
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVinylene carbonate reactivity at lithium metal surface: first-principles insights into the early steps of SEI formation†
Francesca
Fasulo
 a,
Ana B.
Muñoz-García
b,
Arianna
Massaro
a,
Orlando
Crescenzi
a,
Chen
Huang
c and
Michele
Pavone
*a
a,
Ana B.
Muñoz-García
b,
Arianna
Massaro
a,
Orlando
Crescenzi
a,
Chen
Huang
c and
Michele
Pavone
*a
aDepartment of Chemical Sciences, University of Naples Federico II, Napoli, Italy. E-mail: michele.pavone@unina.it
bDepartment of Physics “E. Pancini”, University of Naples Federico II, Napoli, Italy
cDepartment of Scientific Computing, Florida State University, Tallahassee, Florida, USA
First published on 1st February 2023
Abstract
Engineering a solid-electrolyte interphase (SEI) with purposely designed molecules represents a promising strategy to achieve durable and effective anodes for lithium metal batteries (LMBs). The use of vinylene carbonate (VC) as an additive in conventional electrolytes has been shown to promote the formation of a stable and protective SEI at the Li metal interface. The fine-tuning and control of the underlying reactions still represent a major issue, due to complex VC decomposition and polymerization processes that may occur upon battery cycling. To dissect the tangled VC reactivity, here we present new atomistic insights into VC-induced SEI formation at the Li(001) interface: Density Functional Embedding Theory (DFET) is employed to combine the best feasible computational approaches to treat molecular species with localized charge (i.e., VC derivatives upon the reductive decomposition process) and the Li metal surface by means of hybrid DFT and semi-local GGA-based methods, respectively. Exploring VC adsorption and dissociation paths, our DFET investigation reveals that the thermodynamically accessible mechanisms for the VC ring-opening reductive reaction on Li(001) feature energy barriers in the range of 0.29–0.34 eV. Dissociation via cleavage at vinylic sites (i.e., CV–OV) is more likely to occur and leads to a highly reactive intermediate that can undergo either further decomposition towards C2H2 and Li2CO3 formation or a polymerization process, in close agreement with experimental observations. By setting solid scientific foundations for advanced understanding of initial SEI formation, our theoretical results can drive future experimental efforts towards the rational design of Li/electrolyte interfaces with tailored properties for high-performing LMB devices.
1 Introduction
Lithium Metal Batteries (LMBs) are among the most promising next-generation energy storage technologies.1–3 The use of Li metal as an anode material is the object of great interest in both academia and industry: the low electrochemical potential (−3.047 V vs. standard hydrogen electrode, SHE), the ultrahigh theoretical specific capacity (∼3860 mA h g−1), and the low density (0.534 g cm−3) are some of the attractive features for the employment of Li metal anodes in high-energy and efficient devices, from electric vehicles to space applications.4,5 However, major challenges including long-term stability and performance issues need to be addressed to boost the LMB development as a viable solution for electric energy storage (EES) applications.Dendrite formation at the metal electrode/electrolyte interface and the related side reactions can increase the generation of electrically “dead” Li, with detrimental effects on coulombic efficiency and mechanical properties.6,7 In the worst scenario, uncontrolled dendritic growth upon cycling can lead to severe safety hazards. A well-known effective strategy to mitigate those problems is given by protective and stable layers between the Li metal and the electrolyte, namely the solid electrolyte interphase (SEI).8 An effective SEI allows Li+ transport and blocks electrons in order to prevent further electrolyte decomposition and ensure continued electrochemical reactions. This nanometer thick film generally consists of organic and inorganic species deriving from the degradation of electrolyte components that are redox-active within the operating voltage range. The resulting intricate mixture covers the highly reductive Li metal anode and thus directly determines its morphology as well as the Li+ deposition-dissolution mechanisms.9–12 Intense research efforts are currently devoted to minimizing the dendritic growth and maintaining the protective nature of the SEI: very promising results have been achieved by modifying the electrolytes with small amounts of electrochemically inactive materials.12–15 Recent experiments proved that the addition of vinylene carbonate (VC) can assist the development of an effective SEI on Li metal either with common organic (e.g., ethylene and diethyl/dimethyl carbonate, EC and DEC/DMC) or protic ionic liquid electrolytes (PILs).16–28 Enhanced performances can be achieved with an electrolyte formulation with a small amount of VC as additive: the optimal concentration of ∼1–2 wt% can successfully limit the uncontrolled electrolyte decomposition not only on graphite or Li metal anodes, but also on LiNi1/3Mn1/3Co1/3O2 cathode material, suggesting promising capability towards both SEI and CEI (cathode electrolyte interphase) formations that can directly benefit anodic and cathodic stability windows, battery lifecycle and coulombic efficiency.16,17,21
From the microscopic point of view, the chemical composition and morphology of the SEI produced in VC-containing electrolytes largely differ from those of VC-free ones, the former showing a mosaic-like nanostructure comprised of organic species, Li2O and Li2CO3 and the latter a multilayered SEI involving Li2O and Li domains.22 The VC passivating properties seem to be remarkable and they are generally ascribed to its peculiar reactivity towards multiple decomposition pathways that can occur during the first charge cycle. In contrast to other carbonates, e.g., ethylene (EC) and propylene (PC) carbonates, VC can also undergo polymerization in the presence of a radical initiator. Poly(vinylene carbonate) (poly(VC)) has been identified on Li metal anodes and the resulting SEI is shown to improve the electrochemical performance at higher working temperature and to successfully resist possible thermal damage.18–20,23–28 As reported by ex situ investigations on Li metal anodes and gas evolution analysis, VC polymerization can change the primary gas generated and it turns out to be mostly accompanied by CO2 release, with rather low or even neglectable development of CO and C2H2.20,21,23–26 As a matter of fact, the development of other reductive decomposition gas components, such as ethylene or methane, is noticeably suppressed in the presence of VC, being mainly related to EC-DMC reduction reactions.28
Despite these remarkable advances, the mechanisms responsible for VC reductive decomposition and polymerization are still little understood, a knowledge gap that can be traced back to intrinsically demanding characterization and modelling of LMB electrode/electrolyte interfaces. On the one hand, the complexity of in situ observation at the electrode–electrolyte interface upon battery operation makes the VC reactivity and subsequent SEI growth hard to unveil. On the other hand, the application of multiscale simulations in a wide range of spatial and temporal scales necessary to unravel intricate reaction mechanisms can be challenging.29 The theoretical investigation performed with atomistic details and direct access to electronic structures can be helpful towards the fundamental understanding of reductive decomposition mechanisms. In general, the first step toward the reduction of carbonate-based molecules is a ring-opening reaction. Different decomposition paths can be envisaged, involving an initial single-bond cleavage occurring either at the –O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– vinyl or at the –O–CO carbonyl group. Computational studies can complement the experimental efforts to gain in-depth understanding and mechanistic insights from an atomistic perspective. To the best of our knowledge, previous DFT-based studies have addressed the VC ring-opening reaction mechanisms in conventional electrolyte solvents,18,30–35 but only different carbonate-based additives have been investigated at the Li metal interface.36,37 On one hand, reductive cleavage at the carbonyl site (formally breaking a vinyl ester C–O bond of the O–CO moiety) seems to be kinetically favored compared to a cleavage at the vinyl site (formally breaking a vinyl ether C–O bond of the O–CC moiety), and the former mechanism can be further promoted in electrolyte formulations with a high content of reducing agents.25,30,32 On the other hand, the less-likely ˙CHCH–O–COO– radical anion derived from the reductive cleavage at the vinyl site is considered as a probable radical initiator toward VC polymerization, despite showing an energy barrier of ∼0.8 eV.33–35 Online Electrochemical Mass Spectrometry Analysis (OEMS) results on the CO2 release would also support this mechanism and thus suggest that radical addition by the initial VC reduction product ˙CHCH–O–COO– could be the dominant pathway toward poly(VC) formation.21
C– vinyl or at the –O–CO carbonyl group. Computational studies can complement the experimental efforts to gain in-depth understanding and mechanistic insights from an atomistic perspective. To the best of our knowledge, previous DFT-based studies have addressed the VC ring-opening reaction mechanisms in conventional electrolyte solvents,18,30–35 but only different carbonate-based additives have been investigated at the Li metal interface.36,37 On one hand, reductive cleavage at the carbonyl site (formally breaking a vinyl ester C–O bond of the O–CO moiety) seems to be kinetically favored compared to a cleavage at the vinyl site (formally breaking a vinyl ether C–O bond of the O–CC moiety), and the former mechanism can be further promoted in electrolyte formulations with a high content of reducing agents.25,30,32 On the other hand, the less-likely ˙CHCH–O–COO– radical anion derived from the reductive cleavage at the vinyl site is considered as a probable radical initiator toward VC polymerization, despite showing an energy barrier of ∼0.8 eV.33–35 Online Electrochemical Mass Spectrometry Analysis (OEMS) results on the CO2 release would also support this mechanism and thus suggest that radical addition by the initial VC reduction product ˙CHCH–O–COO– could be the dominant pathway toward poly(VC) formation.21
Notwithstanding the broad literature and the great advances in this field, we believe that atomistic investigation on VC decomposition mechanisms should not disregard the role of the Li interface. While recent studies investigated the influence of Li+ in the electrolyte medium,18,30–34 we now consider the role of the Li metal anode surface representing the physical contact at the electrode/electrolyte interface in LMB devices and acting as a key player in the VC reductive decomposition. To this end, we hereby report a thorough study on the adsorption and dissociation of VC molecule on the most stable Li metal surface, i.e., the (001) lattice termination.36,38 The higher stability suggests that the (001) surface is predominant in real samples, as Li metal will preferentially expose the termination requiring minor energy expenditure (i.e., lowest surface energy, see Table S1 in the ESI†). Other Li metal surfaces may still be present and could draw interest due to different electron densities probably leading to different activity towards VC decomposition. Nevertheless, addressing the multitude of surface reactivity was beyond the scope of this work, where we choose to focus on the main VC/Li(001) interactions by application of the recently proposed Density Functional Embedding Theory (DFET).39–43 Even though Density Functional Theory (DFT) provides a great balance between computational effort and accuracy, the widely used density functionals within the general gradient approximation (GGA) (i.e., without including Hartree–Fock-like exchange) are not able to describe the correct physics of bond breaking processes and strongly correlated systems.44 The development of density-based embedding strategies has successfully yielded localized features in periodic metallic systems.45 By employing the unique embedding potential (Vemb), we model the VC/Li(001) interface as a system that can be partitioned into a cluster and its environment.40 In this way, we can overcome the current limitation of semi-local DFT methods in modeling localized interactions between reacting molecules and metal surfaces. On one hand, the choice of hybrid functionals (in which the conventional LDA or GGA exchange-correlation functional is mixed with a fraction of the Hartree–Fock exchange energy) to describe confined charge transfer phenomena is well-suited for the VC decomposition products with localized electron density.46 On the other hand, the GGA level of theory enables reliable predictions on the electronic structure of metallic systems, e.g., the Li metal surface.47 In this work, the environment (i.e., lithium surface) is treated with the Perdew–Burke–Ernzerhof (PBE) functional,48 and the clusters are treated with the Heyd–Scuseria–Ernzerhof (HSE06) hybrid functional.49,50 All in all, the application of the HSE06-in-PBE approach allows the decoupling of the localized electronic nature of organic reactive species from the metallic background. Our results show that the VC decomposition can take place on the Li(001) metal surface provided that an energy barrier in the range of 0.29–0.34 eV is overcome. The reductive ring-opening mechanism via cleavage at the vinyl ether C–O site (CV–OV) is more likely to occur, and in principle can proceed to further decomposition towards the formation of Li2CO3 and acetylene or even towards polymerization, in close agreement with experimental observations.22 The theoretical characterization of species from VC ring-opening, enabled by DFET, allows us to highlight their potentially high reactivity on the Li metal surface and their key role as chain linkers or radical initiators. While the general picture on the SEI composition and the effects induced by VC-containing electrolytes is taking shape, this work sheds light on the origin of VC reactivity at the Li metal interface, which is essential to exploit rational design and engineering strategies for a safe and durable SEI. Beyond these results, our study paves the route for future applications of DFET-based methods for advanced investigation on electrocatalytic processes occurring at heterogeneous interfaces.
2 Structural models and computational details
The structural model for the vinylene carbonate (VC) interfaced to the Li(001) metal surface is based on the periodic slab approach.51 We cleave up the Li bulk structure along the (001) lattice plane and then introduce a vacuum slab of 15 Å along the c direction (see Fig. S1 in the ESI†). A 4 × 4 supercell containing 4 layers of Li atoms (i.e., 64 atoms) is adopted to host the VC molecule as well as all the reaction intermediates upon VC dissociation pathways.Spin-polarized DFT calculations are performed within periodic boundary conditions (PBCs) by employing plane-wave (PW) basis sets as implemented in the Vienna Ab initio Simulation Package (VASP, ver. 5.4.1).52–56 We use the PBE exchange-correlation functional within GGA and add the D3 a posteriori correction to account for van der Waals (vdW) forces at the interface.48,57,58 Core electrons are described by projector-augmented wave (PAW) potentials obtained from the VASP repository,59 while the valence/outer-core electrons that are included in the self-consistent-field calculations are: [1s22s1] for Li, [2s22p2] for C, [2s22p4] for O, and [1s1] for H atoms. Pseudo-wave functions are expanded in a PW basis set with a kinetic energy of 600 eV and a 4 × 4 × 1 Monkhorst–Pack k-point mesh for sampling the Brillouin zone. The Methfessel-Paxton method of the first order with a 0.2 eV smearing width is used for the Li surface.60 Geometry optimization is carried out by relaxing the atomic positions within the two topmost layers of the Li(001) slab until the maximum forces acting on each atom are below 30 meV Å−1, while all the other coordinates are kept constant. For all the calculations, the convergence threshold for energy is set to 10−5 eV.
DFET is applied to address the VC reactivity and improve the description of the molecule–metal interface.40–43 By employing the unique embedding potential (Vemb), we model the Li(001)/VC interface as a cluster system embedded in the extended Li-surface environment (see the ESI† for further details on method validation). Vemb can be solved by maximizing an extended Wu-Yang functional known from an optimized effective potential (OEP) approach and then can be used in combination with higher-level methods on the cluster subsystem to achieve a more accurate description of localized electronic structures.61 In our study, the HSE06 hybrid functional is employed, and the HSE06-in-PBE energies within DFET are calculated as:
| E = EPBELi−VC,PBC − EPBELi−VC,cluster[Vemb] + EHSE06Li−VC,cluster[Vemb] | (1) |
VC reaction mechanisms are characterized via calculations of minimum energy paths (MEPs) for the VC dissociation reactions, and the corresponding barrier heights are determined by the Climbing Image-Nudged Elastic Band (CI-NEB) approach.66
3 Results and discussion
3.1. VC adsorption at Li metal (001) surface termination
The first step for SEI formation at the electrode/electrolyte interface is represented by the interaction and adsorption of VC molecules at the Li metal surface. Here, we address the adsorption of a single molecule on the Li(001) surface termination. Several interacting modes exist, and we explore most of them by considering side, parallel and perpendicular arrangements of the VC molecule with respect to the Li surface (see Fig. S3†): (i) the side position would expose two oxygen atoms (i.e., the carbonyl and vinylic oxygen, OC and OV) towards the Li surface and actually leads to a stable bidentate configuration that is named Li[OCOV] hereafter; (ii) in the parallel arrangement, the VC molecule could in principle be stabilized by stacking interactions; in practice, parallel arrangements are found to evolve to either of two tilted configurations, forming one or two Li–O bonds via the carbonyl oxygen atom, OC, and thus corresponding to “end-on tilted” or “bridged tilted” configurations, respectively (Li[OC]e,t and Li[OC]b,t); (iii) when placed perpendicularly to the surface, the OC directly points towards the Li surface and binds either in an “end-on perpendicular” or “bridged perpendicular” coordination modes (Li[OC]e,p and Li[OC]b,p) according to the number of established linkages. The corresponding adsorption energies can be calculated as:| Eads = EVC−Li − ELi − EVC | (2) |
 | ||
| Fig. 1 VC adsorption on Li metal: minimum-energy structures of VC/Li(001) adsorbed states in different configurations and corresponding adsorption energies calculated according to eqn (2) at the PBE-D3 level of theory. Insets: charge density difference plots computed according to eqn (3) (isosurface: 3 meV Å−3). Color code: Li (green), C (black), O (red), and H (white); electron gain (yellow) and electron loss (cyan). | ||
Adsorption energy results show that the most favorable configuration is given by the bidentate mode, Li[OCOV], followed by the two bridged states, Li[OC]b,t and Li[OC]b,p, and then by the end-on ones, Li[OC]e,t and Li[OC]e,p. The number of specific molecule–surface interactions explains this trend, with an extra stabilization occurring in the two-bond configurations (bidentate and bridged states) compared to the one-bond ones (end-on coordinations). Detailed structural analysis reveals that Li metal undergoes a slight surface reorganization, with a displacement of Li atoms of about 0.2–0.3 Å toward the VC molecule in all the considered adsorbed states (see Fig. S3†). Moreover, there is an elongation of the carbonyl CO bond of about 0.02 Å compared to the value in the free VC molecule (see Fig. S2 and S3†), while no significant pyramidalization at the carbonyl site is observed (evaluated as variation of the dihedral angle OV,OV–CO from 180°, i.e., the planar configuration, see Table S2†). The largest dihedral variation is detected for the bidentate configurations, namely Li[OC]b,t (∼5°). Overall, the Li–O bonds that are established at the interface lie in the range of 1.86–2.19 Å, which is comparable to those in the Li2O solid phase, i.e., 1.99 Å.67 These results would suggest that VC adsorption on the Li(001) surface exhibits an ionic-like character, that can be unveiled from electronic structure analysis. The charge density (CD) difference upon VC adsorption can be calculated as:
| Δρ = ρVC−Li − ρLi − ρVC | (3) |
We also consider the Bader charges variation upon adsorption on both VC and Li sides of the interface. The overall negative charge variation on the whole VC molecule, i.e., ΔqVC, suggests that the adsorbed VC is reduced compared to the isolated molecule (first row in Table 1), while the positive/negative variations calculated on Li/O atoms (ΔqLi > 0 and ΔqO < 0, second and third rows in Table 1) confirm the surface-to-molecule charge transfer occurring along the Li–O bonds and accounting for electron depletion/accumulation on the Li/VC side. The comparison of Bader charges among the different configurations reveals that the lowest-energy bidentate Li[OCOV] state displays a smaller extent of electron reduction (less negative ΔqVC and ΔqO) compared to the bridged Li[OC]b,t and Li[OC]b,p configurations. However, the electron transfers in these cases are delocalized on the two oxygen atoms (OC and OV) bonded to the surface, which could explain the higher stability. These results highlight the direct correlation between the thermodynamic stability of VC/Li interfaces and the related charge transfer degree.
| Li[OCOV] | Li[OC]b,t | Li[OC]b,p | Li[OC]e,t | Li[OC]e,p | ||
|---|---|---|---|---|---|---|
| ΔqVC | −0.26 | −0.36 | −0.31 | −0.25 | −0.19 | |
| ΔqLi | +0.86 (OC) | +0.23 (OV) | +0.70/+0.87 | +0.48/+0.59 | +1.13 | +0.81 |
| ΔqO | −0.13 (OC) | −0.08 (OV) | −0.28 | −0.23 | −0.14 | −0.11 |
| q O | −1.24 (OC) | −1.11 (OV) | −1.34 | −1.30 | −1.26 | −1.22 |
Similar results have been reported for similar cyclic carbonates, i.e., EC and PC, by taking implicit solvent into account, thus suggesting that the unsaturated site in the ester ring does not affect the adsorption on the Li metal anode and that solvation effects can be neglected in such a first attempt to model VC/Li interactions.36,37
We calculate the projected Density of States (pDOS) for an unbound state, that is modelled by placing the VC molecule far away from the surface (i.e., ∼5 Å), and all the adsorbed configurations. As confirmed by the comparative pDOS in Fig. S4,† the distance of ∼5 Å is adequate to represent an unbound system and avoid any VC-surface interaction.
Following similar approaches employed for EC- and PC-Li systems,36,37 the effect of VC/Li interactions on the electronic structure can be evaluated from the energy gap between the LUMO (i.e., lowest unoccupied molecular orbital) and the Fermi energy (ELUMO − EF, highlighted by the black arrows in Fig. 2). Comparison between the pDOS of unbound and each bound states reveals that the VC adsorption on the Li(001) surface significantly lowers the LUMO energy, the effect being more remarkable for the two bridged configurations, Li[OC]b,p and Li[OC]b,t (ΔE = ELUMO − EF = 0.66 and 0.74 eV, respectively). One requirement to enable the SEI formation at the electrolyte/anode interface is that the LUMO energy of the electrolyte should lie across or just above the Fermi level of the anode material.6 The two bidentate states exhibit the lowest energy gaps (slightly differing for ∼0.08 eV) and could be considered as the most likely ones to initiate the reductive decomposition. However, we do believe that the extent of surface reconstruction also describes the most prone configuration to undergo dissociation reactions. As anticipated before, the highest variation of the dihedral angle observed for Li[OC]b,t (see Table S2†) couples to an enhanced charge transfer (see Table 1) and thus motivates our choice to select this configuration for the overall balance between thermodynamic stability and electronic features.
 | ||
| Fig. 2 Projected Density of States (pDOS) of the VC-adsorbed configurations on Li(001). From top to bottom: unbound state Li + VC; bidentate Li[OCOV]; bridged tilted Li[OC]b,t and perpendicular Li[OC]b,p; end-on tilted Li[OC]e,t and perpendicular Li[OC]e,p. Black arrows show the energy gap, ΔE = ELUMO − EF. Color code: Li (green lines) and VC molecule (red solid pattern). The corresponding structures are shown to the side for clarity. | ||
3.2. Reductive dissociation pathways towards VC decomposition
Inspired by previous mechanistic studies on bare molecular systems, we consider two possible reduction pathways, differing for the ring-opening site along CC–OV or CV–OV bonds (see Fig. 3 in light blue and orange, respectively).25,30,32–34 The so-obtained initial ring-opening products (open-VC) are hereafter designated as DISS(CCOV) and DISS(CVOV) dissociated states and can undergo further reductive decomposition: the former would proceed to the formation of an acetylene molecule (C2H2) and lithium oxide cluster (Li2O) via a two-step reaction with CO evolution; the latter would involve the release of C2H2 coupled to the formation of a lithium carbonate cluster (Li2CO3) in only one step. From the open debate in the literature concerning the dominant pathway, we gather that the CC–OV breaking is kinetically more accessible and that CO diffusion into the Li metal structure can take place; however, low acetylene release would conflict with this hypothesis.25,30,32,37 On the other hand, cleaving the CV–OV bond would lead to the 2-(carboxylatooxy)ethenyl radical anion (˙CHCH–O–COO–), which has been proposed as a radical initiator for VC polymerization.21,33,34 We must note that the whole picture on VC reactivity could be noticeably affected by the Li metal environment, which has been neglected in previous reports.18,30–35 Hence, we first address the thermodynamic stability of each reaction intermediate along the two decomposition pathways occurring on the Li(001) surface using standard semi-local DFT calculations. In Fig. 3, the resulting minimum-energy structures of each reaction step are reported together with the corresponding energetics. The energy variation upon each reaction step is calculated at the PBE-D3 level of theory as:| ΔE = En − En−1 | (4) |
CH–O–COO– radical anion debated in the literature.21,33,34 The potentially high reactivity of this species may be exerted towards several processes beyond the reductive ring-opening decomposition, which makes its chemical nature key to unveiling the overall VC role in the SEI formation on the Li metal anode.
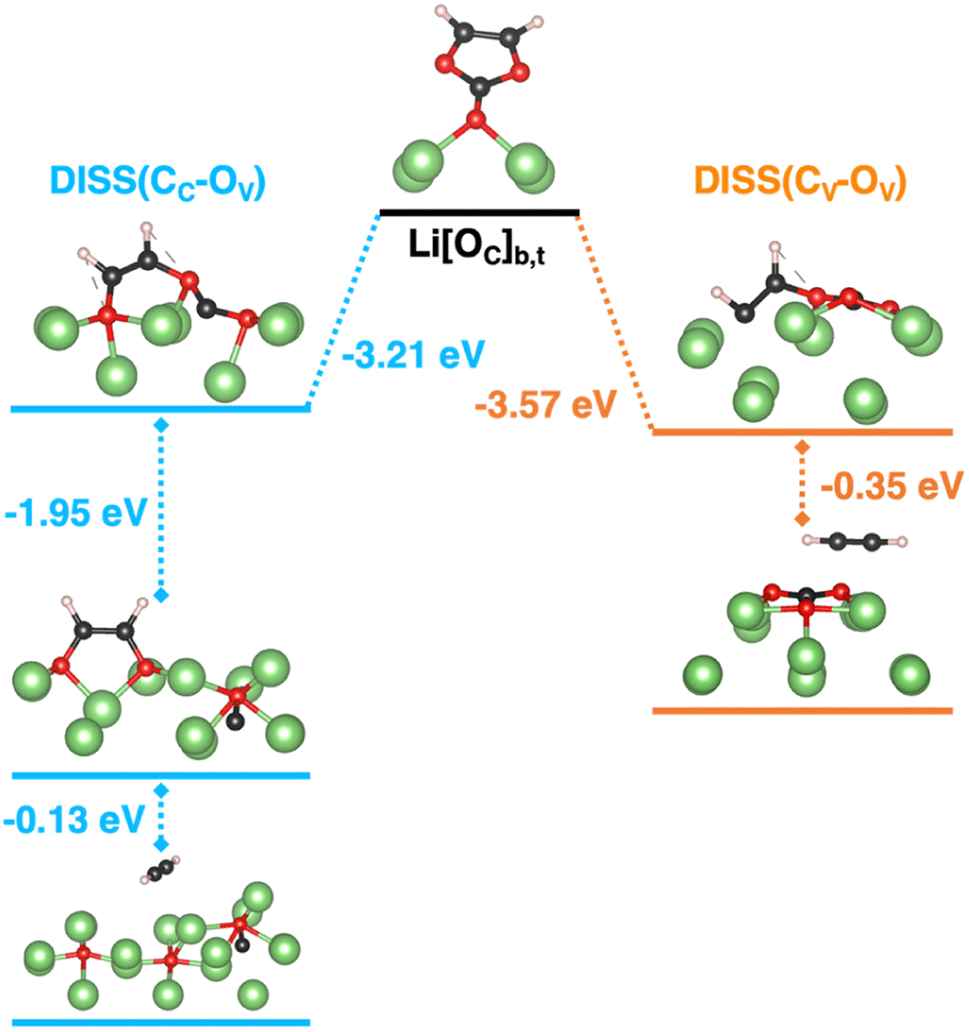 | ||
| Fig. 3 Reductive VC ring-opening dissociation on the Li(001) surface: minimum-energy structures along two possible reaction pathways obtained at the PBE-D3 level of theory with their corresponding energetics, ΔE, calculated according to eqn (4). Color code as in Fig. 1, CC–OV (light blue) and CV–OV cleavage (orange). | ||
On one hand, more accurate theoretical methods beyond DFT are required to achieve reliable and useful predictions of highly correlated systems with localized electron density. On the other hand, it is important to preserve the suitable description for the metal surface without losing accuracy. To this end, we propose the mechanistic study of ring-opening reactions by means of DFET. In this approach, the total system is partitioned into two subsystems sharing the embedding potential Vemb: the cluster, which contains the localized phenomena under study, and the environment. Usually, the cluster can be treated at the most accurate level of theory, or the most suited also for the property of interest. In our case, we perform HSE06-in-PBE calculations, that is, we describe the cluster (the VC reactive site at the Li interface) by using the hybrid HSE06 functional and account for the environment (the remaining Li metal surface) at the PBE level of theory. From the method validation based on different-sized clusters (see the ESI† for further details), we choose to solve the Vemb for a 12-atom Li cluster, which represents an optimal compromise for modelling both the local interactions and the reactivity at the VC/Li(001) interface with an affordable computational cost (Fig. S5 and S6 in the ESI†). The corresponding DFET energetics for adsorption/dissociation properties are computed according to eqn (5):
| Eads/dis = Eads/dis-VC@Li − ELi − EVC | (5) |
| E ads/dis (eV) | Li[OCOV] | Li[OC]b,t | DISS(CCOV) | DISS(CVOV) |
|---|---|---|---|---|
| DFT-PBE(PAW) | −0.511 | −0.556 | −3.581 | −3.970 |
| DFT-PBE(NC) | −0.471 | −0.530 | −3.521 | −3.886 |
| DFET-HSE06-in-PBE | −0.266 | −0.419 | −3.227 | −3.737 |
From the values listed in Table 2, we can see that both adsorption and dissociation energies at the HSE06-in-PBE level of theory are lower than DFT-PBE results. However, the overall energy trend remains unchanged, and the relative stabilities can be confirmed with higher accuracy. It is noteworthy that the localized anionic nature in DISS(CVOV) is also revealed by the Bader charges computed at the HSE06-in-PBE level of theory (qC = −1.04 vs. −0.85 from DFET and DFT, respectively), thus confirming the before-mentioned potential reactivity of this carbanionic-like species.
To gain deeper insights into the two ring-opening mechanisms, we investigate the MEPs along the VC dissociation process. By applying the CI-NEB method between the adsorbed Li[OC]b,t state (as the initial state, i.e., i00), and a given dissociated structure, DISS(CCOV) or DISS(CVOV), (final state, i.e., i06), we find that both the MEPs can be described by the i01-i05 images reported in the top and bottom panels of Fig. 4. The corresponding energy variations are derived at the DFT-PBE and DFET-HSE06-in-PBE levels of theory and plotted in the middle panel of Fig. 4. We can clearly see that the HSE06-in-PBE method predicts an energy barrier of about 0.3 eV, which is associated with the formation of the high-energy i01 images along both MEPs and is not detectable with semi-local PBE-DFT approaches. The results listed in Table S2† show that both PBE(PAW) and PBE(NC) pseudo-potentials provide a negligible barrier for both the degradation mechanisms, well below the barrier heights found at the DFET-HSE06-in-PBE level of theory.
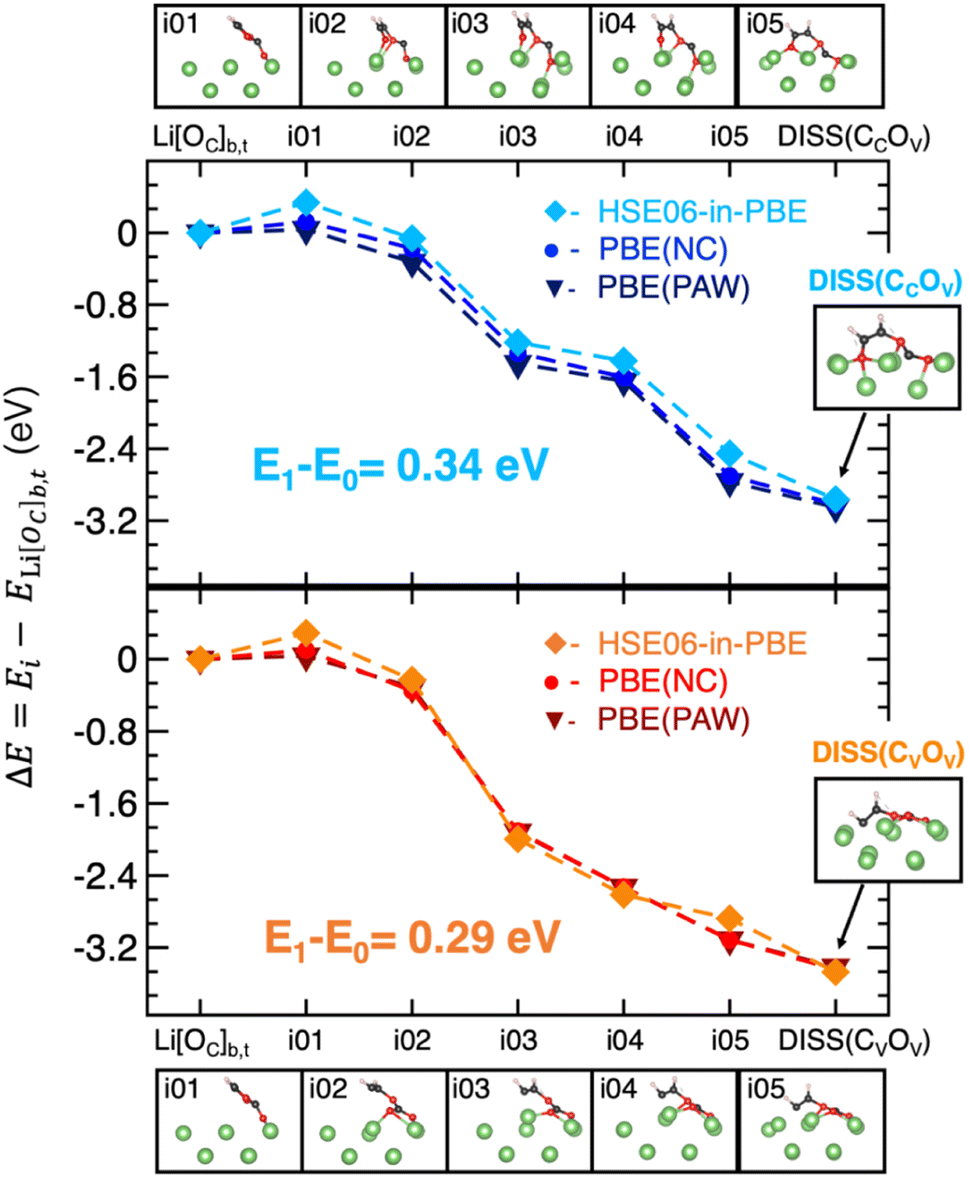 | ||
| Fig. 4 VC migration on Li metal: minimum energy paths (MEPs) for the migration of the VC molecule from its adsorbed to dissociated states, (top) DISS(CCOV) and (bottom) DISS(CVOV), computed by means of the CI-NEB approach. The corresponding energetics obtained at the DFT-PBE(PAW) (dark-blue and burgundy lines), DFT-PBE(NC) (blue and red lines) and DFET-HSE06-in-PBE (light-blue and orange lines) levels of theory are plotted along the MEPs. The related reaction intermediates (images i01-i05) are also displayed. | ||
Fig. 5a shows the structural and electronic analysis on the highest energy structures, i.e., the i01-DISS(CCOV) and i01-DISS(CVOV) transition states. We have quantified the structural reconstruction as the geometrical distortion on the carbonyl carbon atom from its original planar configuration: the further the OV, OV–CO dihedral angle from 180° value, the larger the rearrangement at the CC atom. As shown in left Fig. 5a, the dihedral angle decreases by about 38° and 30° for i01-DISS(CCOV) and i01-DISS(CVOV), respectively, suggesting that the VC ring undergoes a structural reorganization from an approximately trigonal planar to a trigonal pyramidal geometry. This pyramidalization at the CC atom in both structures comes with the elongation of CC–OV and CC–OC bonds (Fig. 5a). Bader charges analysis on the VC molecule (qVC), combined with the spin density of the i00-ground state (Li[OC]b,t) and the two i01-transition states reveals, that the ring-opening mechanisms are coupled to the reduction of the VC molecule to a ketyl-like radical anion (see Fig. 5a). The energy barriers predicted by the HSE06-in-PBE approach are therefore associated with the VC pyramidalization and formation of the radical anion. The minor extent of structural and electronic reorganization in i01-DISS(CVOV) could account for the lower energy barrier and thus suggest a more accessible pathway leading to a more stable dissociated product (ΔE = −3.57 vs. −3.21 eV, see Fig. 3). This conclusion is in line with previous studies reporting the preferential breaking of the CV–OV bond.33,34 While proving experimentally the thermodynamics of reaction profiles and the existence of an energy barrier associated with VC pyramidalization is extremely difficult, our theoretical findings can directly relate to existing experimentally observed phenomena. The predicted dissociation products and related stability can be compared to the composition and morphology of the VC-induced SEI. In particular, the formation of a Li2O cluster, following bond cleavage at the CC–OV site and the decomposition to acetylene and Li2CO3, resulting from CV–OV bond breaking, relates to the mosaic-like nanostructure comprising organic species, Li2O and Li2CO3 reported by Xu et al.22 Moreover, our computed barrier heights are slightly lower than those reported by Wang et al. (∼0.3 vs. 0.8 eV), owing to the presence of the Li metal surface acting as an electron donor and thus favoring the reductive reaction.32,33 By following the Bader charge trend along the MEPs reported in Fig. S7,† we observe a slight decrease of net charge on the vinylic oxygen, while the charge lowering on both carbonyl and vinylic carbons is more significant. This clearly suggests that a reductive mechanism is taking place, as a result of the before-mentioned charge transfer from the Li(001) surface. The qC values along DISS(CVOV) are more negative (up to −1.04) and would account for an even more anionic nature of the reaction intermediates compared to DISS(CCOV). This result is confirmed by the electronic analysis in Fig. 5b: DISS(CVOV) and DISS(CCOV) correspond, respectively, to a carbanion and a radical species, as revealed by the spin density and the extent of charge transfer associated with their formation (qVC = −3.16 vs. −2.48 for the radical DISS(CCOV) and the carbanion DISS(CVOV), respectively).
 | ||
| Fig. 5 (a) Structural details (CCOV/CVOV bond length and dihedral (OV, OV–CO)), spin density (isosurface 5 × 10−4 e−/bohr3) and total Bader charge on the VC molecule of the i00- (Li[OC]b,t adsorbed state) and i01-images (DISS(CCOV) and DISS(CVOV) dissociated states) along the MEPs. (b) Projected Density of States (pDOS), spin density (isosurface 0.05 e−/bohr3) and total Bader charge on the VC molecule of Li[OC]b,t, DISS(CCOV) and DISS(CVOV). Color code as in Fig. 2. | ||
In both cases, the ring-opening reaction proceeds with further electron transfer from Li to VC, leading to two highly reactive products that can exert different activities towards SEI formation on the Li metal surface. Recent scientific discussions highlight that open-VC (i.e., the ˙CH–CH–O–COO– radical anion arising from CV–OV bond breaking) can initiate complex polymerization reactions via radical attack on other VC molecules.21,24–26,35 The preferential reactivity of open-VC to other VC molecules rather than EC also emphasizes that considering explicit solvent molecules would not be key to VC reactivity on the Li metal anode.35 The presence of poly(VC) or cross-linked polyacetylene has been confirmed by IR, NMR and XRD analysis performed on VC-containing electrolytes employed in LMBs, and it is associated with good Li passivation and effective inhibition of further electrolyte degradation.21,24–26,35 Although the formation of polymer chains or complex superstructures was beyond the scope of this work, we attain the identification of the reactive species responsible for VC decomposition that can in principle play the role of a potential chain linker or a polymer initiator. By proving the high reactivity of the dissociated states, i.e., the DISS(CVOV) and DISS(CCOV) intermediates with anionic and radical nature, respectively, we show that the activity towards polymerization (as a result of the Li surface-to-VC electron transfer) rather than further decomposition to fully reduced moieties would also provide feasible explanation of the low acetylene release observed in the experiments.20,21,23–26
Our model is capable of representing the main VC/Li(001) surface interactions and the first steps towards reductive decomposition. We must note that VC-induced SEI formation and growth can be sensibly influenced by other interfacial reactions and solvent molecules and association with dissolved Li+ departing from the anode upon cycling. As an early attempt to characterize the origin and underlying mechanism of VC decomposition, here we have neglected the role of electrolyte elements to determine the critical effect of the Li metal surface in promoting the VC degradation reactions in VC-induced SEI formation. As a matter of fact, adsorption trends, reaction profiles and predicted polymerization activity discussed in this work are in line with former mechanistic studies considering similar systems with implicit solvation as well as with VC/Li+ association.30,31,35,36 Moreover, our study shows how an advanced DFET approach is able to properly describe the VC dissociation mechanisms and the related intermediates featuring localized charges. DFET represents an innovative computational tool capable of providing a reliable picture for electrocatalytic reactions occurring at heterogeneous interfaces. By elucidating the VC reductive mechanism on the Li surface, we believe that the theoretical efforts reported so far will empower future studies aiming to unveil the key aspects in the formation of a safe and stable SEI, thus guiding the rational engineering of efficient Li metal anodes and providing value to upcoming battery design principles.
4 Conclusions
We hereby present new atomistic insights into the first steps of VC-induced SEI formation at the Li metal anode surface as derived from first principles. Despite the broad literature on the beneficial use of VC-containing electrolytes for an effective and durable SEI, an in-depth understanding of reductive mechanisms that are responsible for SEI formation is still required. We have performed a thorough investigation on VC adsorption and reactivity at the Li(001) surface termination, which can be helpful for future design and engineering strategies towards a stable and protective SEI for efficient LMBs.Density Functional Embedding Theory is employed to combine hybrid-DFT and semi-local GGA-based methods, which are accurate and reliable for molecular species and metal surfaces, respectively. Our aim is to unveil the VC reactivity at the Li surface and the underlying decomposition mechanisms and dissect the roles of VC and the Li background in the formation of a stable and durable SEI. Our main findings can be summarized as follows:
- Concerning the adsorption of a VC molecule on the Li(001) surface, several explored VC orientations lead to stable metal electrode/electrolyte interfaces, the binding being stronger when more Li–O interactions are established. The pDOS calculations and Bader charge analysis are consistent with the adsorption trend and reveal surface-to-molecule charge transfers for all the configurations.
- The reductive decomposition pathway is tracked along two possible ring-opening mechanisms, following the bond cleavage at the carbonyl C–vinylic O (CC–OV) and vinylic C–vinylic O (CV–OV) sites. The first pathway consists in a two-step decomposition, leading to release of acetylene and CO as well as to formation of a Li2O cluster; along the second pathway, a single reaction step leads to simultaneous development of both acetylene and Li2CO3. While being thermodynamically feasible, the VC decomposition features either way an energy barrier of ∼0.3 eV associated with the VC pyramidalization due to further reduction favored by the Li metal surface. Because of the ketyl-like anion radical nature of the transition states, the detection of such barriers is only possible with HSE06-in-PBE DFET calculations. In particular, the ring-opening at the CV–OV site turns out to be easier and leads to a highly anionic intermediate, namely DISS(CVOV), with a net negative charge on the C atom of −1.04, while the less favourable DISS(CCOV) has a radical nature. In both cases, the reaction proceeds with further electron transfer from the Li surface to VC, leading to two products that can have different reactivities towards SEI formation processes, e.g., polymerization. By directly undergoing polymerization rather than further decomposition to fully reduced moieties, the VC reactivity unveiled so far would also provide feasible explanation of the low acetylene release observed in the experiments. Regardless of whether such species will act as chain linkers or polymer initiators, their stability is key to the VC reactivity as it can outline rational strategies to tune and design artificial SEI layers containing tailored macro-molecular systems or polymers.
Beyond these specific results, our study also highlights the suitability of DFET-based methods in modelling complex electrocatalytic reactions at heterogeneous interfaces with affordable computational costs, thanks to the combination of suitable computational approaches for different cluster/environment systems. The application of DFET, a new yet established theoretical method beyond standard semi-local DFT, allows new insights into the mechanisms of VC decomposition at the LMB electrode to be obtained, providing new fundamental knowledge for the fine-tuning of VC-based SEI engineering. As the first study to attain the description of early VC reactivity at the Li metal interface, this work will enable further investigations aiming to include realistic interfacial features and electrolyte elements, such as solvation effects and association with Li+ departing from the Li anode upon cycling, and thus addressing the many subtle features of the complex SEI formation process and growth.
Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
Part of this work was carried out within the EU-funded “PSIONIC” project and the activities “Ricerca Sistema Elettrico” funded through contributions to research and development by the Italian Ministry of Economic Development. The computing resources and the related technical support used for this work have been provided by CRESCO/ENEA-GRID High Performance Computing infrastructure and its staff.68 CRESCO/ENEAGRID High Performance Computing infrastructure is funded by ENEA, Italy, the Italian National Agency for New Technologies, Energy and Sustainable Economic Development and by Italian and European research programs; see https://www.cresco.enea.it/ for information.References
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef.
- J. Ma, Y. Li, N. S. Grundish, J. B. Goodenough, Y. Chen, L. Guo, Z. Peng, X. Qi, F. Yang, L. Qie, C.-A. Wang, B. Huang, Z. Huang, L. Chen, D. Su, G. Wang, X. Peng, Z. Chen, J. Yang, S. He, X. Zhang, H. Yu, C. Fu, M. Jiang, W. Deng, C.-F. Sun, Q. Pan, Y. Tang, X. Li, X. Ji, F. Wan, Z. Niu, F. Lian, C. Wang, G. G. Wallace, M. Fan, Q. Meng, S. Xin, Y.-G. Guo and L.-J. Wan, J. Phys. D: Appl. Phys., 2021, 54, 183001 CrossRef CAS.
- K. Kerman, A. Luntz, V. Viswanathan, Y.-M. Chiang and Z. Chen, J. Electrochem. Soc., 2017, 164, A1731–A1744 CrossRef CAS.
- R. Wang, W. Cui, F. Chu and F. Wu, J. Energy Chem., 2020, 48, 145–159 CrossRef.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- C. Fang, X. Wang and Y. S. Meng, Trends Chem., 2019, 1, 152–158 CrossRef CAS.
- Z. Yu, Y. Cui and Z. Bao, Cell Rep. Phys. Sci., 2020, 1, 100119 CrossRef.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao, F. Wei, J.-G. Zhang and Q. Zhang, Adv. Sci., 2016, 3, 1500213 CrossRef PubMed.
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047–2051 CrossRef CAS.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, npj Comput. Mater., 2018, 4, 15 CrossRef.
- J. He, H. Wang, Q. Zhou, S. Qi, M. Wu, F. Li, W. Hu and J. Ma, Small Methods, 2021, 5, 2100441 CrossRef CAS PubMed.
- Y. Zhang, D. Krishnamurthy and V. Viswanathan, J. Electrochem. Soc., 2020, 167, 070554 CrossRef.
- H. Bin Son, M.-Y. Jeong, J.-G. Han, K. Kim, K. H. Kim, K.-M. Jeong and N.-S. Choi, J. Power Sources, 2018, 400, 147–156 CrossRef.
- Y. Qian, C. Schultz, P. Niehoff, T. Schwieters, S. Nowak, F. M. Schappacher and M. Winter, J. Power Sources, 2016, 332, 60–71 CrossRef CAS.
- G. Lingua, M. Falco, T. Stettner, C. Gerbaldi and A. Balducci, J. Power Sources, 2021, 481, 228979 CrossRef CAS.
- L. El Ouatani, R. Dedryvère, C. Siret, P. Biensan, S. Reynaud, P. Iratçabal and D. Gonbeau, J. Electrochem. Soc., 2009, 156, A103 CrossRef CAS.
- J. Chai, Z. Liu, J. Ma, J. Wang, X. Liu, H. Liu, J. Zhang, G. Cui and L. Chen, Adv. Sci., 2017, 4, 1600377 CrossRef PubMed.
- H. Ota, K. Shima, M. Ue and J. Yamaki, Electrochim. Acta, 2004, 49, 565–572 CrossRef CAS.
- B. Zhang, M. Metzger, S. Solchenbach, M. Payne, S. Meini, H. A. Gasteiger, A. Garsuch and B. L. Lucht, J. Phys. Chem. C, 2015, 119, 11337–11348 CrossRef CAS.
- Y. Xu, H. Wu, Y. He, Q. Chen, J.-G. Zhang, W. Xu and C. Wang, Nano Lett., 2019, 20, 418–425 CrossRef PubMed.
- D. Pritzl, S. Solchenbach, M. Wetjen and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, A2625–A2635 CrossRef CAS.
- A. L. Michan, B. S. Parimalam, M. Leskes, R. N. Kerber, T. Yoon, C. P. Grey and B. L. Lucht, Chem. Mater., 2016, 28, 8149–8159 CrossRef CAS.
- S. Grugeon, P. Jankowski, D. Cailleu, C. Forestier, L. Sannier, M. Armand, P. Johansson and S. Laruelle, J. Power Sources, 2019, 427, 77–84 CrossRef CAS.
- H. Zhou, H. Liu, Y. Li, X. Yue, X. Wang, M. Gonzalez, Y. S. Meng and P. Liu, J. Mater. Chem. A, 2019, 7, 16984–16991 RSC.
- H.-H. Lee, Y.-Y. Wang, C.-C. Wan, M.-H. Yang, H.-C. Wu and D.-T. Shieh, J. Appl. Electrochem., 2005, 35, 615–623 CrossRef CAS.
- H. Ota, Y. Sakata, A. Inoue and S. Yamaguchi, J. Electrochem. Soc., 2004, 151, A1659 CrossRef CAS.
- S. Shi, J. Gao, Y. Liu, Y. Zhao, Q. Wu, W. Ju, C. Ouyang and R. Xiao, Chin. Phys. B, 2016, 25, 018212 CrossRef.
- Y.-K. Han and S.-U. Lee, Bull. Korean Chem. Soc., 2005, 26, 43–46 CrossRef CAS.
- Y. Kamikawa, K. Amezawa and K. Terada, J. Phys. Chem. C, 2020, 124, 19937–19944 CrossRef CAS.
- Y. Wang and P. B. Balbuena, J. Phys. Chem. B, 2002, 106, 4486–4495 CrossRef CAS.
- Y. Wang, S. Nakamura, K. Tasaki and P. B. Balbuena, J. Am. Chem. Soc., 2002, 124, 4408–4421 CrossRef CAS PubMed.
- K. Ushirogata, K. Sodeyama, Y. Okuno and Y. Tateyama, J. Am. Chem. Soc., 2013, 135, 11967–11974 CrossRef CAS PubMed.
- F. A. Soto, Y. Ma, J. M. Martinez de la Hoz, J. M. Seminario and P. B. Balbuena, Chem. Mater., 2015, 27, 7990–8000 CrossRef CAS.
- M. Ebadi, D. Brandell and C. M. Araujo, J. Chem. Phys., 2016, 145, 204701 CrossRef PubMed.
- M. D. Brennan, M. Breedon, A. S. Best, T. Morishita and M. J. S. Spencer, Electrochim. Acta, 2017, 243, 320–330 CrossRef CAS.
- J. Wang and S.-Q. Wang, Surf. Sci., 2014, 630, 216–224 CrossRef CAS.
- C. Huang and E. A. Carter, J. Chem. Phys., 2011, 135, 194104 CrossRef PubMed.
- C. Huang, M. Pavone and E. A. Carter, J. Chem. Phys., 2011, 134, 154110 CrossRef PubMed.
- F. Libisch, C. Huang and E. A. Carter, Acc. Chem. Res., 2014, 47, 2768–2775 CrossRef CAS PubMed.
- C. Huang, A. B. Muñoz-García and M. Pavone, J. Chem. Phys., 2016, 145, 244103 CrossRef PubMed.
- Y.-C. Chi, M. Shaban Tameh and C. Huang, J. Chem. Theory Comput., 2021, 17, 2737–2751 CrossRef CAS PubMed.
- V. B. Oyeyemi, J. A. Keith, M. Pavone and E. A. Carter, J. Phys. Chem. Lett., 2012, 3, 289–293 CrossRef CAS PubMed.
- P. Huang and E. A. Carter, J. Chem. Phys., 2006, 125, 084102 CrossRef PubMed.
- O. A. Vydrov, G. E. Scuseria and J. P. Perdew, J. Chem. Phys., 2007, 126, 154109 CrossRef PubMed.
- W. Gao, T. A. Abtew, T. Cai, Y.-Y. Sun, S. Zhang and P. Zhang, Solid State Commun., 2016, 234–235, 10–13 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- W. Sun and G. Ceder, Surf. Sci., 2013, 617, 53–59 CrossRef CAS.
- W. Koch and M. C. Holthausen, A Chemist's Guide to Density Functional Theory, Wiley-VCH, II., 2001 Search PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- M. Methfessel and A. T. Paxton, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616–3621 CrossRef CAS PubMed.
- W. Yang and Q. Wu, Phys. Rev. Lett., 2002, 89, 143002 CrossRef PubMed.
- C. Huang, J. Chem. Phys., 2016, 144, 124106 CrossRef PubMed.
- M. Fuchs and M. Scheffler, Comput. Phys. Commun., 1999, 119, 67–98 CrossRef CAS.
- D. R. Hamann, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 2980–2987 CrossRef PubMed.
- N. Troullier and J. L. Martins, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 1993–2006 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- K. C. Lau, L. A. Curtiss and J. Greeley, J. Phys. Chem. C, 2011, 115, 23625–23633 CrossRef CAS.
- G. Ponti, F. Palombi, D. Abate, F. Ambrosino, G. Aprea, T. Bastianelli, F. Beone, R. Bertini, G. Bracco, M. Caporicci, B. Calosso, M. Chinnici, A. Colavincenzo, A. Cucurullo, P. Dangelo, M. De Rosa, P. De Michele, A. Funel, G. Furini, D. Giammattei, S. Giusepponi, R. Guadagni, G. Guarnieri, A. Italiano, S. Magagnino, A. Mariano, G. Mencuccini, C. Mercuri, S. Migliori, P. Ornelli, S. Pecoraro, A. Perozziello, S. Pierattini, S. Podda, F. Poggi, A. Quintiliani, A. Rocchi, C. Scio, F. Simoni and A. Vita, in 2014 International Conference on High Performance Computing & Simulation (HPCS), IEEE, 2014, pp. 1030–1033 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary Material is available online and contains a structural model for the Li(001) surface, convergence tests for kinetic energy cutoff, structural and electronic analysis of adsorbed and dissociated states for the VC/Li(001) interface, DFET method validation, and structural and electronic analysis along the MEPs. See DOI: https://doi.org/10.1039/d2ta08772c |
| This journal is © The Royal Society of Chemistry 2023 |