
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltramicroporous iron-isonicotinate MOFs combining size-exclusion kinetics and thermodynamics for efficient CO2/N2 gas separation†
Isabel
Abánades Lázaro‡
 a,
Eleni C.
Mazarakioti‡
a,
Eduardo
Andres-Garcia
a,
Bruno J. C.
Vieira
b,
João C.
Waerenborgh
b,
Iñigo J.
Vitórica-Yrezábal
c,
Mónica
Giménez-Marqués
a and
Guillermo
Mínguez Espallargas
*a
a,
Eleni C.
Mazarakioti‡
a,
Eduardo
Andres-Garcia
a,
Bruno J. C.
Vieira
b,
João C.
Waerenborgh
b,
Iñigo J.
Vitórica-Yrezábal
c,
Mónica
Giménez-Marqués
a and
Guillermo
Mínguez Espallargas
*a
aInstituto de Ciencia Molecular (ICMol), Universitat de València, Catedrático José Beltrán Martínez No 2, 46980 Paterna, Valencia, Spain. E-mail: guillermo.minguez@uv.es
bCentro de Ciências e Tecnologias Nucleares, DECN, Instituto Superior Técnico, Universidade de Lisboa, 2695-066 Bobadela, LRS, Portugal
cSchool of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 17th February 2023
Abstract
Two ultramicroporous 2D and 3D iron-based Metal–Organic Frameworks (MOFs) have been obtained by solvothermal synthesis using different ratios and concentrations of precursors. Their reduced pore space decorated with pendant pyridine from tangling isonicotinic ligands enables the combination of size-exclusion kinetic gas separation, due to their small pores, with thermodynamic separation, resulting from the interaction of the linker with CO2 molecules. This combined separation results in efficient materials for dynamic breakthrough gas separation with virtually infinite CO2/N2 selectivity in a wide operando range and with complete renewability at room temperature and ambient pressure.
Introduction
CO2 gas separation is one of the main goals of the scientific community due to the hazardous effects of this greenhouse gas on global warming.1–3 At an industrial level, CO2 separation from combustion sources is still an expensive process mainly based on amine scrubbers (corrosive) or cryogenic distillation.4,5 Membranes or microorganisms such as microbial or algae have also been proposed as a solution to reduce the energy cost of the separation process, but membranes are still mechanically fragile and microorganisms can only operate under certain conditions,6 hindering the industrial application of these technologies.Metal–Organic Frameworks (MOFs)7–9 are versatile materials formed by organic molecules (linkers) and metal ions or clusters (nodes) that have received much attention for a variety of applications due to their intrinsic properties.10–13 Thus, as a consequence of the large porosity of MOFs, significant interest has been directed toward their gas storage and separation applications.14–17 Interestingly, the chemical versatility of this type of porous materials allows not only the modification of the pore size, but also the decoration of the pores with functional groups that enhance the interaction with the gas molecules.
In this context, despite the continuous interest in increasing the porosity of MOFs,18,19 those bearing small pores (<7 Å) or constricted pore windows, the so-called ultramicroporous MOFs, have emerged as promising materials due to their benefits regarding gas separation.20–25 For instance, reducing the pore size to a certain dimension allows only CO2 molecules, with a slightly smaller kinetic diameter than N2, to be adsorbed,26 resulting in size-exclusion kinetic gas separation with some of the best-reported selectivities,15,16,20,27 surpassing by far micro- and mesoporous MOFs. Size-exclusion kinetic separation has also the advantage of mild material renewability due to the weak gas-adsorbent interactions,15,16,28 in contrast to the high energy required upon regenerating materials with large gas-adsorbent interactions.27 Thus, the use of ultramicroporous MOFs could have a considerable reduction in the energy cost of gas separation technologies, which account for 70% of the total energy in a typical chemical plant.29
Recently, computational and experimental structure–performance studies for CO2 gas separation with over 3000 MOFs28 have shown that MOFs with a pore-limiting diameter between 3.8 Å and 5.0 Å, a largest cavity diameter between 5.0 Å and 7.5 Å and surface area <1000 m2 g−1, are the best candidates for selective separation of CO2. Therefore, despite the inconvenience of reduced gas adsorption capacity in comparison with mesoporous MOFs,14,28,30,31 their improved selectivity and eased renewability14,28 place ultramicroporous materials among the most promising candidates for gas separation applications.
In addition, the combination of both size-exclusion kinetics with thermodynamic separation based on the introduction of functional units that selectively interact with certain gas molecules should increase the selectivity and working capacity of ultramicroporous MOFs, while maintaining efficient desorption processes. Hence, MOFs composed of a small linker that has a functional group that weakly interacts with CO2 molecules are very propitious for CO2/N2 gas separation.14,16 This dual kinetic-thermodynamic performance has been recently demonstrated with the use of an anilato-based ultramicroporous MOF that exhibits efficient CO2/N2 gas separation under a range of conditions and is easily regenerated at room temperature.16 However, the synthesis of functional linkers may increase the cost of the materials hindering its industrial application.
Herein, by using the commercially available linker isonicotinic acid (HINA) and iron as the metal source, we report the synthesis and separation capacity of two ultramicroporous iron-isonicotinate MOFs.
Results and discussion
Synthesis and characterization
Aiming to combine both favourable kinetics and thermodynamics for CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N2 gas separation, we selected the commercially available isonicotinic acid as the linker (Fig. 1), in an attempt to promote effective interactions between the pendant pyridine and the CO2 gas molecules.14,32,33 This affinity has been proved in different porous materials formed by isonicotinic acid, which exhibit effective CO2/N2 separation despite the absence of a size exclusion mechanism.34–37
:N2 gas separation, we selected the commercially available isonicotinic acid as the linker (Fig. 1), in an attempt to promote effective interactions between the pendant pyridine and the CO2 gas molecules.14,32,33 This affinity has been proved in different porous materials formed by isonicotinic acid, which exhibit effective CO2/N2 separation despite the absence of a size exclusion mechanism.34–37
 | ||
| Fig. 1 Synthetic scheme of MUV-26 MOFs. | ||
Two materials, denoted MUV-26α(DMF) and MUV-26β(DMF) (MUV = Materials of the University of Valencia), were obtained as crystalline powders (Fig. S12†) (yield ca. 45%, ca. 400 mg) by solvothermal reaction of isonicotinic acid with the trimeric iron(III) oxo acetate cluster in DMF at 120 °C for 48 hours by varying the ratios of the starting materials and their initial concentration to selectively target the desired MOF product (see Section S1 in the ESI†).
