
High-efficiency overall alkaline seawater splitting: using a nickel–iron sulfide nanosheet array as a bifunctional electrocatalyst†
Jie
Chen
a,
Longcheng
Zhang
 a,
Jun
Li
a,
Xun
He
a,
Yinyuan
Zheng
b,
Shengjun
Sun
c,
Xiaodong
Fang
d,
Dongdong
Zheng
a,
Yongsong
Luo
a,
Yan
Wang
a,
Jing
Zhang
d,
Lisi
Xie
d,
Zhengwei
Cai
c,
Yuntong
Sun
c,
Abdulmohsen Ali
Alshehri
e,
Qingquan
Kong
*d,
Chengwu
Tang
*b and
Xuping
Sun
*ac
a,
Jun
Li
a,
Xun
He
a,
Yinyuan
Zheng
b,
Shengjun
Sun
c,
Xiaodong
Fang
d,
Dongdong
Zheng
a,
Yongsong
Luo
a,
Yan
Wang
a,
Jing
Zhang
d,
Lisi
Xie
d,
Zhengwei
Cai
c,
Yuntong
Sun
c,
Abdulmohsen Ali
Alshehri
e,
Qingquan
Kong
*d,
Chengwu
Tang
*b and
Xuping
Sun
*ac
aInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 610054, Sichuan, China. E-mail: xpsun@uestc.edu.cn; xpsun@sdnu.edu.cn
bHuzhou Key Laboratory of Translational Medicine, First People's Hospital Affiliated to Huzhou University, Huzhou 313000, Zhejiang, China. E-mail: dr_tcw@zjhu.edu.cn
cCollege of Chemistry, Chemical Engineering and Materials Science, Shandong Normal University, Jinan 250014, Shandong, China
dInterdisciplinary Materials Research Center, Institute for Advanced Study, Chengdu University, Chengdu 610106, China. E-mail: kongqingquan@cdu.edu.cn
eChemistry Department, Faculty of Science, King Abdulaziz University, P. O. Box 80203, Jeddah 21589, Saudi Arabia
First published on 22nd December 2022
Abstract
The use of large-current-density electrolysis of seawater is promising for a massive hydrogen (H2) production. This process, however, requires high-performance and cost-effective bifunctional catalysts for the oxygen evolution reaction (OER) and the hydrogen evolution reaction (HER). Herein, a nickel–iron sulfide nanosheet array on nickel foam (NiFeS/NF) is demonstrated to be a superb bifunctional electrocatalyst for seawater splitting, delivering the industrially demanded current density of 500 mA cm−2 at overpotentials of 300 and 347 mV for OER and HER in alkaline seawater, respectively. Moreover, its corresponding two-electrode electrolyzer only requires a cell voltage of 1.85 V to drive 500 mA cm−2 and shows a strong stability for at least 50 h of electrolysis in alkaline seawater, outperforming the most recently reported seawater-splitting catalyst electrodes.
Hydrogen (H2) is a carbon-neutral energy carrier alternative to fossil fuels, and is an inspired, ideal choice for future energy applications.1,2 Among the currently pursued hydrogen generation technologies, water electrolysis provides a promising, environmentally friendly, and sustainable route for high-purity H2 preparation.3,4 The large energy consumption of industrial level electrolysis of water is attributed to the overpotential of the cathodic hydrogen evolution reaction (HER) and the anodic oxygen evolution reaction (OER).5–8 Using highly active catalysts can greatly lower the overpotentials, and accelerate the kinetics of OER or HER. Currently, IrO2/RuO2 and Pt-based compounds exhibit the benchmark electrocatalytic activity for OER and HER, respectively, but the drawbacks of exorbitant price, rarity, and instability seriously restrict their widespread utilization. Therefore, it is vital to develop high efficiency and earth-abundant electrocatalysts. In particular, considerable attention has been given to the bifunctional electrocatalysts for both HER and OER because of their merits of use of simplified devices and lower costs.9,10
Due to the freshwater shortage, electrochemical freshwater splitting is difficult to apply at a large scale, especially in coastal arid areas and islands. Considering the abundance of seawater resources on earth, it is highly advantageous to directly produce H2 by electrochemical seawater splitting, using seawater as a starting feedstock.11–15 However, the wide implementation of seawater electrolysis has serious challenges, including the occurrence of an anodic chlorine evolution reaction (CER), resulting in a compromised faradaic efficiency of the OER process.16–18 The two-electron oxidation reaction of chloride to hypochlorite stands out when compared to the OER in terms of reaction kinetics,19,20 but the thermodynamic standard potential of OER is ∼480 mV lower than that of CER in alkaline electrolytes.21,22 In this context, there is no interfering chlorine chemistry when a catalyst used as a water oxidization electrode can operate at an overpotential of not more than 480 mV to obtain a high current density during alkaline seawater electrolysis. In addition, the issues of catalyst poisoning and the degradation of catalytic activity will also occur due to the Cl− corrosion and deposition of insoluble precipitates (Ca(OH)2 and Mg(OH)2) over the electrode.23–25
Recently, transition-metal compound catalysts are an area of interest for applications in alkaline seawater electrolysis due to their high abundance and comparable activities to noble-metal catalysts.26–28 Among these, the NiFe-based catalysts, especially NiFe layered double hydroxide (LDH) ones have demonstrated excellent OER activity in alkaline seawater.29–31 Nevertheless, NiFe-LDH suffers from intrinsically poor electrical conductivity and corrosion resistance.32–34 The fabrication of NiFeS has been considered as a promising strategy to enhance the OER catalytic activity of NiFe-LDH due to its superior electrochemical conductivity and intrinsic activity.35,36 In addition, the present S-species can be beneficial for seawater electrolysis because they generate a negatively charged anion-rich surface, which can effectively repel Cl− in seawater, and therefore enhance the resistance for Cl− corrosion.37–39
Herein, a NiFeS nanosheet array grown in situ on Ni foam (NiFeS/NF) via a two-step hydrothermal approach (see ESI† for preparative detail) is proposed as a superb bifunctional catalyst for seawater electrolysis, capable of driving a high current density of 500 mA cm−2 for the OER and HER process, with the need for overpotentials of only 300 and 347 mV in alkaline seawater electrolyte, respectively. In addition, this NiFeS/NF-assembled two-electrode electrolyzer operates at a low cell voltage of 1.85 V at 500 mA cm−2 with robust stability in alkaline seawater.
