
Electrochemical graphitization transformation of deposited carbon for Li-ion storage: sustainable energy utilization from coke oven solid waste†
Tao
Rong
a,
Wei
Guan
a,
Weili
Song
b,
Haibin
Zuo
 *a,
Jingxiu
Wang
a,
Qingguo
Xue
a and
Shuqiang
Jiao
*a
*a,
Jingxiu
Wang
a,
Qingguo
Xue
a and
Shuqiang
Jiao
*a
aState Key Laboratory of Advanced Metallurgy, University of Science and Technology Beijing, Beijing, 100083, China. E-mail: zuohaibin@ustb.edu.cn; sjiao@ustb.edu.cn
bInstitute of Advanced Structure Technology, Beijing Institute of Technology, Beijing, 100081, China
First published on 21st November 2022
Abstract
Developing new precursors of synthetic graphite from harmful carbon-containing solid waste generated by coking enterprises has been an important topic in graphitization research for the world's green development and creating new energy. In this study, coke oven deposited carbon was used as a precursor to prepare graphite by a molten salt electrolysis method. The structure transformation law of deposited carbon and the changes of the O, N, and S atomic contents on the surface during the electrolysis process were explored. The results showed that deposited carbon is transformed into a graphitic structure with a graphitization degree of 0.74 and the conversion rate of O, N and S exceeds 50% in the molten CaCl2 salt under 2.6 V at 900 °C for 8 h. The low-temperature graphitization transition process of deposited carbon assisted by an electric field was investigated by molecular simulation calculations. Deposited carbon-derived graphite was demonstrated as a useful negative electrode material for lithium-ion batteries and delivered a high reversible capacity of 325 mA h g−1 and an excellent coulombic efficiency of 99.5% at 1C after 600 cycles, which could provide a reference for value-added utilization of carbon-containing solid waste in coking enterprises.
1 Introduction
As a key and strategic material, graphite has been widely demanded in technologies such as lithium-ion batteries,1–3 graphene4,5 and other structural materials.6–9 However, there are severe challenges faced in the application of natural graphite, such as limited supply, instability in natural product quality and environmental pressure from mining operations.10,11 Synthetic graphite with a relatively simple synthesis process, low cost, and superior electrochemical properties has attracted insights and efforts from a large number of researchers.12–14 In order to cope with the global energy crisis and environmental pressure, it is hence necessary to develop useful synthetic graphite.15Currently, researchers attempt to prepare synthetic graphite from a variety of materials, including semi-coke,16 bituminous coal,12 waste PET plastic,13 poly vinyl alcohol17 and lignin.18 However, excellent precursors should be sustainable, inexpensive, and easily accessible. Due to the above characteristics, deposited carbon (DC) in coke ovens, which is a kind of coking by-product, has been paid more and more attention and studied as a potential precursor. DC is generated by the pyrolysis of crude gas in the coking process and commonly found in coking chambers and ascension pipes.19,20 DC that causes uneven temperature in the coking chamber and hinders coke pushing19 is harmful to coking production. The potential output of DC in coke ovens is relatively large, which is directly related to the operating system of coking enterprises. Taking the production data of a coking enterprise in China as a calculation reference,21 the potential production of DC in China and worldwide is about 1 million tons (Mt) per year and 1.5 Mt per year, respectively. It is worth noting that in China, the potential DC amount has even far exceeded the natural graphite production in 2017.22 However, there have been no efficient and valuable utilization methods available for treating DC.19,23 Therefore, it is of great significance to achieve the application of DC graphitization using DC as a precursor. Compared with other precursors, DC is not inferior in terms of chemical composition with a carbon content of not less than 90% and a low content of impurity elements in previous research.24,25 Hence, it is very important to perform further purification and utilization of DC. In addition, DC originates from coke oven emissions and contains polycyclic aromatic hydrocarbons, which seriously endanger the health of operators.26,27 Therefore, a harmless treatment of DC is urgently needed and DC can be a promising precursor for preparing graphite.
However, the current mainstream high-temperature (3000 °C) method to produce graphite is highly energy-intensive, and it has detrimental effects on the environment.15,28 Although the addition of various new and efficient catalysts can reduce the temperature of graphitization,29,30 the catalyst remaining in the graphite product generally limits the application of the catalytic graphite.30 In order to further reduce the processing energy consumption and the adverse effects of production methods, researchers have made many attempts for graphitization. Jin et al.31,32 brought surprises to other researchers by graphitizing carbon black at 800–950 °C through molten salt electrolysis and laid the foundation for graphitization transformation of solid carbon materials by molten salt electrolysis. Later, Zhu et al.33 prospectively studied the behavior of the oxygen element and the electrolytic graphitization process of hyper coal in molten CaCl2. The optimized electrolysis product was applied as an anode for lithium-ion batteries with a good electrochemical performance. Thapaliya et al.34,35 expanded the types of precursors and further verified the possibility of molten salt electrolysis graphitization by studying the graphitization transition behaviors of biomass-derived carbon and gasified coal chars under different electrolysis conditions. However, the carbon structure transformation law and graphitization mechanism of other carbon materials in the molten salt electrolysis process were not clearly presented, which were important guides for realizing the synergistic regulation of the structure and functions of carbon materials and further optimizing the process parameters, with the aim to achieve the application of carbon materials with low cost and high added value.
In order to achieve the high-value utilization of DC in coking enterprises, DC collected from ascension pipes in coking enterprises is used as a potential precursor to form a graphite based composite anode in this study. Through the method of molten salt electrolysis, DC was directly converted into flake graphite in molten salt at 900 °C for 8 h, when the cell voltage was 2.6 V. Molten salt low-temperature graphitization is more competitive in terms of energy consumption and production cost compared to the traditional Acheson process. The structure transformation law of DC and the mechanism of graphitization were explored, and the influence of the electric field on the molecular structure and electronic properties of the precursor was investigated. It was found that the electric field could affect electron injection and local charge redistribution during electrochemical graphitization. Graphitized electrolysis products were used in Li-ion batteries and the electrochemical performance was systematically evaluated in terms of reversible capacity and coulombic efficiency.
2 Experimental
2.1 Materials
Deposited carbon (DC) was provided by Baoshan Iron & Steel Co., Ltd and it was produced from the ascension pipe of a coke oven in a division coking plant. The DC was crushed into powder samples with a size less than 100 μm for the molten salt electrolysis experiment. The composition and particle size distribution of DC are shown in Table S1 and Fig. S1,† respectively. The contents of C, O, N and S in the DC are 90.21 wt%, 3.72 wt%, 0.84 wt% and 0.58 wt%, respectively. The average particle size (Dv(50)) of the DC is 34.7 μm. The true density of DC is measured as 1.76 g cm−3, according to the gas volume method.36Anhydrous calcium chloride (AR, 96%) was purchased from Shanghai Macklin Biochemical Co., Ltd. Before the electrolysis experiments, it was dehydrated at a temperature of 300 °C according to a reported method37 to prevent the adverse effect of crystal water remaining in CaCl2 on electrolysis experiment.38
2.2 Molten salt electrolysis experiments
The molten salt electrolysis experiments were conducted following the procedures developed and described in our previous study.33 First, about 1 g of the crushed DC powder was wrapped with a stainless steel mesh to obtain a layered structure (2.5 cm × 2.5 cm) as the cathode. This is not like previous reports in which the cathode material was fabricated into agglomerates,31 and the loose layered structure helps to facilitate the infiltration of molten salts. A high-purity graphite rod with a diameter of 6 mm was used as the anode. The electrolysis experiments were performed in a high temperature tube furnace (SKL10-BY, Baotou Yunjie, China), and 200 g of pre-dried CaCl2 was placed in an alumina crucible with an internal diameter of 64 mm and a height of 120 mm. Next, the temperature of the reaction crucible in the heating furnace was raised to a desired temperature (850–950 °C) at a heating rate of 5 °C min−1 to ensure the molten state of CaCl2, under a flow of 200 cm3 min−1 of Ar. After the temperature was stabilized, the electrodes were immersed in the molten salt to a depth of 30 mm. Constant cathodic polarization was carried out using an electrochemical workstation (Versa STAT 3, Princeton Applied Research, USA). Finally, electrolysis products (EPs) were collected by acid washing (1 mol per L HCl) and distilled water washing to get neutral powder after removing the cathode from the reactor.2.3 Characterization of electrolysis products
X-ray diffraction (XRD) patterns were recorded using a SMARTLAB-9 diffractometer (Rigaku, Japan) with a CuKα source. Structural parameters including interlayer spacing (d002), crystallite diameter (La), crystallite height (Lc), stacking layer number (N) and degree of graphitization (G) were calculated.35 Raman spectra were recorded on a Lab RAM HR Evolution Raman spectrometer (HORIBA, France) with an excitation wavelength of 532 nm. Peak splitting and curve fitting operations were performed on the spectra.39,40 Morphologies and microstructure of the electrolysis products were investigated by field emission scanning electron microscopy (FESEM, Zeiss SIGMA 300, Zeiss, Germany) and transmission electron microscopy (TEM, Titan3™, FEI, USA), respectively. Fourier transform infrared (FTIR) spectra of the electrolysis products obtained with a NEXUS670FT-IR spectrometer (THERMO NICOIET, USA) within a wavenumber range of 400 cm−1 to 4000 cm−1 were adopted to investigate functional groups. X-ray photoelectron spectroscopy (XPS) spectra were obtained with an ESCALAB250Xi spectrometer (Thermo Fisher Scientific, USA) to investigate the valence changes in elements. Low temperature nitrogen adsorption–desorption isotherms were obtained to analyze the specific surface area and pore size distribution of samples (ASAP2020, Micromeritics, USA).2.4 Electrochemical measurements
The electrochemical performances of electrolysis products (EP) were evaluated using CR2032 cells assembled in a glove box filled with argon gas. The working electrode was prepared by combining EP, carbon black and polyvinyl difluoride (PVDF) in N-methyl-2-pyrrolidone (NMP) with a weight ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1.5 and casting the obtained slurry on copper foil. The mass loading of EP was 0.45 mg cm−2. A polypropylene microporous membrane (Celgard 2500) and lithium metal sheet were employed as the separator and counter electrode, respectively. 1.0 mol per L LiPF6 solution in a mixed solvent of ethylene carbonate and diethyl carbonate (1:1 vol%) with 5 wt% fluoroethylene carbonate was used as the electrolyte. Cyclic voltammetry (CV) tests were performed using an electrochemical workstation (CHI66E, CH Instruments, China). The cycling stability and rate performances of EPs were evaluated using a NEWARE-BTS (NEWARE, America) at different rates of 0.1C, 0.2C, 0.5C and 1C (1C = 372 mA g−1) in the potential range of 0.01–2 V.
:1:1.5 and casting the obtained slurry on copper foil. The mass loading of EP was 0.45 mg cm−2. A polypropylene microporous membrane (Celgard 2500) and lithium metal sheet were employed as the separator and counter electrode, respectively. 1.0 mol per L LiPF6 solution in a mixed solvent of ethylene carbonate and diethyl carbonate (1:1 vol%) with 5 wt% fluoroethylene carbonate was used as the electrolyte. Cyclic voltammetry (CV) tests were performed using an electrochemical workstation (CHI66E, CH Instruments, China). The cycling stability and rate performances of EPs were evaluated using a NEWARE-BTS (NEWARE, America) at different rates of 0.1C, 0.2C, 0.5C and 1C (1C = 372 mA g−1) in the potential range of 0.01–2 V.
3 Results and discussion
As a typical solid waste from the coking industry, the annual production of DC in China has exceeded 1 Mt (Fig. 1a). The current main methods to treat DC mainly include mechanical cleaning – coal blending coking and combustion (Fig. 1b), which can cause serious environmental pollution problems and greenhouse gas emissions (Fig. 1c). This poses a huge challenge to the green and low-carbon sustainable development of coking enterprises. Electrochemical graphitization gives a new sustainable pathway for utilizing DC. | ||
| Fig. 1 a) Production of DC in China in 2017–2021; (b) comparison of the processing capacity of mechanical cleaning – coal blending coking and combustion; (c) gas emissions from complete combustion of DC produced in a coking plant with a design coke capacity of 1.05 Mt per year; (d) schematic diagram of the molten salt electrolysis process using DC in coke ovens as a precursor. | ||
In order to verify the possibility of graphitization transformation of coke oven DC, the molten salt electrolysis process was conducted as shown in Fig. 1d. The electrolysis products shown in Table S2† were obtained under different electrolysis temperatures (850–950 °C), cell voltages (2.2–2.6 V) and electrolysis times (0–12 h). Through a variety of characterization methods, the effects of different experimental conditions on the structure of electrolysis products were investigated to clarify the mechanism of carbon structure transformation. In addition, the electrolysis products obtained under optimized conditions were used as electrodes in lithium-ion batteries to prove that DC-based electrolytic graphite can be used as an energy storage material.
3.1 Structural evolution of electrolysis products
Fig. 2a shows the XRD patterns of DC and electrolysis products. Obviously, DC and individual electrolytic products prepared under the conditions of low electrolysis temperature, small cell voltage or short electrolysis time including EP-850 °C, EP-2.2 V, EP-2.4 V and EP-4 h exhibit strong background intensity and wide peaks near 25° indicating the ubiquitous presence of disordered or amorphous carbon. However, EP-8 h (EP-2.6 h and EP-900 °C), EP-12 h and EP-950 °C show similar graphite structures to the pattern of PDF#99-0057-Graphite. It can be concluded that the phase transformation from DC to graphite was realized through molten salt electrolysis under low-temperature conditions. By developing the molten salt low-temperature graphitization process, temperature is no longer a key limiting factor for the graphitization of carbon materials. | ||
| Fig. 2 a) XRD patterns of DC and EPs; (b) curve-fitting of the XRD pattern of DC; (c) stacking layer number (N) and degree of graphitization (G) of products with different electrolysis times; (d) Raman spectra of the samples; (e) ID/IG values of DC and EPs; (f) curve-fitting of the Raman spectrum of EP-8 h; (g) ID1/IG values of products with different electrolysis times. | ||
XRD patterns were de-convoluted to reveal different carbon structures. Taking DC as an example (Fig. 2b), there are four curves in the range of 15–30° and 40–50° representing the γ band, 002 band, 100 band and 101 band.41,42 Then the structural parameters are calculated and listed in Table S3.† The degree of graphitization (G) is used as an important indicator to evaluate the graphitization transformation effect of DC in molten salt electrolysis. When the electrolysis temperature is 900 °C and the cell voltage is 2.6 V, the G value of the electrolysis product increases from −0.81 of DC to 0.74 of EP-12 h (Fig. 2c). It seems that the electrolysis time is positively correlated with the graphitization degree of the electrolysis product. The relationship between the G value and temperature is shown in Fig. S2.† The G value of EP-850 °C is only −1.06, which is even lower than the G value of DC. When the electrolysis temperature was raised to 900 °C, the G value of EP-8 h is greatly increased. The DC is transformed from an amorphous structure to a graphitic structure with an interlayer spacing of 0.3379 nm and stacking layer number of 51.40 (Fig. 2c). The electrolysis temperature can directly affect the viscosity of molten CaCl2,43 which affects the infiltration process of molten salt inside the working electrode. In a previous study, the graphitization of the carbon material did not occur when the carbon material was soaked in molten salt at 900 °C for 8 h.33 In this work, when the cell voltage was 2.2 V or 2.4 V, there was no graphitization of the carbon material. When the cell voltage was further increased to 2.6 V, the G value of EP-8 h was 0.74, suggesting that the cell voltage was the determining factor affecting the graphitization of DC in molten salt. Raman spectroscopy was widely used for studying subtle structural changes of carbon materials. As shown in Fig. 2d, it is obvious that the two peaks are the D peak at 1350 cm−1 and the G peak at 1580 cm−1 for all samples. The ratio of ID/IG is usually used as a criterion for determining the degree of structural ordering of carbon materials. Fig. 2e shows the ID/IG value of different samples. It was found that ID/IG values of EP-850 °C, EP-2.2 V and EP-2.4 V were higher than that of DC. This phenomenon could be caused by the condensation and polymerization of DC in the early stage of electrolysis. The breakage of chemical bonds in macromolecular compounds leads to the formation of defects and amorphous structures.44 According to the method reported,44–46 the Raman spectral bands ranging from 800 cm−1 to 2000 cm−1 were curve-fitted into five bands (D1, D2, D3, D4, and G). Raman spectral parameters such as peak position, full width at half maximum (FWHM), and intensity ratios were obtained (Table S4†). Fig. 2f shows the Raman spectrum of EP-8 h. The disappearance of the D3 and D4 bands means that the content of graphite carbon (sp2) increases significantly.46 Combined with XRD results, it is confirmed that EP-8 h was successfully transformed into a graphite structure. In Fig. 2g, ID1/IG decreases with the increase of electrolysis time, which suggests that the order degree of samples increases. ID1/IG of EP-12 h is 0.20 within the range of 0.11 to 0.30 as reported in the literature.32,33
In previous studies, sheet-like structures were prevalent in electrolysis products.31–33 However, in this study, SEM and TEM images of DC and electrolysis products also revealed the transformation of DC to a highly crystalline graphitic structure during molten salt electrolysis. Fig. 3a is a schematic diagram of the significant structural evolution of the DC surface during electrochemical graphitization. Specifically, as shown in Fig. 3b, the surface of the DC matrix is relatively flat, and there are some tiny spacings embedded in the substrate. These spacings may originate from natural growth during DC generation in coke ovens or from DC fragmentation prior to electrolysis.20 At the early stage of electrolysis, the surface of EP-4 h is densely wrinkled. These complex morphological changes are accompanied by the generation of local fractures in the samples, which was probably ascribed to the differences in the mechanical properties of different components under complex physical and chemical conditions. When the electrolysis time was further extended to 8 h, some obvious small-sized flakes appeared due to the structural breakage, and the pores were caused by irregular distribution of these flakes. These seem to be related to the stress release of the wrinkles in EP-4 h. It is noted that the individual flake appears to be flat at the micrometer scale. Obviously, the flakes are further enlarged to form flake clusters in EP-12 h compared with EP-8 h. It is concluded from both the XRD and Raman results that the key feature for the formation of the graphitic structure during electrochemical conversion is the appearance of flakes in EP-8 h and EP-12 h. The TEM image of the flakes in EP-8 h is shown in Fig. 3c. The high-resolution TEM image of EP-8 h (Fig. 3d) shows that the interlayer spacing is 0.3391 nm which is close to the value for natural graphite.47 Although the interlayer spacing of the petaloid flakes reported in our former study (0.3285 nm) is smaller than the flake structure in EP-8 h,33 EP-8 h appears to be competitive due to its high degree of graphitization and short production time. During the electrochemical transformation, the change of the sample microstructure affects its specific surface area and pore size distribution. Fig. 3e shows the N2 adsorption–desorption curves of DC and EP-8 h at −195.8 °C. The correspondingly calculated BET surface areas are 4.1161 m2 g−1 and 7.4926 m2 g−1, respectively. The lower surface area of amorphous carbon than graphite seems to be related to the difference in their structural unit size. After molten salt electrolysis, the specific surface area and the average pore size of the sample increased significantly. Fig. S3† shows the pore size distribution of DC and EP-8 h. The average adsorption pore size of the latter is much larger than that of the former (43.86969 nm vs. 16.64374 nm).
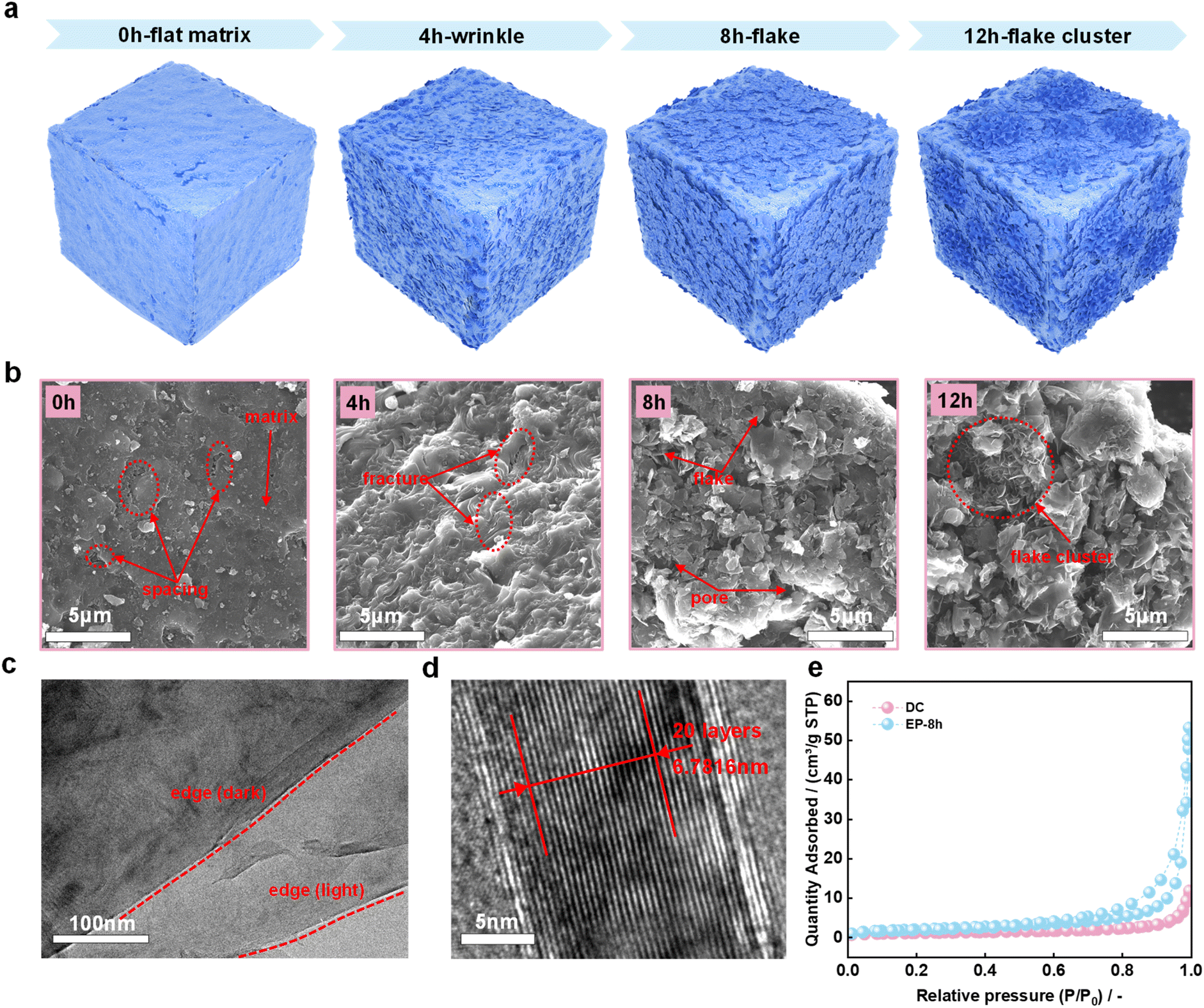 | ||
| Fig. 3 Microstructure evolution during molten salt electrolysis. (a) Schematic diagram of the significant structural evolution of the DC surface; (b) SEM images of DC, EP-4 h, EP-8 h and EP-12 h; (c) TEM image of EP-8 h; (d) high-resolution TEM image of EP-8 h; (e) N2 adsorption–desorption curves of DC and EP-8 h at −195.8 °C. | ||
3.2 Mechanism of structure conversion
To reveal the carbon structure transformation mechanism of DC during molten salt electrolysis, the functional groups and chemical bonds of DC and electrolysis products at different times were investigated by FTIR as shown in Fig. S4.†Fig. 4a shows the FTIR spectra of DC, EP-4 h, EP-8 h and EP-12 h at 1000–1800 cm−1. The absorption band located near 1100 cm−1 is caused by the stretching vibration of SO (1080–1120 cm−1) bonds and C–O–C (1120–1160 cm−1) bonds,48 and the broad absorption band around 1620 cm−1 is assigned to the stretching vibration of CC bonds in the aromatic structure.49 The intensity of the absorption peak is related to the amount of chemical bonds in the sample. Previous studies32,35 have shown that the electrolytic graphitization process of molten salts is accompanied by the occurrence of deoxidation reactions. As shown in Fig. 4b, the relative intensities (SO/CC and C–O–C/CC) decreased with the extension of electrolysis time in the current study. It can be speculated that the content of oxygen-containing chemical bonds in the DC decreased with the prolongation of electrolysis time. In Fig. 4c, the absorption bands in the wavenumber range of 2800–3000 cm−1 are ascribed to the stretching vibrations of aliphatic structures.50 Four peaks can be fitted including antisymmetric CH3 stretching, antisymmetric CH2 stretching, symmetric CH3 stretching and symmetric CH2 stretching.51 In the study of carbon materials, the aliphatic structural parameter A(CH2)/A(CH3) can be used to characterize the length of aliphatic chains and the degree of branching aliphatic side-chains.42,46,51,52 It can be calculated from eqn (1), where A2920 and A2960 represent the area of –CH2 and –CH3 groups, respectively.
 | (1) |
 | ||
| Fig. 4 (a) FTIR spectra (1000–1800 cm−1) of DC and products with different electrolysis times; (b) content of oxygen-containing chemical bonds in different samples; (c) curve-fitting of the FTIR spectra (2000–3000 cm−1); (d) A(CH2)/A(CH3) varies with electrolysis time; (e) the XPS spectra of DC and products with different electrolysis times; (f) O/C, N/C and S/C atomic ratios obtained by XPS using by high resolution scans; (g) the deconvoluted C 1s XPS spectra of DC and EP-8 h; (h) relative areas of sp2 and sp3 in different samples; (i) current response at a cell voltage of 2.6 V, 900 °C. | ||
As shown in Fig. 4d, A(CH2)/A(CH3) decreases with the increase of electrolysis time, which implies that the aliphatic chains of the aromatics ring are gradually shortened and structures between aromatic rings become more compact in DC during the electrochemical conversion of molten salt.
XPS is the most sensitive method for probing the surface of carbon materials.53 The XPS spectra (Fig. 4e) show the presence of carbon, oxygen, sulfur and nitrogen in DC, EP-4 h, EP-8 h and EP-12 h. With the progress of the electrochemical conversion of DC in molten salt, the atomic contents of elements such as O, N, and S in the surface layer of the electrolysis product decrease with the increase in electrolysis time as shown in Table S5† and Fig. 4f.
Atomic conversion rate (R) was defined to represent the degree of removal of oxygen, nitrogen and sulfur atoms on the surface of DC during the electrochemical conversion in molten salts. It can be calculated using eqn (2), where X represent atomic species, including O, N and S; A[X],0 represents the atomic percentage of X in DC, %; A[X],t represents the atomic percentage of X in products prepared after electrolysis for t h, %.
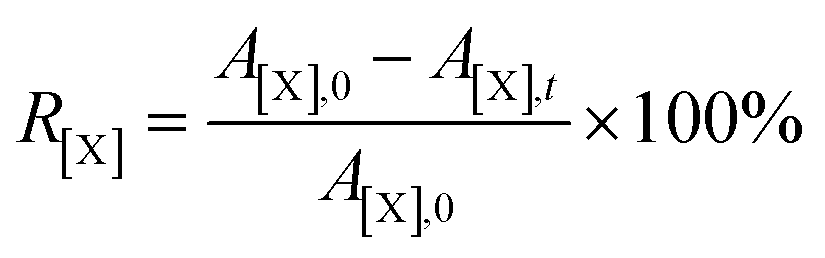 | (2) |
Under the current electrolysis conditions, the atomic conversion rate of N atoms on the surface of DC is highest after 12 h of electrolysis (Fig. S5†). Normalizing the data in Fig. S5,† the atomic conversion rate of S atoms on the surface of DC is smallest in the range of 8–12 h as shown in Fig. S6,† which indicates the complete removal of S atoms, but the removal process of O and N atoms is in progress.
The removal of O, N, and S during electrolysis is accompanied by the evolution of the chemical structure of C in DC. Fig. 4g shows the high resolution XPS spectra of C 1s for DC and EP-8 h, and the deconvolution of the C 1s peaks showed 3 different peaks: sp2, sp3 and π–π* shake-up feature.54 The sp2 represents carbon in CC bonds in aromatic rings and in aliphatic chains,53 sp3 represents carbon in CH2 and CH3 (ref. 54) and π–π* transitions are common in aromatic ring structures.55 Table S6† shows the results of chemical bonds calculated from XPS measurements. Fig. 4h shows the relative area of sp2 and sp3 in DC, EP-4 h, EP-8 h and EP-12 h. With the increase of electrolysis time, the content of graphite carbon (sp2) in the DC increases. In summary, the curve of relationship between the working current and time is shown in Fig. 4i. In the early stage of electrolysis, a large current response is associated with double layer charging.31 Then, the current quickly drops to below 0.45 A. In this study, the O, N, and S atoms in DC were gradually removed, with the S atom being first stabilized. During the electrolysis process, the disordered C atoms in DC are subsequently rearranged with a long distance, and stacked into a highly ordered graphitic structure. The current response stabilizes at about 0.15 A at the later stage of electrolysis.
The energy efficiency (e) of the molten salt process is specified in eqn (3).
 | (3) |
More and more studies56–59 have shown that the electric field could cause a series of physical and chemical changes in materials, affecting the molecular structure and electronic properties of materials. In order to explore the effect of electric field on the molecular structure of DC during molten salt electrolysis, DC was simplified into a structure with seven benzene rings attached to one functional group. At room temperature, the experimental structure with –CH2CH3 was optimized as shown in Fig. 5a. When electric fields of different strengths (0, 0.001 eV Å−1 e−1 and 0.01 eV Å−1 e−1) were applied in the X-axis direction, the bond lengths d1 and d2 in the –CH2CH3 structure increase with the increase of the electric field (Fig. 5b and c), and the bond angle ∠A decreases with increasing electric field (Fig. 5d). In the absence of an applied electric field, the degree of ordering of the precursors immersed in molten salt shows almost no variation.33 After the application of an electric field, the stable molecular structure of the DC accelerates the transformation to a highly ordered structure under the action of the electric field force. Furthermore, as shown in Fig. 5e, the electric field significantly affects the local charge density in the molecular structure. The charge density of a specific structure correlates with the catalytic activity,60 specifically, the reactivity of functional groups containing O, N, and S. Due to the occurrence of deoxidation, denitrification, and desulfurization reactions of DC, it is concluded that the changes of the charge density of a specific structure are related to electrochemical graphitization.
 | ||
| Fig. 5 Effect of the electric field on the molecular structure and electronic properties of DC. (a) Simplified hydrogen-free structure with seven benzene rings attached to one –CH2CH3 group; (b–d) effect of electric field strength on bond lengths d1, d2 and bond angle ∠A; (e) effect of electric field on the charge density of the molecular structure in (a); (f) charge population of atoms of different simplified groups after applying the electric field. | ||
DC contains various elements such as S, C, and O, implying the presence of various functional groups, including –COOH, –CHO, –OH, –NH2, –SH, etc. During molten salt electrolysis, the electric field changes the number of the charge population of O, N, and S atoms in functional groups (Fig. 5f). The electronegativity of atoms in the local molecular structure can be changed under the electric field,61,62 which represents the electron attracting ability of functional groups containing O, N, and S, and affects the transfer of electrons between atoms.
In this study, it was found that the electric field accelerates the transformation of DC to the graphitic structure, affects electron injection and local charge redistribution and promotes the deoxidation, denitrification, and desulfurization reactions of DC during electrochemical graphitization.
3.3 Electrochemical performances of electrolysis products
To explore the electrochemical performance of electrolysis products as potential anode materials for Li-ion batteries, EP-8 h was applied in EP|Li half cells because of its moderate preparation conditions and high degree of graphitization.Fig. 6a shows the CV curves of EP-8 h. In the first cycle, there was an obvious wide reduction peak at 0.7 V but it disappeared in the second and third cycles, which is related to the generation of irreversible SEI films.14,63 This phenomenon also occurs in other investigated electrolytic carbon materials, such as biochar-based electrolytic graphite34 and coal char-based electrolytic graphite.35 In addition, the reduction peak near 0.1 V represents the process of lithium ion intercalation into graphite, and the oxidation peak at 0.3 V represents the process of lithium ion extraction from graphite.14
 | ||
| Fig. 6 Electrochemical performance of EP-8 h|Li half cells. (a) The CV curves of EP-8 h at a scan rate of 0.2 mV s−1; (b) 1st and 2nd charge–discharge capacity at 1C; (c) rate performance of EP-8 h at 0.1C to 1C, (d) cycling performance of EP-8 h at 1C; (e) comparison of the initial charging specific capacity and initial coulombic efficiency of products based on different precursors in the literature. | ||
The initial discharge capacity of EP-8 h is 452 mA h g−1 at 1C as shown in Fig. 6b, which is significantly higher than that of commercial graphite (313 mA h g−1). Besides, the reversible capacities in the second cycle of electrolytic carbon and commercial graphite are almost the same. The initial irreversible capacity and initial coulombic efficiency of EP-8 h are 154 mA h g−1 and 65%, respectively. The initial irreversible capacity of EP-8 h is related to the formation of dead Li and a stable SEI.64 After molten salt electrolysis, the specific surface area of DC increases and the mesoporous structure is more developed, which can provide more intercalation sites for lithium ions and cause soft short circuits and severe dendrite formation, resulting in capacity attenuation.64
At the rates of 0.1C, 0.2C, 0.5C and 1C, the average discharge capacities of EP-8 h are 302.62 mA h g−1, 297.11 mA h g−1, 289.60 mA h g−1 and 279.09 mA h g−1 (Fig. 6c), and when the current density was restored to 0.2C, the capacity of DC could still be maintained at 295.41 mA h g−1 (average capacity of 20 cycles), similar to the commercial graphite reported in the literature.65 This demonstrated that the electrolytic graphite produced by low-temperature graphitization exhibits a good rate capability due to the complex flake structure and prepared mesoporous structure.
Fig. 6d shows the good long-term cycling stability of EP-8 h at 1C. After 600 cycles, EP-8 h can still maintain a high reversible capacity of 325 mA h g−1 and an excellent coulombic efficiency of 99.5%. During cycling, the specific capacity of EP-8 h increases slightly, which is related to the activation of lithium ions and is a common performance of porous carbon-based materials.66
The reversible capacity and coulombic efficiency are important indicators for evaluating the electrochemical performance of batteries.64 In previous studies, activated coconut (ACC),34 gasified coal char (GCC),35 hyper coal (HPC)33 and amorphous hard carbon (HC)67 were converted into graphite materials by molten salt electrolysis, and the optimized electrolysis products were applied in lithium-ion batteries. Comparing EP-8 h with these precursors, the electrolysis products based on DC exhibit a good performance with a stable reversible capacity and a common initial coulombic efficiency (Fig. 6e), which illustrates the promising application prospect of DC-based electrolytic graphite materials in lithium-ion batteries.
4 Conclusion
The molten salt electrolysis method was used to graphitize coke oven DC. It was found that DC was successfully transformed into a graphitic structure with a graphitization degree of 0.74 under optimized conditions at a cell voltage of 2.6 V and a molten pool temperature of 900 °C for 8 h. For the DC precursor, a graphite yield of about 90% was achieved with an energy efficiency of about 6.085 kW h kg−1. During the electrochemical transformation of DC, the cell voltage is a key factor for the structural transformation, and increasing the temperature is conducive to the formation of the graphitic structure. The appearance of flake structures in the electrolysis product is a typical signal of the graphitization transition, which is accompanied by a significant increase in the specific surface area of the sample. Characterization of DC and its derivatives produced at different times by FTIR and XPS analyses showed a decreased content of oxygen-containing chemical bonds, gradually shortened aliphatic chains of aromatic rings, and more compact structures between aromatic rings during the molten salt electrolysis process. By molecular simulation calculations, it is found that the effect of electric field on the molecular structure and electronic properties of DC may lead to the heteroatom removal of DC and the rearrangement of carbon atoms to form a graphitic structure. DC derivatives produced from the low-temperature molten salt electrochemical graphitization process, with a porous structure, exhibited good rate performance and cycle performance for use as cathode materials in lithium-ion batteries. The results provide a new type of synthetic graphite precursor, and could pave the way for treating hazardous wastes including DC from coking enterprises worldwide.Author contributions
Tao Rong: writing – original draft, data curation, formal analysis; Wei Guan: investigation; Weili Song: writing – review & editing; Haibin Zuo: conceptualization, project administration; Jingxiu Wang: writing – review & editing; Qingguo Xue: conceptualization; Shuqiang Jiao: writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant numbers U1960205) and Minmetals Science and Technology Special Plan (grant number 2020ZXZ01).References
- X. Zhou, L. J. Wan and Y. G. Guo, Adv. Mater., 2013, 25, 2152–2157 CrossRef CAS.
- J. Xu, Y. Xu, C. Lai, T. Xia, B. Zhang and X. Zhou, Sci. China: Chem., 2021, 64, 1267–1282 CrossRef CAS.
- F. Wu, Q. Li, L. Chen, Z. Wang, G. Chen, L. Bao, Y. Lu, S. Chen and Y. Su, Acta Phys.-Chim. Sin., 2022, 38, 2007017 Search PubMed.
- J. Xu, J. Liao, Y. Xu, J. Li, C. Zhu, J. Lin and X. Zhou, J. Energy Chem., 2022, 68, 284–292 CrossRef CAS.
- Y. Du, Z. Yi, Z. Zhang, J. Liao, Y. Xu, J. Bao and X. Zhou, Chem. Eng. J., 2022, 435, 131500 Search PubMed.
- T. Xiao, X. Yang, K. Hooman, L. Jin, C. Yang and T. J. Lu, Int. J. Therm. Sci., 2022, 179, 107706 CrossRef CAS.
- Z. Yuan, Z. Hu, P. Gao, W. Zhang, Y. Tang, L. Li, K. Shi, S. Han, C. Fan, J. Liu and J. Liu, Electrochim. Acta, 2022, 409, 139985 CrossRef CAS.
- W. Zhang, C. Liao and X. Ning, Chem. Eng. J., 2022, 435, 135110 CrossRef CAS.
- C. Cavallari, M. Brunelli, S. Radescu, M. Dubois, N. Batisse, G. B. M. Vaughan, H. E. Fischer and V. Pischedda, Carbon, 2019, 147, 1–8 CrossRef CAS.
- T. J. Robshaw, D. Atkinson, J. R. Howse, M. D. Ogden and D. J. Cumming, Cleaner Prod. Lett., 2022, 2, 100004 CrossRef CAS.
- Q. Q. Zhang, X. Z. Gong and X. C. Meng, Mater. Sci. Forum, 2018, 913, 1011–1017 Search PubMed.
- M. Shi, C. Song, Z. Tai, K. Zou, Y. Duan, X. Dai, J. Sun, Y. Chen and Y. Liu, Fuel, 2021, 292, 120250 CrossRef CAS.
- S. Ko, Y. J. Kwon, J. U. Lee and Y.-P. Jeon, J. Ind. Eng. Chem., 2020, 83, 449–458 CrossRef CAS.
- B. Xing, C. Zhang, Y. Cao, G. Huang, Q. Liu, C. Zhang, Z. Chen, G. Yi, L. Chen and J. Yu, Fuel Process. Technol., 2018, 172, 162–171 CrossRef CAS.
- N. A. Banek, K. R. McKenzie Jr, D. T. Abele and M. J. Wagner, Sci. Rep., 2022, 12, 8080 CrossRef CAS.
- M. Zhong, J. Yan, L. Wang, Y. Huang, L. Li, S. Gao, Y. Tian, W. Shen, J. Zhang and S. Guo, Sustainable Mater. Technol., 2022, 33, e00476 CrossRef CAS.
- N. Ohta, K. Nagaoka, K. Hoshi, S. Bitoh and M. Inagaki, J. Power Sources, 2009, 194, 985–990 CrossRef CAS.
- N. A. Karim, M. M. Ramli, C. M. R. Ghazali and M. N. Mohtar, Mater. Today: Proc., 2019, 16, 2088–2095 CAS.
- Y. Abe and M. Sugiura, ISIJ Int., 2022, 62, 64–73 CrossRef CAS.
- M. Piechaczek, Ł. Smędowski and S. Pusz, Int. J. Coal Geol., 2015, 139, 40–48 CrossRef CAS.
- Y. Wang, Y. Xu and D. Hu, Baosteel Technol., 2019, 37–44 Search PubMed.
- A. D. Jara, A. Betemariam, G. Woldetinsae and J. Y. Kim, Int. J. Min. Sci. Technol., 2019, 29, 671–689 CrossRef CAS.
- S. S. Makgato, R. M. S. Falcon and E. M. N. Chirwa, J. Cleaner Prod., 2019, 221, 684–694 CrossRef CAS.
- V. Zymla and F. Honnart, ISIJ Int., 2007, 47, 1422–1431 CrossRef CAS.
- H. Wang, B. Jin, X. Wang and G. Tang, Processes, 2019, 7, 508 CrossRef CAS.
- K. Zou, S. Wang, P. Wang, X. Duan, Y. Yang, M. D. Yazdi, J. Stowell, Y. Wang, W. Yao and W. Wang, Environ. Pollut., 2021, 273, 116434 CrossRef CAS PubMed.
- W. Wang, P. Wang, S. Wang, X. Duan, T. Wang, X. Feng, L. Li, Y. Zhang, G. Li, J. Zhao, L. Li, Y. Wang, Z. Yan, F. Feng, X. Zhou, W. Yao, Y. Zhang and Y. Yang, Ecotoxicol. Environ. Saf., 2019, 182, 109453 CrossRef CAS PubMed.
- Y. Lan, X. Zhao, W. Zhang, L. Mu and S. Wang, Energy, 2022, 245, 123292 CrossRef CAS.
- L. Tang, Q. Mao, Z. You, Z. Yao, X. Zhu, Q. Zhong and J. Xiao, Carbon, 2022, 188, 336–348 CrossRef CAS.
- X. Zhao, K. Liu, F. Guo, Y. Zhang and J. Wu, Colloids Surf., A, 2022, 636, 128142 CrossRef CAS.
- X. Jin, R. He and S. Dai, Chemistry, 2017, 23, 11455–11459 CrossRef CAS PubMed.
- J. Peng, N. Chen, R. He, Z. Wang, S. Dai and X. Jin, Angew. Chem., Int. Ed. Engl., 2017, 56, 1751–1755 CrossRef CAS PubMed.
- Z. Zhu, H. Zuo, S. Li, J. Tu, W. Guan, W.-L. Song, J. Zhao, D. Tian and S. Jiao, J. Mater. Chem. A, 2019, 7, 7533–7540 RSC.
- B. P. Thapaliya, H. Luo, P. Halstenberg, H. M. Meyer 3rd, J. R. Dunlap and S. Dai, ACS Appl. Mater. Interfaces, 2021, 13, 4393–4401 CrossRef CAS PubMed.
- B. P. Thapaliya, H. Luo, M. Li, W.-Y. Tsai, H. M. Meyer, J. R. Dunlap, J. Nanda, I. Belharouak and S. Dai, J. Electrochem. Soc., 2021, 168, 046504 CrossRef CAS.
- J. Zhu, K. Komatsu, H. Li, Y. Kudo, R. Okuda, Y. Ishibashi, T. Yamada, I. Toda, S. Ohshio, Y. Tuda and H. Saitoh, Measurement, 2021, 173, 108627 CrossRef.
- J. Tu, J. Wang, S. Li, W. L. Song, M. Wang, H. Zhu and S. Jiao, Nanoscale, 2019, 11, 12537–12546 RSC.
- A. Mukherjee, R. Kumaresan and S. Ghosh, J. Electroanal. Chem., 2021, 902, 115778 CrossRef CAS.
- M. Baysal, A. Yürüm, B. Yıldız and Y. Yürüm, Int. J. Coal Geol., 2016, 163, 166–176 CrossRef CAS.
- X. He, X. Liu, B. Nie and D. Song, Fuel, 2017, 206, 555–563 CrossRef CAS.
- G. N. Okolo, H. W. J. P. Neomagus, R. C. Everson, M. J. Roberts, J. R. Bunt, R. Sakurovs and J. P. Mathews, Fuel, 2015, 158, 779–792 CrossRef CAS.
- J. Yan, Z. Lei, Z. Li, Z. Wang, S. Ren, S. Kang, X. Wang and H. Shui, Fuel, 2020, 268, 117038 CrossRef CAS.
- Z. Rong, G. Pan, J. Lu, S. Liu, J. Ding, W. Wang and D.-J. Lee, Renewable Energy, 2021, 163, 579–588 CrossRef CAS.
- J. Yu, Q. Guo, L. Ding, Y. Gong and G. Yu, Fuel, 2020, 270, 117603 CrossRef CAS.
- K. Li, Q. Liu, H. Cheng, M. Hu and S. Zhang, Spectrochim. Acta, Part A, 2021, 249, 119286 CrossRef CAS PubMed.
- C. Chen, Y. Tang and X. Guo, Fuel, 2022, 310, 122362 CrossRef CAS.
- S. Lee, S. Y. Cho, Y. S. Chung, Y. C. Choi and S. Lee, Carbon, 2022, 199, 70–79 CrossRef CAS.
- S. Lin, Z. Liu, E. Zhao, J. Qian, X. Li, Q. Zhang and M. Ali, Process Saf. Environ. Prot., 2019, 130, 48–56 CrossRef CAS.
- O. O. Sonibare, T. Haeger and S. F. Foley, Energy, 2010, 35, 5347–5353 CrossRef CAS.
- J. Jiang, S. Zhang, P. Longhurst, W. Yang and S. Zheng, Spectrochim. Acta, Part A, 2021, 255, 119724 CrossRef CAS PubMed.
- Y. Liu, B. Sun, L. Tajcmanova, C. Liu and J. Wu, Spectrochim. Acta, Part A, 2022, 272, 120947 CrossRef CAS PubMed.
- L. Zhao, N. Guanhua, W. Hui, S. Qian, W. Gang, J. Bingyou and Z. Chao, Fuel, 2020, 272, 117705 CrossRef.
- G. Levi, O. Senneca, M. Causà, P. Salatino, P. Lacovig and S. Lizzit, Carbon, 2015, 90, 181–196 CrossRef CAS.
- M. Varga, T. Izak, V. Vretenar, H. Kozak, J. Holovsky, A. Artemenko, M. Hulman, V. Skakalova, D. S. Lee and A. Kromka, Carbon, 2017, 111, 54–61 CrossRef CAS.
- A. M. Puziy, O. I. Poddubnaya, R. P. Socha, J. Gurgul and M. Wisniewski, Carbon, 2008, 46, 2113–2123 CrossRef CAS.
- Y. Li, S. Zhang, Z. Bao, N. Sun and S. Lin, Innovative Food Sci. Emerging Technol., 2022, 76, 102918 CrossRef CAS.
- Z. Tao, X. Wang, Y. Wei, L. Lv, D. Wu and M. Yang, Chem. Phys., 2017, 483–484, 122–131 CrossRef CAS.
- Y. Tao, Q. Wang, K. Sun, Q. Zhang, W. Liu, J. Du and Z. Liu, Spectrochim. Acta, Part A, 2020, 231, 118108 CrossRef CAS PubMed.
- Š. Dědičová, J. Dočkal, F. Moučka and J. Jirsák, J. Mol. Liq., 2021, 338, 116622 CrossRef.
- X. Wu, G. Yang, L. Zhou and X. Han, Comput. Theor. Chem., 2013, 1017, 109–116 CrossRef CAS.
- K. Selvaraju, M. Jothi and P. Kumaradhas, Comput. Theor. Chem., 2012, 992, 9–17 CrossRef CAS.
- Y. Bian, W. Zeng, M. He, Y. Ma, Y. Liu, Y. Kong and J. Pan, J. Colloid Interface Sci., 2019, 534, 20–30 CrossRef CAS PubMed.
- W. Fan, J. Zhang, R. Ma, Y. Chen and C. Wang, J. Electroanal. Chem., 2022, 908, 116087 CrossRef CAS.
- C. J. Huang, B. Thirumalraj, H. C. Tao, K. N. Shitaw, H. Sutiono, T. T. Hagos, T. T. Beyene, L. M. Kuo, C. C. Wang, S. H. Wu, W. N. Su and B. J. Hwang, Nat. Commun., 2021, 12, 1452 CrossRef CAS PubMed.
- H. Xiao, G. Ji, L. Ye, Y. Li, J. Zhang, L. Ming, B. Zhang and X. Ou, J. Alloys Compd., 2021, 888, 161593 CrossRef CAS.
- G. Singh, J. Lee, R. Bahadur, A. Karakoti, J. Yi and A. Vinu, Chem. Eng. J., 2022, 433, 134464 CrossRef CAS.
- P. Bagri, B. Thapaliya, Z. Yang, W. Jiang, D. Sulejmanovic, H. Luo and S. Dai, Chem. Commun., 2020, 56, 2783–2786 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta08198a |
| This journal is © The Royal Society of Chemistry 2023 |