Single crystal X-ray diffraction of rhombohedral and square black single crystals (Fig. 1) reveals that MUV-26α(DMF) crystallizes in the orthorhombic Pnma space group, with unit cell parameters a = 20.5173(8) Å, b = 19.3490(5) Å and c = 11.4667(3) Å, whereas MUV-26β(DMF) crystallizes in the monoclinic P21 chiral space group, with unit cell parameters a = 10.3799(7) Å, b = 21.7382(10) Å, c = 20.9849(10) Å and β = 102.392(6)° (see section S2 in the ESI†). Structural analysis shows that both materials contain the trimeric oxo acetate iron moieties and isonicotinate units (Fig. 2). Each iron is octahedrally coordinated with 4 bidentate carboxylate groups bridging the iron centres in the equatorial positions, as commonly found in other MOFs such as MIL-100 (ref. 38) and MUV-2.39 The coordination sphere of the iron is completed by an additional INA ligand via the pyridine group or water molecules. Thus, MUV-26α(DMF) has as composition [Fe3O(INA)6]·DMF, whereas MUV-26β(DMF) has as composition [(Fe3O)2(INA)12(H2O)]·DMF.
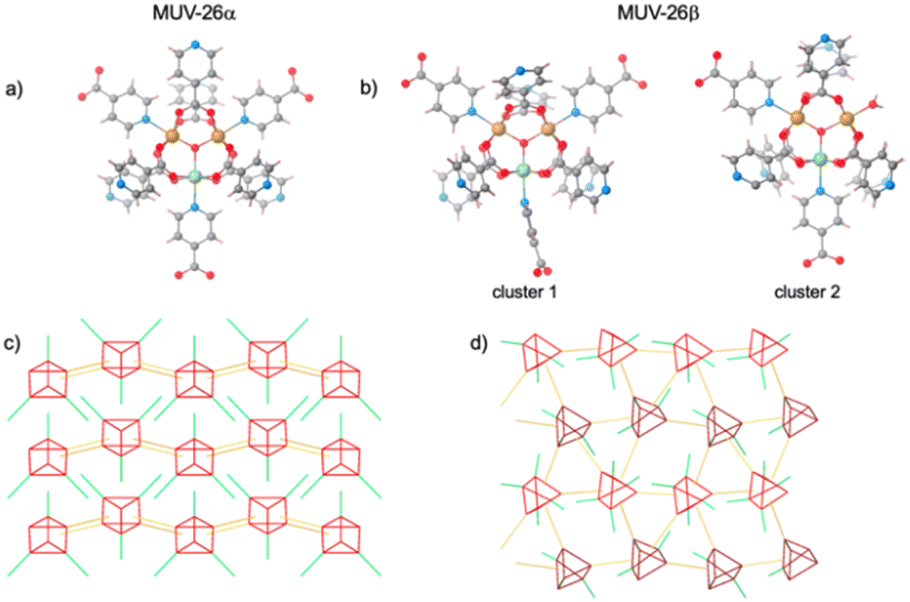 | ||
| Fig. 2 Crystal structures of MUV-26. (a) Trimetallic cluster of MUV-26α(DMF), indicating the Fe(III) in orange and the Fe(II) in light green. (b) Two different clusters from MUV-26β(DMF), indicating the Fe(III) in orange and the Fe(II) in light green. (c) Connectivity between clusters in MUV-26α(DMF), resulting in a 2D framework. (d) Connectivity between clusters in MUV-26 β(DMF), resulting in a 3D framework. In (c) and (d) the bridging isonicotinates are shown in gold, and the pendant pyridyl groups are in green. | ||
In both MUV-26α(DMF) and MUV-26β(DMF), each cluster has two chemically different iron centres, corresponding to Fe(III) and Fe(II) oxidation states, according to bond valence sum calculations43,44 (see Tables S3–S5 in the ESI†). The presence of different oxidation states in iron trimers has been previously observed for other MOFs such as MIL-100 upon heating.45 This stable mixed-valence situation in MUV-26α(DMF) and MUV-26β(DMF) has been confirmed with Mössbauer spectroscopy, revealing that 1/3 of the irons are Fe(II) in both systems (see Section 3.4 in the ESI†).
Despite the large structural similarities between MUV-26α(DMF) and MUV-26β(DMF), close inspection of the structures reveals notable differences. Thus, MUV-26α(DMF) is formed solely by one type of cluster, in which all the apical positions of the Fe centres coordinate to pyridine moieties, with a total of 9 linkers connected to each cluster (6 via carboxylate groups and 3 via pyridine groups). 3 of these linkers connected via carboxylate groups are pendant pyridine linkers, i.e. unconnected to other clusters, behaving as pyridyl functional groups. The resulting extended framework is 2D, with pyridine molecules pointing to the interlayer space (see Fig. 3). Quite differently, MUV-26β(DMF) has two inequivalent clusters (see Fig. 2b). One of them, denoted “cluster 1” in Fig. 2, is quite similar to that found in MUV-26α(DMF), connected via 6 carboxylate and 3 pyridine groups, although one of the latter has the ring rotated 90° with respect to the other two (note the isonicotinate group bound to Fe(II) in cluster 1 of Fig. 2b). The second cluster possesses a coordinated water molecule in the axial position of one of the irons, thus being formed by 6 carboxylates, 2 pyridines and 1 water molecule. Interestingly, MUV-26β(DMF) compound similarly exhibits pendant pyridyl groups (See S2.3†), although the connectivity of the framework is three-dimensional.
 | ||
| Fig. 3 Crystal structure of MUV-26α(DMF) along the different axis. The pendant pyridyl groups are shown in green (omitted in (a) to clearly show the 2D network), whereas the bridging isonicotinate ligands are shown in white and the trimetallic SBU is shown in red. | ||
Both compounds possess ultra-small pores (<4 Å) and large void space (24.7% in MUV-26α(DMF) and 19.4% in MUV-26β(DMF)), which suggests its possible application in CO2/N2 gas separation.27,28,31 Specifically, MUV-26α(DMF) has 1D channels that run parallel to the b axis (Fig. 3), and are delimited by pyridyl groups, thus making this pore very hydrophilic. In contrast, MUV-26β(DMF) has a more complex porous structure (see Fig. S6†), but also with the pyridyl groups pointing towards the pores. These pores are filled with DMF molecules in the as-synthetized materials.
Before further characterisation, the materials were thoroughly washed with DMF and MeOH, and then activated at 150 °C for 24 hours. The integrity and phase purity of the activated bulk materials, denoted MUV-26α and MUV-26β, were assessed by a number of techniques, including PXRD, acid-digested 1H-NMR, FT-IR and TGA40 (see Section S4.1 in the ESI†). The robustness of the single crystals allowed for collecting single crystal X-ray diffraction data on the fully (MUV-26α) and partially (MUV-26β) activated structures, unequivocally showing the retention of the crystal structures upon activation.
Single component gas adsorption
Single component N2 and CO2 volumetric gas adsorption isotherms were carried out to initially evaluate the potential of these materials for gas sorption (See Section S4.2 in the ESI†). The CO2 adsorption capacity of MUV-26α and MUV-26β was respectively 2.2 and 2.0 mmol g−1 at 273 K. These CO2 capacities are in the range of ultramicroporous MOFs, and as expected, below the typically larger CO2 capacities of mesoporous MOFs, up to 9.1 mmol g−1 for PCN-124 under similar conditions.41 Analysis of these isotherms revealed BET surface areas of ca. 267 m2 g−1 and 242 m2 g−1 respectively for MUV-26α and MUV-26β and similar pores of ca. 3.58 Å (see Fig. S21 in the ESI†). In both materials, N2 isotherms revealed negligible uptake at 77 K, which agrees with the pore size of the materials, given that the kinetic diameter of N2 (3.64 Å) is larger than the one of CO2 (3.3 Å). Similar results were obtained upon high pressure studies, which also revealed negligible N2 uptake (Fig. S24 and S25†).Further investigations with higher CO2 pressures carried out at different temperatures revealed no major changes in loading capacity (Fig. 4), exhibiting a maximum of 2.8 mmol g−1 (MUV-26α) and of 2.0 mmol g−1 (MUV-26β) at 6 bar and 283 K (see S4.3†). Importantly, complete regeneration of the materials is obtained upon pressure changes at room temperature, thus reducing regeneration cost in view of industrial applications. The structural robustness of the materials was ascertained after the high-pressure CO2 sorption studies by sorption capacity replication at 25 °C and PXRD analysis (Fig. S25 and S27†). Virial fitting of the isotherms at different temperatures allows calculating the enthalpy of adsorption which resulted in 31.4 and 30.4 kJ mol−1 for MUV-26α and MUV-26β, respectively (see Fig. S26†). These energy values are higher than our previously reported anilato-based ultramicroporous MOF (21.07 kJ mol−1) which exhibited some of the highest reported selectivities.17 Essentially, the presence of pendant linkers with pyridine moieties pointing to the reduced pore space (see Fig. 3) seems to enhance the thermodynamics of CO2 adsorption due to effective interaction with CO2 molecules,42–44 as previously reported for a Ni-isonicotinate MOF.45 Nevertheless, the enthalpy of adsorption values agree with computational and experimental studies showing that 9 out of the 15 best MOFs for CO2 gas separation have enthalpies of adsorption between 30 and 50 kJ mol−1.28
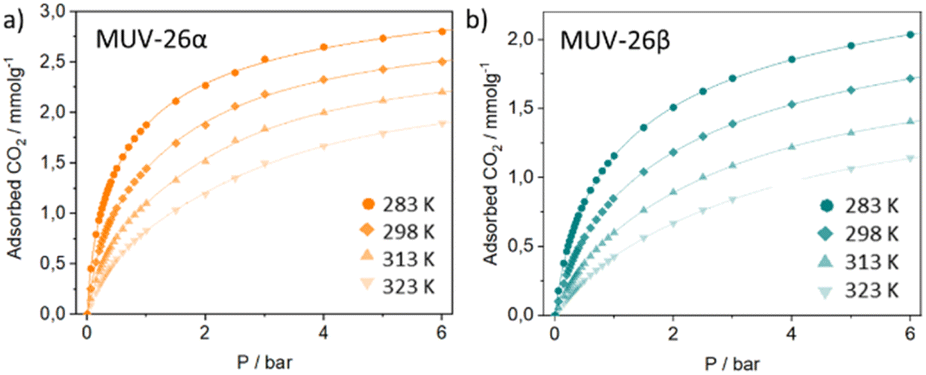 | ||
| Fig. 4 High-pressure gravimetric CO2 adsorption isotherms of MUV-26α (a) and MUV-26β (b) at different temperatures. The lines correspond to the Virial fitting of the data. | ||
CS2 adsorption by MUV-26α
In order to elucidate the interactions of CO2 with MUV-26 compounds, crystals of MUV-26α were soaked for 1 day in CS2, a molecule crystallographically easier to locate due to the larger amount of electron density found at the sulphur atoms. CS2 and CO2 have similar geometric and electronic configurations, allowing to correlate the crystallographic positions of the CS2 with the ones occupied by the CO2 during the sorption process. Single crystal X-ray diffraction analysis of the MUV-26α(CS2) structure revealed the uptake of 1.5 molecules of CS2 per formula unit of MUV-26α. CS2 molecules were crystallographically located in two different positions in the interlayer space of the MUV-26α coordination polymers.The CS2 molecules interact with pyridyl groups of the isonicotinate ligands forming hydrogen S⋯H–C hydrogen bonds and S⋯C–O contacts with the carbons of the carboxylate groups (Fig. S7 and Table S6†). These type of hydrogen bonds with pyridyl groups and electrostatic interactions with the carbon in the carboxylate groups of interactions have been also reported for CO2.46 Occupancy of the CS2 molecules is similar to CO2 adsorption at 1 bar of pressure at room temperature.
Breakthrough CO2/N2 gas separation
The observed differences between N2 and CO2 sorption, together with the experimental CO2 adsorption capacity and complete material renewability at room temperature, brought us to study the separation performance under breakthrough dynamic conditions (see ESI Section S.5† for detailed experimental procedures, breakthrough experiments and adsorption capacities).To evaluate the aforementioned parameters, we have diluted CO2 in three concentrations, namely 5, 20 and 50% in CO2:N2 gas mixtures maintaining constant the total flow (15 mL min−1) and a final pressure of 1 bar (operational CO2 partial pressures 0.05, 0.2 and 0.5 bar, respectively). Helium was added to the gas mixture as a system tracer to evaluate the possible adsorption of N2.
Fig. 5 exemplifies the breakthrough curves at 298 K of the most and least challenging conditions for CO2:N2 separation (5% CO2 and 50% CO2, respectively) for both materials. CO2 break-time is remarkably larger than the He and N2 ones, evidencing a selective CO2 adsorption, as anticipated by single-component isotherms, that results in gas separation. When CO2 finally breaks through the column, the typical roll-up is observed for He and N2 profiles.
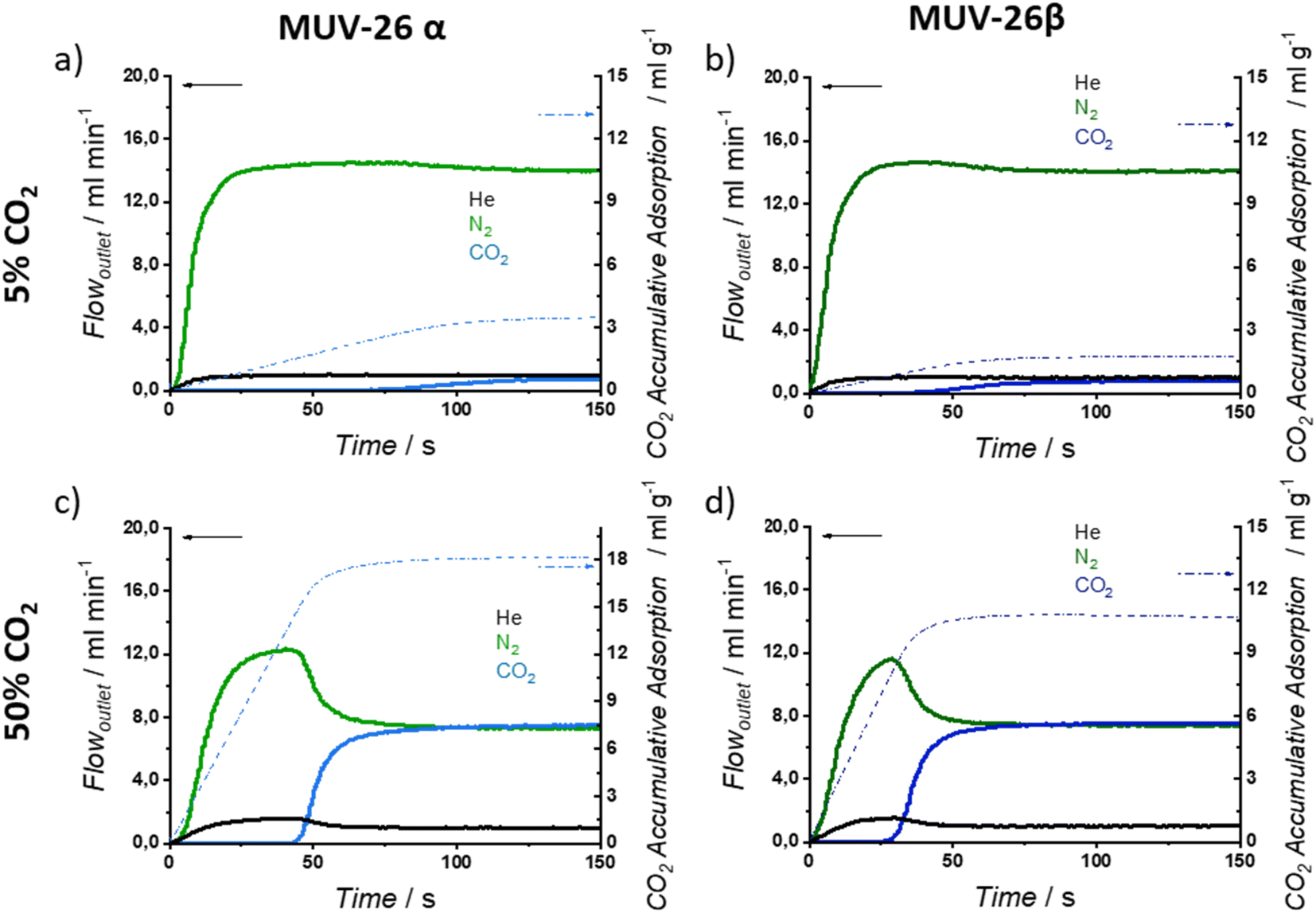 | ||
| Fig. 5 Breakthrough exit flowrates (solid line, left axis) and CO2 accumulative adsorption (dash-dot line, right axis) vs. time at 298 K and 1 bar, on MUV-26. Time zero is set with the first detection of helium (tracer). The total flow rate is 15 mL min−1. Inlet composition corresponds to 5:95 (CO2:N2) for (a) MUV-26α and (b) MUV-26β, whereas the inlet composition corresponds to 50:50 for (c) MUV-26α and (d) MUV-26β. | ||
Under dynamic conditions, CO2 adsorption capacity is slightly higher for MUV-26α, adsorbing up to 18.4 mL g−1 (0.82 mol kg−1) for an inlet composition of 50:50 CO2:N2 at 298 K (see ESI Section S.5 and Tables S11 and S13† for detailed adsorption capacities). It is worth noting that single gas isotherms revealed a ca. 1.44 mol kg−1 CO2 adsorption capacity at 1 bar and 298 K, exemplifying the differences between dynamic and static adsorption capacities and highlighting the need to evaluate the adsorption capacity of materials under out-of-equilibrium dynamic conditions. For both materials, CO2 adsorption capacity increases with its concentration.
In an attempt to gain insights into the (dominating) role in the gas separation performance of thermodynamics (exothermal process favoured with low temperatures) and kinetics (diffusion effects improved at increasing temperatures), studies at different temperatures were carried out.47,48 Three different temperatures (283, 298 and 323 K) have been studied for a 20% CO2 dilution (see Fig. 6), which is more representative of an industrial CO2 stream concentration. Calculation of the dynamic selectivity value (α) was then determined from the different amount of adsorbed gas obtained in the breakthrough profiles. The calculated selectivities are virtually infinite for both materials under all the studied conditions, as no N2 is adsorbed even under the most diluted conditions (see ESI Section S.5†). To the best of our knowledge, there are no reported porous materials with such a high CO2 selectivity for all the studied conditions, as typically ultramicroporous MOFs show reduced selectivity at high temperatures, as a result of an improved diffusion of the gas mixture (see Table 1).15,16
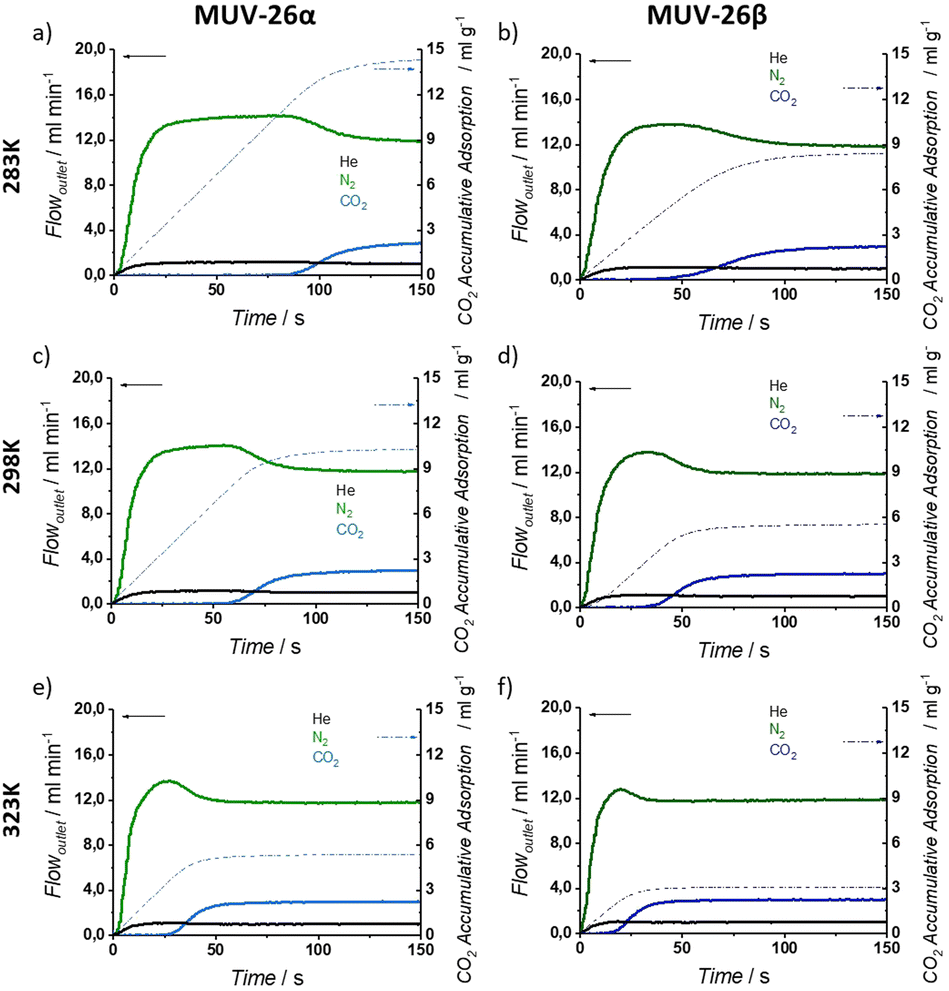 | ||
| Fig. 6 Breakthrough exit flow rates (solid line, left axis) and CO2 accumulative adsorption (dash-dot line, right axis) vs. time for 20:80 (CO2:N2) inlet composition at variable temperatures. 283 K for (a) MUV-26α and (b) MUV-26β; 298 K for (c) MUV-26α and (d) MUV-26β; 323 K for (e) MUV-26α and (f) MUV-26β. | ||
| T/%CO2 in N2 | α | Reference | |
|---|---|---|---|
| a Given that no Nitrogen is adsorbed during the breakthrough experiments, we are reporting the virtually infinite selectivity as >1000. | |||
| MUV-26α | 298 K/5% | >1000 | This work |
| 323 K/5% | >1000 | This work | |
| MUV-26β | 298 K/5% | >1000 | This work |
| 323 K/5% | >1000 | This work | |
| Co[(trz2An)]n·3H2O | 298 K/5% | >500 | 16 |
| 298 K/20% | >1000 | 16 | |
| 323 K/5% | 10 | 16 | |
| ZIF-100 | 298 K/50% | 25 | 49 |
| ZIF-95 | 298 K/50% | 18 | 49 |
| MIL-53(Al)–NH2 | 303 K/50% | 15 | 50 |
| MIL-100 (Fe) | 303 K/50% | 8.6 | 51 |
| 303 K/15% | 4.6 | 51 | |
| MOF-508b | 303 K/50% | 5 | 52 |
| JLU-MOF56 | 298 K/5% | 38.6 | 53 |
| 298 K/10% | 32.9 | 53 | |
| 298 K/15% | 32.8 | 53 | |
| 298 K/50% | 34.7 | 53 | |
| UTSA-120 | 298 K/15% | 600 | 54 |
| Cu(hfipbb) | 298 K | 21.5 | 55 |
| 318 K | 18.3 | 55 | |
| UTSA-16 | 296 K/15% | 329 | 56 |
| IISERP-MOF2 | 313 K/14% | 1853 | 45 |
In fact, our previously reported anilate-based ultramicroporous MOF, which exhibited virtually infinite CO2:N2 separation, had a remarkable decrease in selectivity under diluted conditions.16 Thus, we investigated the gas separation performance of MUV-26α under diluted conditions (5% CO2) and increased temperature (323 K), showing also no N2 gas uptake. This positions MUV-26 materials among the most selective materials for CO2:N2 gas separation.
These results confirm that our materials effectively separate CO2 from N2 not only through size-exclusion kinetics but also through thermodynamic gas-adsorbent interactions, given that although the selectivity is complete in all the cases, the adsorption capacity is higher at low temperatures (see Tables S11 and S13 in the ESI†), where thermodynamic effects are favoured.44
Moreover, MUV-26 materials are easily regenerable at room temperature and ambient pressure by simply flowing Ar for 20 minutes, showing identical selectivity for all the aforementioned conditions after two gas separation adsorption–desorption cycles (see ESI Section S.5†).
Finally, we evaluated the stability of MUV-26α over 10 adsorption–desorption gas separation cycles (20% CO2, 298 K) (Fig. S34†), demonstrating the reusability of the material with complete regeneration at room temperature and 1 bar, with retention of the crystalline structure as confirmed by XRPD (Fig. S38†).
Conclusions
The combination of the commercially available isonicotinic acid with preformed trimeric iron clusters under different solvothermal conditions has yielded two different novel ultramicroporous MOFs, namely MUV-26α and MUV-26β. Both MOFs are composed of the same secondary building units (mixed-valence iron trimers and isonicotinate linkers), have remarkably small pores (<4 Å) and importantly, contain tangling linkers in their structure in which the pyridine unit is pendant and points to the pore space.These MOFs combine both kinetic and thermodynamic separations57 given their small pore window that excludes N2 adsorption and the pendant pyridine groups that point to the small pore cavities and interact with CO2 molecules. The materials exhibit virtually infinite CO2/N2 selectivity under all the studied breakthrough conditions, including challenging gas mixtures unprecedented in the literature with low CO2 concentrations (5%) and high temperatures (323 K).
These materials undergo full regeneration at room temperature and ambient pressure and have proven fully regenerable during 10 consecutive replications, exhibiting relatively high working capacities at 1 bar (up to 0.82 mol kg−1 in dynamic gas separation conditions), and resulting in separation potential values of 58 mol L−1 for MUV-26α and 37 mol L−1 for MUV-26β, which are significantly higher than zeolites46 and most reported MOFs.17,28
It is important to remark that although mesoporous and microporous MOFs exhibit superior adsorption capacities than ultramicroporous MOFs, their poor selectivity hinders their application in the gas separation industry. Moreover, even though highly porous MOFs can be functionalised to increase selectivity through thermodynamic processes, considering the gas molecules adsorbed on a large pore space, the gas-framework interactions around the walls will be stronger than for those gas molecules in the centre of the framework, meaning that the energy for desorption will follow a gradient. In contrast, for ultramicroporous materials in which the pores are so small that only a few gas molecules are adsorbed, the gas-adsorbent interaction will be of equal strength, providing narrow desorption energy beneficial for industrial applications.5,27
All-in-all, the facile synthesis using commercially available precursors, the virtually infinite CO2/N2 selectivity even under diluted CO2 concentrations and high temperatures, the good adsorption capacities and the complete regeneration under ambient conditions leads to one of the highest adsorbent performance scores reported to date, positioning these MOFs among the most promising materials for CO2/N2 gas separation.
Experimental section
Synthesis of MUV-26α(DMF)
450 mg (0.65 mmol) of the preformed cluster [Fe3O(CH3COO)6]ClO4·3H2O was dissolved in 41.25 mL of DMF. Then, 923 mg of isonicotinic acid (7.5 mmol) was added to the solution and sonicated until complete dispersion. The dispersion was placed in an oven and heated to 120 °C for 48 hours. The reaction was cooled down to room temperature and the precipitated crystals were washed with DMF (×5). For the activated sample, further washing with MeOH (×3) was performed, followed by overnight immersion in MeOH and activation at 150 °C under vacuum for 24 hours, yielding ca. 400 mg of crystals, corresponding to an approximate yield of 44%.Synthesis of MUV-26β(DMF)
A similar procedure to the synthesis of MUV-26α(DMF) was performed, but using 691 mg (1 mmol) of the preformed cluster [Fe3O(CH3COO)6]ClO4·3H2O and 1.847 g of isonicotinic acid (15 mmol), yielding ca. 450 mg of crystals, corresponding to an approximate yield of 49%.Single crystal diffraction
Single crystals were mounted on glass fibres using a viscous hydrocarbon oil to coat the crystals and then transferred directly to the cold nitrogen stream for data collection. X-ray data were collected at 120 K on a Supernova diffractometer equipped with a graphite-monochromated enhance (Mo) X-ray source (λ = 0.71073 Å). The program CrysAlisPro, Rigaku, was used for unit cell determinations and data reduction. Empirical absorption correction was performed using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm. The crystal structures were solved with the ShelXT structure solution program and refined against all F2 values using the SHELXL and Olex2 suite of programs.58,59 Non-hydrogen atoms were refined anisotropically, and hydrogen atoms were placed in calculated positions refined using idealized geometries (riding model) and assigned fixed isotropic displacement parameters. Crystallographic data are summarized in Tables S1 and S2.† CCDC 2217199–2217202, 2237339–2237340 contains the supplementary crystallographic data for this paper.Gas sorption
Low-pressure N2 and CO2 volumetric isotherms were carried out in a Tristar II Plus Micromeritics sorptometer, at 77 K and 273 K, respectively. Activation was set at 393 K, under vacuum, for 2 hours. High-pressure gravimetric adsorption isotherms of CO2 were measured at different temperatures, ranging from 283 to 323 K, in an IGA-100 gas sorption analyser (from Hiden Isochema) using approximately 50 mg of sample. Before each adsorption experiment, the sample was outgassed at 393 K under vacuum (10−5 Pa) for two hours. Equilibrium conditions corresponded to 600 s interval, and 0.001 mg min−1 tolerance.Dynamic adsorptive separation measurements
An ABR (HIDEN Isochema) automated breakthrough analyser setup, based on a packed adsorption column, was used to determine the adsorption dynamics of gas mixtures. Pressure, temperature and inlet composition were controlled, and the outlet composition was analysed, by an integrated mass spectrometer (HPR-20 QIC). The fixed-bed column was filled with 374 mg of MUV-26α or with 447 mg of MUV-26β. Before each measurement, the sample was regenerated at atmospheric temperature and pressure, in 40 mL min−1 Ar flow for 20 minutes. Operation conditions ranged 283–323 K, at 1 bar. The inlet mixture was set to a 15 mL min−1 flow of a dilution of carbon dioxide in N2 (5%, 20%, 50%). Time zero was set with the first detection of helium, which was used as a trace (an extra 1 mL min−1 of He in the total feed flow of 16 mL min−1).Mössbauer spectroscopy
Mössbauer spectra were collected at 4 K in transmission mode using a bath cryostat as described in detail in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work has been supported by the European Union (ERC-2016-CoG 724681-S-CAGE), grants PID2020-117177GB-I00, PID2020-118564GA-I00, TED2021-132729A-I00 and CEX2019-000919-M, funded by MCIN/AEI/10.13039/501100011033, and the Generalitat Valenciana (PROMETEO programme and IDIFEDER/2021/075). This study forms part of the Advanced Materials program and was supported by MCIN with funding from European Union NextGenerationEU (PRTR-C17.I1) and by Generalitat Valenciana (project MAF/2022/31). M. G.-M., I. A. L. and E. A.-G. thank MICINN for a Ramón y Cajal fellowship (RYC2019-027902-I), a Juan de la Cierva Incorporación fellowship (IJC2020-044374-I) and a Margarita Salas fellowship (MS21-035), respectively. FCT (Portugal) through contracts UID/Multi/04349/2019 and PTDC/QUI-QIN/32240/2017 is acknowledged.Notes and references
- R. L. Siegelman, E. J. Kim and J. R. Long, Nat. Mater., 2021, 20, 1060–1072 CrossRef CAS PubMed
.
- A. Stips, D. Macias, C. Coughlan, E. Garcia-Gorriz and X. S. Liang, Sci. Rep., 2016, 6, 21691 CrossRef CAS PubMed
- G. S. Callendar and Q. J. Roy, Meteor. Soc., 1938, 64, 223–240 CrossRef
- J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef CAS
- A. A. Olajire, Energy, 2010, 35, 2610–2628 CrossRef CAS
- D. M. Thomas, J. Mechery and S. V. Paulose, Environ. Sci. Pollut. Res., 2016, 23, 16926–16940 CrossRef CAS PubMed
- M. Safaei, M. M. Foroughi, N. Ebrahimpoor, S. Jahani, A. Omidi and M. Khatami, TrAC, Trends Anal. Chem., 2019, 118, 401–425 CrossRef CAS
- P. Z. Moghadam, A. Li, X.-W. Liu, R. Bueno-Perez, S. Wang, S. Wiggin, P. A. Wood and D. Fairen-Jimenez, Chem. Sci., 2020, 11, 8373–8387 RSC
- H.-C. Joe Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC
- Q. Wang and D. Astruc, Chem. Rev., 2020, 120, 1438–1511 CrossRef CAS PubMed
- I. A. Lázaro and R. S. Forgan, Coord. Chem. Rev., 2019, 380, 230–259 CrossRef
- G. Mínguez Espallargas and E. Coronado, Chem. Soc. Rev., 2017, 47, 533–557 RSC
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC
- H. Li, L. Li, R.-B. Lin, W. Zhou, Z. Zhang, S. Xiang and B. Chen, Energychem, 2019, 1, 100006–100045 CrossRef
- E. Miguel-Casañ, E. Andres-Garcia, J. Calbo, M. Giménez-Marqués and G. Mínguez Espallargas, Chem. – Eur. J., 2021, 27, 4653–4659 CrossRef PubMed
- N. Monni, E. Andres-Garcia, K. Caamaño, V. García-López, J. M. Clemente-Juan, M. Giménez-Marqués, M. Oggianu, E. Cadoni, G. Mínguez Espallargas, M. Clemente-León, M. L. Mercuri and E. Coronado, J. Mater. Chem. A, 2021, 9, 25189–25195 RSC
- R.-B. Lin, S. Xiang, W. Zhou and B. Chen, Chem, 2020, 6, 337–363 CAS
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 3749–3761 CrossRef CAS
- X. Zhang, Z. Chen, X. Liu, S. L. Hanna, X. Wang, R. Taheri-Ledari, A. Maleki, P. Li and O. K. Farha, Chem. Soc. Rev., 2020, 49, 7406–7427 RSC
- T. Wang, E. Lin, Y.-L. Peng, Y. Chen, P. Cheng and Z. Zhang, Coord. Chem. Rev., 2020, 423, 213485–213518 CrossRef CAS
- S. Mukherjee, S. Chen, A. A. Bezrukov, M. Mostrom, V. V. Terskikh, D. Franz, S. Wang, A. Kumar, M. Chen, B. Space, Y. Huang and M. J. Zaworotko, Angew. Chem., Int. Ed., 2020, 59, 16188–16194 CrossRef CAS PubMed
- H. S. Scott, N. Ogiwara, K.-J. Chen, D. G. Madden, T. Pham, K. Forrest, B. Space, S. Horike, J. J. P. IV, S. Kitagawa and M. J. Zaworotko, Chem. Sci., 2016, 7, 5470–5476 RSC
- K. Chen, D. G. Madden, T. Pham, K. A. Forrest, A. Kumar, Q. Yang, W. Xue, B. Space, J. J. Perry, J. Zhang, X. Chen and M. J. Zaworotko, Angew. Chem., Int. Ed., 2016, 55, 10268–10272 CrossRef CAS PubMed
- P. M. Bhatt, Y. Belmabkhout, A. Cadiau, K. Adil, O. Shekhah, A. Shkurenko, L. J. Barbour and M. Eddaoudi, J. Am. Chem. Soc., 2016, 138, 9301–9307 CrossRef CAS PubMed
- Z. Zhang, S. B. Peh, R. Krishna, C. Kang, K. Chai, Y. Wang, D. Shi and D. Zhao, Angew. Chem., Int. Ed., 2021, 60, 17198–17204 CrossRef CAS PubMed
- K. Adil, Y. Belmabkhout, R. S. Pillai, A. Cadiau, P. M. Bhatt, A. H. Assen, G. Maurin and M. Eddaoudi, Chem. Soc. Rev., 2017, 46, 3402–3430 RSC
- S. Shalini, S. Nandi, A. Justin, R. Maity and R. Vaidhyanathan, Chem. Commun., 2018, 54, 13472–13490 RSC
- C. Altintas, G. Avci, H. Daglar, A. N. V. Azar, S. Velioglu, I. Erucar and S. Keskin, ACS Appl. Mater. Interfaces, 2018, 10, 17257–17268 CrossRef CAS PubMed
- M. T. Ho, G. W. Allinson and D. E. Wiley, Ind. Eng. Chem. Res., 2008, 47, 1562–1568 CrossRef CAS
- H. Li, K. Wang, Y. Sun, C. T. Lollar, J. Li and H.-C. Zhou, Mater. Today, 2018, 21, 108–121 CrossRef CAS
- C. G. Piscopo and S. Loebbecke, Chempluschem, 2020, 85, 538–547 CrossRef CAS PubMed
- A. Henley, M. J. Lennox, T. L. Easun, F. Moreau, M. Schröder and E. Besley, J. Phys. Chem. C, 2016, 120, 27342–27348 CrossRef CAS
- X. Li, Q. Su, K. Luo, H. Li, G. Li and Q. Wu, Mater. Lett., 2020, 282, 128704–128708 CrossRef
- D.-M. Chen, N.-N. Zhang, C.-S. Liu, Z.-H. Jiang, X.-D. Wang and M. Du, Inorg. Chem., 2017, 56, 2379–2382 CrossRef CAS PubMed
- H. D. Singh, S. Nandi, D. Chakraborty, K. Singh, C. P. Vinod and R. Vaidhyanathan, Chem. – Asian J., 2022, 17, e202101305–e202101311 CrossRef CAS PubMed
- S. Nandi, S. Collins, D. Chakraborty, D. Banerjee, P. K. Thallapally, T. K. Woo and R. Vaidhyanathan, J. Am. Chem. Soc., 2017, 139, 1734–1737 CrossRef CAS PubMed
- L. Zhou, H. Fan, B. Zhou, Z. Cui, B. Qin, X. Zhang, W. Li and J. Zhang, Dalton Trans., 2018, 48, 296–303 RSC
- H. Leclerc, A. Vimont, J.-C. Lavalley, M. Daturi, A. D. Wiersum, P. L. Llwellyn, P. Horcajada, G. Férey and C. Serre, Phys. Chem. Chem. Phys., 2011, 13, 11748–11756 RSC
- M. Souto, A. Santiago-Portillo, M. Palomino, I. J. Vitórica-Yrezábal, B. J. C. Vieira, J. C. Waerenborgh, S. Valencia, S. Navalón, F. Rey, H. García and G. Mínguez Espallargas, Chem. Sci., 2018, 9, 2413–2418 RSC
- I. A. Lázaro, Eur. J. Inorg. Chem., 2020, 2020, 4284–4294 CrossRef
- J. Park, J.-R. Li, Y.-P. Chen, J. Yu, A. A. Yakovenko, Z. U. Wang, L.-B. Sun, P. B. Balbuena and H.-C. Zhou, Chem. Commun., 2012, 48, 9995–9997 RSC
- X. Luo, Y. Guo, F. Ding, H. Zhao, G. Cui, H. Li and C. Wang, Angew. Chem., Int. Ed., 2014, 53, 7053–7057 CrossRef CAS PubMed
- S. P.-M. Ung and C.-J. Li, RSC Sustain., 2023, 1, 11–37 RSC
- X.-J. Wang, P.-Z. Li, Y. Chen, Q. Zhang, H. Zhang, X. X. Chan, R. Ganguly, Y. Li, J. Jiang and Y. Zhao, Sci. Rep., 2013, 3, 1149–1154 CrossRef PubMed
- S. Nandi, S. Collins, D. Chakraborty, D. Banerjee, P. K. Thallapally, T. K. Woo and R. Vaidhyanathan, J. Am. Chem. Soc., 2017, 139(5), 1734–1737 CrossRef CAS PubMed
-
(a) H. M. Lee, S. Youn, M. Saleh, J. W. Lee and K. S. Kim, Phys. Chem. Chem. Phys., 2015, 17, 10925 RSC
- M. M. Zagho, M. K. Hassan, M. Khraisheh, M. A. A. Al-Maadeed and S. Nazarenko, Chem. Eng. J. Adv., 2021, 6, 100091–100111 CrossRef CAS
- A. N. Ebelegi, N. Ayawei and D. Wankasi, Open J. Phys. Chem., 2020, 10, 166–182 CrossRef CAS
- B. Wang, A. P. Côté, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nature, 2008, 453, 207–211 CrossRef CAS PubMed
- E. Stavitski, E. A. Pidko, S. Couck, T. Remy, E. J. M. Hensen, B. M. Weckhuysen, J. Denayer, J. Gascon and F. Kapteijn, Langmuir, 2011, 27, 3970–3976 CrossRef CAS PubMed
- S. Xian, J. Peng, Z. Zhang, Q. Xia, H. Wang and Z. Li, Chem. Eng. J., 2015, 270, 385–392 CrossRef CAS
- M. Palomino, A. Corma, J. L. Jordá, F. Rey and S. Valencia, Chem. Commun., 2011, 48, 215–217 RSC
- S. Liu, S. Yao, B. Liu, X. Sun, Y. Yuan, G. Li, L. Zhang and Y. Liu, Dalton Trans., 2019, 48, 1680–1685 RSC
- H.-M. Wen, C. Liao, L. Li, A. Alsalme, Z. Alothman, R. Krishna, H. Wu, W. Zhou, J. Hu and B. Chen, J. Mater. Chem. A, 2019, 7, 3128–3134 RSC
- X. Wu, B. Yuan, Z. Bao and S. Deng, J. Colloid Interface Sci., 2014, 430, 78–84 CrossRef CAS PubMed
- S. Xiang, Y. He, Z. Zhang, H. Wu, W. Zhou, R. Krishna and B. Chen, Nat. Commun., 2012, 3, 954–1003 CrossRef PubMed
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Containing experimental conditions, detailed characterisation and breakthrough gas separation measurements available. CCDC 2217199–2217202, 2237339–2237340. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ta08934c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |