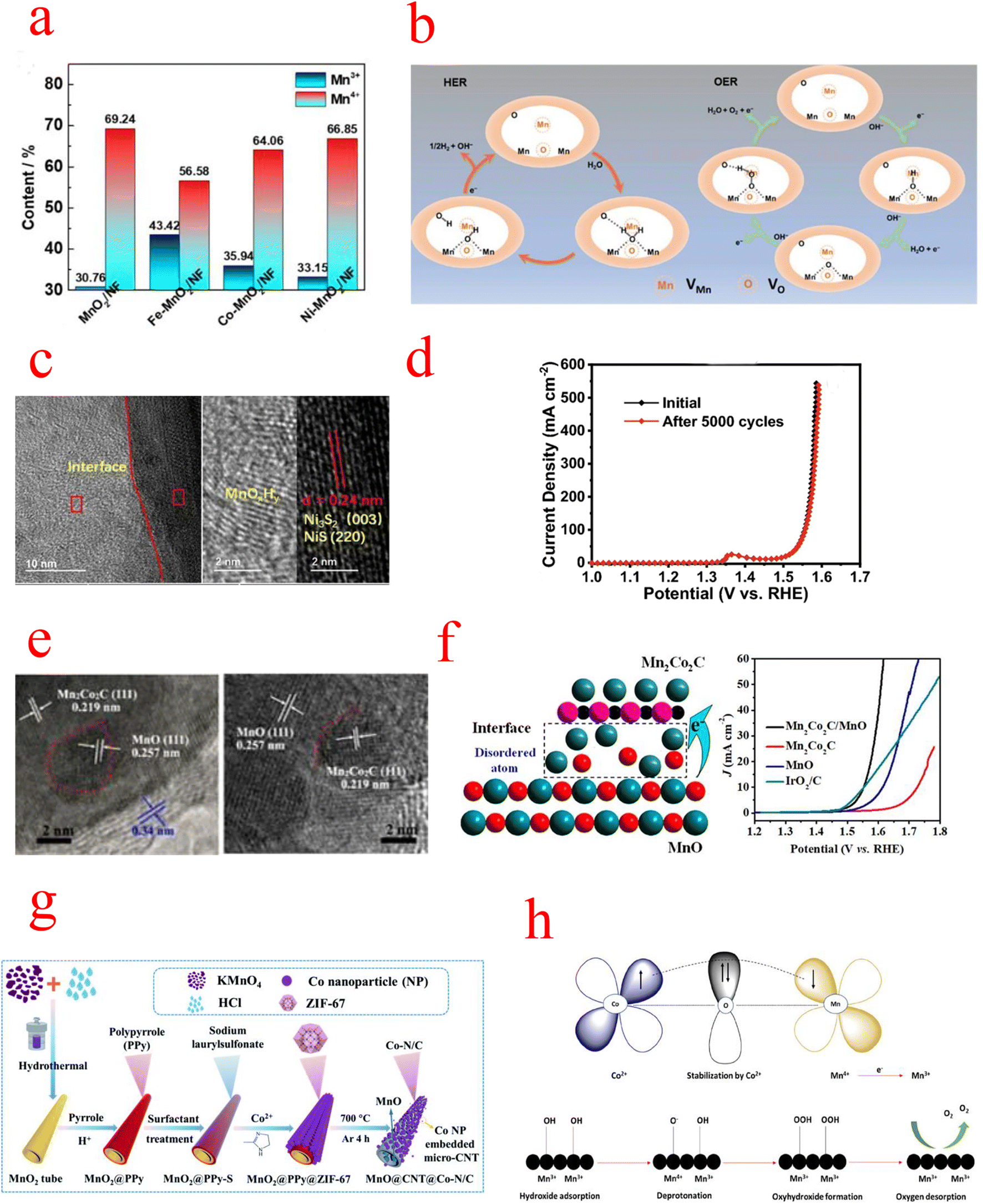 | ||
| Fig. 8 (a) Schematic illustration of the formation of Fe–MnO2/NF. (b) Schematic illustration of the HER and OER processes of DV–MnO2 under alkaline conditions. (c) HRTEM images of NixSy@MnOxHy nanorods. (d) Comparison of LSV curves for NixSy@MnOxHy before and after the 5000-cycle stability test. (e) HRTEM images (f) schematic catalytic mechanism of Mn2Co2C/MnO. (g) Schematic illustrating the preparation of the 1D MnO@CNT@Co–N/C hierarchical nanostructure. (h) Probable OER mechanistic pathway (a) Reproduced with permission from ref. 103. Copyright 2021, Elsevier. (b) Reproduced with permission from ref. 115. Copyright 2021, John Wiley and Sons. (c and d) Reproduced with permission from ref. 120. Copyright 2022, Springer Nature. (e and f) Reproduced with permission from ref. 122. Copyright 2020, American Chemical Society. (g) Reproduced with permission from ref. 123. Copyright 2021, Royal Society of Chemistry. (h) Reproduced with permission from ref. 124. Copyright 2022, American Chemical Society. | ||
NixSy@MnOxHy/NF constructed by Wang exhibits excellent activity and stability in alkaline solutions. Since the NixSy electrocatalyst is easily oxidized to the corresponding metal oxides/(oxygen) hydroxyl oxides during the OER, the MnOx shell protection strategy can effectively improve its catalyst stability. The integration of MnOxHy and NixSy forms a heterostructure with abundant Mn–S bonds(Fig. 8c), leading to stronger electronic interactions, and its activity may be the conversion of NiS into amorphous Ni species (NiOxHy), while the synergistic interaction between MnOxHy and NixSy significantly accelerates the kinetics of HER and OER processes and enhances the charge transfer, and the OER performance remains intact after 5000 CV cycles (Fig. 8d).120 Niu et al. constructed nanowire porous structures using low nitrogen acetic acid (NTA) as a chelating agent to stabilize Mn2+ and Co2+ metal ions. MnOx alone is not effective as a bifunctional catalyst due to its insufficient OER activity and electron transfer rate limitation, so the synergistic effect between the metal Co and MnOX was exploited to design bifunctional electrophiles for OER and ORR catalysts. In particular, a strong charge exchange between Co (200) and MnOX (200) occurs at the heterogeneous interface between metal Co and MnOX. This means that the O atoms can easily gain electrons from the Co atoms, which leads to a change in the electronic structure of pristine manganites, and charge redistribution facilitates the formation of abundant catalytically active sites on the OER and ORR surfaces.121 Meng prepared Mn2Co2C/MnO catalysts with cubic porous structures by pyrolysis, and TEM revealed the presence of abundant heterogeneous interfaces between Mn2Co2C and MnO (Fig. 8e), while XPS confirmed that MnO has an elevated oxidation state [Mn(2+δ)+], which is favorable for OH− adsorption and subsequent desorption processes. In addition, extended EXAFS measurements further revealed the presence of disordered Mn/O atoms and dangling bonds at the heterogeneous interfaces, which act as additional active sites for adsorption/desorption (Fig. 8f).122 Interfacial engineering is not a combination of any two catalysts. As mentioned above, there are interfaces that increase the conductivity of a single component, others that increase the active site, and a combination that achieves small reaction activation energies by modulating the energy band structure. Shell–core interfaces have also been designed to take advantage of the durability of MnOx. However, the active sites of MnOx heterojunctions are mostly catalysts of another set of components, and how to design catalysts with MnOx as the active center is a future trend.