Fig. 1a shows the fabrication process of NiFeS/NF, which involves the following two steps: (1) in situ hydrothermal growth of a NiFe-LDH nanosheet array on NF (NiFe-LDH/NF), and (2) ion exchange of OH− with S2−. The X-ray diffraction (XRD) and scanning electron microscopy (SEM) results suggest that the NF (Fig. S1, ESI†) is fully overlaid with a densely packed NiFe-LDH nanosheet array (Fig. S2 and S3, ESI†). After sulfurization treatment, the XRD pattern (Fig. 1b) shows diffraction peaks characteristic of a FeNi2S4 crystal phase (JCPDS no. 47-1740)40 and a metallic Ni substrate (JCPDS no. 04-0850), which indicated the successful conversion of NiFe-LDH/NF to NiFeS/NF. The NiFeS/NF (Fig. 1c and d) still maintains the nanosheet-like structure of NiFe-LDH/NF. The transmission electron microscopy (TEM) image (Fig. 1e) of NiFeS further validates its nanosheet feature. The lattice spacing, from the lattice fringes, of 0.287 nm, corresponding to the (311) plane of FeNi2S4, is seen clearly from the high-resolution TEM (HRTEM) image of one NiFeS nanosheet (Fig. 1f). The SEM and corresponding energy-dispersive X-ray (EDX) elemental mapping images of NiFeS/NF confirm the co-existence of elemental Fe, Ni, and S with a homogeneous distribution in the nanosheet array (Fig. 1g). As shown in the high-resolution X-ray photoelectron spectroscopy (XPS) spectrum of Ni 2p (Fig. 1h), the characteristic peaks located at 856.3 and 874.1 eV correspond to Ni2+ 2p3/2 and Ni2+ 2p1/2, respectively.41 The peak centered at 853.0 eV can be indexed to metallic Ni.42 The presence of two satellite peaks at 862.0 and 879.7 eV is also observed. The Fe 2p spectrum (Fig. 1i) shows one spin–orbit doublet peak of Fe 2p3/2 at 712.7 eV and Fe 2p1/2 at 725.4 eV, suggesting the existence of the Fe3+ oxidation state.36 As shown in Fig. 1j, there are three peaks at 161.5, 163.0, and 169.0 eV corresponding to S 2p3/2, S 2p1/2, and sulfate (SO42−) possibly contributed to the surface oxidation of sulfide in air in the S 2p region of NiFeS/NF, respectively.43,44 The previously mentioned analyses confirm the successful synthesis of NiFeS/NF.
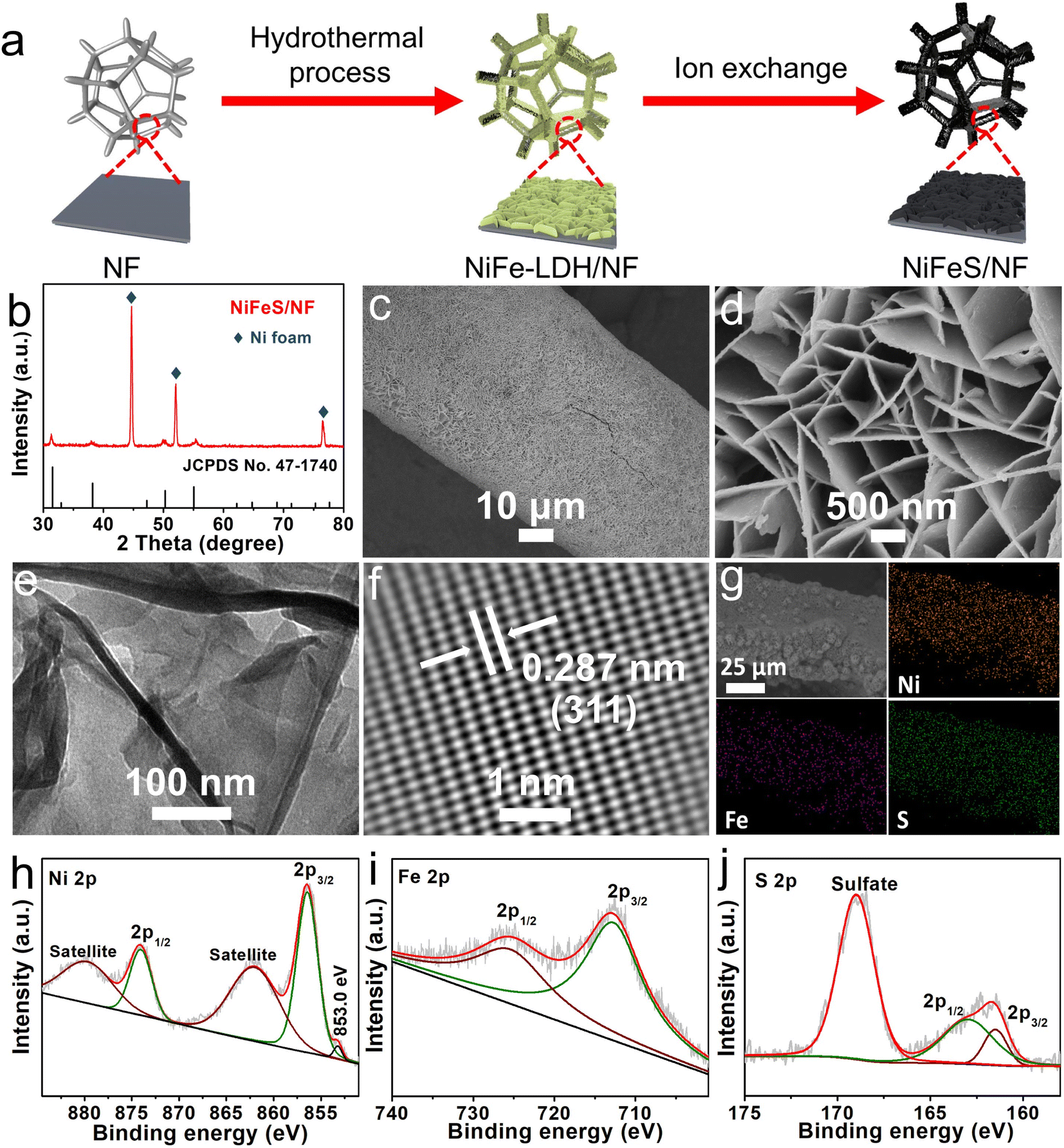 | ||
| Fig. 1 (a) Schematic illustration of the fabrication process for NiFeS/NF. (b) The XRD pattern of NiFeS/NF. (c) Low- and (d) high-magnification SEM images of NiFeS/NF. (e) The TEM and (f) HRTEM images of the NiFeS nanosheet. (g) The SEM and the corresponding EDX elemental mapping images of NiFeS/NF. The XPS spectra of NiFeS for the (h) Ni 2p, (i) Fe 2p, and (j) S 2p regions. | ||
The OER electrocatalytic activity of NiFeS/NF was first examined in 1 M KOH. As controls, NiFe-LDH/NF, bare NF, and commercial RuO2 loaded on NF (RuO2/NF) were also tested under the same conditions. Fig. 2a shows the relevant linear sweep voltammetry (LSV) curves. As can be seen, the NiFeS/NF exhibits a superior catalytic activity compared to the NiFe-LDH/NF and benchmark RuO2/NF, whereas the bare NF has an almost undetectable OER activity. The NiFeS/NF demands overpotentials of only 215 and 272 mV to give the current densities (j) of 100 and 500 mA cm−2, respectively, which were much smaller than those of NiFe-LDH/NF (280 and 422 mV) and RuO2/NF (333 and 495 mV). The Tafel slope is a key criterion in evaluating the reaction kinetics. As shown in Fig. 2b, the NiFeS/NF has the lowest Tafel slope value of 56.37 mV dec−1 of all four electrodes (NiFe-LDH/NF: 80.47 mV dec−1, RuO2/NF: 98.01 mV dec−1, and NF: 138.33 mV dec−1), indicating that NiFeS/NF had the best OER dynamics. The electrochemical impedance spectroscopy (EIS) results (Fig. 2c) also confirm enhanced reaction kinetics and charge transfer capability on NiFeS/NF, as shown by its much smaller charge transfer resistance (Rct, ∼4.36 Ω) than that of NiFe-LDH/NF (∼16.70 Ω, Table S1, ESI†). In addition, the double-layer capacitance (Cdl) value of NiFeS/NF (4.19 mF cm−2) is 2.85 times higher than that of NiFe-LDH/NF (1.47 mF cm−2), suggesting that the NiFeS/NF surpasses the NiFe-LDH/NF in terms of electrochemically active surface area, which is positively related to the Cdl (Fig. S4, ESI†). The multi-step chronopotentiometry curve (Fig. S5, ESI†) shows the rapid stabilization of potential in each step, indicating the outstanding mass transport ability of this NiFeS/NF. To further widen the practical application circumstances of NiFeS/NF, the NiFeS/NF was applied as the anode to catalyze water oxidation in alkaline simulated seawater (1 M KOH + 0.5 M NaCl) and alkaline seawater (1 M KOH + Seawater). The NiFeS/NF exhibits two nearly overlapping LSV curves recorded in both 1 M KOH and 1 M KOH + 0.5 M NaCl, and the high catalytic activity of NiFeS/NF was also well-maintained with only a slight attenuation even when tested in the complex alkaline seawater (Fig. 2d). The LSV curves of NiFeS/NF without iR compensation for OER in the three studied electrolytes are shown in Fig. S6 (ESI).†Fig. 2e shows the overpotentials from Fig. 2d at 100, 200, and 500 mA cm−2 in the different electrolytes. The NiFeS/NF electrode only needed 219 and 226 mV overpotentials to drive 100 mA cm−2 in 1 M KOH + 0.5 M NaCl and 1 M KOH + Seawater, respectively, which suggesting its higher corrosion resistance to Cl−. This overpotential was lower than that of all the self-supported catalysts reported in alkaline seawater (Fig. 2f and Table S2, ESI†). Stability is another essential standard for evaluating the electrocatalyst. To test the stability of the NiFeS/NF electrocatalyst in 1 M KOH + Seawater solution, the LSV curves after 1000 and 5000 cyclic voltammetry (CV) scans were obtained (Fig. 2g), and they showed no noticeable loss in j compared with the previous one before cycling. In addition, the chronopotentiometry measurements performed at j of 100 and 500 mA cm−2 were also used to assess the long-term stability of the NiFeS/NF electrocatalyst (Fig. 2h). Only a slight fluctuation in potential could be found, even at a j of 500 mA cm−2 during 24 h operation, demonstrating the robust stability of the NiFeS/NF under a high current density. After the long-term stability test, the position of the diffraction peaks shown in the XRD pattern of post-OER NiFeS/NF (Fig. S7, ESI†) is consistent with that of the initial catalyst, but the attenuation of peak intensity after the OER indicates the formation of amorphous substances. The nanosheet structure is also retained, which could be observed from the SEM images of post-OER NiFeS/NF (Fig. S8, ESI†). The HRTEM image of post-OER NiFeS/NF in Fig. S9 (ESI)† shows an amorphous layer on the surface of the nanosheet, which agrees with the XRD results. The corresponding high-resolution XPS spectrum in the Ni 2p region of the post-OER NiFeS/NF catalyst (Fig. S10, ESI†) suggests the occurrence of small peaks located at 857.5 and 874.8 eV which correspond to Ni3+ in NiOOH that is demonstrated as an active phase for OER.45,46 No apparent change can be found from the Fe 2p region. Only the peak of SO42− at 169.0 eV can be observed from the S 2p spectrum for the post-OER NiFeS/NF catalyst, indicating the oxidation of S2− under a high anodic potential. The in situ Raman spectroscopy was used to monitor phase evolution during the OER process in alkaline seawater. As shown in Fig. S11 (ESI),† the Raman spectra show primary sharp peaks characteristic of NiFeS in the region from 200 to 400 cm−1.47 With the potential increase, the peaks at 472 and 543 cm−1, corresponding to NiOOH which is demonstrated the real OER active phase,47,48 occur at 1.5 V. According to the HRTEM, XPS, and in situ Raman results, the NiFeS deconstructs into NiOOH and SO42− during the OER process. In addition, our electrochemical tests found that the LSV curve recorded in 1 M KOH + Seawater with the addition of 0.1 M SO42− shows a higher j than that recorded in 1 M KOH + Seawater (Fig. S12, ESI†), suggesting that the SO42− promotes the OER activity.49 In addition, we used colorimetric test papers to determine the possible presence of hypochlorite products in the electrolyte after the long-term stability tests. As shown in Fig. S13 (ESI),† there was no color change in the test papers, suggesting that hypochlorite was not produced during the seawater oxidation stability tests.
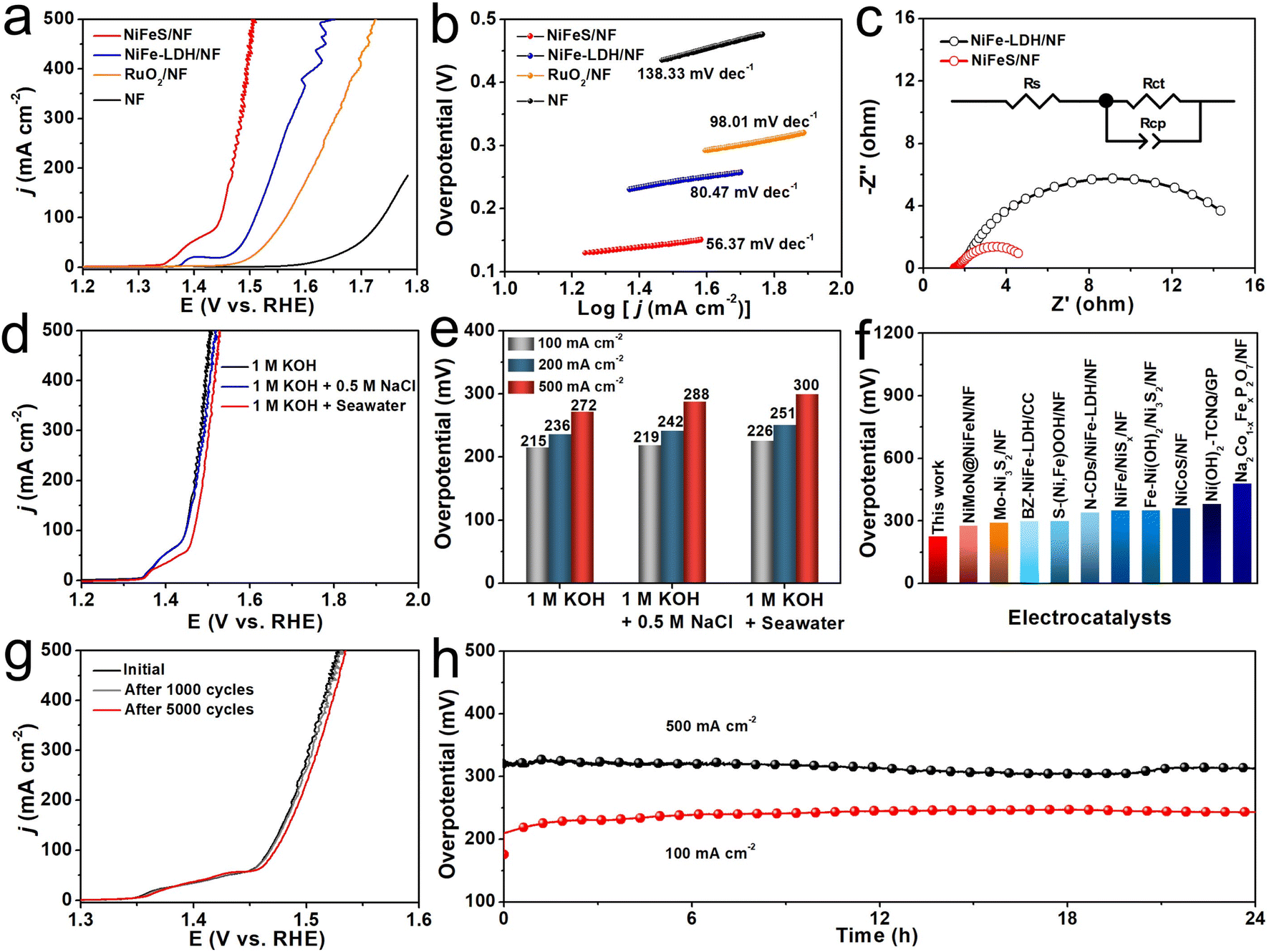 | ||
| Fig. 2 (a) The LSV curves, and (b) the corresponding Tafel plots of different electrocatalysts in 1 M KOH. (c) The Nyquist plots. (d) The LSV curves, and (e) the corresponding overpotentials for the NiFeS/NF electrode tested in different electrolytes. (f) Comparison of overpotentials at 100 mA cm−2 for NiFeS/NF and other seawater OER electrocatalysts. (g) The LSV curves before and after 1000 (5000) CV cycles in 1 M KOH + Seawater electrolyte. (h) Chronopotentiometry curves of NiFeS/NF at 100 and 500 mA cm−2 for OER in 1 M KOH + Seawater electrolyte. | ||
The HER processes catalyzed by NiFeS/NF, NiFe-LDH/NF, Pt/C loaded on NF, and bare NF were performed in 1 M KOH. As shown in Fig. 3a, the NiFeS/NF electrode performs efficiently for the HER process with an overpotential demand of 196 mV to yield a j of −100 mA cm−2, much lower than that of NiFe-LDH/NF (322 mV) and bare NF (345 mV) but larger than that of Pt/C loaded on NF (125 mV). The corresponding Tafel slope (Fig. 3b) shows the same trend with the LSV results (NiFeS/NF: 102.93 mV dec−1, NiFe-LDH/NF: 186.26 mV dec−1, NF: 204.11 mV dec−1, and Pt/C: 47.17 mV dec−1), proving that there is a faster HER kinetic reaction on NiFeS/NF. We also tested the HER activity of the NiFeS/NF in different electrolytes and Fig. 3c shows the corresponding LSV curves. The outstanding HER activity remains in 1 M KOH + 0.5 M NaCl, delivering j of −100, −200, and −500 mA cm−2 at overpotentials of 198, 213, and 287 mV. In the 1 M KOH + Seawater solution, the activity shows some degradation, which is attributed to the deposition of insoluble precipitates on the cathode surface covering some active sites. In this electrolyte, the overpotentials at the j of −100, −200, and −500 mA cm−2 are 217, 262, and 347 mV, as shown in Fig. 3d. The LSV curves of NiFeS/NF without iR compensation for HER in the three studied electrolytes are shown in Fig. S14 (ESI).† The excellent HER performance of NiFeS/NF is competitive with most of the reported HER electrocatalysts (Fig. 3e and Table S3, ESI†). Yet another essential criterion for an HER catalyst is its operational stability. Fig. 3f shows nearly completely overlapping LSV curves before and after 1000 consecutive CV cycles. Even after 5000 CV cycles, only a slight decrease in j can be observed. In addition, we also performed long-term stability tests under j of −100 and −500 mA cm−2 in alkaline seawater. As shown in Fig. 3g, the potential remains highly stable with a negligible increase throughout 25 h of operation under both −100 and −500 mA cm−2. The results of the XRD, SEM, and TEM analyses indicate that the catalyst's crystalline phase and morphology remain preserved after the HER stability test (Fig. S15–S17, ESI†). These results suggest the superior stability of the NiFeS/NF. In order to gain further insight into the catalytic mechanism of the NiFeS electrode during the HER process, density functional theory (DFT) calculations were conducted. Using the HRTEM results, a FeNi2S4 (311) slab model was built. Notably, as displayed in Fig. S18 (ESI),† the FeNi2S4 (311) has six different terminated faces, named S1–S6. Among these surfaces, S3 is mostly stable. Therefore, we chose S3 for further calculations. We first calculated the charge density difference of this model catalyst. As shown in Fig. 3h, the S species are enriched with electrons, which can repel Cl− during the process and in turn are beneficial for seawater splitting. The density of states (DOS) was also determined for the NiFeS electrode. The DOS increases near the Fermi level, indicating that the material has excellent electronic conductivity, thus enhancing its electrocatalytic performance (Fig. 3i). Furthermore, the Gibbs free energy diagrams of the HER on the model catalyst built were also calculated. The free energy of the adsorbed states (ΔGH*) were also calculated based on previous studies.50 The ΔGH* for the adsorbed H atoms are 0.12 eV, −0.35 eV, and 0.34 eV, respectively (Fig. 3j). As shown in Fig. S19 (ESI),† H atoms are preferentially adsorbed on the S atom sites, rather than Fe and Ni. In particular, for the H1 configuration, where the H atom is adsorbed on the S atom that bonded with one Fe atom and two Ni atoms, a considerably lowered adsorption free energy of 0.12 eV can be achieved, and this is much closer to the thermoneutral value, giving an outstanding HER performance.
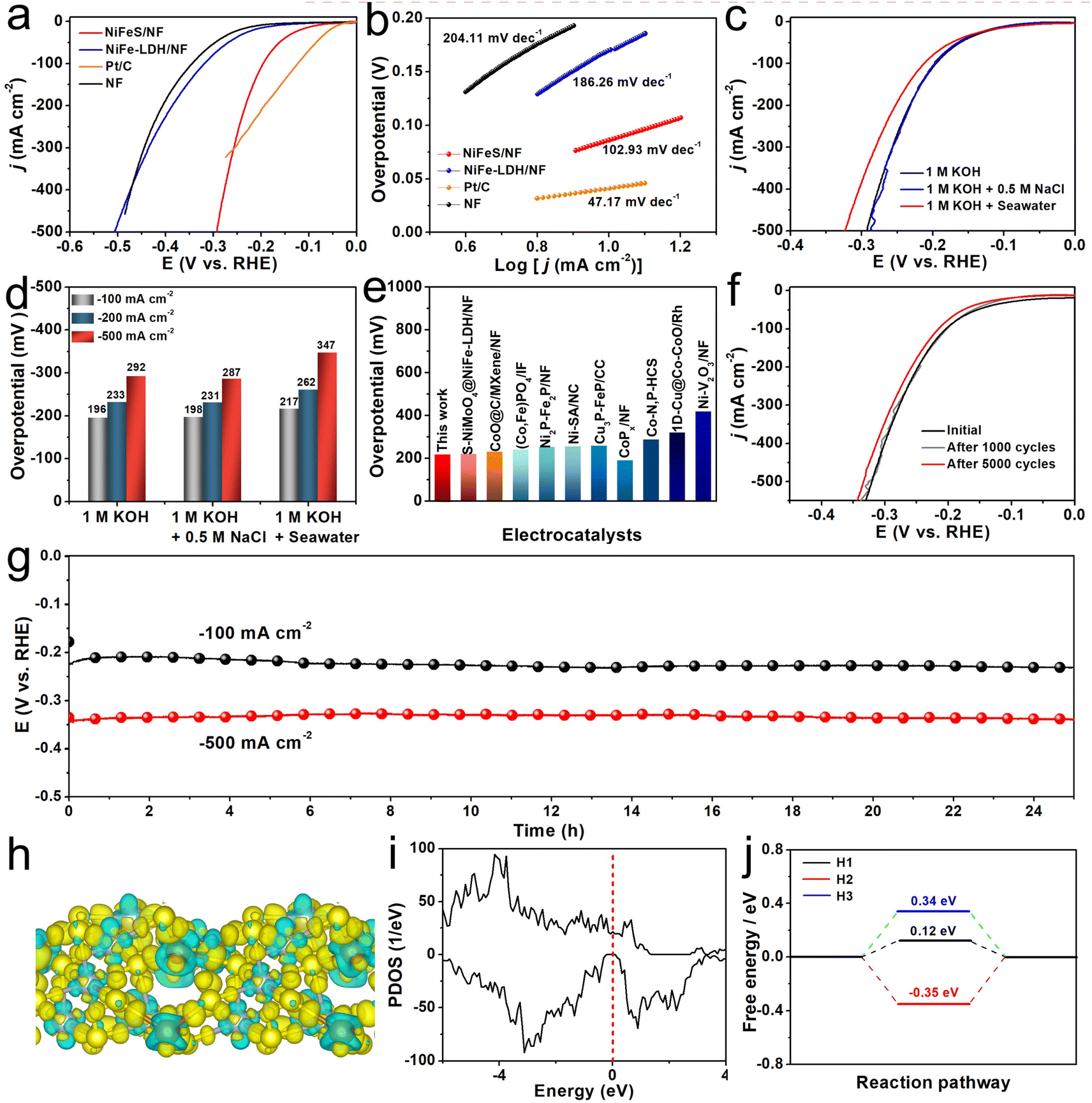 | ||
| Fig. 3 (a) The LSV curves, and (b) the corresponding Tafel plots of different electrocatalysts in 1 M KOH. (c) The LSV curves, and (d) the corresponding overpotentials for a NiFeS/NF electrode tested in different electrolytes. (e) Comparison of overpotentials at −100 mA cm−2 for NiFeS/NF and other seawater HER electrocatalysts. (f) The LSV curves before and after 1000 (5000) CV cycles in 1 M KOH + Seawater electrolyte. (g) The chronopotentiometry curves of NiFeS/NF at −100 and −500 mA cm−2 for HER in 1 M KOH + Seawater electrolyte. (h) The charge density difference plot, and (i) the computed projected density of states (PDOS) of NiFeS. (j) The free energy diagrams of the HER process over NiFeS. | ||
Given the exceptional OER and HER processes when catalyzed by NiFeS/NF, a self-assembled electrolyzer using NiFeS/NF both as anode and cathode was used to explore the overall water/seawater electrolysis (Fig. 4a). As shown in Fig. 4b, the cell voltages of NiFeS/NF needed to achieve j of 100 and 500 mA cm−2 are 1.65 and 1.82 V, respectively, in 1 M KOH, and are 40 and 300 mV lower than the voltages for the RuO2‖Pt/C pair (1.69 and 2.12 V), respectively. Moreover, the electrolyzer requires voltages of only 1.67 and 1.85 V to deliver j of 100 and 500 mA cm−2 in 1 M KOH + Seawater electrolyte, outperforming most of the as-reported bifunctional electrocatalysts for overall seawater electrolysis (Fig. 4c and Table S4, ESI†). The long-term stability of the electrolyzer for overall water/seawater splitting was confirmed by the chronopotentiometry measurement. As shown in Fig. 4d, the electrolyzer achieved a steady operation of 60 h at 500 mA cm−2 in 1 M KOH. Very impressively, the electrolyzer also worked successively at j of 100 and 500 mA cm−2 for 50 h in 1 M KOH + Seawater electrolyte, during which no obvious voltage change can be found even at a j of 500 mA cm−2 (Fig. 4e), which suggests the strong durability of this system under a high applied current density.
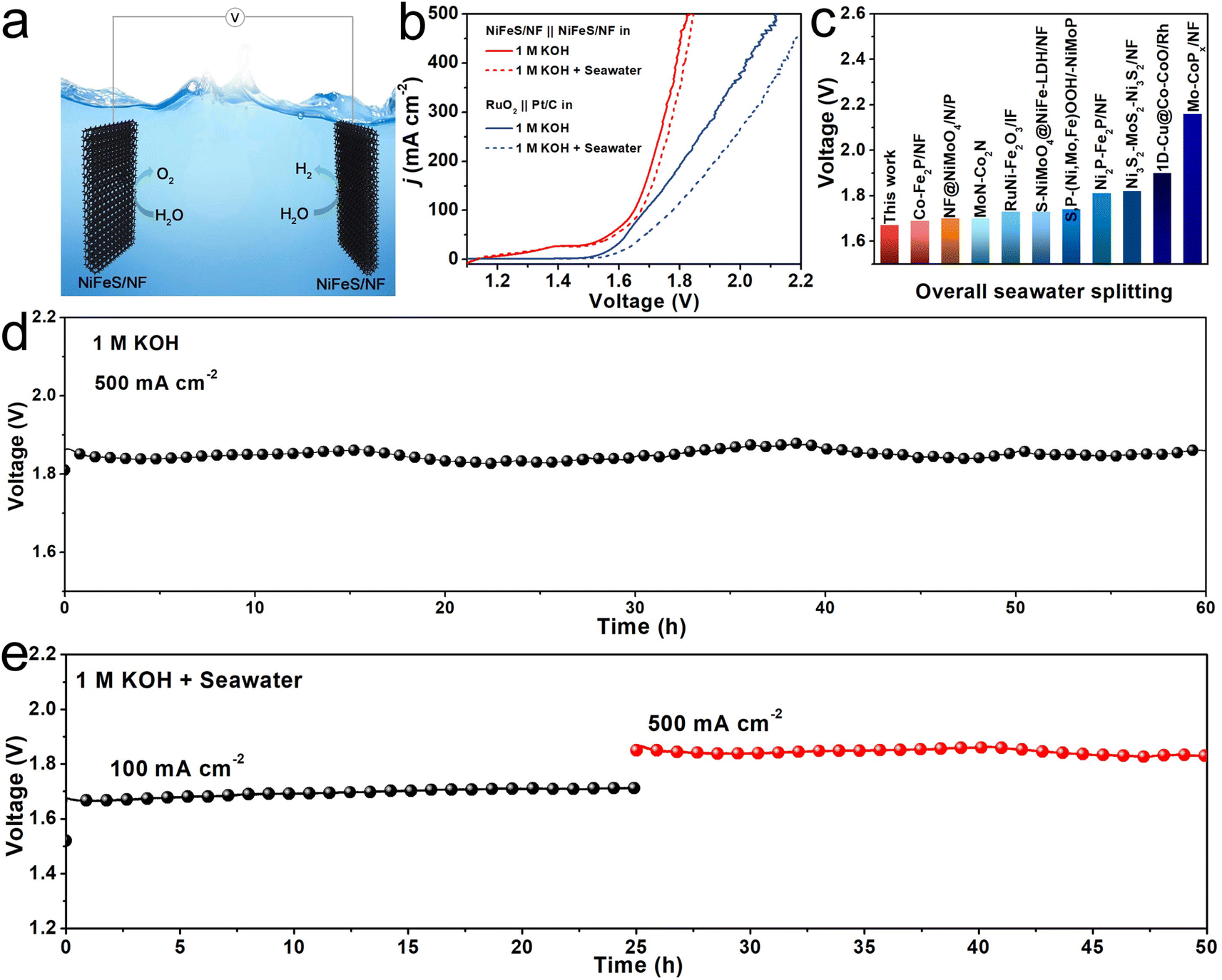 | ||
| Fig. 4 (a) A schematic illustration of an overall water splitting electrolyzer using NiFeS/NF as both anode and cathode. (b) The overall water/seawater splitting performance of NiFeS/NF‖NiFeS/NF and the RuO2‖Pt/C pairs in 1 M KOH and 1 M KOH + Seawater. (c) Comparison of overpotentials at 100 mA cm−2 for NiFeS/NF with other bifunctional electrocatalysts. Chronopotentiometry curves of the NiFeS/NF‖NiFeS/NF pair in (d) 1 M KOH, and (e) 1 M KOH + Seawater electrolyte. | ||
In summary, the NiFeS nanoarray performs highly efficiently and robustly in seawater electrolysis and requires low overpotentials of only 300 and 347 mV for the OER and HER, respectively, in alkaline seawater, to generate 500 mA cm−2. The two-electrode seawater electrolyzer utilizing NiFeS/NF as both anode and cathode can deliver 500 mA cm−2 at a cell voltage of 1.85 V with superior stability. All these remarkable features, together with the flexible features of NiFeS/NF, promise its potential practical use in technological devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Free Exploration Project of Frontier Technology for Laoshan Laboratory (Grant No. 16-02), and the National Natural Science Foundation of China (Grant No. 22072015).References
- K. Zhang, X. Liang, L. Wang, K. Sun, Y. Wang, Z. Xie, Q. Wu, X. Bai, M. S. Hamdy, H. Chen and X. Zou, Nano Research Energy, 2022, 1, e9120032 CrossRef.
- Q. Liu, S. Sun, L. Zhang, Y. Luo, Q. Yang, K. Dong, X. Fang, D. Zheng, A. A. Alshehri and X. Sun, Nano Res., 2022, 15, 8922–8927 CrossRef CAS.
- H. Jin, H. Yu, H. Li, K. Davey, T. Song, U. Paik and S. Qiao, Angew. Chem., Int. Ed., 2022, 61, e202203850 CAS.
- S. Xu, H. Zhao, T. Li, J. Liang, S. Lu, G. Chen, S. Gao, A. M. Asiri, Q. Wu and X. Sun, J. Mater. Chem. A, 2020, 8, 19729–19745 RSC.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed.
- W. Du, Y. Shi, W. Zhou, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 7051–7055 CrossRef CAS.
- C. Tang, R. Zhang, W. Lu, L. He, X. Jiang, A. M. Asiri and X. Sun, Adv. Mater., 2017, 29, 1602441 CrossRef.
- Z. Jin, P. Li and D. Xiao, Green Chem., 2016, 18, 1459–1464 RSC.
- L. Wu, L. Yu, F. Zhang, B. McElhenny, D. Luo, A. Karim, S. Chen and Z. Ren, Adv. Funct. Mater., 2021, 31, 2006484 CrossRef CAS.
- C. Tang, N. Cheng, Z. Pu, W. Xing and X. Sun, Angew. Chem., Int. Ed., 2015, 54, 9351–9355 CrossRef CAS.
- L. Yu, Q. Zhu, S. Song, B. McElhenny, D. Wang, C. Wu, Z. Qin, J. Bao, Y. Yu, S. Chen and Z. Ren, Nat. Commun., 2019, 10, 5106 CrossRef.
- X. Lu, J. Pan, E. Lovell, T. H. Tan, Y. H. Ng and R. Amal, Energy Environ. Sci., 2018, 11, 1898–1910 RSC.
- T. Ma, W. Xu, B. Li, X. Chen, J. Zhao, S. Wan, K. Jiang, S. Zhang, Z. Wang, Z. Tian, Z. Lu and L. Chen, Angew. Chem., Int. Ed., 2011, 60, 22740–22744 CrossRef.
- X. Wu, S. Zhou, Z. Wang, J. Liu, W. Pei, P. Yang, J. Zhao and J. Qiu, Adv. Energy Mater., 2019, 9, 1901333 CrossRef.
- L. Zhang, J. Wang, P. Liu, J. Liang, Y. Luo, G. Cui, B. Tang, Q. Liu, X. Yan, H. Hao, M. Liu, R. Gao and X. Sun, Nano Res., 2022, 15, 6084–6090 CrossRef CAS.
- C. Wang, M. Zhu, Z. Cao, P. Zhu, Y. Cao, X. Xu, C. Xu and Z. Yin, Appl. Catal., B, 2021, 291, 120071 CrossRef CAS.
- T. Cui, X. Zhai, L. Guo, J. Chi, Y. Zhang, J. Zhu, X. Sun and L. Wang, Chin. J. Catal., 2022, 43, 2202–2211 CrossRef CAS.
- L. Zhuang, J. Li, K. Wang, Z. Li, M. Zhu and Z. Xu, Adv. Funct. Mater., 2022, 32, 2201127 CrossRef CAS.
- M. A. Khan, T. Al-Attas, S. Roy, M. M. Rahman, N. Ghaffour, V. Thangadurai, S. Larter, J. Hu, P. M. Ajayan and M. G. Kibria, Energy Environ. Sci., 2021, 14, 4831–4839 RSC.
- S. Dresp, F. Dionigi, M. Klingenhof and P. Strasser, ACS Energy Lett., 2019, 4, 933–942 CrossRef CAS.
- F. Dionigi, T. Reier, Z. Pawolek, M. Gliech and P. Strasser, ChemSusChem, 2016, 9, 962–972 CrossRef CAS PubMed.
- H. Zhang, S. Geng, M. Ouyang, H. Yadegari, F. Xie and D. J. Riley, Adv. Sci., 2022, 9, 2200146 CrossRef CAS.
- L. Zhang, J. Liang, L. Yue, K. Dong, J. Li, D. Zhao, Z. Li, S. Sun, Y. Luo, Q. Liu, G. Cui, A. A. Alshehri, X. Guo and X. Sun, Nano Research Energy, 2022, 1, e9120028 CrossRef.
- W. Tong, M. Forster, F. Dionigi, S. Dresp, R. S. Erami, P. Strasser, A. J. Cowan and P. Farràs, Nat. Energy, 2020, 5, 367–377 CrossRef CAS.
- H. Chen, Y. Zou, J. Li, K. Zhang, Y. Xia, B. Hui and D. Yang, Appl. Catal., B, 2021, 293, 120215 CrossRef CAS.
- Z. Yu, Y. Li, V. Martin-Diaconescu, L. Simonelli, J. R. Esquius, I. Amorim, A. Araujo, L. Meng, J. L. Faria and L. Liu, Adv. Funct. Mater., 2022, 32, 2206138 CrossRef CAS.
- F. Ma, S. Wang, X. Gong, X. Liu, Z. Wang, P. Wang, Y. Liu, H. Cheng, Y. Dai, Z. Zheng and B. Huang, Appl. Catal., B, 2022, 307, 121198 CrossRef CAS.
- Y. Yu, J. Li, J. Luo, Z. Kang, C. Jia, Z. Liu, W. Huang, Q. Chen, P. Deng, Y. Shen and X. Tian, Mater. Today Nano, 2022, 18, 100216 CrossRef CAS.
- Z. Wang, C. Wang, L. Ye, X. Liu, L. Xin, Y. Yang, L. Wang, W. Hou, Y. Wen and T. Zhan, Inorg. Chem., 2022, 61, 15256–15265 CrossRef CAS.
- P. Ding, H. Song, J. Chang and S. Lu, Nano Res., 2022, 15, 7063–7070 CrossRef CAS.
- H. Wang, L. Chen, L. Tan, X. Liu, Y. Wen, W. Hou and T. Zhan, J. Colloid Interface Sci., 2022, 613, 349–358 CrossRef CAS.
- P. Ding, C. Meng, J. Liang, T. Li, Y. Wang, Q. Liu, Y. Luo, G. Cui, A. M. Asiri, S. Lu and X. Sun, Inorg. Chem., 2021, 60, 12703–12708 CrossRef CAS.
- B. Zhang, S. Liu, S. Zhang, Y. Cao, H. Wang, C. Han and J. Sun, Small, 2022, 18, 2203852 CrossRef CAS.
- Q. Tu, W. Liu, M. Jiang, W. Wang, Q. Kang, P. Wang, W. Zhou and F. Zhou, ACS Appl. Energy Mater., 2021, 4, 4630–4637 CrossRef CAS.
- Y. Yang, H. Meng, C. Kong, S. Yan, W. Ma, H. Zhu, F. Ma, C. Wang and Z. Hu, J. Colloid Interface Sci., 2021, 599, 300–312 CrossRef CAS.
- W. Zhu, Z. Yue, W. Zhang, N. Hu, Z. Luo, M. Ren, Z. Xu, Z. Wei, Y. Suo and J. Wang, J. Mater. Chem. A, 2018, 6, 4346–4353 RSC.
- Y. Kuang, M. J. Kenney, Y. Meng, W. Hung, Y. Liu, J. E. Huang, R. Prasanna, P. Li, Y. Li, L. Wang, M. Lin, M. D. McGehee, X. Sun and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6624–6629 CrossRef CAS.
- S. Y. Jung, S. Kang, K. M. Kim, S. Mhin, J. C. Kim, S. J. Kim, E. Enkhtuvshin, S. Choi and H. Han, Appl. Surf. Sci., 2021, 568, 150965 CrossRef CAS.
- L. Tan, J. Yu, C. Wang, H. Wang, X. Liu, H. Gao, L. Xin, D. Liu, W. Hou and T. Zhan, Adv. Funct. Mater., 2022, 32, 2200951 CrossRef CAS.
- F. Wang, X. Zhang, J. Zhou, Z. Shi, B. Dong, J. Xie, Y. Dong, J. Yu and Y. Chai, Inorg. Chem. Front., 2022, 9, 2068–2080 RSC.
- B. Dong, X. Zhao, G. Han, X. Li, X. Shang, Y. Liu, W. Hu, Y. Chai, H. Zhao and C. Liu, J. Mater. Chem. A, 2016, 4, 13499–13508 RSC.
- M. C. Biesinger, B. P. Payne, L. W. M. Lau, A. Gerson and R. S. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- Y. Jin, X. Yue, H. Du, K. Wang, S. Huang and P. K. Shen, J. Mater. Chem. A, 2018, 6, 5592–5597 RSC.
- F. Wu, X. Guo, G. Hao, Y. Hu and W. Jiang, Adv. Mater. Interfaces, 2019, 6, 1900788 CrossRef CAS.
- C. Tang, Z. L. Zhao, J. Chen, B. Li, L. Chen and C. M. Li, Electrochim. Acta, 2017, 248, 243–249 CrossRef CAS.
- C. Liu, D. Jia, Q. Hao, X. Zheng, Y. Li, C. Tang, H. Liu, J. Zhang and X. Zheng, ACS Appl. Mater. Interfaces, 2019, 11, 27667–27676 CrossRef CAS.
- Y. Wu, Y. Li, M. Yuan, H. Hao, X. San, Z. Lv, L. Xu and B. Wei, Chem. Eng. J., 2022, 427, 131944 CrossRef CAS.
- Y. Zhao, X. Jia, G. Chen, L. Shang, G. I. N. Waterhouse, L. Wu, C. Tung, D. O'Hare and T. Zhang, J. Am. Chem. Soc., 2016, 138, 6517–6524 CrossRef CAS.
- Y. Shi, W. Du, W. Zhou, C. Wang, S. Lu, S. Lu and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 22470–22474 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and supplementary figures. See DOI: https://doi.org/10.1039/d2ta08568b |
| This journal is © The Royal Society of Chemistry 2023 |