MnOx can be used not only as a framework for metal–organic structures but also as a catalyst for the ORR/OER, which is commonly combined with conductive materials (conductive polymers or carbon materials) to enhance their electrical conductivity and catalytic properties. Li grew polypyrrole (PPy) nanotubes (NTs) and zeolite imidazole framework-67 (ZIF-67) sequentially on MnO2 NTs as the substrate and oxidant, and subsequently formed a one-dimensional (1D) layered ternary nanocomposite by sintering, as shown in the flow chart in Fig. 8g. The MnOx particles separated in carbon nanotubes (CNTs) have a mixed valence of Mn2+/4+, which can greatly facilitate the diffusion of electrolytes and electron transfer in redox reactions. In addition, the Co–N/C formed on CNTs provides multiple catalytically active sites (e.g., Co–Nx, Co–O, and C–N molecules), and the MnO@CNT@Co–N/C catalysts exhibit better activity and durability compared with commercial catalysts of noble metals.123,130 Wang introduced Mn species into the zeolite-imidazole frameworks (ZIFs) and then further pyrolyzed the Mn-containing bimetallic ZIFs to synthesize Co@Co4N and MnO embedded in porous N-doped carbon nanocubes. The introduction of Mn species not only increased the surface content of pyridine/graphitized N and Co4N in Co@Co4N/MnO-NC but also converted during calcination to the MnO phase. These high pyridine/graphitic N and moderate MnO species provide efficient catalytic performance for the ORR, while the uniformly distributed Co@Co4N nanoparticles ensure an abundance of active sites for the OER.131,132 Selvasundarasekar et al. constructed a bimetallic ZIF catalyst containing Co and Mn metal ions and formed the fibers by the electrospinning technique. Molecular orbital studies evaluated the active centers of the bimetals and found that the Co2+ concentration inhibits the Jahn–Teller distortion produced by Mn3+, while OER activity is mainly attributed to Mn3+ ions and a moderate amount of Co2+ leads to a stronger overlap between the anti-bound Mn3+ eg orbitals and oxygen adsorbed O 2p orbitals, enhancing the shorter binding nature of the OER intermediates on the catalytic surface. And a detailed OER mechanistic pathway was hypothesized (Fig. 8h). First, Co2+ is initially stabilized by the high oxidation of Mn(IV), then it is reduced to Mn3+, after which OH− ions are adsorbed on the Mn3+ catalytic surface and subsequently it is converted to oxides and oxyhydroxides. Finally, O2 is desorbed from the catalytic surface. The work of these individuals provides ideas for the search for active centers in bimetallic site catalysts.124
| MnOx composite catalysts | Overpotential (mV) | Tafel slope (mV dec−1) | Catalyst morphology/support template/catalyst loading | Synthetic method | Ref. |
|---|---|---|---|---|---|
| a CFP: carbon fiber paper; FTO: fuorine-doped tin oxide; NF: nickel foam; GCE: glassy carbon electrode; RDE: rotating disk electrode. | |||||
| CoO/MnO | 280 | 67.9 | Porous nanowires and nanosheets/NF/— | Hydrothermal and vacuum annealing | 139 |
| CoP/MnO | 230 | 95 | One-dimensional hollow nanofibers/carbon paper/2 mg cm−2 | Electrospinning technology, thermal annealing, and phosphating process | 140 |
| CoFe@CNT/MnO | 193 | 82 | Nanoparticles/CFP/— | Hydrothermal and CVD pyrolysis | 141 |
| MnOx/NiFe-LDH | 174 | 48 | Nanosheets/FTO/— | Atomic layer deposition (ALD) and hydrothermal | 142 |
| MnOx/NiFe-LDH/NF | 265 | 73 | Nanosheets/NF/— | Hydrothermal and electrodeposition | 143 |
| Fe3O4@MnOx | 188 | 77.6 | Hierarchical flower-like core–shell microsphere/NF/— | Hydrothermal | 144 |
| MnOx/NiFeP/NF | 247 | 59.8 | Nanosheets/NF/— | Hydrothermal phosphating and electrodeposition | 145 |
| MnO/Co-CNTs | 420 | 59 | 1-D hollow tube and particles/glassy carbon/— | High-temperature sintering and growth on carbon nanotubes | 146 |
| MnO/Co/PGC | 300 | 77 | Nanoparticles/rotating disk electrode RDE/— | Hydrothermal and high-temperature calcination | 147 |
| Co/MnO/NC | 379 | 101 | Nanometric grain size/GCE/30 μg cm−2 | Solvothermal and calcination annealing | 148 |
| Mn2Co2C/MnO | 320 | 80 | Nanocubes and nanoparticles/glassy carbon electrode/— | Coprecipitation and pyrolysis | 122 |
| Co/MnO@NC | 235 | 53 | Nanowire and nanoparticles/— | Solvothermal and pyrolysis | 121 |
| Mn3O4@CS/CP | 290 | 95 | Spheres/glassy carbon/204 μg cm−2 | Hydrothermal and high-temperature calcination | 149 |
| Mn3O4/CoP | 306 | 51.8 | Nanorod and nanoparticles/NF/1 mg cm−2 | Hydrothermal | 150 |
| MnFe2O4/NF | 310 | 65 | Particles/NF/40 mg cm−2 | Hydrothermal | 151 |
| Mn3O4/NiCo2S4 | 320 | 79 | 1D rod-like structure and sheet-like/glassy carbon/0.38 mg cm−2 | Hydrothermal and annealing | 152 |
| Co3O4/Mn3O4 | 360 | 78.2 | Nanoparticles/carbon paper/0.382 mg cm−2 | Hydrothermal | 153 |
| RuO2/(Co, Mn)3O4 | 270 | 77 | Nanoparticles/commercial carbon cloth/— | Hydrothermal | 154 |
| N–C/Mn2O3 | 347 | 75.5 | Star-like structure with rough surface/NF/— | Hydrothermal and annealing | 155 |
| MnO/CoMn alloy@N | 300 | — | Nanoparticles/glassy carbon/0.28 mg cm−2 | Annealing | 156 |
| Co/MnO@N–C | 350 | 84.5 | The loosened foam-like/glassy carbon/0.6 mg cm−2 | Hydrothermal and pyrolysis | 157 |
| MnO/Co@NGC | 326 | 90 | Cubical/glassy carbon/— | Pyrolysis | 158 |
5. Summary and perspectives
This feature article first explains the conventional adsorption mechanism of the OER in alkaline environments, then presents the progress of different MnOx studies on the OER, and presents the catalytic mechanism of MnOx with several strategies for improvement. As far as the current study is concerned, MnOx as an anode catalyst can reduce the OER excess potential and is an electrocatalyst that could be developed for the electrolysis of water. For MnO2, the catalytic activity depends primarily on the crystal structure, morphology, size, pore structure, and surface oxidation state of the manganese atoms. It has been found that the catalytic activity of α-MnO2 in the OER is higher than that of β-MnO2 and δ-MnO2 in early research. More importantly, the large interlayer spacing of MnO2 allows for the insertion of additional ions, which affects its catalytic performance. Next, for Mn2O3, the presence of distorted octahedra occupied by the Mn3+(d4) center elongates the Mn–O bond due to the Jahn–Teller effect in Mn2O3, and the Mn3+–O octahedra are activated to participate in the OER. Due to its special d-electron structure, Mn2O3 has a much higher overpotential than other transition metal oxides. In this respect, the poor electronic conductivity and relatively low OER dynamics of Mn2O3 are mostly improved by combining them with other catalysts with higher OER activity. Mn3O4 with a spinel structure has tetrahedral and octahedral centers occupied by Mn3+and Mn2+ ions, respectively, and it has been demonstrated that Mn3+ and Mn2+ in Mn3O4 affect its OER and ORR performance. In addition, the spinel oxide MnFe2O4, also with significant OER catalytic activity, is mentioned in this paper.Current research applications of MnOx in electrochemistry are mainly in the direction of lithium batteries and capacitors, while in the direction of electrocatalysis, their activity is still distant from that of noble metals and transition metals such as Fe, and Ni. In this paper, while describing the current research progress, we propose the following points to further improve the electrochemical properties of MnOx. (1) Continue to deepen the research on the catalytic mechanism of MnOx. The mixing of multiple valence states of Mn leads to a catalytic center that is still not well defined for MnOx, and the difference in OER activity may be due to differences in the surface electronic states of Mn3+. Recent studies have proposed that the eg orbital of Mn3+ is involved in the σ-bonding with the anionic adsorbent; the higher the Mn3+ content in the manganese group, the more favorable it is to enhance the adsorption of –OH on the catalyst surface. As a consequence, increasing the content of Mn3+ ions is an effective strategy to improve its OER performance. (2) On the other hand, most of the research on MnOx has been done in alkaline environments as the Mn3+ ion can be unstable due to disproportionation reactions in acidic environments. However, the development of MnOx for the OER in acidic media has made great progress and the obtained MnOx has good catalytic properties. Therefore, we believe that the development of MnOx in acidic media is a positive direction.
In terms of enhancing the catalytic activity of MnOx, this can be done by introducing vacancies and forming heterogeneous interfaces. First, for the problem of insignificant catalytic activity, vacancies can activate adjacent metal sites as fresh active sites for electrocatalysis and promote the OER by synergistic interaction with Mn ions; meanwhile, the presence of a large number of oxygen vacancies in MnOx promotes the formation of Mn3+ active sites, which leads to their semiconducting nature and optimizes the local electronic configuration. Second, for low conductivity, the preparation of atomic heterojunctions can effectively modulate the electronic and band structure of the catalysts to provide moderate adsorption/desorption behavior, and MnOx can form heterojunctions that effectively prevent the recombination of electrons and holes and thus improve their conductivity. MOFs can be used as catalysts due to their tunable pore structure, high specific surface area, and multiple coordination sites, while MnOx can be used as a metal backbone for organic structures and also as a catalyst for the OER. Finally, we propose that recombining MnOx catalysts with other electrically active materials or conducting substrates is also an effective strategy for fabricating high-performance electrocatalysts.
In summary, the development of MnOx materials with high stability and good catalytic properties is our objective. We believe that with the continued maturation of material preparation techniques, the gradual evolution of characterization methods, the exploration of reasonable structures, and the breakthroughs in related electrocatalytic reactions, commercial applications of MnOx can eventually be realized.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 51971058 and 52071072).References
- G. Semieniuk, L. Taylor, A. Rezai and D. K. Foley, Nat. Clim. Change, 2021, 11, 313–342 CrossRef.
- D. Gernaat, H. S. de Boer, V. Daioglou, S. G. Yalew, C. Muller and D. P. van Vuuren, Nat. Clim. Change, 2021, 11, 362 CrossRef.
- T. Yi, H. M. K. Sari, X. Li, F. Wang, Y. Zhu, J. Hu, J. Zhang and X. Li, Nano Energy, 2021, 85, 105955 CrossRef CAS.
- X. Xie, L. Du, L. Yan, S. Park, Y. Qiu, J. Sokolowski, W. Wang and Y. Shao, Adv. Funct. Mater., 2022, 32, 2110036 CrossRef CAS.
- L. W. Chen and H. W. Liang, Catal. Sci. Technol., 2021, 11, 4673–4689 RSC.
- H. J. Song, H. Yoon, B. Ju and D. W. Kim, Adv. Energy Mater., 2021, 11, 2002428 CrossRef CAS.
- W. Zhang, M. Yin, Q. Zhao, C. Jin, N. Wang, S. Ji, C. L. Ritt, M. Elimelech and Q. An, Nat. Nanotechnol., 2021, 16, 337–343 CrossRef CAS PubMed.
- Y. Zhang, F. Gao, D. Wang, Z. Li, X. Wang, C. Wang, K. Zhang and Y. Du, Coord. Chem. Rev., 2023, 475, 214916 CrossRef CAS.
- Q. Chen, N. Gong, T. Zhu, C. Yang, W. Peng, Y. Li, F. Zhang and X. Fan, Small, 2022, 18, 2105696 CrossRef CAS PubMed.
- T. T. Wei, P. P. Peng, Y. R. Ji, Y. R. Zhu, T. F. Yi and Y. Xie, J. Energy Chem., 2022, 71, 400–410 CrossRef CAS.
- Y. T. Yan, J. H. Lin, J. Cao, S. Guo, X. H. Zheng, J. C. Feng and J. L. Qi, J. Mater. Chem. A, 2019, 7, 24486–24492 RSC.
- W. Shi, W. Lee and J. M. Xue, Chemsuschem, 2021, 14, 1634–1658 CrossRef CAS PubMed.
- Q. Zhao, A. Song, S. Ding, R. Qin, Y. Cui, S. Li and F. Pan, Adv. Mater., 2020, 32, 2002450 CrossRef CAS PubMed.
- T. Xiong, Y. Zhang, W. S. V. Lee and J. Xue, Adv. Energy Mater., 2020, 10, 2001769 CrossRef CAS.
- F. Y. Chen, Z. Y. Wu, Z. Adler and H. T. Wang, Joule, 2021, 5, 1704–1731 CrossRef CAS.
- Z. Lei, T. Wang, B. Zhao, W. Cai, Y. Liu, S. Jiao, Q. Li, R. Cao and M. Liu, Adv. Energy Mater., 2020, 10, 2000478 CrossRef CAS.
- Y. Qin, Y. Liu, Y. Zhang, Y. Gu, Y. Lian, Y. Su, J. Hu, X. Zhao, Y. Peng, K. Feng, J. Zhong, M. H. Rummeli and Z. Deng, ACS Catal., 2023, 13, 256–266 CrossRef CAS.
- J. Yu, F. Yu, M. F. Yuen and C. D. Wang, J. Mater. Chem. A, 2021, 9, 9389–9430 RSC.
- C. L. Hu, L. Zhang and J. L. Gong, Energy Environ. Sci., 2019, 12, 2620–2645 RSC.
- Q. Liang, G. Brocks and A. Bieberle-Hütter, JPhys Energy, 2021, 3, 26001 CrossRef CAS.
- J. O. Olowoyo and R. J. Kriek, Small, 2022, 18, 2203125 CrossRef CAS PubMed.
- K. Zhang and R. Zou, Small, 2021, 17, 2100129 CrossRef CAS PubMed.
- M. Tahir, L. Pan, F. Idrees, X. W. Zhang, L. Wang, J. J. Zou and Z. L. Wang, Nano Energy, 2017, 37, 136–157 CrossRef CAS.
- H. Y. Chang, Z. J. Liang, L. Wang and C. Wang, Nanoscale, 2022, 14, 5639–5656 RSC.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, Chemcatchem, 2010, 2, 724–761 CrossRef CAS.
- C. X. Zhao, J. N. Liu, J. Wang, D. Ren, B. Q. Li and Q. Zhang, Chem. Soc. Rev., 2021, 50, 7745–7778 RSC.
- Z. Y. Yu, Y. Duan, X. Y. Feng, X. X. Yu, M. R. Gao and S. H. Yu, Adv. Mater., 2021, 33, 2007100 CrossRef CAS PubMed.
- L. K. Gao, X. Cui, C. D. Sewell, J. Li and Z. Q. Lin, Chem. Soc. Rev., 2021, 50, 8428–8469 RSC.
- Y. Zhang, C. Han, J. Gao, L. Pan, J. Wu, X. Zhu and J. Zou, ACS Catal., 2021, 11, 12485–12509 CrossRef CAS.
- Z. Xue, X. Y. Zhang, J. Q. Qin and R. P. Liu, J. Mater. Chem. A, 2019, 7, 23091–23097 RSC.
- X. W. Liu, J. J. Xu, Z. Y. Ni, R. C. Wang, J. H. You and R. Guo, Chem. Eng. J., 2019, 356, 22–33 CrossRef CAS.
- M. I. Jamesh and X. M. Sun, J. Power Sources, 2018, 400, 31–68 CrossRef CAS.
- H. Osgood, S. V. Devaguptapu, H. Xu, J. Cho and G. Wu, Nano Today, 2016, 11, 601–625 CrossRef CAS.
- M. M. Najafpour, M. Holynska and S. Salimi, Coord. Chem. Rev., 2015, 285, 65–75 CrossRef CAS.
- J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, Science, 2011, 333, 733–736 CrossRef CAS PubMed.
- M. M. Najafpour, B. Haghighi, D. J. Sedigh and M. Z. Ghobadi, Dalton Trans., 2013, 42, 16683 RSC.
- M. M. Najafpour and D. J. Sedigh, Dalton Trans., 2013, 42, 12173–12178 RSC.
- A. Konarov, N. Voronina, J. H. Jo, Z. Bakenov, Y. K. Sun and S. T. Myung, ACS Energy Lett., 2018, 3, 2620–2640 CrossRef CAS.
- M. H. Alfaruqi, V. Mathew, J. Gim, S. Kim, J. Song, J. P. Baboo, S. H. Choi and J. Kim, Chem. Mater., 2015, 27, 3609–3620 CrossRef CAS.
- D. Wang, L. Wang, G. Liang, H. Li, Z. Liu, Z. Tang, J. Liang and C. Zhi, ACS Nano, 2019, 13, 10643–10652 CrossRef CAS PubMed.
- V. B. R. Boppana, S. Yusuf, G. S. Hutchings and F. Jiao, Adv. Funct. Mater., 2013, 23, 878–884 CrossRef CAS.
- R. Yang, Y. Fan, R. Ye, Y. Tang, X. Cao, Z. Yin and Z. Zeng, Adv. Mater., 2021, 33, 2004862 CrossRef CAS PubMed.
- X. Peng, H. Y. Peng, K. N. Zhao, Y. X. Zhang, F. J. Xia, J. H. Lyu, G. Van Tendeloo, C. L. Sun and J. S. Wu, ACS Appl. Mater. Interfaces, 2021, 13, 33644–33651 CrossRef CAS PubMed.
- D. M. Robinson, Y. B. Go, M. Mui, G. Gardner, Z. J. Zhang, D. Mastrogiovanni, E. Garfunkel, J. Li, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2013, 135, 3494–3501 CrossRef CAS PubMed.
- Y. T. Meng, W. Q. Song, H. Huang, Z. Ren, S. Y. Chen and S. L. Suib, J. Am. Chem. Soc., 2014, 136, 11452–11464 CrossRef CAS PubMed.
- X. Zhang, P. Yu, D. Wang and Y. Ma, J. Nanosci. Nanotechnol., 2010, 10, 898–904 CrossRef CAS PubMed.
- L. M. Housel, L. Wang, A. Abraham, J. Huang, G. D. Renderos, C. D. Quilty, A. B. Brady, A. C. Marschilok, K. J. Takeuchi and E. S. Takeuchi, Acc. Chem. Res., 2018, 51, 575–582 CrossRef CAS PubMed.
- Z. Yang, D. C. Ford, J. S. Park, Y. Ren, S. Kim, H. Kim, T. T. Fister, M. K. Y. Chan and M. M. Thackeray, Chem. Mater., 2017, 29, 1507–1517 CrossRef CAS.
- Y. Gu, G. Yan, Y. Lian, P. Qi, Q. Mu, C. Zhang, Z. Deng and Y. Peng, Energy Storage Mater., 2019, 23, 252–260 CrossRef.
- Y. Zhou, Z. Zhou, L. Hu, R. Tian, Y. Wang, H. Arandiyan, F. Chen, M. Li, T. Wan, Z. Han, Z. Ma, X. Lu, C. Cazorla, T. Wu and D. Chu, Chem. Eng. J., 2022, 438, 135561 CrossRef CAS.
- S. Ni, H. Zhang, Y. Zhao, X. Li, Y. Sun, J. Qian, Q. Xu, P. Gao, D. Wu, K. Kato, M. Yamauchi and Y. Sun, Chem. Eng. J., 2019, 366, 631–638 CrossRef CAS.
- D. A. Kitchaev, S. T. Dacek, W. H. Sun and G. Ceder, J. Am. Chem. Soc., 2017, 139, 2672–2681 CrossRef CAS PubMed.
- X. C. Duan, J. Q. Yang, H. Y. Gao, J. M. Ma, L. F. Jiao and W. J. Zheng, Crystengcomm, 2012, 14, 4196–4204 RSC.
- S. L. Suib, J. Mater. Chem., 2008, 18, 1623–1631 RSC.
- Y. Gu, Y. Min, L. Li, Y. Lian, H. Sun, D. Wang, M. H. Rummeli, J. Guo, J. Zhong, L. Xu, Y. Peng and Z. Deng, Chem. Mater., 2021, 33, 4135–4145 CrossRef CAS.
- W. Yao, G. M. Odegard, Z. Huang, Y. Yuan, H. Asayesh-Ardakani, S. Sharifi-Asl, M. Cheng, B. Song, R. Deivanayagam, F. Long, C. R. Friedrich, K. Amine, J. Lu and R. Shahbazian-Yassar, Nano Energy, 2018, 48, 301–311 CrossRef CAS.
- S. Park, Y. H. Lee, S. Choi, H. Seo, M. Y. Lee, M. Balamurugan and K. T. Nam, Energy Environ. Sci., 2020, 13, 2310–2340 RSC.
- Y. Tang, S. Zheng, S. Cao, H. Xue and H. Pang, J. Mater. Chem. A, 2020, 8, 18492–18514 RSC.
- B. Cheng, K. Kong, L. Zhang, R. Sa, T. Gu, Y. Rui and R. Wang, Chem. Eng. J., 2022, 441, 136122 CrossRef CAS.
- Z. W. Chen, M. Ju, M. Z. Sun, L. Jin, R. M. Cai, Z. Wang, L. Dong, L. M. Peng, X. Long, B. L. Huang and S. H. Yang, Angew. Chem., Int. Ed., 2021, 60, 9699–9705 CrossRef CAS PubMed.
- I. G. McKendry, L. J. Mohamad, A. C. Thenuwara, T. Marshall, E. Borguet, D. R. Strongin and M. J. Zdilla, ACS Energy Lett., 2018, 3, 2280–2285 CrossRef CAS.
- Y. Y. Pu, M. J. Lawrence, V. Celorrio, Q. Wang, M. Gu, Z. Z. Sun, L. A. Jacome, A. E. Russell, L. M. Huang and P. Rodriguez, J. Mater. Chem. A, 2020, 8, 13340–13350 RSC.
- G. Gupta, K. Selvakumar, N. Lakshminarasimhan, S. Kumar and M. Mamlouk, J. Power Sources, 2020, 461, 228131 CrossRef CAS.
- X. F. Lu, A. L. Wang, H. Xu, X. J. He, Y. X. Tong and G. R. Li, J. Mater. Chem. A, 2015, 3, 16560–16566 RSC.
- X. F. Lu, Z. X. Huang, Y. X. Tong and G. R. Li, Chem. Sci., 2016, 7, 510–517 RSC.
- N. N. Xu, J. W. Liu, J. L. Qiao, H. T. Huang and X. D. Zhou, J. Power Sources, 2020, 455, 227992 CrossRef CAS.
- B. Z. Jiang, C. J. Xu, C. L. Wu, L. B. Dong, J. Li and F. Y. Kang, Electrochim. Acta, 2017, 229, 422–428 CrossRef CAS.
- M. Mao, X. X. Wu, Y. Hu, Q. H. Yuan, Y. B. He and F. Y. Kang, J. Energy Chem., 2021, 52, 277–283 CrossRef CAS.
- N. Parveen, S. A. Ansari, M. Z. Ansari and M. O. Ansari, Environ. Chem. Lett., 2022, 20, 283–309 CrossRef CAS.
- S. E. Balaghi, C. A. Triana and G. R. Patzke, ACS Catal., 2020, 10, 2074–2087 CrossRef CAS.
- P. Liu, Y. Zheng, H. Zhu and T. Li, ACS Appl. Nano Mater., 2019, 2, 744–749 CrossRef CAS.
- G. Zhao, Y. Yao, W. Lu, G. Liu, X. Guo, A. Tricoli and Y. Zhu, Nano Lett., 2021, 21, 7012–7020 CrossRef CAS PubMed.
- L. Bigiani, C. Maccato, T. Andreu, A. Gasparotto, C. Sada, E. Modin, O. I. Lebedev, J. R. Morante and D. Barreca, ACS Appl. Nano Mater., 2020, 3, 9889–9898 CrossRef CAS.
- K. R. Yoon, G. Y. Lee, J. W. Jung, N. H. Kim, S. O. Kim and I. D. Kim, Nano Lett., 2016, 16, 2076–2083 CrossRef CAS PubMed.
- B. Yang, X. Chang, X. Y. Ding, X. Z. Ma and M. Y. Zhang, J. Colloid Interface Sci., 2022, 623, 196–204 CrossRef CAS PubMed.
- P. R. Garcês Gonçalves, H. A. De Abreu and H. A. Duarte, J. Phys. Chem. C, 2018, 122, 20841–20849 CrossRef.
- Y. Shi, P. F. Ndione, L. Y. Lim, D. Sokaras, T. Weng, A. R. Nagaraja, A. G. Karydas, J. D. Perkins, T. O. Mason, D. S. Ginley, A. Zunger and M. F. Toney, Chem. Mater., 2014, 26, 1867–1873 CrossRef CAS.
- C. Chowde Gowda, A. Mathur, A. Parui, P. Kumbhakar, P. Pandey, S. Sharma, A. Chandra, A. K. Singh, A. Halder and C. S. Tiwary, Ind. Eng. Chem., 2022, 113, 153–160 CrossRef CAS.
- Y. Gorlin, B. Lassalle-Kaiser, J. D. Benck, S. Gul, S. M. Webb, V. K. Yachandra, J. Yano and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 8525–8534 CrossRef CAS PubMed.
- M. Huynh, C. Shi, S. J. L. Billinge and D. G. Nocera, J. Am. Chem. Soc., 2015, 137, 14887–14904 CrossRef CAS PubMed.
- C. Guo, Y. Zheng, J. Ran, F. Xie, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2017, 56, 8539–8543 CrossRef CAS PubMed.
- F. B. Wang, G. D. Li, X. G. Meng, S. J. Xu and W. Q. Ma, J. Power Sources, 2020, 462, 228162 CrossRef CAS.
- Q. Huang, X. Zhong, Q. Zhang, X. Wu, M. Jiao, B. Chen, J. Sheng and G. Zhou, J. Energy Chem., 2022, 68, 679–687 CrossRef CAS.
- J. S. Hong, H. Seo, Y. H. Lee, K. H. Cho, C. Ko, S. Park and K. T. Nam, Small Methods, 2020, 4, 1900733 CrossRef CAS.
- K. H. Cho, H. Seo, S. Park, Y. H. Lee, M. Y. Lee, N. H. Cho and K. T. Nam, Adv. Funct. Mater., 2020, 30, 1910424 CrossRef CAS.
- M. Y. Lee, H. Ha, K. H. Cho, H. Seo, S. Park, Y. H. Lee, S. J. Kwon, T. W. Lee and K. T. Nam, ACS Catal., 2020, 10, 1237–1245 CrossRef CAS.
- J. Kim, J. Lee, C. Liu, S. Pandey, S. Woo Joo, N. Son and M. Kang, Appl. Surf. Sci., 2021, 546, 149124 CrossRef CAS.
- D. Zhang, J. Cao, X. Zhang, N. Insin, S. Wang, J. Han, Y. Zhao, J. Qin and Y. Huang, Adv. Funct. Mater., 2021, 31, 2009412 CrossRef CAS.
- Q. Tan, X. Li, B. Zhang, X. Chen, Y. Tian, H. Wan, L. Zhang, L. Miao, C. Wang, Y. Gan, J. Jiang, Y. Wang and H. Wang, Adv. Energy Mater., 2020, 10, 2001050 CrossRef CAS.
- M. Augustin, O. Yezerska, D. Fenske, I. Bardenhagen, A. Westphal, M. Knipper, T. Plaggenborg, J. Kolny-Olesiak and J. Parisi, Electrochim. Acta, 2015, 158, 383–389 CrossRef CAS.
- R. Pokhrel, M. K. Goetz, S. E. Shaner, X. X. Wu and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 8384–8387 CrossRef CAS PubMed.
- J. M. Lee, S. B. Patil, B. Kang, S. Lee, M. G. Kim and S. J. Hwang, J. Mater. Chem. A, 2018, 6, 12565–12573 RSC.
- H. Li, W. Zhang, K. Sun, J. Guo, K. Yuan, J. Fu, T. Zhang, X. Zhang, H. Long, Z. Zhang, Y. Lai and H. Sun, Adv. Energy Mater., 2021, 11, 2100867 CrossRef CAS.
- K. Jin, H. Seo, T. Hayashi, M. Balamurugan, D. Jeong, Y. K. Go, J. S. Hong, K. H. Cho, H. Kakizaki, N. Bonnet-Mercier, M. G. Kim, S. H. Kim, R. Nakamura and K. T. Nam, J. Am. Chem. Soc., 2017, 139, 2277–2285 CrossRef CAS PubMed.
- Y. Tong, Y. Q. Guo, P. Z. Chen, H. F. Liu, M. X. Zhang, L. D. Zhang, W. S. Yan, W. S. Chu, C. Z. Wu and Y. Xie, Chem, 2017, 3, 812–821 CAS.
- H. Y. An, Z. Chen, J. X. Yang, Z. C. Feng, X. L. Wang, F. T. Fan and C. Li, J. Catal., 2018, 367, 53–61 CrossRef CAS.
- B. Kang, X. Jin, S. M. Oh, S. B. Patila, M. G. Kim, S. H. Kim and S. J. Hwang, Appl. Catal., B, 2018, 236, 107–116 CrossRef CAS.
- F. Yang, J. Xie, D. Rao, X. Liu, J. Jiang and X. Lu, Nano Energy, 2021, 85, 106020 CrossRef CAS.
- P. Plate, C. Höhn, U. Bloeck, P. Bogdanoff, S. Fiechter, F. F. Abdi, R. van de Krol and A. C. Bronneberg, ACS Appl. Mater. Interfaces, 2021, 13, 2428–2436 CrossRef CAS PubMed.
- H. Feizi, S. M. Hosseini, Z. Zand and M. M. Najafpour, J. Hydrogen Energy, 2022, 47, 7813–7822 CrossRef CAS.
- T. Takashima, K. Hashimoto and R. Nakamura, J. Am. Chem. Soc., 2012, 134, 18153–18156 CrossRef CAS PubMed.
- J. Park, H. Kim, K. Jin, B. J. Lee, Y. S. Park, H. Kim, I. Park, K. D. Yang, H. Y. Jeong, J. Kim, K. T. Hong, H. W. Jang, K. Kang and K. T. Nam, J. Am. Chem. Soc., 2014, 136, 4201–4211 CrossRef CAS PubMed.
- J. Lu, H. Wang, Y. Sun, X. Wang, X. Song and R. Wang, Chem. Eng. J., 2021, 417, 127894 CrossRef CAS.
- H. Kakizaki, H. Ooka, T. Hayashi, A. Yamaguchi, N. Bonnet Mercier, K. Hashimoto and R. Nakamura, Adv. Funct. Mater., 2018, 28, 1706319 CrossRef.
- Z. M. Chan, D. A. Kitchaev, J. N. Weker, C. Schnedermann, K. Lim, G. Ceder, W. Tumas, M. F. Toney and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5261–E5268 CAS.
- C. Wei, Z. Feng, G. G. Scherer, J. Barber, Y. Shao-Horn and Z. J. Xu, Adv. Mater., 2017, 29, 1606800 CrossRef PubMed.
- T. Takashima, K. Hashimoto and R. Nakamura, J. Am. Chem. Soc., 2012, 134, 1519–1527 CrossRef CAS PubMed.
- M. Huynh, D. K. Bediako and D. G. Nocera, J. Am. Chem. Soc., 2014, 136, 6002–6010 CrossRef CAS PubMed.
- K. X. Wang, Y. L. Wang, B. Yang, Z. J. Li, X. T. Qin, Q. H. Zhang, L. C. Lei, M. Qiu, G. Wu and Y. Hou, Energy Environ. Sci., 2022, 15, 2356–2365 RSC.
- H. Xue, A. Meng, T. Yang, Z. Li and C. Chen, J. Energy Chem., 2022, 71, 639–651 CrossRef CAS.
- K. Ham, S. Hong, S. Kang, K. Cho and J. Lee, ACS Energy Lett., 2021, 6, 364–370 CrossRef CAS.
- Y. Yan, P. Wang, J. Lin, J. Cao and J. Qi, J. Energy Chem., 2021, 58, 446–462 CrossRef CAS.
- Y. Yan, J. Lin, T. Xu, B. Liu, K. Huang, L. Qiao, S. Liu, J. Cao, S. C. Jun, Y. Yamauchi and J. Qi, Adv. Energy Mater., 2022, 12, 2200434 CrossRef CAS.
- Y. Zhao, C. Chang, F. Teng, Y. Zhao, G. Chen, R. Shi, G. I. N. Waterhouse, W. Huang and T. Zhang, Adv. Energy Mater., 2017, 7, 1700005 CrossRef.
- Y. Liu, H. T. D. Bui, A. R. Jadhav, T. Yang, S. Saqlain, Y. Luo, J. Yu, A. Kumar, H. Wang, L. Wang, V. Q. Bui, M. G. Kim, Y. D. Kim and H. Lee, Adv. Funct. Mater., 2021, 31, 2010718 CrossRef CAS.
- Z. Zhang, H. Sun, J. Li, Z. Shi, M. Fan, H. Bian, T. Wang and D. Gao, J. Power Sources, 2021, 491, 229584 CrossRef CAS.
- Q. C. Xu, J. H. Zhang, H. X. Zhang, L. Y. Zhang, L. Chen, Y. J. Hu, H. Jiang and C. Z. Li, Energy Environ. Sci., 2021, 14, 5228–5259 RSC.
- Z. X. Li, M. L. Hu, P. Wang, J. H. Liu, J. S. Yao and C. Y. Li, Coord. Chem. Rev., 2021, 439, 213953 CrossRef CAS.
- J. Zhu, M. Sun, S. J. Liu, X. H. Liu, K. Hu and L. Wang, J. Mater. Chem. A, 2019, 7, 26975–26983 RSC.
- P. Wang, Y. Luo, G. Zhang, Z. Chen, H. Ranganathan, S. Sun and Z. Shi, Nano-Micro Lett., 2022, 14, 117–120 CrossRef PubMed.
- Y. L. Niu, X. Teng, S. Q. Gong, X. Liu, M. Z. Xu and Z. F. Chen, Energy Storage Mater., 2021, 43, 42–52 CrossRef.
- T. Meng and M. H. Cao, ACS Sustainable Chem. Eng., 2020, 8, 13271–13281 CrossRef CAS.
- F. Li, T. T. Qin, Y. P. Sun, R. J. Jiang, J. F. Yuan, X. Q. Liu and A. P. O'Mullane, J. Mater. Chem. A, 2021, 9, 22533–22543 RSC.
- S. S. Selvasundarasekar, T. K. Bijoy, S. Kumaravel, A. Karmakar, R. Madhu, K. Bera, S. Nagappan, H. N. Dhandapani, G. A. M. Mersal, M. M. Ibrahim, D. Sarkar, S. M. Yusuf, S. Lee and S. Kundu, ACS Appl. Mater. Interfaces, 2022, 14, 46581–46594 CrossRef CAS PubMed.
- S. Wu, H. Min, W. Shi and P. Cheng, Adv. Mater., 2019, 32, 1805871 CrossRef PubMed.
- B. Li, J. Ma and P. Cheng, Angew. Chem., Int. Ed., 2018, 57, 6834–6837 CrossRef CAS PubMed.
- J. Wang, J. Wang, M. Zhang, S. Li, R. Liu and Z. Li, J. Alloys Compd., 2020, 821, 153463 CrossRef CAS.
- X. Wei, S. Cao, H. Xu, C. Jiang, Z. Wang, Y. Ouyang, X. Lu, F. Dai and D. Sun, ACS Mater. Lett., 2022, 4, 1991–1998 CrossRef CAS.
- T. F. Yi, L. Y. Qiu, J. Mei, S. Y. Qi, P. Cui, S. H. Luo, Y. R. Zhu, Y. Xie and Y. B. He, Sci. Bull., 2020, 65, 546–556 CrossRef CAS PubMed.
- Y. Chen, Y. Guo, H. Cui, Z. Xie, X. Zhang, J. Wei and Z. Zhou, J. Mater. Chem. A, 2018, 6, 9716–9722 RSC.
- F. Wang, H. Zhao, Y. Ma, Y. Yang, B. Li, Y. Cui, Z. Guo and L. Wang, J. Energy Chem., 2020, 50, 52–62 CrossRef.
- C. Chen, Z. J. Tang, J. Y. Li, C. Y. Du, T. Ouyang, K. Xiao and Z. Q. Liu, Adv. Funct. Mater., 2023, 33, 2210143 CrossRef CAS.
- B. Liu, Y. Sun, L. Liu, S. Xu and X. Yan, Adv. Funct. Mater., 2018, 28, 1704973 CrossRef.
- Y. W. Li, S. K. Su, C. Z. Yue, J. Shu, P. F. Zhang, F. H. Du, S. N. Wang, H. Y. Ma, J. Yin and X. Shao, Dalton Trans., 2021, 50, 17265–17274 RSC.
- R. Guo, A. Yan, J. Xu, B. Xu, T. Li, X. Liu, T. Yi and S. Luo, J. Alloys Compd., 2020, 817, 153246 CrossRef CAS.
- K. H. Cho, H. Seo, S. Park, Y. H. Lee, M. Y. Lee, N. H. Cho and K. T. Nam, Adv. Funct. Mater., 2020, 30, 1910424 CrossRef CAS.
- Y. T. Yan, J. Q. Liu, K. K. Huang, J. L. Qi, L. Qiao, X. H. Zheng and W. Cai, J. Mater. Chem. A, 2021, 9, 26777–26787 RSC.
- J. Qi, Y. Yan, Y. Cai, J. Cao and J. Feng, Adv. Funct. Mater., 2021, 31, 2006030 CrossRef CAS.
- Y. Lin, G. Yang, Y. Fu, B. Zhu, J. Zhao and J. Li, J. Phys. Chem. Solids, 2022, 160, 110373 CrossRef CAS.
- X. Chang, B. Yang, X. Ding, X. Ma and M. Zhang, J. Colloid Interface Sci., 2022, 610, 663–670 CrossRef CAS PubMed.
- S. Chen, Y. Huang, M. Li, P. Sun, X. Lv, B. Li, L. Fang and X. Sun, J. Electroanal. Chem., 2021, 895, 115513 CrossRef CAS.
- Y. D. Xue, Z. S. Fishman, J. A. Rohr, Z. H. Pan, Y. T. Wang, C. H. Zhang, S. L. Zheng, Y. Zhang and S. Hu, J. Mater. Chem. A, 2018, 6, 21918–21926 RSC.
- Z. Wang, C. Wang, L. Ye, X. Liu, L. Xin, Y. Yang, L. Wang, W. Hou, Y. Wen and T. Zhan, Inorg. Chem., 2022, 61, 15256–15265 CrossRef CAS PubMed.
- Y. W. Li, S. K. Su, C. Z. Yue, J. Shu, P. F. Zhang, F. H. Du, S. N. Wang, H. Y. Ma, J. Yin and X. Shao, Dalton Trans., 2021, 50, 17265–17274 RSC.
- P. Wang, Y. Luo, G. Zhang, M. Wu, Z. Chen, S. Sun and Z. Shi, Small, 2022, 18, 2105803 CrossRef CAS PubMed.
- W. Zhang, L. Zong, K. Fan, L. Cui, Q. Zhang, J. Zhao, L. Wang and S. Feng, J. Alloys Compd., 2021, 874, 159965 CrossRef CAS.
- X. F. Lu, Y. Chen, S. Wang, S. Gao and X. W. D. Lou, Adv. Mater., 2019, 31, 1902339 CrossRef PubMed.
- W. Wang, H. Wang, Z. Wu, Y. Yu, M. Asif, Z. Wang, X. Qiu and H. Liu, Electrochim. Acta, 2018, 281, 486–493 CrossRef CAS.
- W. Q. Li, H. Q. Fu, Y. H. Cao, H. J. Wang, H. Yu, Z. W. Qiao, H. Liang and F. Peng, Chemelectrochem, 2019, 6, 359–368 CrossRef.
- R. Dong, A. Zhu, W. Zeng, L. Qiao, L. Lu, Y. Liu, P. Tan and J. Pan, Appl. Surf. Sci., 2021, 544, 148860 CrossRef CAS.
- J. Kim, J. Lee, C. Liu, S. Pandey, S. Woo Joo, N. Son and M. Kang, Appl. Surf. Sci., 2021, 546, 149124 CrossRef CAS.
- F. Wang, G. Li, X. Meng, S. Xu and W. Ma, J. Power Sources, 2020, 462, 228162 CrossRef CAS.
- Q. Huang, X. Zhong, Q. Zhang, X. Wu, M. Jiao, B. Chen, J. Sheng and G. Zhou, J. Energy Chem., 2022, 68, 679–687 CrossRef CAS.
- S. Niu, X. Kong, S. Li, Y. Zhang, J. Wu, W. Zhao and P. Xu, Appl. Catal., B, 2021, 297, 120442 CrossRef CAS.
- X. Zhai, X. Wang, X. Pang, J. Zhang, Q. Wu, W. Na, Y. Zhou and L. Tian, J. Taiwan Inst. Chem. Eng., 2021, 126, 383–391 CrossRef CAS.
- C. Deng, K. Wu, J. Scott, S. Zhu, R. Amal and D. Wang, J. Mater. Chem. A, 2019, 7, 20649–20657 RSC.
- C. Zhang, F. Kong, Y. Qiao, Q. Zhao, A. Kong and Y. Shan, Chem. - Asian J., 2020, 15, 3535–3541 CrossRef CAS PubMed.
- T. Meng, B. Mao and M. Cao, Inorg. Chem., 2021, 60, 10340–10349 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |